

Abstract
The generation of new cells is one of the most fundamental aspects of cell biology. Proper regulation of the cell cycle is critical for human health, as underscored by many diseases associated with errors in cell cycle regulation, including both cancer and hereditary diseases. A large body of work has identified regulatory mechanisms and checkpoints that ensure accurate and timely replication and segregation of chromosomal DNA. However, few studies have evaluated the extent to which similar checkpoints exist for the division of cytoplasmic components, including organelles. Such checkpoint mechanisms might be crucial for compartments that cannot be generated de novo, such as the endoplasmic reticulum (ER). In this review, we highlight recent work in the model organism Saccharomyces cerevisiae that led to the discovery of such a checkpoint that ensures that cells inherit functional ER into the daughter cell.
Evidence for cell cycle checkpoints for dividing the cytoplasm
When a eukaryotic cell undergoes division, it faces the challenge of generating genetically identical daughter cells (Ciccia and Elledge, 2010; Matellan and Monje-Casas, 2020; Musacchio and Desai, 2017). The cell must copy the entire genome—which consists of over three billion base pairs in human—without making major mistakes. This process, called DNA replication, must occur within a specific length of time. Handling three billion base pairs alone also imposes spatial and temporal constrains to the cell. In order to ensure that the genome is separated into two dividing cells, these events are assisted by several “checkpoints” at the heart of the cell cycle operation. If mistakes are found, the cell temporally halts cell division, providing an opportunity for fixing mistakes. Upon recovery, the cell resumes the cell cycle at the point at which it stopped to finish generating a new daughter cell. The importance of cell cycle checkpoints has been underscored by many human diseases, such as cancer, that occur due to the failure of the cell to recognize or fix mistakes (Holland and Cleveland, 2012). Each year, incredible resources are poured into efforts to better understand cell cycle mechanisms and checkpoints with the hope of generating more effective treatments for cancer and other diseases.
A unique feature of eukaryotic cells is the compartmentalization of specific cellular functions into organelles that are surrounded by unique sets of membranes (Figure 1). This framework allows cellular functions to fine-tune themselves discretely, but also requires these organelles to be divided during cell division. In contrast to genome replication and separation, however, we know relatively little about the rules that govern the division of cytoplasmic components or organelles (Mascanzoni et al., 2019; Vevea et al., 2014; Weisman, 2003). Historically, almost all cell division studies have focused on issues associated with the genome and the cell cycle checkpoints ensuring that all dividing cells end up with completely and accurately replicated DNA. Some experimental results suggest that the division of non-genomic components, such as proteins and organelles, might be regulated during the cell cycle. For example, treating mammalian cells with DNA-damaging agents results in a massive reorganization of the Golgi, revealing an intimate link between cell cycle DNA replication and division of Golgi (Farber-Katz et al., 2014). Furthermore, these results hint at the presence of cell cycle checkpoints for the proper division of other organelles, such as the endoplasmic reticulum (ER), mitochondria, and endosomes, although relatively little is known about the molecular mechanisms underlying their regulation. In contrast, a recent study reported that the inheritance of a vacuole from the mother cell can fail without halting the cell cycle, as long as a new functional vacuole can be generated in the daughter cell (Jin and Weisman, 2015), revealing that there may be different strategies for ensuring the presence of functional organelles in newly generated cells.
Figure 1:
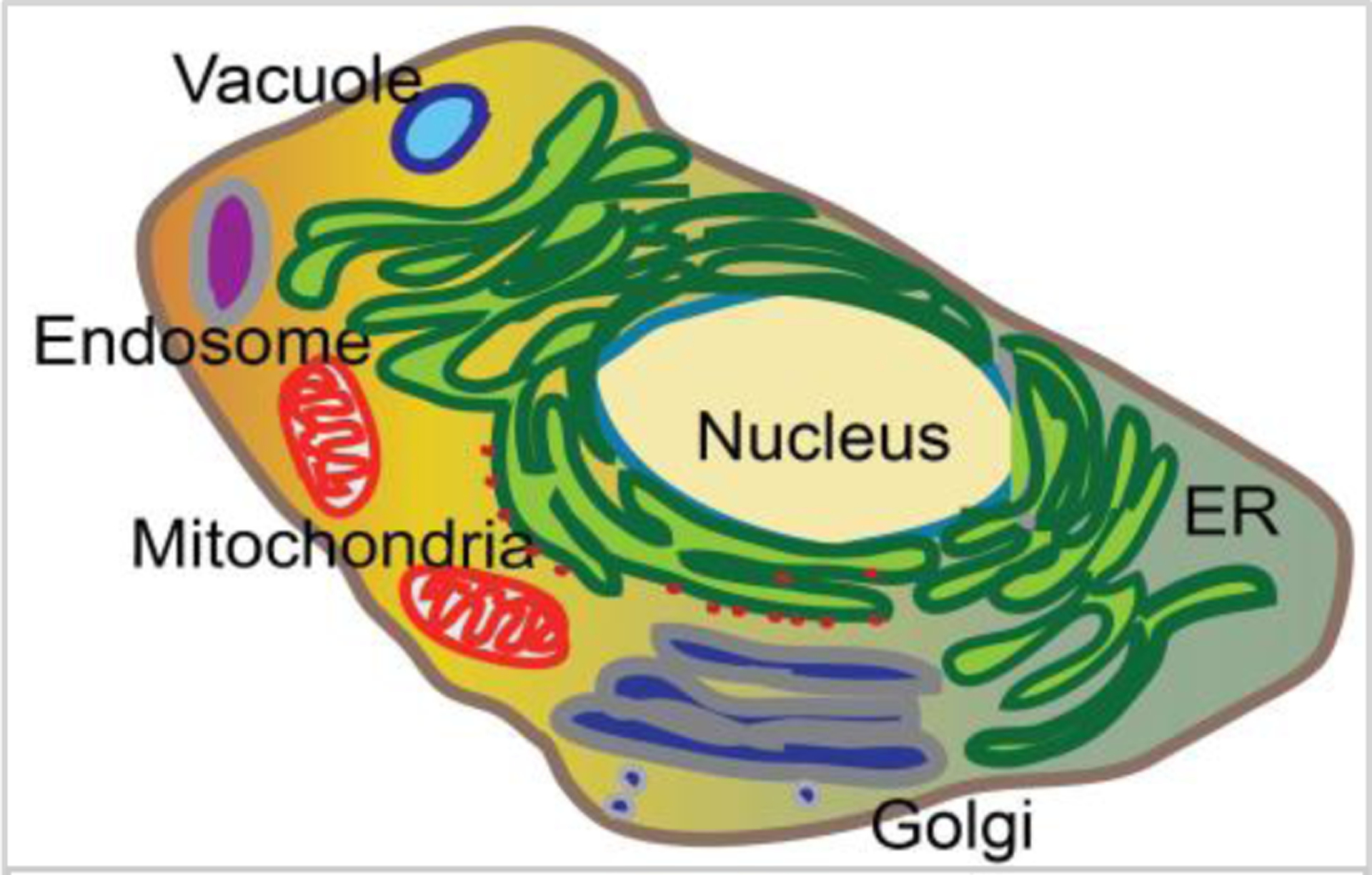
Cellular functions are compartmentalized in organelles in eukaryotic cells
In recent years, studies have begun to unravel the molecular mechanisms that ensure the inheritance of functional ER to a daughter cell. Here, we provide an overview of an exciting discovery of a cell cycle regulatory checkpoint—termed the ER stress surveillance (ERSU) cell cycle checkpoint—in the budding yeast Saccharomyces cerevisiae (S. cerevisiae) (Babour et al., 2010; Chao et al., 2019; Pina et al., 2018; Pina et al., 2016; Pina and Niwa, 2015). Since the initial discovery of the ERSU pathway, subsequent work has provided intriguing mechanistic insights on the ERSU cell cycle checkpoint. Specifically, a recent finding surprisingly revealed that increased levels of phytosphingosine (PHS), an early biosynthetic intermediate of sphingolipids, triggers the ERSU cell cycle checkpoint. As the ERSU pathway is one of the few cell cycle regulatory checkpoints for non-genomic components with significant mechanistic understanding, the principles that operate the ERSU checkpoint may provide fundamental insights into cell cycle checkpoints for other components.
The endoplasmic reticulum is a gateway of the secretory pathway
The ER is one of the biggest organelles in eukaryotic cells, with an extensive reticular structure that can spread throughout large portions of the cytoplasm. Indeed, approximately half of the total membrane of a cell consists of the ER (Alberts et al., 2002). The ER membrane is contiguous with the outer nuclear membrane, which is connected to the inner nuclear membrane via the nuclear pore complex. In addition, recent studies have revealed that the ER membrane establishes contact sites with other organelles, including mitochondria, endosomes, and peroxisomes, with certain functional significance (Cohen et al., 2018; Farre et al., 2019; Helle et al., 2013; Kvam and Goldfarb, 2006; Tamura et al., 2019; Wu et al., 2018). Whether the contact sites remain during cell division and how they affect cell cycle division are still unclear.
Beyond interacting with other essential organelles, the ER plays critical roles in various cellular functions. For example, the ER generates mature, fully functional proteins like growth factors and hormones, which are destined to be secreted outside of the cell. Similarly, the ER produces properly folded, mature transmembrane cell-surface receptors and receptor kinases. As they emerge from the polyribosomes, these proteins, collectively called secretory pathway proteins, are targeted and translocated into the ER lumen as linear unmodified polypeptides for folding, modification, and other maturation steps (Aviram and Schuldiner, 2017; Brown et al., 1995; Walter et al., 1984). Secretory pathway proteins consist of one-third of the total proteome of eukaryotic cells, making this one of the primary tasks of the ER. Upon association with ER-resident chaperones and other protein-folding components, nascent polypeptides undergo maturation steps including folding, modification, and complex formation in order to generate fully folded functional proteins (Ma and Hendershot, 2001; Matlack et al., 1998; McMaster, 2001; Meldolesi and Pozzan, 1998; Voeltz et al., 2002). The folding processes are regulated in such a way that only properly folded proteins can exit from the ER, heading towards the Golgi and their final destinations. Somehow, proteins that have not completed the folding steps are recognized and remain in the ER lumen to complete the process before exiting to the Golgi. Any permanently misfolded proteins are also recognized by the ER and are ultimately degraded by a mechanism specific to permanently misfolded proteins; this process is called ER-associated degradation (ERAD) (Berner et al., 2018; Goder et al., 2019; Hampton and Sommer, 2012; Johnson and DeBose-Boyd, 2018; McCaffrey and Braakman, 2016; Mehrtash and Hochstrasser, 2019; Pobre et al., 2019; Sun and Brodsky, 2019; Wu and Rapoport, 2018). During its lifetime, a cell encounters a series of conditions necessary to produce high levels of secreted proteins. Collectively, the increased need for producing secretory pathway proteins or the accumulation of unfolded proteins is termed “ER stress”. When a cell recognizes ER stress, it triggers an intracellular signal-transduction pathway called the unfolded protein response (UPR), which helps the cell keep up with the increased demands of ER functions (Mori et al., 2000; Ron and Walter, 2007; Rutkowski and Kaufman, 2004). In addition to producing secretory pathway proteins, the UPR regulates ERAD. Thus, via the UPR, the ER regulates and meets the overall composition of the cellular proteome (Hampton and Sommer, 2012; Pobre et al., 2019; Sun and Brodsky, 2019; Wu and Rapoport, 2018). The ER also plays vital roles in lipid biosynthesis and metabolism. Nearly all of the initial steps of cellular lipid synthesis occur on the ER membrane. The ER also plays other important functions, such as regulating cellular Ca2+ levels or detoxifying unwanted chemicals, and thus it is a multi-faced functional compartment (Karagas and Venkatachalam, 2019; Mittal et al., 2015; Pierro et al., 2019; Preuss et al., 1991). Importantly, organelles such as the ER cannot be generated de novo; therefore, the ER must be inherited from the mother cell.
Division of yeast ER during the cell cycle
Cell cycle division of the ER is better described in yeast, partly because the yeast ER is less complex than the ER in human cells: the tubular ER surrounds the cortex of the cell and is termed cortical ER (cER). The ER also surrounds the nucleus, comprising the perinuclear ER (pnER), which is contiguous with the outer nuclear membrane (Voeltz et al., 2002). The nuclear pore complexes, large macromolecular complexes, in essence, physically separate the inner nuclear membrane from the outer nuclear membrane and the yeast pnER (Forbes et al., 2015; Schwartz et al., 2015). The tubular ER structures (or ER tubules) are approximately 50–100 nm in diameter and form a network of membranous tubules that connect the cER to the pnER (Koning et al., 2002; Preuss et al., 1991). Under normal growth conditions, a tubular ER emanating from the mother pnER starts to grow and enters the daughter cell by passing through the septin ring at the bud neck during the G1/S phase of the cell cycle (Du et al., 2001; Estrada de Martin et al., 2005; Estrada et al., 2003). Here, we refer to this tubular ER as the “initial ER tubule” to distinguish from other tubular ER linking both the pnER and the cER (Figure 2). The growth of the initial ER tubule is directed (Figure 2, step I & II) and eventually anchored to the bud tip by its interactions with proteins or protein complexes localized at the bud tip (step III), including the exocyst complex (Heider and Munson, 2012; Munson and Novick, 2006). Upon anchoring to the bud tip, the initial ER tubule changes the direction of its growth, and spreads the tubular ER along the cortex of the daughter cell (step IV). Studies have shown that the phosphatase Ptc1, which is localized at the bud tip, play important roles in re-directing tubular ER growth to the lateral direction (Du et al., 2006), leading to the generation of the cER in the daughter cell. Once the cER is inherited in the daughter cell, the pnER and nucleus enter the daughter cells (Figure 2, step V), followed by nuclear division (step VI). The movement of the cER into the daughter cell has been reported to occur on the actin cable; Myo4 and She3 connect the cER to the actin cable. By contrast, the movement of the pnER is mediated by microtubules (Estrada et al., 2003). Currently, we lack a detailed understanding of the specific movements of the tubular ER linking the cER and the pnER. In addition, it remains unknown whether the movement of the Initial ER tubule (IET) depends on cables of actin or microtubules. Furthermore, the factors determining the number or location of such tubular ER remain elusive. Regardless of the tubular ER, it is unclear how the movement of the cER depends on actin cables while that of the pnER occurs in a microtubule-dependent manner, when the lumen of both types of ER is connected. Additionally, the movement of the pnER must be coordinated with that of the nucleus and the nuclear components. Further work is required to determine how such maneuvers are achieved in a coordinated manner with the division of the genome. Ultimately, beyond the ER and the nuclear transport, transit of other organelles through the bud neck will also have to take place before committing to undergo cytokinesis (Figure 2).
Figure 2: Stages of ER inheritance in yeast:
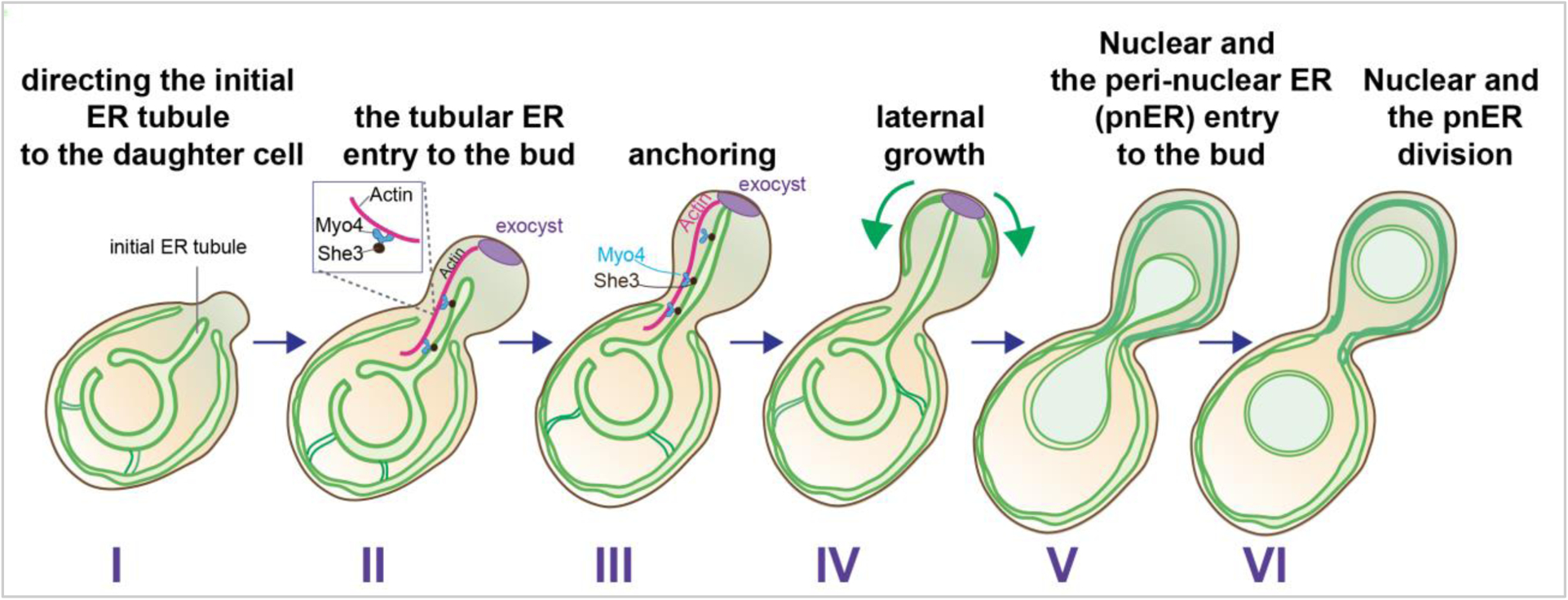
ER inheritance of the yeast cell starts with tubular ER (initial tubular ER) entry into the daughter cell on actin cables in association with Myo4 and She3 proteins (step I and II). The initial tubular ER reaches and is anchored by components localized at the bud tip such as the exocyst (in purple) (step III), which allows for lateral growth of the ER, generating the cortical ER (cER) (step IV). The nucleus and the peri-nuclear ER move into the daughter cell (step V), followed by nuclear division (step VI). Ultimately, cytokinesis separates the two cells.
The functional importance of the ER and the fact that the ER cannot be synthesized de novo suggest that the inheritance of the functional ER must be tightly regulated. This begs interesting questions: for example, what happens if the size or functional capacity of the ER is not adequate enough to divide into two cells? If the ER does not meet the functional demands of a cell or if the ER is too small to divide into two daughter cells, these conditions might impact the progression of the cell cycle. Moreover, it is unclear whether such conditions cause the cell cycle to stop at a specific point. Given that inheriting an accurate and complete set of chromosomal DNA is fundamental to the life of a cell, most studies on cell cycles, thus far, have focused on how cells maintain accurately replicated DNA and how sister chromatids are separated in a faithful and timely manner.
A cell cycle checkpoint for ER division
From the late 1980s through the 1990s, the use of various DNA damage agents and the isolation of conditional cell cycle mutants in yeast opened the door for studying cell cycle mechanisms. These methods brought about a flurry of fascinating discoveries on cell cycle checkpoints for ensuring the inheritance and division of genomic information (Elledge and Harper, 1994; Liu et al., 2000; Morgan, 1995; Zhou and Elledge, 2000). Given the major mechanistic differences between yeast and mammalian cells in terms of cell cycle modes, researchers initially predicted that the regulatory checkpoints for the major cell cycle events would be different for yeast and mammalian cells. However, it turned out that several key regulatory events and the timing for ensuring accurate chromosomal DNA replication and sister chromatid separation are remarkably conserved across yeast and mammalian cells.
Motivated by the discovery of cell cycle checkpoints for the genome, new approaches of using well-characterized chemical agents to diminish ER function in yeast cells have created opportunities for (1) learning how cells handle functionally stressed ER during the cell cycle and (2) identifying cell-cycle checkpoints for inheriting functional ER. The chemical agents tunicamycin (Tm) and 1,4-Dithiothreitol (DTT) stress ER function by inhibiting glycosylation and disrupting disulfide bonds, respectively. Indeed, previous studies have shown that triggering ER stress by treatment of yeast cells with either of these agents blocks cells in the G1/S phase (Arnold and Tanner, 1982; Vai et al., 1987) or leads to a chromosome maintenance defect (Henry et al., 2010). Similar experiments in mammalian cells also revealed that ER stress leads to a G1/S phase block (Cullinan and Diehl, 2006). Subsequent studies, however, found that this G1/S phase arrest stemmed from the lack of components required for budding and/or bud growth. For example, the emergence and growth of the daughter cell requires properly localized polarisome components at the incipient bud site (McMillan et al., 1998). The subcellular localization of the polarisome subunits (e.g., EPO1, SPA2, BEM3, PEA1, BNI1, BUD6, and MSB3/MSB4) to the incipient bud site depends on the early secretory pathway and the functional capacity of the ER, which is the gatekeeper of the secretory pathway (Bidlingmaier and Snyder, 2004; Li et al., 2013). Tm treatment of unbudded cells with unfolding agents at the beginning of the cell cycle causes polarisome subunits to unfold, thus preventing them from exiting the ER and heading towards the incipient bud site. Ultimately, the diminished localization of the polarisome subunit contributes to blocking bud emergence and bud growth. Thus, the primary cause of cell cycle block is the lack of the functional polarisome components required for the budding step. Although these studies uncovered that the lack of bud emergence ultimately induces a “morphogenesis block”, they did not reveal a direct impact of the ER or ER function itself on the cell cycle (Bonilla and Cunningham, 2003). The disrupted localization of the polarisome was overcome by an ER stress cell cycle assay that introduced a delay in the timing of ER stress induction until polarisome components could reach the incipient bud site and no longer induce the morphogenesis checkpoint (Bicknell et al., 2007). This delay, introduced to alpha factor-synchronized cells successfully induces ER stress without bud emergence block, making it possible to investigate if and how the steps in the cell cycle are impacted by ER stress (Bicknell et al., 2007). Use of this ER stress cell cycle assay led to the finding that a functionally stressed ER causes cytokinesis block.
Subsequent work investigated how ER stress induces cytokinesis block. Successful cytokinesis requires that cortical actin patches become polarized to either side of the bud neck late in the cell cycle (Doyle and Botstein, 1996; Kilmartin and Adams, 1984; Mulholland et al., 1994; Novick and Botstein, 1985; Waddle et al., 1996). Actin visualized upon staining with Fluor546-phalloidin revealed that actin patches are localized to the cortex of the cells throughout the cell cycle and become redistributed just prior to cytokinesis, regardless of ER stress (Bicknell et al., 2007). Thus, the ER stress-induced cytokinesis defect is not caused by a delay or an alteration in the actin patch redistribution. Instead, further evidence suggests that ER stress-induced cytokinesis block is caused, at least in part, by the mislocalization of the septin ring. The next section summarizes the studies that have investigated the consequences of having functionally stressed ER on the budding yeast cell cycle, ultimately defining the ERSU cell cycle checkpoint.
The ERSU cell cycle checkpoint hallmark event 1: cER inheritance block
The finding that ER stress causes cell cycle block underscores the importance of stopping cell division if ER function is compromised. Based on the yeast ER inheritance mechanism described, there might be at least two ways to prevent the daughter cell generation if the ER is functionally stressed: First, (1) ER inheritance into the daughter cell is not sensitive to ER stress, but ER stress-induced cytokinesis block somehow ensures that the generation of a daughter cell with non-functional ER is prevented. Alternatively, (2) the inheritance of the stressed ER is blocked and the lack of a functional ER in the daughter cell prevents cytokinesis to separate mother and daughter cells. To distinguish between these possibilities, the impact of ER stress on ER inheritance was initially visualized by an ER reporter, a fusion protein between GFP and the N-terminal transmembrane domain (amino acids 1–702) of HMG-CoA reductase isozyme 1 (Babour et al., 2010; Du et al., 2001; Hampton et al., 1996). In either case, demonstrating that stressed ER directly impacts cell cycle progression would support the idea that ER functional homeostasis is ensured during the cell cycle.
Monitoring the impact of ER stress on ER behaviors revealed that ER stress induction leads to daughter cells without the cER (Babour et al., 2010). This impact was most prominent in class of cells with a small bud index (ratio between mother and daughter cell size) (Figure 3). In contrast, cells with larger bud sizes, with or without a divided pnER (class II or III cells, respectively), showed minimal impact of ER stress on ER inheritance. Based on the mechanism of ER entry into the daughter cell (Figure 2), cells with medium/larger buds most likely have had (1) the cER inherited prior to ER stress exposure or (2) the initial ER tubule already committed to remain in the daughter. In those cases, the committed cER in the daughter cell would be unlikely to be released and returned back to the mother cell. Indeed, researchers demonstrated this experimentally using synchronized cells (Bicknell et al., 2007): ER stress at the early stage of the cell cycle and prior to commitment of the cER’s presence in the daughter cell (Figure 3) blocked the cER from entering the daughter cell (Pina and Niwa, 2015). This finding indicates that cells can (1) recognize the functional capacity of the ER at an early point of the cell cycle and (2) block the inheritance of a stressed ER into the daughter cell. Interestingly, if cells encountered ER stress after cER establishment in the daughter cell (Figure 2, Steps IV-VI), cells underwent cytokinesis to separate into mother and daughter cells. In the subsequent round of the cell cycle, however, cER inheritance was blocked, ultimately resulting in cytokinesis block. These results revealed that cytokinesis block occurs in ER-stressed cells to prevent the generation of cells without the presence of an ER.
Figure 3: Hallmark events of the ERSU checkpoint.
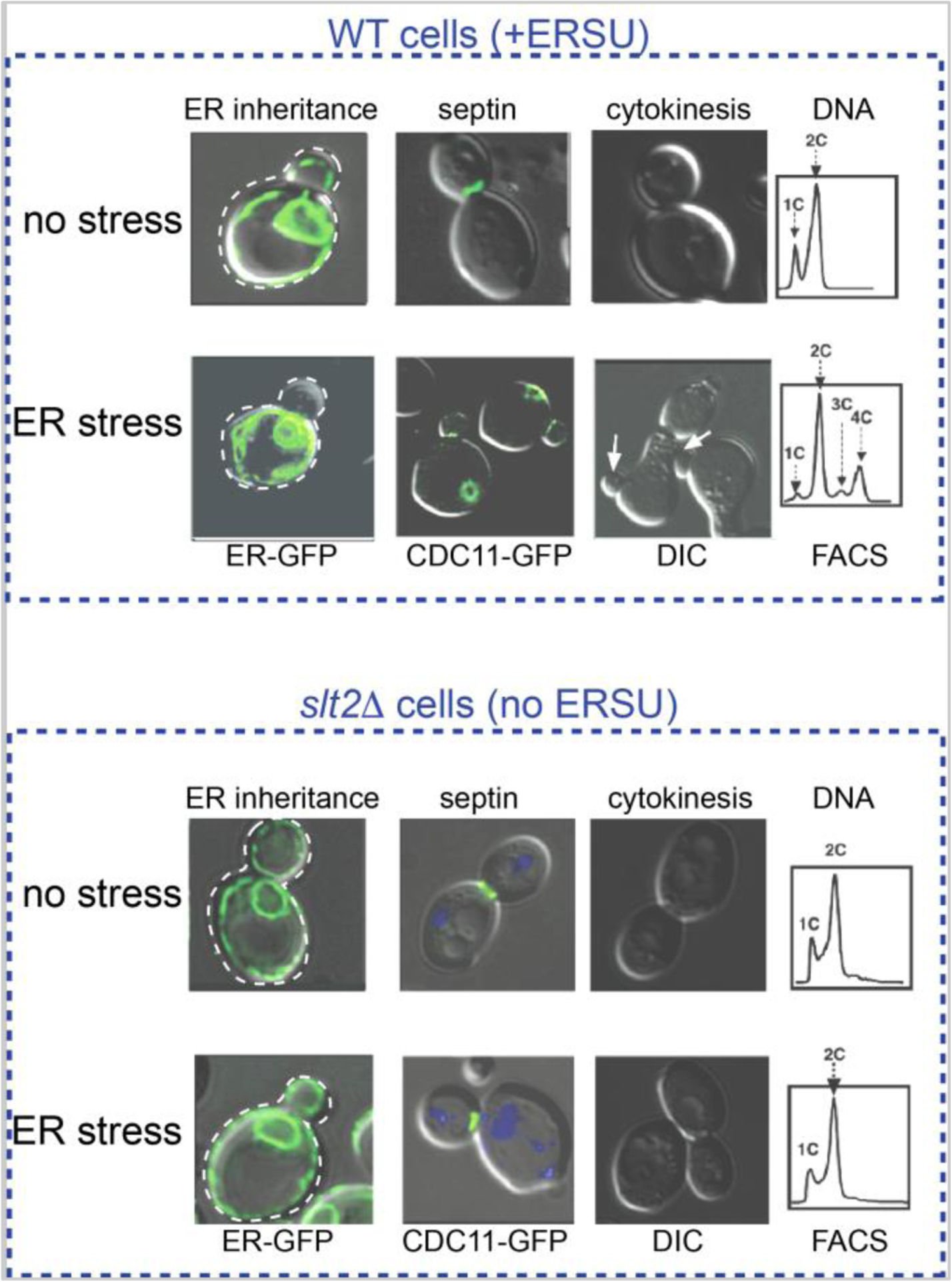
Under ER stress, the cortical ER (cER) visualized by HMG1-GFP reporter is blocked from entering the daughter cell. The septin ring, visualized by CDC11-GFP, moves away from the bud neck. These events result in cytokinesis block. In ERSU-deficient slt2Δ cells, the cER enters into the daughter cell and the septin ring stays at the bud neck even under ER stress. The inability to induce the ERSU pathway in response to ER stress ultimately results in cell death.
ERSU checkpoint hallmark event 2: Septin ring transfer from the bud neck
Septin has been reported to establish the diffusion barrier between mother and daughter cells. Studies using N-terminally green fluorescent protein (GFP)-tagged individual septin subunits (CDC3, SHS1, CDC10, CDC11, and CDC12) in unstressed or ER-stressed yeast cells found that the multi-subunit septin ring moves away from the bud neck and re-localizes elsewhere in response to ER stress (Figure 3) (Babour et al., 2010; Chao et al., 2019). At the beginning of the cell cycle – even prior to bud emergence – five septin subunits start to assemble at the incipient bud site to generate septin rings in yeast. Once assembled, the septin ring functions as a diffusion barrier of cellular components/organelles from the mother to the daughter cell (Caudron and Barral, 2009; Chao et al., 2014; Clay et al., 2014). Towards the end of the cell cycle, the septin ring splits vertically into two rings, leading to the division of the cytoplasm. Finally, after cell division the septin ring disassembles into each subunit, prior to reassembly at the new bud site (Field and Kellogg, 1999; Versele and Thorner, 2005).
ER stress does not seem to affect the initial targeting of septin subunits: the formation of septin rings at the bud neck appears as a ring-like structure with similar kinetics during ER stress, rather than being dispersed throughout the cytoplasm in unstressed cells. Upon ER stress induction, the septin ring is ‘misplaced’ or moves away from the bud neck to elsewhere on the cell surface. Interestingly, the ring-like appearance is retained at the new location, even though it is no longer localized at the bud neck. As the septin ring is a major player in cytokinesis, its absence at the bud neck under ER stress should contribute significantly to cytokinesis block. Furthermore, mislocalized septin ring persists once it moves away from the bud neck to the new location, until ER function is re-established and the cell prepares to re-enter the cell cycle. A more recent study reported the surprising finding that the septin ring is misplaced to the bud scars in response to ER stress. Furthermore, among five subunits, mislocalization of the Shs1 subunit to the bud scar is a key determinant of outcome, with failure to move Shs1 to the bud scar diminishing the ability of the cells to re-enter the cell cycle upon re-establishment of ER homeostasis (Chao et al., 2019). These findings revealed an unprecedented feature of the ERSU checkpoint. The mechanistic details and functional implications of ER stress-induced septin ring mislocalization will be discussed elsewhere (a review in preparation).
The ERSU pathway is independent of the UPR
In S. cerevisiae, ER stress induces: (1) ER inheritance block, (2) septin ring mislocalization, resulting in (3) cytokinesis block. However, a major question remained as to whether these events represent a part of the cell cycle checkpoint mechanism for ensuring that a daughter cell receives a functional ER. Another possibility is that these events happen to occur upon ER stress but are unrelated events and do not necessarily represent a cell cycle checkpoint. A signaling pathway known to be activated by ER stress is the UPR (Ron and Walter, 2007; Schroder and Kaufman, 2005). The UPR in yeast cells is initiated by a single-span ER transmembrane protein called Ire1, which contains an ER luminal domain that functions as an ER stress sensor (Cox et al., 1993; Mori et al., 1993). Mechanistically, ER stress is sensed by dissociation of the ER chaperone Kar2/BiP from the ER luminal domain of Ire1 (Bertolotti et al., 2000; Kimata et al., 2007; Oikawa et al., 2007), and the Ire1 ER luminal domain recognizes certain features of unfolded proteins (Credle et al., 2005; Zhou et al., 2006). These events lead to Ire1 dimerization/oligomerization and autophosphorylation through its Ire1 kinase domain (Shamu and Walter, 1996) and activation of the sequence-specific endoribonuclease (RNase) domain. Activated Ire1 RNase cleaves the UPR intron of HAC1 mRNA, encoding for an UPR-specific transcription factor (Cox et al., 1996; Mori et al., 2000), followed by ligation of two HAC1 mRNA exons by tRNA ligase (Sidrauski et al., 1996). The splicing of HAC1 mRNA is a key regulatory step for the UPR, as it causes a frameshift, removing a stop codon within the intron and allowing the second exon coding sequences in frame with the coding sequence of the first exon. The second exon contains a transcriptional activation domain and, thus, Ire1-dependent splicing of HAC1 mRNA is required for the generation of a UPR-specific transcription factor. Spliced Hac1 protein activates the transcription of UPR target genes that ultimately help to re-establish ER functional homeostasis. The UPR, particularly IRE1, is an ancient pathway conserved in all eukaryotic cells (Walter and Ron, 2011).
Given the functional roles of the UPR (Back and Kaufman, 2012; Mori, 2009; Ron and Walter, 2007), it was reasonable to consider that the UPR plays a role in inducing ER inheritance block, septin ring transfer, and cytokinesis block in response to ER stress. It was, therefore, a great surprise when these cell cycle events turned out to not require IRE1 function. In ire1Δ cells, ER stress caused ER inheritance block, septin ring transfer, and cytokinesis block at levels similar to wild-type (WT) cells. The lack of UPR involvement suggested the presence of a new pathway that induces these events in response to ER stress.
Identifying the first ERSU component
The lack of IRE1 involvement in the ERSU checkpoint suggested the presence of an independent pathway parallel to the UPR that coordinates the functional status of the ER with its inheritance during the cell cycle, leading to a quest for the ERSU components. The lack of an ERSU component should prevent ER inheritance block, septin ring mislocalization, and temporary cytokinesis block, having deleterious consequences on cell growth. Thus, yeast cells carrying the deletion of a gene coding for a protein either physically or functionally linked to the ER were screened for their ability to (1) grow on media containing ER stress-inducing agents such as Tm or DTT, (2) block cER inheritance, and (3) mislocalize the septin ring under ER stress. This screen identified SLT2, which encodes a MAP kinase (6). ER-stressed slt2 knockout (slt2Δ) cells were unable to block cER inheritance, mislocalize septin ring, or grow on Tm or DTT plates. Importantly, slt2Δ cells were able to activate the UPR pathway, since HAC1 mRNA was spliced normally with similar kinetics as in WT cells, revealing that the functional homeostasis of the ER was disrupted in slt2Δ cells. The inability of slt2Δ cells to mount all the ERSU phenotypes demonstrated that the ERSU cell cycle phenotypes are not individual phenotypes that happen to occur under ER stress, but rather are linked under a specific cell cycle regulatory event. Despite the normal appearance of septin rings and cER inheritance, slt2Δ cells were incapable of cell growth under ER stress, illustrating the importance of SLT2 in cell cycle progression. Additionally, these findings revealed that cER inheritance block and septin ring transfer are critical for survival during ER stress. Taken together, a SLT2-dependent, but UPR-independent, ERSU cell cycle regulatory pathway is critical for cells to respond to ER stress (Figure 4).
Figure 4: ERSU checkpoint pathway.
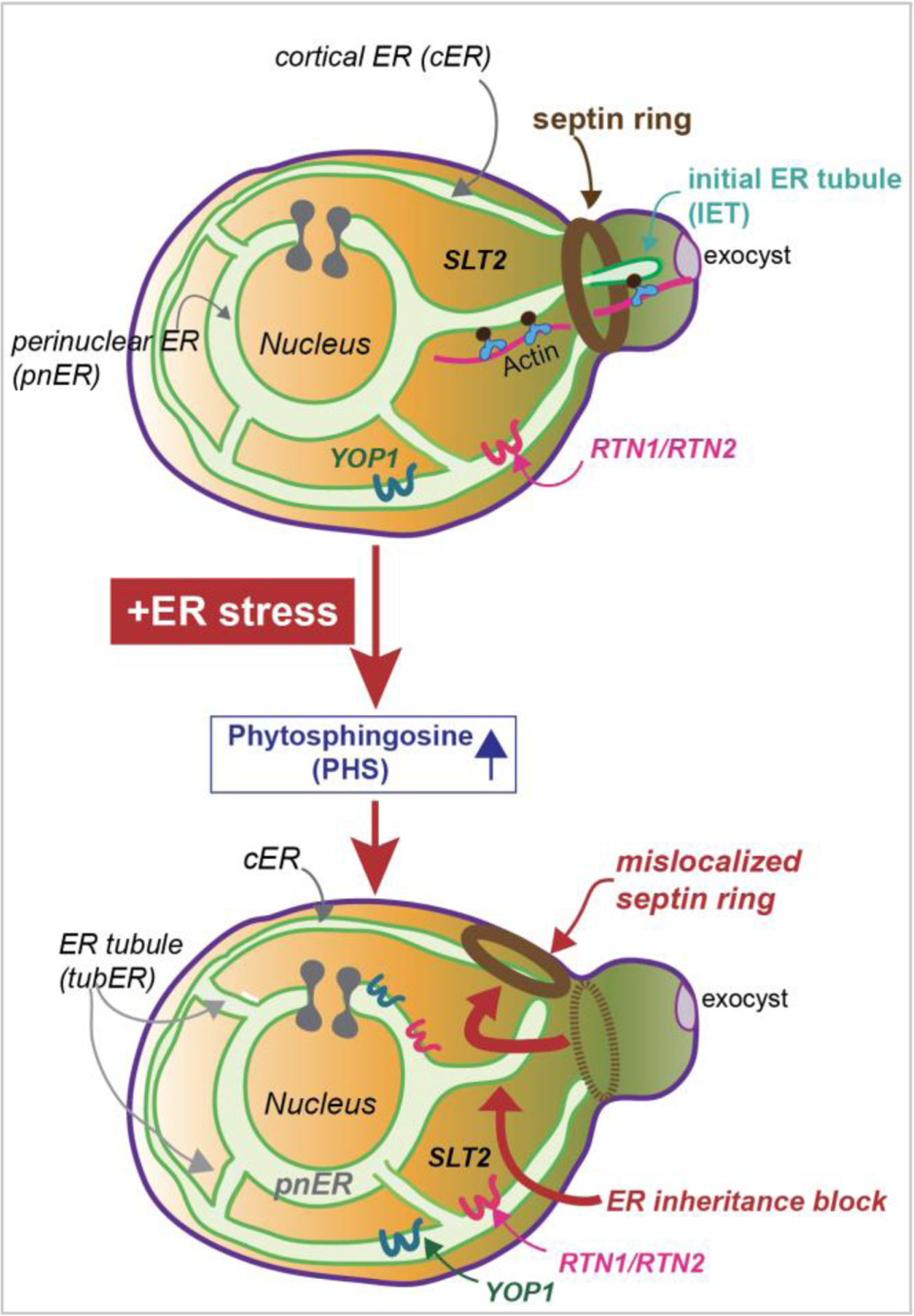
In response to ER stress, (1) the cellular levels of phytosphingosine (PHS) become elevated, initiating the hallmark events of the ERSU: (2) the initial ER tubule (IET) entry into the daughter cell is blocked and (3) the septin ring mislocalizes from the bud neck, ultimately, (4) resulting in cytokinesis cell cycle block. These events are mediated by the reticulon family proteins RTN1, RTN2, YOP1, and MAP kinase, SLT2, and SLT2’s upstream kinases such as PKC1. Levels of Rtn1 and Yop1 increase under ER stress. Currently, the detailed molecular mechanisms by which these components activate the ERSU hallmark events have not yet been investigated. Cytokinesis block continues until ER functional homeostasis is recovered by activation of the UPR, which is also activated by ER stress independent of the ERSU events. Upon recovery, ER inheritance and the cell cycle resume. Recent results revealed that septin ring mislocalization plays a critical role in timely re-entry into the cell cycle when ER functional homeostasis is re-established (Chao et al. 2019).
The ERSU cell cycle checkpoint is independent of the cell wall integrity pathway
In addition to SLT2, a MAP kinase, its upstream kinases PKC1, BCK1, MKK1, and MKK2 were also found to be involved in the ERSU pathway (Babour et al., 2010). Furthermore, the cell surface component Wsc1 – but not other isoforms of Wsc proteins such as Wsc2, 3, and 4 – plays a role in the ERSU pathway. Some of these components are also involved in the cell wall integrity (CWI) pathway, which is induced by excess turgor pressure against the cell wall. For example, the CWI pathway in S. cerevisiae involves PKC1-SLT2 activation via Wsc1 and Wsc2. However, several experimental results clearly distinguish the ERSU checkpoint from the CWI pathway: (1) Wsc1 but not Wsc2 is involved in ERSU pathway events, such as ER inheritance block, septin ring transfer, and cytokinesis block. This is in contrast to the CWI pathway, which depends on both Wsc1 and Wsc2. (2) Treating cells with a known CWI pathway agonist, such as calcofular white, does not activate the ERSU pathway. (3) A Wsc1-AAA mutation that disrupts the internalization of Wsc1 from the cell surface has no impact on ERSU checkpoint activation, despite blocking the induction of the CWI pathway. Taken together, the ER stress-induced ERSU checkpoint is distinct from the previously described CWI pathway.
Loss of the ability of ire1Δ cells to sustain their cell growth under ER stress provided the functional significance of the UPR signaling pathway in response to ER stress. Without the UPR pathway, cells are incapable of handling the accumulation of unfolded proteins in the ER lumen. The lack of slt2Δ cell growth on Tm-containing media also reveals the functional importance of the ERSU pathway. Importantly, the cell growth of ER-stressed slt2Δ cells can be effectively rescued by preventing the inheritance of the stressed ER into the daughter cell. This has been achieved, for example, by the addition of a small quantity of nocodazol, an agent that prevents microtubule polymerization, by the use of actin1–1, a temperature-sensitive Actin mutation, or by the use of myo4Δ cells. Both Actin and Myo4 are required for the entry of the ER into the daughter cell (Estrada et al., 2003). Results of these experiments revealed that the presence of stressed ER in the daughter cell leads to cell death.
Initiation of the ERSU cell cycle checkpoint
The discovery that the ERSU is an independent signaling pathway induced by ER stress beyond the well-studied UPR pathway, and the identification of SLT2 as a key ERSU player, was rather unexpected and generated excitement. However, the initiators of the ERSU cell cycle checkpoint remained a mystery. SLT2 plays roles in a wide range of signaling events, including the CWI response and gene silencing events (Jimenez-Gutierrez et al., 2020; Levin and Errede, 1995). Consistent with being a multi-functional protein, SLT2 is localized throughout the cytoplasm under both normal growth and ER stress conditions. However, SLT2 has not been reported to associate physically with the ER membrane. Since the ERSU hallmark events such as cER inheritance block, septin ring mislocalization, and cytokinesis block, are initiated in response to ER stress, the initiation step is expected to occur on the ER membrane.
Another unanticipated finding led to the identification of ERSU-initiating components on the ER membrane (Pina et al., 2016). This began with the puzzling observation of a differential impact of ER stress on the inheritance of the pnER and cER: ER stress induction blocked cER inheritance, whereas the pnER, contiguous with the cER and the inner nuclear membrane, was inherited normally into the daughter cell even under ER stress (Babour et al., 2010). Since the lumen of the cER and the pnER are contiguous via the presence of the tubular ER, the different responses between the cER and pnER were rather difficult to reconcile. A potential explanation for the observed differences between the cER and the pnER might stem from the spatial differences in the extent of ER stress. Surprisingly, this effort to provide a molecular explanation for these differences led to the identification of ERSU components.
During ER stress, Kar2/BiP binding to the elevated levels of unfolded client proteins increases, resulting in the reduction of Kar2/BiP mobility within the ER lumen (Lai et al., 2010; Lajoie et al., 2012). This finding was demonstrated by a fluorescent recovery after photobleach (FRAP) experiment using a GFP-tagged form of Kar2/BiP (Kar2/BiP-GFP). As anticipated from the contiguous nature of the pnER and cER, fluorescence recovery of Kar2/BiP-GFP was similar throughout the ER. In response to ER stress, the rate of the fluorescence recovery of Kar2/BiP GFP in the pnER was significantly decreased as anticipated. However, the decrease in the fluorescence recovery was asymmetric, differing significantly between the pnER and cER. This result thus revealed differences in Kar2/BiP mobility in the pnER versus cER, which might reflect differences in the level of ER stress (Pina et al., 2016).
Further investigation provided a basis for the use of asymmetric FRAP for identifying ERSU components. In UPR-deficient ire1Δ cells, similar magnitudes of FRAP differences between the cER and pnER were also observed. Surprisingly, however, in ERSU-deficient slt2Δ cells, the extent of the reduction in the mobility of KAR2/BiP-GFP of the pnER became similar in the cER (Pina et al., 2018). In ER-stressed slt2Δ cells, both cER and pnER entered the daughter cell; by contrast, in ER-stressed WT or ire1Δ cells, only pnER entered the daughter cell. Further investigation confirmed that the loss of the asymmetric difference between the cER and pnER provides a means to identify ERSU-deficient cells like slt2Δ, although the molecular bases of these differences between the cER and pnER were unclear.
Indeed, based on the FRAP behavior, a search for ER components that establish the differential ER chaperone behaviors under ER stress identified bona fide ERSU components (Pina et al., 2016). Specifically, rtn1Δrtn2Δyop1Δ cells lacking all three reticulon family proteins, RTN1, RTN2, and YOP1, displayed a phenotype with diminished asymmetric behaviors of the KAR2/BiP-GFP in response to ER stress (Figure 5). Reticulons are known to play important roles in the generation of ER membrane curvature, and therefore affect the overall architecture of the ER including the cER (Hu et al., 2011; Westrate et al., 2015). Thus, the cER of rtn1Δrtn2Δyop1Δ cells was significantly altered compared to that of WT cells and the FRAP behaviors of rtn1Δrtn2Δyop1Δ were similar to those of ERSU-deficient slt2Δ cells (Figure 5). Indeed, ER stress induction of rtn1Δrtn2Δyop1Δ cells displayed all of the hallmark phenotypes of ERSU-deficient cells (Pina et al., 2016).
Figure 5: ER stress has a greater effect on BiP/Kar2-sfGFP mobility in the cER than in the pnER and the lack of the ERSU component SLT2 causes significant changes in BiP/Kar2-sfGFP mobility in the pnER.
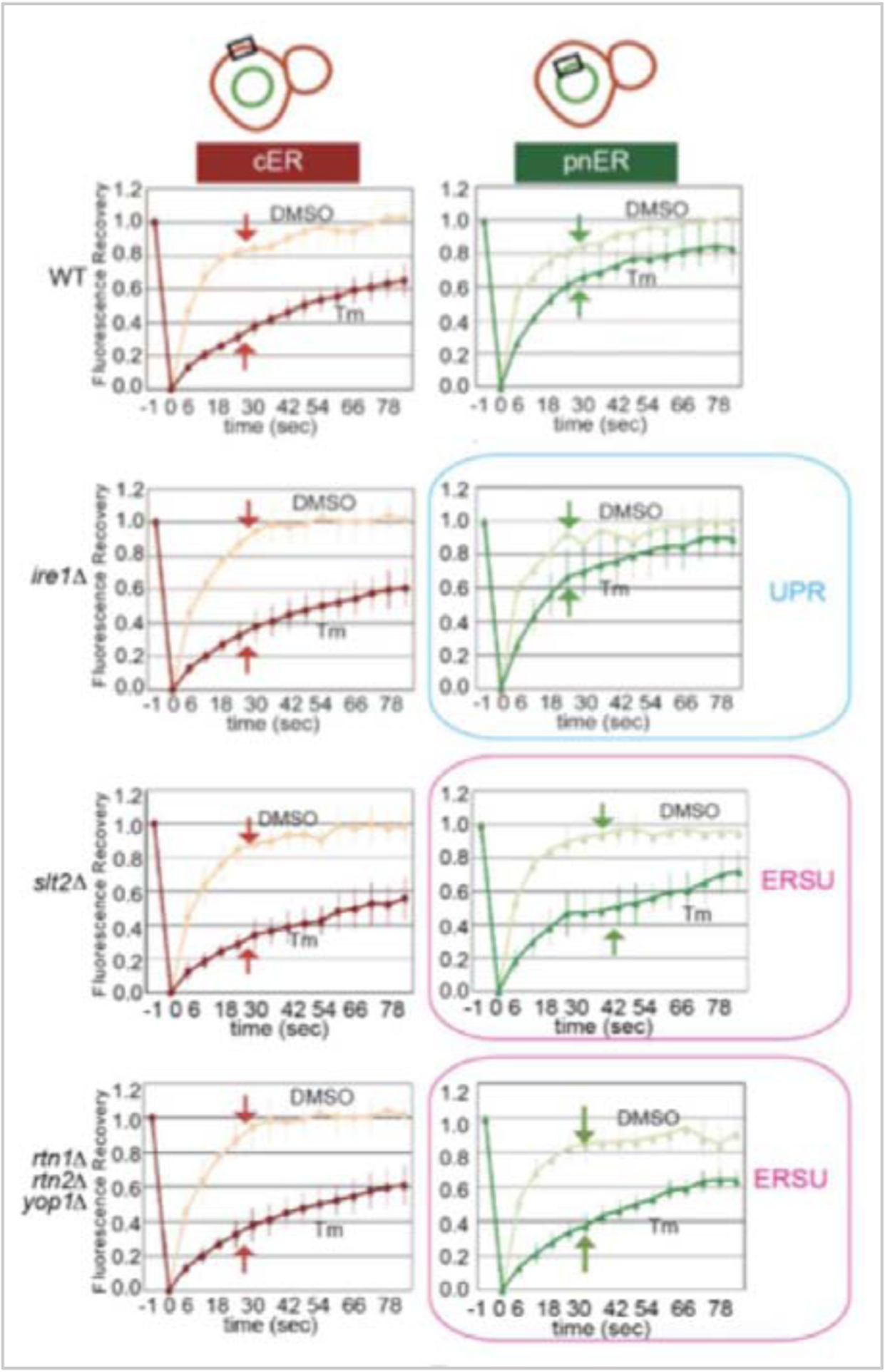
Representative FRAP profiles of BiP/Kar2-sfGFP in either the pnER or cER of WT cells. Upon ER stress induction, fluorescence recovery was slowed down in the cER to a greater degree than in the pnER. WT and ire1Δ cells showed a similar mobility of BiP/Kar2-sfGFP. In contrast, the mobility was altered upon ER stress in ERSU-deficient slt2Δ cells. Interestingly, BiP/Kar2-sfGFP mobility in rtn1Δrtn2Δyop1Δ cells showed a similar change as in slt2Δ cells, suggesting the possibility that RTN1, RTN2, and YOP1 are ERSU components. Subsequent experiments confirmed that this was the case. (the FRAP profiles were taken from figures in (Pina et al., 2016))
The discovery that the asymmetric differences in the luminal conditions of the cER and pnER and that reticulon family proteins such as Rtn1 and Yop1 are involved in the ERSU pathway suggested that the specific shape of the ER contributes to the ERSU-initiating signals. Although the detailed architectural and functional relationships of the ER are not fully understood, the local distributions or functions of the ER-resident or transmembrane proteins are known to dictate specific ER shapes. For example, an ER sheet-like structure is enriched in the pnER region, whereas an ER tubule structure is concentrated in the cER region (Schwarz and Blower, 2016; Wang and Rapoport, 2019; Westrate et al., 2015). Furthermore, ribosomes are bound to the surface of the ER sheets, marking the area of the ER that generates secretory pathway proteins and constituting the rough ER. The smooth ER that lacks surface-associated ribosomes consists of the tubular ER and is involved in lipid biosynthesis. The distributions of ER sheets and tubules are regulated by a balance of the activities of two functionally antagonistic ER structural proteins, Lunapark1 (Lnp1) and Sey1/Atlastin (Chen et al., 2013; Goyal and Blackstone, 2013; Wang and Rapoport, 2019). These proteins are associated with three-way ER junctions, impact ER structure upon association with Rtn1, Rtn2, and Yop1 proteins, and act antagonistically (Anwar et al., 2012; Chen et al., 2015; Hu et al., 2008). Loss of the LNP1 gene causes the Sey1 protein to localize to the cER and pnER and generates a more densely reticulated ER structure in yeast and a more sheet-like ER in mammalian cells (Hu et al., 2009; Shemesh et al., 2014). The loss of SEY1 and YOP1 reduces branched tubular ER, resulting in an accumulation of Lnp1 at the cER and pnER. Given such relationships, altering the proportions of the reticular structure vs. ER sheets upon disruption of the LNP1, but not SEY1, gene in rtn1Δrtn2Δyop1Δ cells indeed restored the asymmetry of KAR2/BiP-GFP FRAP behavior between the cER and pnER. Furthermore, the lack of the LNP1, but not SEY1, gene restored the ERSU-deficient phenotype in rtn1Δrtn2Δyop1Δ cells. Finally, recent studies identified mutant forms of either Rtn1 or Yop1, both carrying a single amino acid change, that diminish the PHS-dependent activation of the ERSU cell cycle checkpoint (unpublished results from the Niwa lab), revealing that RTN1, RTN2, and YOP1 respond to ER stress-induced PHS to activate the ERSU events. Yop1 is independent of the role of these proteins in proper ER structure. Importantly, cells carrying a mutant Rtn1 with several amino acid changes that alter the asymmetric FRAP behaviors of KAR2/BiP-GFP (Shibata et al., 2008) was found to be ERSU defective (unpublished results from the Niwa lab), providing further confirmation that RTN1, RTN2, and YOP1 play vital roles in ER inheritance block during ER stress.
Identifying the ERSU activating signal: phytosphingosine
The next important knowledge gap is the identity of the activating signal(s) of the ERSU pathway. Since the UPR sensor Ire1 is not involved in ERSU pathway activation, it is unlikely that signals activating IRE1, for example the accumulation of unfolded proteins, are involved in the ERSU. However, the activating signal(s) still must be induced by ER stress, which can be triggered by well-characterized ER stress-inducing agents, such as Tm or DTT, or by using a temperature-sensitive allele of ero1–1 that causes unfolded proteins to accumulate.
A number of key findings suggested that sphingolipid levels increase in response to treatment with an ER stress-inducing agent such as Tm in WT yeast cells. The increase is relatively small and transient, but similar changes in sphingolipid levels have been reported during heat shock (Jenkins, 2003; Meier et al., 2006). The early steps of sphingolipid biosynthesis are initiated on the ER membrane; thus, the ER stress-induced increase in their production might act as an ERSU initiating signal. Indeed, exogenous addition of an ER-localized sphingolipid, PHS, to unstressed cells activates all of the ERSU cell cycle checkpoint hallmark events including (1) cER inheritance, (2) septin ring mislocalization, (3) Slt2 phosphorylation, and (4) cytokinesis arrest. The involvement of PHS is specific, as other sphingolipids/ceramides, such as dihydroceramide (DHC) and ceramide, do not robustly activate the ERSU pathway (Figure 6A).
Figure 6: Sphingolipid biosynthetic pathways:
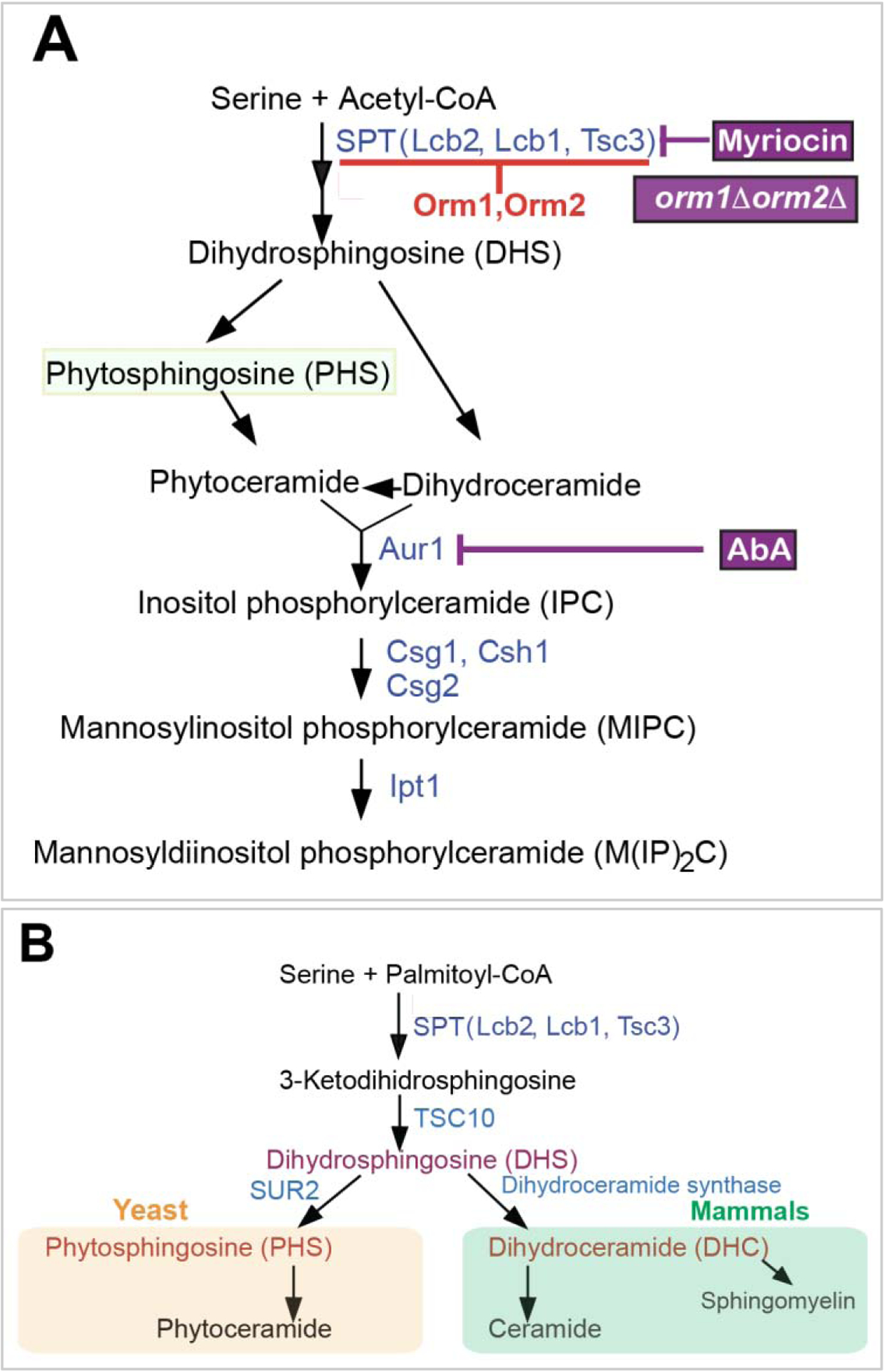
A. An abbreviated yeast sphingolipid biosynthetic pathway. Phytosphingosine (PHS) is highlighted. Treatment of cells with myriocin reduces PHS levels, while orm1Δ orm2Δ cells or Aureobasidin A (AbA) treatment of cells increases the PHS levels. B. A comparison of the human and yeast sphingolipid biosynthetic pathways.
Subsequent experiments using pharmacological drugs such as myriocin or aureobasidin A (AbA) also provided confirmation of the role of PHS as an activating signal (Pina et al., 2018). For example, treatment of cells with AbA, which blocks the conversion of PHS to further downstream sphingolipids, led to an increase in the levels of PHS and activated the ERSU pathway, even in the absence of ER stress-inducing agents. Treating cells with an ER stress-inducing agent, such as Tm or DTT, further increased the extent of ERSU pathway activation. Additionally, treating cells with myriocin, which blocks serine palmitoyltransferase (SPT), diminished the level of ERSU pathway induction. As SPT generates 3-ketohydrosphingosine from serine and palmitoyl-CoA in the first step of the biosynthetic pathway, treating cells with myriocin reduces cellular levels of sphingolipids including PHS (Figure 6A).
The involvement of PHS in ERSU pathway activation is also supported by genetic experiments in yeast (Schuldiner et al., 2005; Schuldiner and Weissman, 2013). For example, an epistatic miniarray profile study identified a series of genes in the sphingolipid biosynthetic pathway as genetic interactors of the ERSU component Slt2. For example, ORM1 and ORM2 encode subunits of an inhibitory complex of SPT; cells lacking ORM1 and ORM2 (orm1Δorm2Δ double knockout cells) have elevated levels of cellular sphingolipids (Breslow et al., 2010; Han et al., 2010). Upon treatment with Tm, orm1Δorm2Δ double knockout cells further increased the frequency of ERSU events. Importantly, adding PHS exogenously did not activate IRE1 or the UPR pathway.
Interestingly, recent studies also reported the involvement of sphingolipids in the establishment of the nuclear envelope and the ER diffusion barrier (Clay et al., 2014; Megyeri et al., 2019). While multiple sphingolipids and ceramides have been reported to function for the ER diffusion barrier, the involvement of PHS in the ERSU checkpoint is specific, as DHC does not support the hallmark events of the ERSU. Furthermore, recent preliminary work has revealed that PHS functions in the ERSU checkpoint independent of changes in the ER diffusion barrier (unpublished results in Niwa lab). Taken together, these observations are consistent with the idea that elevated PHS in response to ER stress acts as an activating signal for the ERSU pathway, leading to ER inheritance block, septin ring mislocalization, Slt2 phosphorylation, and cytokinesis block.
Sphingolipids and ER homeostasis in mammalian cells
The sphingolipid biosynthetic pathway is conserved in eukaryotic cells (Gault et al., 2010; Riezman, 2006), although some details differ depending upon the specific species. Interestingly, a study found that ER stress induction also increases dihydrosphingosine (DHS) and DHC levels in mammalian cells (Tam et al., 2018). Like PHS in yeast, both DHS and DHC are early intermediates of the sphingolipid biosynthetic pathway in mammalian cells (Figure 6B) (Riezman, 2006). The increased levels of these sphingolipids are transient. The molecular mechanisms of the UPR pathway are more complex in mammalian cells than those in yeast cells: in yeast, IRE1 is the only ER transmembrane UPR signaling component, whereas the mammalian UPR pathway is activated by two additional ER transmembrane components, PERK and ATG6 (Schroder and Kaufman, 2005). While the mammalian ERSU checkpoint has yet to be defined, the additional mammalian UPR components (i.e., ATF6 and PERK) might provide means to respond to ER stress-induced DHS and DHC. Exogenously added DHS or DHC indeed activated ATF6α, but did not affect PERK or IRE1 (Tam et al., 2018). Further studies revealed a unique DHS/DHC binding motif within the transmembrane domain of ATF6α. A single amino acid substitution within this motif resulted in inactivation of ATF6α by DHS and DHC. However, these ATF6α mutants were still activated by increased levels of unfolded proteins in the ER. Conversely, mutations within the ER luminal domain of ATF6α failed to respond to the increased levels of unfolded proteins but remained active to respond to exogenously added DHS and DHC. These results indicate that ATF6α has two distinct stress-sensing domains, one through the ER luminal domain and the other via the transmembrane motif. Currently, it remains to be determined if the mammalian ERSU checkpoint exists and how such responses are wired, but it is possible that ATF6α is a dual component acting on the UPR pathway and the ERSU cell cycle checkpoint for the ER in mammalian cells by two distinct domains. Furthermore, the identification of the ATF6α DHS/DHC binding motif provides hints about the activation mechanisms of ERSU components, such as Rtn1 or Yop1, via PHS.
Many more exciting questions about the yeast ERSU cell cycle checkpoint
The discovery of the ERSU pathway has led to many interesting questions. For example, why is PHS the activating signal, instead of the more abundant sphingolipids or ceramides? Production of sphingolipids and ceramide begins at the ER and synthesis of PHS occurs at the ER (Riezman, 2006). Once converted, ceramide is transported to the Golgi where further downstream lipid biosynthetic steps continue (Funato and Riezman, 2001; Liu et al., 2017). Finally, some of these lipids reach the plasma membrane. For example, glycerosphingolipids and cholesterol (ergosterol in yeast) generate a subdomain within the plasma membrane, the lipid rafts (Hurst and Fratti, 2020; Lingwood and Simons, 2010). The use of more abundant lipids as signals might require more major cellular changes before the ERSU pathway is turned on. For the ERSU pathway to work as a temporary halt of the cell cycle during ER stress, such dramatic changes might not be suitable.
How are PHS levels increased during ER stress? How are increased levels of PHS recognized? Do Rtn1, Rtn2, and Yop1 proteins somehow measure the PHS levels? Or, do alternative components exist, ultimately leading to ER inheritance block, septin ring mislocalization, and Slt2 phosphorylation via Rtn1, Rtn2, and Yop1? Do PHS levels contribute to the establishment of the asymmetry between the cER and the pnER? Interestingly, a mutagenesis study of Rtn1 reported that certain mutations within the Rtn1 protein caused the FRAP profile of BiP/Kar2–GFP in the cER to differ from that in the pnER, even in the absence of ER stress induction (Shibata et al., 2008), revealing that Rtn1 is sensitive to ER functional homeostasis.
ER stress induces both the ERSU and UPR pathways. While UPR activation re-establishes ER homeostasis, the ERSU pathway halts both the cell cycle and ER inheritance. In order for cells to re-enter the cell cycle in a timely manner, certain components such as those that re-evaluate ER functional status during cell cycle block and those that establish the release from cytokinesis block, must be involved. Furthermore, after release from cytokinesis block, how do cells re-enter the cell cycle? If ER functional homeostasis does not recover within a certain period of time, what happens to the cell? Are there time limits for cell cycle recovery? Answering these questions will be critical to fully understanding the yeast ERSU cell cycle checkpoint, a mechanism to ensure all dividing cells will have sufficient levels of the ER.
Potential for cell cycle regulation of the ER in mammalian cells
Given the complexity of the mammalian ER network, examining the effect of ER stress on the ER distribution in dividing cells is challenging. During prophase, the nuclear membranes detach from the lamins and chromatin to disassemble. In fact, the nuclear membrane components are retracted into and become dispersed throughout the ER (Anderson et al., 2009). During the metaphase-to-anaphase transition, however, the nuclear envelope (NE) re-forms from ER tubules and sheets to enclose each set of daughter chromosomes (Anderson and Hetzer, 2008). Thus, the ER is intimately involved in NE disassembly and reassembly. Furthermore, during NE disassembly, nuclear membrane proteins such as POM121, an essential transmembrane protein of the nuclear pore, have to become dispersed throughout the ER and relocate to the newly reassembled NE and nuclear pores as the NE reassembles. Similarly, studies have reported that RTN3 first is associated with chromatin-bound membranes, but becomes increasingly dispersed throughout the surrounding ER by anaphase (Anderson and Hetzer, 2008). Thus, such processes might be impacted by ER stress. Indeed, induction of ER stress resulted in the mislocalization of septin ring subunits (unpublished results from the Niwa lab). Understanding the mechanisms by which the ER is divided in normal cells and how this is perturbed under various stress conditions will contribute to our understanding of human disease. Indeed, dysregulated ER function is a prominent feature of many disorders, including diabetes, Alzheimer’s disease, and Parkinson’s disease, which are increasing public health concerns. Thus, studies on ER function may ultimately lead to the development of new treatments for such diseases.
Summary
The discovery of the ERSU cell cycle checkpoint pathway unveiled the exciting possibility that the division of cytoplasmic components such as organelles or protein complexes is regulated in order to generate fully functional eukaryotic cells. Further studies in this area may unveil new strategies for designing novel drugs for human diseases that are caused by improper cell cycle regulation.
Highlights.
The ER stress surveillance (ERSU) checkpoint ensures inheritance of functional ER in yeast.
The ERSU temporally halts the cell cycle, while the UPR re-establishes ER function. The ERSU is mediated by the reticulon family proteins RTN1, RTN2, and YOP1, and the MAP kinase SLT2.
The ERSU checkpoint is activated by phytosphingosine, an early biosynthetic sphingolipid.
Septin ring transfer from the bud neck to the bud scar is a hallmark event of the ERSU in response to ER stress
Transfer of the septin ring is required for timely re-entry into the cell cycle. Age impacts cell-cycle re-entry upon ER stress recovery
Acknowledgements
The author apologizes for absence of citations within the space limit of this review. The author thanks members of the laboratory for critical reading of the manuscript. The author thanks members of the Niwa lab for careful reading of the review. The work is supported by NIH (RO1 GM087415) and Cancer Research Coordinating Committee fund (CRR-20-632388).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest Statement:
The author declare no competing or financial interest on the review.
References
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, and Walter P (2002). Molecular Biology of the Cell, 4th edition Molecular Biology of the Cell, 4th edition. [Google Scholar]
- Anderson DJ, and Hetzer MW (2008). Reshaping of the endoplasmic reticulum limits the rate for nuclear envelope formation. J Cell Biol 182, 911–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ, Vargas JD, Hsiao JP, and Hetzer MW (2009). Recruitment of functionally distinct membrane proteins to chromatin mediates nuclear envelope formation in vivo. J Cell Biol 186, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold E, and Tanner W (1982). An obligatory role of protein glycosylation in the life cycle of yeast cells. FEBS Lett 148, 49–53. [DOI] [PubMed] [Google Scholar]
- Aviram N, and Schuldiner M (2017). Targeting and translocation of proteins to the endoplasmic reticulum at a glance. J Cell Sci 130, 4079–4085. [DOI] [PubMed] [Google Scholar]
- Babour A, Bicknell AA, Tourtellotte J, and Niwa M (2010). A surveillance pathway monitors the fitness of the endoplasmic reticulum to control its inheritance. Cell 142, 256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SH, and Kaufman RJ (2012). Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem 81, 767–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berner N, Reutter KR, and Wolf DH (2018). Protein Quality Control of the Endoplasmic Reticulum and Ubiquitin-Proteasome-Triggered Degradation of Aberrant Proteins: Yeast Pioneers the Path. Annu Rev Biochem 87, 751–782. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang YH, Hendershot LM, Harding HP, and Ron D (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2, 326–332. [DOI] [PubMed] [Google Scholar]
- Bicknell AA, Babour A, Federovitch CM, and Niwa M (2007). A novel role in cytokinesis reveals a housekeeping function for the unfolded protein response. J Cell Biol 177, 1017–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidlingmaier S, and Snyder M (2004). Regulation of polarized growth initiation and termination cycles by the polarisome and Cdc42 regulators. J Cell Biol 164, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla M, and Cunningham KW (2003). Mitogen-activated protein kinase stimulation of Ca(2+) signaling is required for survival of endoplasmic reticulum stress in yeast. Mol Biol Cell 14, 4296–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, and Weissman JS (2010). Orm family proteins mediate sphingolipid homeostasis. Nature 463, 1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JD, Ng DT, Ogg SC, and Walter P (1995). Targeting pathways to the endoplasmic reticulum membrane. Cold Spring Harb Symp Quant Biol 60, 23–30. [DOI] [PubMed] [Google Scholar]
- Caudron F, and Barral Y (2009). Septins and the lateral compartmentalization of eukaryotic membranes. Dev Cell 16, 493–506. [DOI] [PubMed] [Google Scholar]
- Chao JT, Pina F, Onishi M, Cohen Y, Lai YS, Schuldiner M, and Niwa M (2019). Transfer of the Septin Ring to Cytokinetic Remnants in ER Stress Directs Age-Sensitive Cell-Cycle Re-entry. Dev Cell 51, 173–191 e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao JT, Wong AK, Tavassoli S, Young BP, Chruscicki A, Fang NN, Howe LJ, Mayor T, Foster LJ, and Loewen CJ (2014). Polarization of the endoplasmic reticulum by ER-septin tethering. Cell 158, 620–632. [DOI] [PubMed] [Google Scholar]
- Chen S, Novick P, and Ferro-Novick S (2013). ER structure and function. Curr Opin Cell Biol 25, 428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay L, Caudron F, Denoth-Lippuner A, Boettcher B, Buvelot Frei S, Snapp EL, and Barral Y (2014). A sphingolipid-dependent diffusion barrier confines ER stress to the yeast mother cell. Elife 3, e01883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Valm AM, and Lippincott-Schwartz J (2018). Interacting organelles. Curr Opin Cell Biol 53, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JS, Shamu CE, and Walter P (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206. [DOI] [PubMed] [Google Scholar]
- Cox JS, Sidrauski C, Shamu C, and Walter P (1996). Regulation of the unfolded protein response pathway. Molecular Biology of the Cell 7, 2944–2944. [Google Scholar]
- Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, and Walter P (2005). On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci U S A 102, 18773–18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, and Diehl JA (2006). Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol 38, 317–332. [DOI] [PubMed] [Google Scholar]
- Doyle T, and Botstein D (1996). Movement of yeast cortical actin cytoskeleton visualized in vivo. Proc Natl Acad Sci U S A 93, 3886–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Pypaert M, Novick P, and Ferro-Novick S (2001). Aux1p/Swa2p is required for cortical endoplasmic reticulum inheritance in Saccharomyces cerevisiae. Molecular Biology of the Cell 12, 2614–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Walker L, Novick P, and Ferro-Novick S (2006). Ptc1p regulates cortical ER inheritance via Slt2p. EMBO J 25, 4413–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ, and Harper JW (1994). Cdk inhibitors: on the threshold of checkpoints and development. Curr Opin Cell Biol 6, 847–852. [DOI] [PubMed] [Google Scholar]
- Estrada de Martin P, Novick P, and Ferro-Novick S (2005). The organization, structure, and inheritance of the ER in higher and lower eukaryotes. Biochem Cell Biol 83, 752–761. [DOI] [PubMed] [Google Scholar]
- Estrada P, Kim J, Coleman J, Walker L, Dunn B, Takizawa P, Novick P, and Ferro-Novick S (2003). Myo4p and She3p are required for cortical ER inheritance in Saccharomyces cerevisiae. J Cell Biol 163, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber-Katz SE, Dippold HC, Buschman MD, Peterman MC, Xing M, Noakes CJ, Tat J, Ng MM, Rahajeng J, Cowan DM, et al. (2014). DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell 156, 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre JC, Mahalingam SS, Proietto M, and Subramani S (2019). Peroxisome biogenesis, membrane contact sites, and quality control. EMBO Rep 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CM, and Kellogg D (1999). Septins: cytoskeletal polymers or signalling GTPases? Trends Cell Biol 9, 387–394. [DOI] [PubMed] [Google Scholar]
- Forbes DJ, Travesa A, Nord MS, and Bernis C (2015). Nuclear transport factors: global regulation of mitosis. Curr Opin Cell Biol 35, 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, and Riezman H (2001). Vesicular and nonvesicular transport of ceramide from ER to the Golgi apparatus in yeast. J Cell Biol 155, 949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault CR, Obeid LM, and Hannun YA (2010). An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol 688, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goder V, Alanis-Dominguez E, and Bustamante-Sequeiros M (2019). Lipids and their (un)known effects on ER-associated protein degradation (ERAD). Biochim Biophys Acta Mol Cell Biol Lipids. [DOI] [PubMed] [Google Scholar]
- Goyal U, and Blackstone C (2013). Untangling the web: mechanisms underlying ER network formation. Biochim Biophys Acta 1833, 2492–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, and Rine J (1996). Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Molecular Biology of the Cell 7, 2029–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, and Sommer T (2012). Finding the will and the way of ERAD substrate retrotranslocation. Curr Opin Cell Biol 24, 460–466. [DOI] [PubMed] [Google Scholar]
- Han S, Lone MA, Schneiter R, and Chang A (2010). Orm1 and Orm2 are conserved endoplasmic reticulum membrane proteins regulating lipid homeostasis and protein quality control. Proc Natl Acad Sci U S A 107, 5851–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heider MR, and Munson M (2012). Exorcising the exocyst complex. Traffic 13, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helle SC, Kanfer G, Kolar K, Lang A, Michel AH, and Kornmann B (2013). Organization and function of membrane contact sites. Biochim Biophys Acta 1833, 2526–2541. [DOI] [PubMed] [Google Scholar]
- Henry KA, Blank HM, Hoose SA, and Polymenis M (2010). The unfolded protein response is not necessary for the G1/S transition, but it is required for chromosome maintenance in Saccharomyces cerevisiae. PLoS One 5, e12732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, and Cleveland DW (2012). Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep 13, 501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Prinz WA, and Rapoport TA (2011). Weaving the web of ER tubules. Cell 147, 1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Zhu PP, Voss C, Rismanchi N, Prinz WA, Rapoport TA, and Blackstone C (2009). A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 138, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LR, and Fratti RA (2020). Lipid Rafts, Sphingolipids, and Ergosterol in Yeast Vacuole Fusion and Maturation. Front Cell Dev Biol 8, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GM (2003). The emerging role for sphingolipids in the eukaryotic heat shock response. Cell Mol Life Sci 60, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Gutierrez E, Alegria-Carrasco E, Sellers-Moya A, Molina M, and Martin H (2020). Not just the wall: the other ways to turn the yeast CWI pathway on. Int Microbiol 23, 107–119. [DOI] [PubMed] [Google Scholar]
- Jin Y, and Weisman LS (2015). The vacuole/lysosome is required for cell-cycle progression. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BM, and DeBose-Boyd RA (2018). Underlying mechanisms for sterol-induced ubiquitination and ER-associated degradation of HMG CoA reductase. Semin Cell Dev Biol 81, 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagas NE, and Venkatachalam K (2019). Roles for the Endoplasmic Reticulum in Regulation of Neuronal Calcium Homeostasis. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin JV, and Adams AE (1984). Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. J Cell Biol 98, 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, and Kohno K (2007). Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol 179, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koning AJ, Larson LL, Cadera EJ, Parrish ML, and Wright RL (2002). Mutations that affect vacuole biogenesis inhibit proliferation of the endoplasmic reticulum in Saccharomyces cerevisiae. Genetics 160, 1335–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvam E, and Goldfarb DS (2006). Nucleus-vacuole junctions in yeast: anatomy of a membrane contact site. Biochem Soc Trans 34, 340–342. [DOI] [PubMed] [Google Scholar]
- Lai CW, Aronson DE, and Snapp EL (2010). BiP availability distinguishes states of homeostasis and stress in the endoplasmic reticulum of living cells. Mol Biol Cell 21, 1909–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie P, Moir RD, Willis IM, and Snapp EL (2012). Kar2p availability defines distinct forms of endoplasmic reticulum stress in living cells. Mol Biol Cell 23, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin DE, and Errede B (1995). The proliferation of MAP kinase signaling pathways in yeast. Curr Opin Cell Biol 7, 197–202. [DOI] [PubMed] [Google Scholar]
- Li X, Ferro-Novick S, and Novick P (2013). Different polarisome components play distinct roles in Slt2p-regulated cortical ER inheritance in Saccharomyces cerevisiae. Mol Biol Cell 24, 3145–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D, and Simons K (2010). Lipid rafts as a membrane-organizing principle. Science 327, 46–50. [DOI] [PubMed] [Google Scholar]
- Liu LK, Choudhary V, Toulmay A, and Prinz WA (2017). An inducible ER-Golgi tether facilitates ceramide transport to alleviate lipotoxicity. J Cell Biol 216, 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Ma Y, and Hendershot LM (2001). The unfolding tale of the unfolded protein response. Cell 107, 827–830. [DOI] [PubMed] [Google Scholar]
- Mascanzoni F, Ayala I, and Colanzi A (2019). Organelle Inheritance Control of Mitotic Entry and Progression: Implications for Tissue Homeostasis and Disease. Front Cell Dev Biol 7, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matellan L, and Monje-Casas F (2020). Regulation of Mitotic Exit by Cell Cycle Checkpoints: Lessons From Saccharomyces cerevisiae. Genes (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlack KE, Mothes W, and Rapoport TA (1998). Protein translocation: tunnel vision. Cell 92, 381–390. [DOI] [PubMed] [Google Scholar]
- McCaffrey K, and Braakman I (2016). Protein quality control at the endoplasmic reticulum. Essays Biochem 60, 227–235. [DOI] [PubMed] [Google Scholar]
- McMaster CR (2001). Lipid metabolism and vesicle trafficking: more than just greasing the transport machinery. Biochem Cell Biol 79, 681–692. [DOI] [PubMed] [Google Scholar]
- McMillan JN, Sia RA, and Lew DJ (1998). A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J Cell Biol 142, 1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megyeri M, Prasad R, Volpert G, Sliwa-Gonzalez A, Haribowo AG, Aguilera-Romero A, Riezman H, Barral Y, Futerman AH, and Schuldiner M (2019). Yeast ceramide synthases, Lag1 and Lac1, have distinct substrate specificity J Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrtash AB, and Hochstrasser M (2019). Ubiquitin-dependent protein degradation at the endoplasmic reticulum and nuclear envelope. Semin Cell Dev Biol 93, 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier KD, Deloche O, Kajiwara K, Funato K, and Riezman H (2006). Sphingoid base is required for translation initiation during heat stress in Saccharomyces cerevisiae. Mol Biol Cell 17, 1164–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldolesi J, and Pozzan T (1998). The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem Sci 23, 10–14. [DOI] [PubMed] [Google Scholar]
- Mittal B, Tulsyan S, Kumar S, Mittal RD, and Agarwal G (2015). Cytochrome P450 in Cancer Susceptibility and Treatment. Adv Clin Chem 71, 77–139. [DOI] [PubMed] [Google Scholar]
- Morgan DO (1995). Principles of CDK regulation. Nature 374, 131–134. [DOI] [PubMed] [Google Scholar]
- Mori K (2009). Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem 146, 743–750. [DOI] [PubMed] [Google Scholar]
- Mori K, Ma W, Gething MJ, and Sambrook J (1993). A transmembrane protein with a cdc2+/CDC28−related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756. [DOI] [PubMed] [Google Scholar]
- Mori K, Ogawa N, Kawahara T, Yanagi H, and Yura T (2000). mRNA splicing-mediated C-terminal replacement of transcription factor Hac1p is required for efficient activation of the unfolded protein response. Proc Natl Acad Sci U S A 97, 4660–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland J, Preuss D, Moon A, Wong A, Drubin D, and Botstein D (1994). Ultrastructure of the yeast actin cytoskeleton and its association with the plasma membrane. J Cell Biol 125, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson M, and Novick P (2006). The exocyst defrocked, a framework of rods revealed. Nat Struct Mol Biol 13, 577–581. [DOI] [PubMed] [Google Scholar]
- Musacchio A, and Desai A (2017). A Molecular View of Kinetochore Assembly and Function. Biology (Basel) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, and Botstein D (1985). Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell 40, 405–416. [DOI] [PubMed] [Google Scholar]
- Oikawa D, Kimata Y, and Kohno K (2007). Self-association and BiP dissociation are not sufficient for activation of the ER stress sensor Ire1. J Cell Sci 120, 1681–1688. [DOI] [PubMed] [Google Scholar]
- Pierro C, Sneyers F, Bultynck G, and Roderick HL (2019). ER Ca(2+) release and store-operated Ca(2+) entry - partners in crime or independent actors in oncogenic transformation? Cell Calcium 82, 102061. [DOI] [PubMed] [Google Scholar]
- Pina F, Yagisawa F, Obara K, Gregerson JD, Kihara A, and Niwa M (2018). Sphingolipids activate the endoplasmic reticulum stress surveillance pathway. J Cell Biol 217, 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina FJ, Fleming T, Pogliano K, and Niwa M (2016). Reticulons Regulate the ER Inheritance Block during ER Stress. Dev Cell 37, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina FJ, and Niwa M (2015). The ER Stress Surveillance (ERSU) pathway regulates daughter cell ER protein aggregate inheritance. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pobre KFR, Poet GJ, and Hendershot LM (2019). The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J Biol Chem 294, 2098–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss D, Mulholland J, Kaiser CA, Orlean P, Albright C, Rose MD, Robbins PW, and Botstein D (1991). Structure of the yeast endoplasmic reticulum: localization of ER proteins using immunofluorescence and immunoelectron microscopy. Yeast 7, 891–911. [DOI] [PubMed] [Google Scholar]
- Riezman H (2006). Organization and functions of sphingolipid biosynthesis in yeast. Biochem Soc Trans 34, 367–369. [DOI] [PubMed] [Google Scholar]
- Ron D, and Walter P (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, and Kaufman RJ (2004). A trip to the ER: coping with stress. Trends Cell Biol 14, 20–28. [DOI] [PubMed] [Google Scholar]
- Schroder M, and Kaufman RJ (2005). The mammalian unfolded protein response. Annu Rev Biochem 74, 739–789. [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF, et al. (2005). Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123, 507–519. [DOI] [PubMed] [Google Scholar]
- Schuldiner M, and Weissman JS (2013). The contribution of systematic approaches to characterizing the proteins and functions of the endoplasmic reticulum. Cold Spring Harb Perspect Biol 5, a013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Travesa A, Martell SW, and Forbes DJ (2015). Analysis of the initiation of nuclear pore assembly by ectopically targeting nucleoporins to chromatin. Nucleus 6, 40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DS, and Blower MD (2016). The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 73, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamu CE, and Walter P (1996). Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J 15, 3028–3039. [PMC free article] [PubMed] [Google Scholar]
- Shemesh T, Klemm RW, Romano FB, Wang S, Vaughan J, Zhuang X, Tukachinsky H, Kozlov MM, and Rapoport TA (2014). A model for the generation and interconversion of ER morphologies. Proc Natl Acad Sci U S A 111, E5243–5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Voss C, Rist JM, Hu J, Rapoport TA, Prinz WA, and Voeltz GK (2008). The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J Biol Chem 283, 18892–18904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Cox JS, and Walter P (1996). tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 87, 405–413. [DOI] [PubMed] [Google Scholar]
- Sun Z, and Brodsky JL (2019). Protein quality control in the secretory pathway. J Cell Biol 218, 3171–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam AB, Roberts LS, Chandra V, Rivera IG, Nomura DK, Forbes DJ, and Niwa M (2018). The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev Cell 46, 327–343 e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Kawano S, and Endo T (2019). Organelle contact zones as sites for lipid transfer. J Biochem 165, 115–123. [DOI] [PubMed] [Google Scholar]
- Vai M, Popolo L, and Alberghina L (1987). Effect of tunicamycin on cell cycle progression in budding yeast. Exp Cell Res 171, 448–459. [DOI] [PubMed] [Google Scholar]
- Versele M, and Thorner J (2005). Some assembly required: yeast septins provide the instruction manual. Trends Cell Biol 15, 414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vevea JD, Swayne TC, Boldogh IR, and Pon LA (2014). Inheritance of the fittest mitochondria in yeast. Trends Cell Biol 24, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz GK, Rolls MM, and Rapoport TA (2002). Structural organization of the endoplasmic reticulum. EMBO Rep 3, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddle JA, Karpova TS, Waterston RH, and Cooper JA (1996). Movement of cortical actin patches in yeast. J Cell Biol 132, 861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Gilmore R, and Blobel G (1984). Protein translocation across the endoplasmic reticulum. Cell 38, 5–8. [DOI] [PubMed] [Google Scholar]
- Walter P, and Ron D (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang N, and Rapoport TA (2019). Reconstituting the reticular ER network - mechanistic implications and open questions. J Cell Sci 132. [DOI] [PubMed] [Google Scholar]
- Weisman LS (2003). Yeast vacuole inheritance and dynamics. Annu Rev Genet 37, 435–460. [DOI] [PubMed] [Google Scholar]
- Westrate LM, Lee JE, Prinz WA, and Voeltz GK (2015). Form follows function: the importance of endoplasmic reticulum shape. Annu Rev Biochem 84, 791–811. [DOI] [PubMed] [Google Scholar]
- Wu H, Carvalho P, and Voeltz GK (2018). Here, there, and everywhere: The importance of ER membrane contact sites. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Rapoport TA (2018). Mechanistic insights into ER-associated protein degradation. Curr Opin Cell Biol 53, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, and Elledge SJ (2000). The DNA damage response: putting checkpoints in perspective. Nature 408, 433–439. [DOI] [PubMed] [Google Scholar]
- Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, and Kaufman RJ (2006). The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci U S A 103, 14343–14348. [DOI] [PMC free article] [PubMed] [Google Scholar]