

SUMMARY
Type II CRISPR-Cas systems defend prokaryotes from bacteriophage infection through the acquisition of short viral DNA sequences known as spacers, that are transcribed into short RNA guides to specify the targets of the Cas9 nuclease. To counter the potentially devastating propagation of escaper phages with target mutations, the host population acquires many different spacer sequences. Whether and how the presence of preexisting targeting spacers in type II systems affects the acquisition of new ones is unknown. Here we demonstrate that previously acquired spacers promote additional spacer acquisition from the vicinity of the target site through the cleavage of the target DNA. Therefore CRISPR immune cells can at the same time destroy the infecting virus and acquire additional spacers. This anticipates the rise of escapers or related viruses that could escape targeting by the first spacer acquired. Our results reveal a new role for Cas9 in the generation of immunological memories.
INTRODUCTION
All immune systems must be able to neutralize rapidly evolving pathogens that diversify to evade them. In bacteria and archaea, clustered regularly interspaced short palindromic repeats (CRISPR) systems and their associated (cas) genes provide immunity against viral (Barrangou et al., 2007) and plasmid infection (Marraffini and Sontheimer, 2008). During infection, short sequences of DNA derived from these foreign nucleic acids are captured and stored within the CRISPR locus as spacers in between the repeats (Barrangou et al., 2007). Spacer sequences are transcribed and processed to generate short CRISPR RNAs (crRNAs) that specify targets of CRISPR immune response (Brouns et al., 2008; Deltcheva et al., 2011; Hale et al., 2008). CrRNAs form ribonucleoprotein complexes with Cas effector nucleases and guide them to foreign nucleic acid targets (protospacers) that are complementary to the spacer sequence (Gasiunas et al., 2012; Jinek et al., 2012; Jore et al., 2011; Kazlauskiene et al., 2016). Cleavage of the invader’s nucleic acids provides immunity to infection (Garneau et al., 2010; Samai et al., 2015; Westra et al., 2012).
Depending on their Cas content, CRISPR-Cas systems have been categorized into six types (I-VI) and 19 subtypes (Koonin et al., 2017). The mechanism that crRNA-Cas complexes utilize to orchestrate the destruction of foreign nucleic acids depends on the CRISPR type. In type II systems, targets must contain a short motif immediately downstream of the protospacer (the protospacer adjacent motif, or PAM) (Bolotin et al., 2005; Deveau et al., 2008); which for the type II-A system of Streptococcus pyogenes used in this study is NGG, where N is any nucleotide (Bikard et al., 2014; Deltcheva et al., 2011). This is because the Cas9 nuclease first searches for this motif in the foreign genome prior to catalyzing the formation of an R-loop structure in which the crRNA guide makes base pair interactions with the complementary strand in the target DNA (Jiang et al., 2016; Sternberg et al., 2014). Complementarity between the 6-12 nucleotides immediately upstream of the PAM, known as the “seed” region of the target, is absolutely required for Cas9 cleavage (Bikard et al., 2014; Jinek et al., 2012). As a consequence of this molecular mechanism, phages with mutations in either the PAM or seed sequences can escape Cas9 destruction (Deveau et al., 2008; Pyenson et al., 2017). However, the many different spacer sequences present in the CRISPR-adapted host population prevent both the propagation of phage escapers (van Houte et al., 2016) as well as the evolution and emergence of new escape mutations (Chabas et al., 2018). In one recent study of the S. pyogenes type II-A CRISPR-Cas system it was shown that all possible targets, defined as any 30-nt region of the phage genome that is followed by an NGG sequence, can be acquired as a new spacer (Heler et al., 2019). Therefore, although a mutant phage can escape targeting and kill a host cell with a particular spacer, its progeny will most likely be destroyed after infection of the next host containing a different spacer (the probability for the presence of two Cas9-inactivating mutations in the same phage genome is very low).
Recent work has investigated the acquisition of new spacers into a minimal type II-A CRISPR locus; i.e. containing a single repeat and no pre-existing spacers (Modell et al., 2017). It revealed the presence of acquisition “hotspots” around linear dsDNA ends, such as the cos site of phages or a dsDNA break (DSB), in the absence of AddAB (the nuclease responsible for the first step in homology-directed DSB repair in gram-positive bacteria (Wigley, 2013)). This led to the hypothesis that the type II-A acquisition machinery uses linear dsDNA ends to extract new spacers. In contrast, in the presence of AddAB the hotspots expanded up to the first chi site (a sequence that halts AddAB DNA degradation (Wigley, 2013)), a result that suggests that AddAB processing of linear DNA ends generates more DNA substrates for the spacer acquisition machinery. Cas9 also participates in spacer selection to ensure that the new sequences are followed by the correct PAM and thus are functional, presumably through the formation of a spacer acquisition complex with Cas1, Cas2 and Csn2 (Heler et al., 2015; Wilkinson et al., 2019). Finally, Cas1 and Cas2 form an integrase complex that is able to incorporate the new spacer DNA into the first position of the CRISPR array, using sequence information located upstream of the first repeat known as the “leader” sequence (McGinn and Marraffini, 2016; Wei et al., 2015a; Wright and Doudna, 2016).
Little is known about the influence of pre-existing spacers in the spacer acquisition process of type II systems. For type I CRISPR-Cas systems, the presence of spacers that match mutated targets (either in the seed or PAM sequences recognized by the Cascade complex) significantly boosts the acquisition of additional spacers (Datsenko et al., 2012; Swarts et al., 2012). Recent bioinformatic analysis suggested that this phenomenon, known as “priming”, also occurs in type II systems (Nicholson et al., 2018); but whether and how the presence of pre-existing spacers affects the type II-A CRISPR-Cas immune response has not been experimentally addressed. Here we show that not only spacers matching mutant targets, but also those providing full immunity, lead to further acquisition of additional spacers. The new sequences originate from the vicinity of the Cas9 target site and protospacer cleavage is required for their acquisition. Finally, we show that priming in type II-A systems prevents the rise of escaper viruses and provides an advantage to the bacterial population. Our results reveal an unexpected role for Cas9 cleavage, beyond the destruction of the target genome, that enables the type II-A CRISPR-Cas immune response to anticipate future threats.
RESULTS
Immune cells acquire additional spacers upon infection
Our lab studies type II-A CRISPR-Cas immunity using an heterologous system, where the CRISPR locus of Streptococcus pyogenes SF370 (Fig. S1A) is cloned into the pC194 plasmid and expressed in Staphylococcus aureus RN4220 [pCRISPR (Heler et al., 2015)]. Recently we discovered that spacer acquisition in this system has a marked preference for linear dsDNA ends (Modell et al., 2017). Since it is well established that these are also the products of Cas9 cleavage both in vivo (Garneau et al., 2010) and in vitro (Gasiunas et al., 2012; Jinek et al., 2012), we decided to investigate if type II-A CRISPR-Cas targeting of phage invaders could lead to spacer acquisition in cultures that are already immune. To test this, the native CRISPR array of pCRISPR was replaced with a single-spacer array containing spacer 174 [pCRISPR(spc174); the sequences of all spacers used in this study are compiled in Supplementary Data File 2], a spacer commonly acquired by this heterologous system following infection with the staphylococcal phage ΦNM4γ4 (Fig. 1A) (Heler et al., 2019). As a control, we utilized a pCRISPR(SR) harboring a single-repeat CRISPR locus; i.e. lacking a pre-existing spacer (we were concerned that possible off-target effects of a “non-targeting” spacer could invalidate it as a negative control). One caveat of this approach is the possibility of a cis effect of the pre-existing spacer on the integration of new ones, which could lead to differences of spacer acquisition due to divergent levels of spacer integration into the “regular” vs. “single-repeat” CRISPR loci. To avoid spacer integration biases, we deleted part of the leader sequence in both plasmids (generating pCRISPR(spc174)ΔL and pCRISPR(SR)ΔL, Fig. S1B-C), a mutation that prevents the integration of new spacers (Fig. S1D) in cis. We then transformed the cells with a second plasmid containing only the leader sequence and a single repeat (pLR), which will enable spacer acquisition in trans into genetically equivalent CRISPR loci (Figs. S1B-D). We corroborated efficient targeting by spc174 in this dual plasmid system (Fig. S1E) and proceeded to test our hypothesis. Cells carrying either sets of plasmids were infected with ΦNM4γ4, collected 30 minutes after infection for plasmid DNA extraction and amplification of its CRISPR locus. In line with previous observations that type II systems acquire spacers preferentially from free DNA ends, we detected an expansion of the CRISPR array (Fig. 1B). This result demonstrated that not only naïve cells, which must acquire new spacers to survive infection, but also immune cells, which a priori do not need extra spacers to destroy the phage, can generate new CRISPR memories.
Figure 1. Immune cells acquire can additional spacers upon infection.
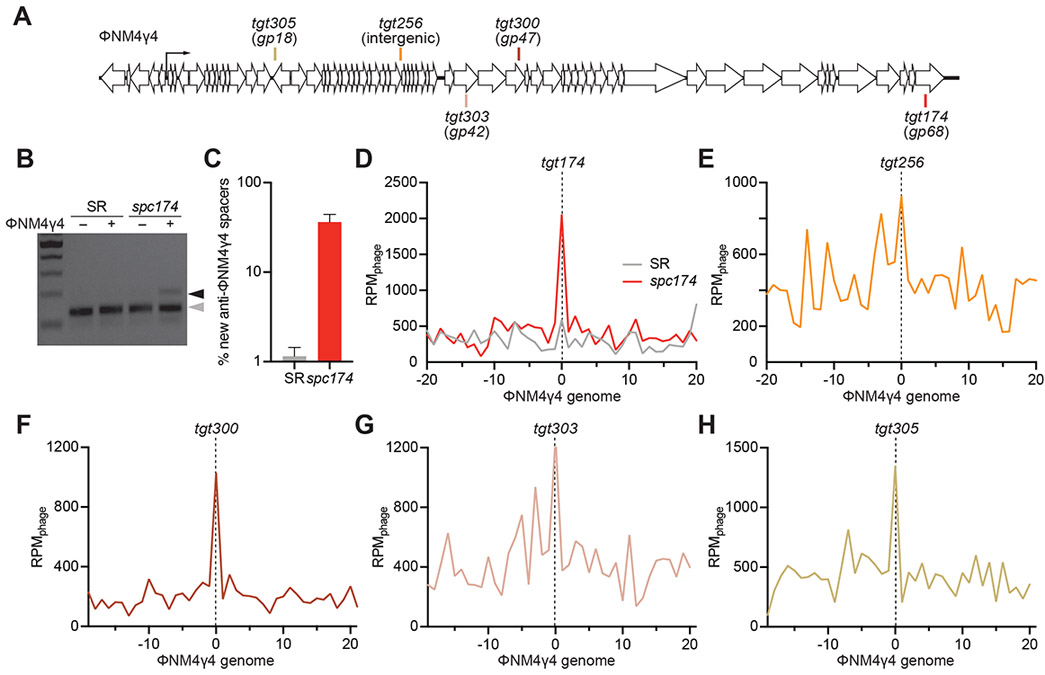
(A) Staphylococcal phage ΦNM4γ4 genome and the Cas9 targets analyzed in this study. (B) Staphylococci harboring pCRISPR(spc174) that targets the phage ΦNM4γ4 and pCRISPR(SR) were infected (+) or mock-infected (−) for 30 minutes and their DNA purified to amplify the CRISPR locus. PCR products were separated by gel electrophoresis to detect the acquisition of new spacers. Grey and black arrows: non-expanded and expanded, respectively, CRISPR arrays. (C) Fraction (%) of spacer sequences matching to the genome of phage ΦNM4γ4 detected in the PCR product of the expanded CRISPR array of staphylococci harboring either pCRISPR(spc174) or pCRISPR(SR). Mean ± StDev values of three independent experiments are shown. (D-H) Distribution of new spacer sequences detected 30 minutes after infection of staphylococci expressing Cas9 programmed to cleave the targets shown in (A). Reads per million of phage reads (RPMphage) are mapped to 1 kb bins of the ΦNM4γ4 genome (shown in linear form, with the specified target in the center). Average curve of three independent experiments is shown. See also Fig. S1.
Pre-existing spacers determine the pattern of spacer acquisition
Next, we wanted to determine how the presence of a targeting spacer influences the acquisition of additional ones. To do this, we compared spacer acquisition in cells carrying pCRISPR(spc174)ΔL or pCRISPR(SR)ΔL. Both strains were infected with ΦNM4γ4, pLR was extracted, the new spacers amplified and the products subject to next-generation sequencing (all next-generation sequencing data presented is compiled in Supplementary Data File 1). Analysis of the data indicated a striking difference in the acquisition of new spacers from the phage genome, with the presence of the pre-existing spacer spc174 resulting in ~ 30-fold more anti-viral spacer acquisition (Fig. 1C). We then mapped the acquired spacers and their abundances along the ΦNM4γ4 genome. We found that, in the presence of spc174, the new spacers clustered around the targeting site (tgt174, Fig. 1D). We corroborated this finding by investigating the sequences acquired in the presence of four different targeting spacers, spc256, spc300, spc303 and spc305 (Fig. 1A). All these spacers provided efficient immunity against ΦNM4γ4 (Fig. S1E) and, although not as striking as the peak generated at the tgt174 site, we observed distinguishable spacer acquisition peaks at the targets of all of them (Figs. 1E-H). Together these results show that the presence of a pre-existing spacer not only increases dramatically the spacer acquisition rate, but also determines the genomic location of the new spacers.
PAM, but not seed, target mutations abrogate spacer-mediated spacer acquisition
To investigate the molecular mechanisms behind the enhanced spacer acquisition observed in immune cells that already contain a functional spacer, we decided to test if its targeting capabilities are required. Therefore we looked for phages that were able to escape spc174 immunity. We analyzed 30 escaper plaques and, as previously described for type II CRISPR-Cas systems (Deveau et al., 2008; Pyenson et al., 2017), we found two types of mutations: within the seed region within the protospacer or within the PAM sequence immediately flanking the protospacer (Fig. S2A-B). We isolated one mutant phage containing each mutation, ΦNM4γ4174-seed and ΦNM4γ4174-PAM (Fig. 2A), and corroborated that indeed they are able to propagate in staphylococci harboring pCRISPR(spc174) (Fig. 2B). Therefore these two phages have partially matching sequences with a pre-existing spacer but cannot be targeted. We infected cells containing pCRISPR(spc174) and pLR plasmids with these two mutant phages, plated the phage-resistant staphylococci and analyzed ten colonies for the presence of new spacers via PCR of the pLR plasmid. As a control, we infected cells lacking the targeting spacer, harboring pCRISPR(SR). We performed this experiment four times and consistently observed that only a few colonies acquired new spacers after infection with ΦNM4γ4174-PAM (Fig. S3A), similar to the no-spacer control (Fig. S3B). In contrast, most of the colonies surviving infection with ΦNM4γ4174-seed acquired new spacers (Fig. S3C), many more than the control strain (Fig. S3D). Quantification of these data showed a significant increase of the frequency of spacer acquisition from the ΦNM4γ4174-seed in the presence of spc174, but not from the ΦNM4γ4174-PAM phage (Fig. 2C). Finally, we amplified the expanded pLR loci in bulk and subjected the resulting PCR products to next-generation sequencing (NGS) to determine both the abundance and the distribution of the newly acquired spacers on the phage genome. Similarly to the colony analysis, deep sequencing results showed a marked increase in the proportion of ΦNM4γ4174-seed-derived spacers compared to ΦNM4γ4174-PAM-derived spacers (Fig. 2D). Mapping of the reads showed that, similarly to the new spacers generated from wild-type ΦNM4γ4, acquired spacers from ΦNM4γ4174-seed cluster around the spc174 target (Fig. 2E). Although qualitatively similar, spacer acquisition from wild-type ΦNM4γ4 and ΦNM4γ4174-seed are quantitatively different, with approximately a 10-fold increase in the acquisition frequency during infections with the wild-type phage. The pattern of spacer capture from the ΦNM4γ4174-PAM phage in the presence of spc174, however, is indistinguishable from the pattern of a no spacer control (Fig. 2F); i.e., is not enhanced by the presence of a matching spacer.
Figure 2. PAM, but not seed, target mutations, abrogate new spacer acquisition in immune cells.
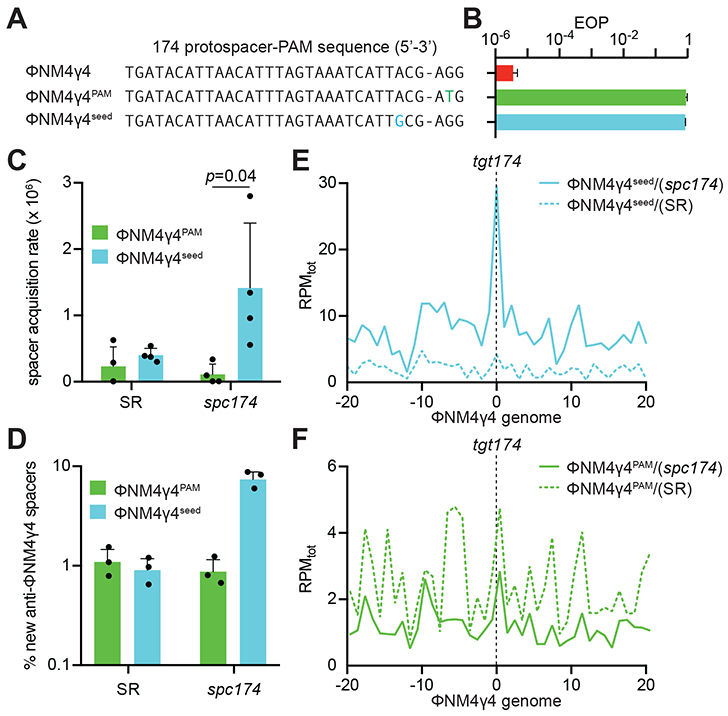
(A) ΦNM4γ4 variants containing different tgt174 sequences: wild-type, PAM (AGG>ATG) and seed (A−3>G−3). (B) Propagation ability of the three ΦNM4γ4 variants described in (A) on staphylococci harboring pCRISPR(spc174), measured as efficiency of plaquing (EOP). Mean ± StDev values of three independent experiments are shown. (C) Spacer acquisition rate after infection with different ΦNM4γ4 phages containing mutations in either the PAM or seed of tgt174, measured as the fraction of cells [harboring either pCRISPR(spc174) or pCRISPR(SR)] that survive infection through the acquisition of a new spacer. Mean ± StDev values of three independent experiments are shown. (D) Fraction (%) of spacer sequences matching to the genome of phage ΦNM4γ4 detected in the PCR product of the expanded CRISPR array of staphylococci harboring either pCRISPR(spc174) or pCRISPR(SR) after infection with different ΦNM4γ4 phages containing mutations in either the PAM or seed of tgt174. Mean ± StDev values of three independent experiments are shown. (E) Distribution of new spacer sequences detected in (D) in cells harboring pCRISPR(spc174), measured as spacer reads per million of total reads (RPMtot), and mapped to 1 kb bins of the ΦNM4γ4 genome (shown in linear form, with tgt174 in the center). Average curve of three independent experiments is shown. (F) Same as (E), but after analysis of bacteria harboring pCRISPR(SR). See also Figs. S2 and S3.
Target orientation does not affect the pattern of spacer acquisition
Our findings resemble the “priming” effect of pre-existing spacers observed during the type I CRISPR-Cas immune response (Datsenko et al., 2012). Similarly to our results, the presence of a pre-existing spacer enhances the rate of acquisition of new spacers, which tend to cluster in the vicinity of the protospacer sequence (Staals et al., 2016). An outstanding feature of type I priming is the orientation bias of the acquired sequences: most of the protospacers that correspond to the new spacers are either located in the same DNA strand as the priming protospacer (defined as the strand that anneals to the crRNA) (Datsenko et al., 2012; Swarts et al., 2012) or in opposite strands at the 5’ or 3’ of the target site (Staals et al., 2016; Westra et al., 2015). Therefore we decided to check whether type II priming displays a similar orientation bias. Analysis of the new spacers acquired as a result of spc174 priming showed a marked strand bias, both during targeting (infection with wild-type ΦNM4γ4, Fig. S4A) and non-targeting (infection with ΦNM4γ4174-seed, Fig. S4B) conditions. If there is orientation bias as in type I priming, relocating the target of spc174 on the opposite strand would reverse the observed strand bias. We unsuccessfully attempted to introduce the spc174 protospacer in the opposite strand of ΦNM4γ4, most likely because the resulting phages are not viable (data not shown). Therefore we decided to investigate the orientation bias during priming by spc256 (Fig. 1E), which targets a non-essential region of ΦNM4γ4 that can be reversed without loss of viral titers (Fig. 3A-B). This spacer targets a protospacer located in the bottom strand of the ΦNM4γ4 genome. Similarly to spc174, spc256 priming showed a strong strand bias, with most new spacers acquired having matching protospacers in the bottom strand (Fig. 3C). When the target was relocated to the top strand in the mutant phage ΦNM4γ4256rev, however, the newly acquired spacers still matched bottom-strand protospacers (Fig. 3D). Therefore, as opposed to priming in type I CRISPR-Cas systems, type II priming lacks an orientation bias.
Figure 3. Target orientation does not affect the pattern of spacer acquisition.
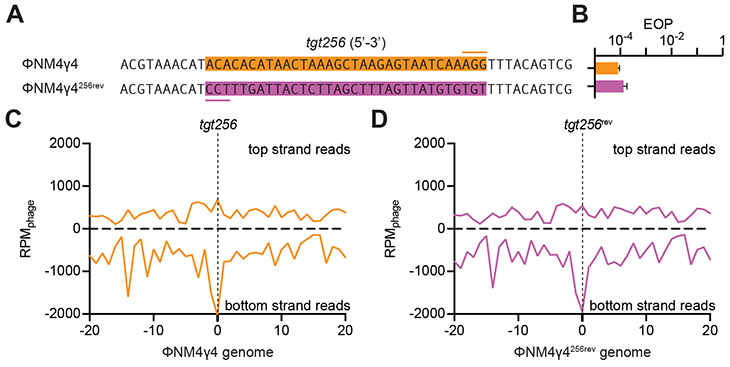
(A) The sequence of tgt256 on the ΦNM4γ4 genome (orange box, the line marks the PAM) was reversed in the ΦNM4γ4256rev mutant virus to allow annealing of the crRNA generated by spc256 to the other strand of the phage genome. (B) Comparison of the efficiency of plaquing (EOP) of ΦNM4γ4 on staphylococci carrying pCRISPR(spc256) of both phages shown in (A). Mean ± StDev values of three independent experiments are shown. (C) Distribution of new spacer sequences acquired after infection of staphylococci carrying pCRISPR(spc256) with ΦNM4γ4, measured as spacer reads per million of total phage reads (RPMphage), and mapped to 1 kb bins of either the top or bottom strands of the phage genome (shown in linear form, with tgt256 in the center). Average curve of three independent experiments is shown. (D) Same as (C) but after infection with ΦNM4γ4256rev. See also Fig. S4.
Target DNA cleavage is required for type II primed spacer acquisition
The distribution of the new spacers acquired through type II priming, narrowly centered at the target site of the priming spacer, suggests that Cas9 binding and/or cleavage of the protospacer are important for the process. To investigate this, we first tested the ability of spc174 to direct the cleavage of the different targets found in ΦNM4γ4, ΦNM4γ4174-seed and ΦNM4γ4174-PAM phages. Biochemical characterization of S. pyogenes Cas9 has shown that binding of the PAM nucleotides is the fundamental first step in target recognition, which then proceeds to the critical pairing of the protospacer DNA and crRNA seed sequences (Sternberg et al., 2014). As a result of this mechanism, PAM mutations that prevent its recognition completely abrogate target cleavage, whereas mismatches within the seed region can lead to low levels of nuclease activity. Indeed, when we tested the cleavage of the different spc174 targets in vitro we found strong cleavage of the wild-type target, lower levels of cleavage of the 174-seed target, and no Cas9 nuclease activity against the 174-PAM target DNA (Figs. 4A-B and S4C). We also tested cleavage of phage DNA in vivo during infection. Cells containing the pCRISPR(spc174) or pCRISPR(SR) plasmids where infected with ΦNM4γ4, ΦNM4γ4174-seed or ΦNM4γ4174-PAM phages, total DNA extracted after 20 minutes and treated with terminal deoxynucleotidyl transferase (TdT) and deoxycytosine to add poly-dC extensions to the 3’ ends of DNA breaks (Fig. S4D). The modified DNA was used as template for amplification with a polyG primer and a second primer annealing upstream of the spc174 target sequence to detect Cas9 cleavage of the phage genome as a PCR product. Whereas no amplification was detected in the non-targeting control samples, strong and weak products were observed for targeting cells infected with wild-type ΦNM4γ4 and ΦNM4γ4174-seed phages, respectively (Fig. 4C). No PCR product was detected for targeting cells infected with mutant phages lacking a functional PAM in the spc174 target. Cloning and sequencing of these PCR products confirmed the specificity of the cleavage (data not shown). Therefore both in vitro and in vivo experiments, as expected, showed a strong cleavage of the wild-type spc174 target, partial cleavage of the target with the seed mutation, and no cleavage of the target with the non-functional PAM. These different levels of Cas9 cleavage are correlated with the level of priming mediated by spc174, as spacer acquisition was highest during infection with wild-type ΦNM4γ4, intermediate with ΦNM4γ4174-seed and very low with ΦNM4γ4174-PAM (Figs. 1B and 2D). As mentioned above, this could be due to a requirement for binding and/or cleavage of the target site. To determine if binding alone can induce type II primed spacer acquisition, we performed short-term infection experiments in staphylococci expressing catalytically dead Cas9 (dCas9), which can bind but not cleave the target specified by the crRNA guide (Bikard et al., 2013; Qi et al., 2013), and has been previously shown to be capable of participating in naïve spacer acquisition (Heler et al., 2015; Wei et al., 2015b). Next-generation sequencing analysis of the acquired spacers in the presence of spc174 targeting showed the reduction of the peak located in the target site to the same background levels observed in the absence of targeting, during infection with both wild-type ΦNM4γ4 (Fig. 4D) and ΦNM4γ4174-seed (Fig. 4E) phages. These data demonstrate that target cleavage is fundamental for type II CRISPR-Cas primed spacer acquisition.
Figure 4. Target DNA cleavage is required for type II primed spacer acquisition.
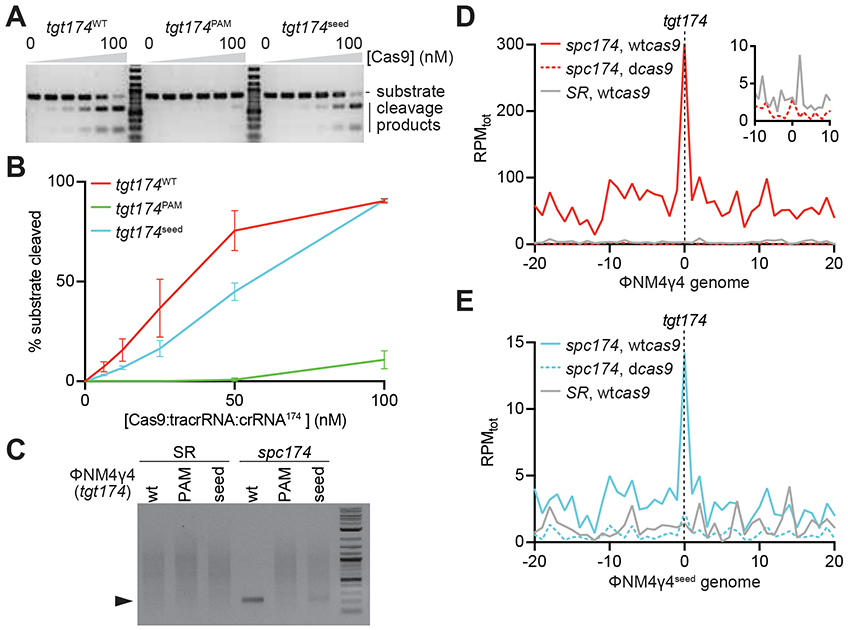
(A) In vitro cleavage assay of a dsDNA oligonucleotide containing the different tgt174 sequences shown in Fig. 2A, incubated with increasing concentrations of a 1:1:1 mix of Cas9:tracrRNA:crRNA174: 0, 6.25, 12.5, 25, 50 and 100 nM. Substrates and cleavage products were separated by agarose gel electrophoresis. A representative image of three replicates is shown. (B) The bands corresponding to the cleavage products in (A) were quantified and plotted against the concentration of the Cas9 ribonucleoprotein complex. Mean ± StDev values of three independent experiments are shown. (C) In vivo cleavage of phage DNA. Cells containing the pCRISPR(spc174) or pCRISPR(SR) plasmids where infected with ΦNM4γ4, ΦNM4γ4174-seed or ΦNM4γ4174-PAM phages and the cleavage products (marked by the black triangle) of the tgt174 sequence were amplified and separated by agarose gel electrophoresis. (D) Distribution of new spacer sequences acquired after infection of staphylococci carrying either pCRISPR(spc174, wtcas9), pCRISPR(spc174, dcas9) or pCRISPR(SR), with ΦNM4γ4, measured as spacer reads per million of total reads (RPMtot), and mapped to 1 kb bins of either the top or bottom strands of the phage genome (shown in linear form, with tgt174 in the center). Average curve of two independent experiments is shown. The inset shows the data from cells harboring pCRISPR(spc174, dcas9) or pCRISPR(SR) around the target site, with a different RPMtot scale. (E) Same as (D) but after infection with ΦNM4γ4seed. See also Fig. S4.
Cleavage-mediated spacer acquisition in Streptococcus thermophilus
In order to confirm our results in a natural experimental system, i.e. as opposed to the heterologous plasmid-based system described above, we explored cleavage-mediated spacer acquisition in Streptococcus thermophilus, the first organism shown to employ CRISPR-Cas systems for anti-phage defense (Barrangou et al., 2007). Strain DGCC7710 harbors two type II-A CRISPR loci (CRISPR1 and CRISPR3) that function independently from one another (Carte et al., 2014) and both are capable of spacer acquisition (Barrangou et al., 2007; Wei et al., 2015a). To eliminate the possibility of competition between these two CRISPR loci for the same substrates during spacer acquisition that might complicate interpretation of our results, we used a mutant containing only CRISPR3 (Fig. S1A), strain CRISPR3-naïve (Varble et al., 2019), since this locus encodes the closest relative to the S. pyogenes SF370 type II-A system used in our previous experiments (Fonfara et al., 2014). We infected CRISPR3-naïve cells with the phage Φ2972 (Fig. 5A) in soft agar to generate ‘bacteriophage-insenstive mutants’ (BIMs) colonies as previously described (Hynes et al., 2017). The resulting BIMs were screened for spacer acquisition by PCR and three of these colonies containing a single, unique spacer targeting Φ2972 were saved for subsequent experiments (CRISPR3α, CRISPR3β, and CRISPR3γ, Fig. 5B). First we confirmed the targeting, and thus cleavage, properties of each spacer (spc-α, spc-β, and spc-γ, Fig. 5A and Supplementary Data File 2) by measuring the ability of Φ2972 to form plaques in lawns of CRISPR-adapted streptococci (Fig. 5C). We then measured cleavage-mediated spacer acquisition after infection of liquid cultures with Φ2972 at a MOI of 10 for thirty minutes. Cells were collected for genomic DNA extraction, amplification of the CRISPR3 locus and NGS of the obtained PCR product. When analyzed quantitatively, sequencing data showed a markedly higher number of new anti-viral spacers for the three targeting strains relative to the CRISPR3-naïve control (~ 80-fold increase on average, Fig. 5D). Finally, qualitative analysis of the distribution pattern of the new spacers revealed a clustering around their respective Cas9 target sites (Fig. 5E). Altogether, these results demonstrate the existence of cleavage-mediated spacer acquisition in the CRISPR3 locus of S. thermophilus, and also show that it shares similar mechanistic features with the S. pyogenes SF370 heterologous system.
Figure 5. Cleavage-dependent spacer acquisition in Streptococcus thermophilus.

(A) Streptococcal phage Φ2972 genome and the Cas9 targets analyzed in this study. (B) Agarose gel electrophoresis of PCR products after amplification of the CRISPR3 array from S. thermophilus CRISPR3-naïve that acquired spacers α, β and γ upon infection with Φ2972. Grey and black arrows: non-expanded and expanded, respectively, CRISPR3 arrays. (C) Comparison of the efficiency of plaquing (EOP) of phage Φ2972 on CRISPR3-α, -β or -γ streptococci. Mean ± StDev values of three independent experiments are shown. (D) Fraction (%) of new spacer sequences matching to the genome of phage Φ2972 detected after infection of CRISPR3-α, -β or -γ streptococci. (−) indicates new spacers acquired in the absence of a targeting spacer by CRISPR-naïve cells. Values for a single experiment are shown. (E) Distribution of new spacer sequences detected 30 minutes after infection of streptococci expressing Cas9 programmed to cleave the targets shown in (A). Reads per million of phage reads (RPMphage) are mapped to 1 kb bins of the Φ2972 genome. Values for a single experiment are shown.
Type II primed spacer acquisition protects against the rise of escapers
A hallmark of CRISPR immunity is the generation of a heterogenous bacterial population with thousands of different spacer sequences, each of them in a different cell (Barrangou et al., 2007; Heler et al., 2015; Paez-Espino et al., 2013). This prevents the rise of phage escapers containing mutations that can avoid the immunity mediated by one of the acquired spacers (van Houte et al., 2016). These phages can kill the cells in the population harboring that particular spacer but will be eliminated upon infection of other host cells that contain different spacers for which they do not have escape mutations. In addition, the expansion of the spacer repertoire defends the host population from infection with related phages that share target sequences with the previous phage that triggered spacer acquisition. Therefore, the enhancement of spacer acquisition promoted by Cas9 cleavage should mediate a more robust type II-A CRISPR-Cas immune response. On one hand, the cleavage of targets with seed escape mutations would allow the host to contain the rise of such escapers after they appear in the population. On the other, the acquisition of additional spacers that occurs after cleavage of perfect targets within immune hosts would enable to anticipate the occurrence of phage (seed and PAM) escapers or related phages; i.e. it would equip the host with a different spacer sequence for defense against phages that cannot be cleaved by the first spacer.
To investigate these scenarios, we first determined whether primed spacer acquisition occurs during infection of staphylococci harboring a naïve pCRISPR, without spacers matching ΦNM4γ4; i.e whether cleavage-mediated spacer acquisition can happen after naïve cells capture their first spacers. We infected naïve cells with ΦNM4γ4 at a multiplicity of infection (MOI) of 1 and allowed the cultures to grow in the presence of phage for 24 hours before amplifying the CRISPR arrays and submitting the resulting PCR products to next-generation sequencing. We then selected reads that contained two new anti-viral spacers and calculated the genomic distance between the first and second acquired sequences (located at the 5’ end of the array (Barrangou et al., 2007; McGinn and Marraffini, 2016)). Consistent with a previous bioinformatic analysis (Nicholson et al., 2018), the histogram of these distances revealed that the majority of the second spacers map within 1 kb of the protospacer targeted by the first spacer (Figs. 6A and S5A-E), without any noticeable strand bias (Fig. S6A). This spacer distribution is similar to that obtained in the spc174 priming experiments (Figs. 1C and 2E), and thus the data strongly suggests primed spacer acquisition follows the naïve type II-A CRISPR-Cas immune response. To determine if primed spacer acquisition can limit the propagation of phage escapers, we selected colonies that acquired a second spacer after priming with spc174 (Fig. S3C) and infected them with wild-type ΦNM4γ4 phage. Whereas the phage population contained a significant number of mutants that were able to propagate in staphylococci equipped with pCRISPR(spc174), we were unable to detect plaques on cultures harboring an additional spacer acquired through priming (Fig. S6B).
Figure 6. Cleavage-dependent spacer acquisition mediates a robust immune response.

(A) Distance between the targets in the ΦNM4γ4 genome specified by the first and second spacers acquired after infection of naïve staphylococci carrying the type II-A CRISPR-Cas system of S. pyogenes with ΦNM4γ4. The number of different spacers within 1-kb bins of the ΦNM4γ4 genome are shown; the position of first spacer acquired in each array is set as 0 kb. (B) Cell survival measured as OD600 after infection of cultures carrying pCRISPR(spc174) with different phages at MOI 10 or none as a control. The average curves of three different replicates are shown, with +/− StDev values shown in lighter colors. (C) Agarose gel electrophoresis of PCR products after amplification of the CRISPR array of cells obtained after the experiment in (B) to detect the integration of new spacers. Grey and black arrows: non-expanded and expanded, respectively, CRISPR arrays. (D) Cell survival measured as OD600 after infection with ΦNM4γ4 at MOI 100 of staphylococci carrying different plasmids. The average curves of three different replicates are shown, with +/− StDev values shown in lighter colors. (E) Same as (C) but for the cells obtained after the experiment in (D). (F) Cell survival measured as OD600 after infection of cultures carrying pCRISPR(spc174) with different phages at MOI 1 or none as a control. “ΦNM4γ4/ΦNM1” indicates infection with ΦNM1γ6PAM 30 minutes of addition of ΦNM4γ4 at MOI 10. “ΦNM1” is an abbreviation for ΦNM1γ6PAM. The average curves of three different replicates are shown, with +/− StDev values shown in lighter colors. (G) Same as (C) but for the cells obtained after the experiment in (F). See also Figs. S5 and S6.
We then tested our first prediction; i.e. that Cas9 partial cleavage would promote the acquisition of new spacers to neutralize seed escaper phages. We infected exponentially-growing staphylococci harboring pCRISPR(spc174) with ΦNM4γ4, ΦNM4γ4174-seed or ΦNM4γ4174-PAM phages (or a mock infection as a control) at a MOI of 10 and we measured the optical density of the culture to monitor cell survival (Fig. 6B). Due to spc174 targeting, cultures infected with wild-type ΦNM4γ4 continue growing, similarly to the control where no phage was added. In contrast, cultures infected with both escaper phages succumbed to infection and stopped growing, and the cells that received ΦNM4γ4174-PAM were not able to recover. Cells infected with ΦNM4γ4174-seed, however, regained growth at 13 hours post-infection. PCR analysis of the three replicas showed that in all cases a second new spacer was acquired (Fig. 6C). Spacer acquisition was absolutely required for the recovery of the culture, as cells expressing the catalytically dead version of Cas1 (dCas1) were incapable of recovery (Figs. S6C-D). In addition, the spacer acquisition that enabled the survival was primed by spc174, since cells harboring pCRISPR(SR) failed to regrow (Figs. S6E-F).
Finally, we tested the second prediction; that Cas9 cleavage within fully immune hosts prepares the cell for future escaper or related phages. First, we infected cells carrying pCRISPR(spc174) with ΦNM4γ4 at a high MOI (100), which increases the concentration of escapers present in the experiment. After the initial decimation of the culture, presumably by escaper phages, resistant cells that acquired new spacers were able to recover (Figs. 6D-E). Cells harboring pCRISPR(SR), lacking the priming spacer, or pCas9(spc174), lacking the Cas1-Cas2-Csn2 acquisition machinery, both failed to regrow (Fig. 6D) or acquire new spacers (Fig. 6E), demonstrating that the survival of staphylococci carrying pCRISPR(spc174) depended on the expansion of the spacer repertoire mediated by spc174-mediated cleavage of wild-type viruses. Second, we infected cells carrying pCRISPR(spc174) with ΦNM4γ4 for 30 minutes and then added the related phage ΦNM1γ6PAM, which shares 73.5 % of sequence identity with ΦNM4γ4 and contains a PAM mutation in tgt174 that prevents Cas9 cleavage. As controls, we also infected staphylococci with either ΦNM4γ4 or ΦNM1γ6PAM alone. Analysis of data from Figures 2B and 2C indicated that, from the 346 different spacer sequences acquired during the first 30 minutes of infection with ΦNM4γ4 in all three replicates, 343 have perfect matches on the ΦNM1γ6PAM genome, and the remaining 3 have imperfect matches that could still enable some level of Cas9 targeting (Fig. S6G and Supplementary Data File 1). Therefore, it is expected that staphylococci carrying pCRISPR(spc174) will use the targeting spacer not only to destroy ΦNM4γ4, but also to prepare the host population with spacers against ΦNM1γ6PAM and thus ensure survival after infection with this phage. As expected, the culture resisted infection with ΦNM4γ4 but not with ΦNM1γ6PAM (Fig. 6F). The latter eventually recovered after about 17 hours, due to the naïve acquisition of spacers (Fig. 6G). In contrast, the culture pre-infected with ΦNM4γ4 recovered from infection with ΦNM1γ6PAM significantly faster, at about 13 hours (Fig. 6F) through the acquisition of new spacers (Fig. 6G). Next generation sequencing of the new spacers showed that, in addition to spacers acquired from the ΦNM1γ6PAM genome, all 346 spacer sequences that were acquired from the ΦNM4γ4 genome after 30 minutes of infection were present at least in one replicate population that survived infection by both phages for 24 hours, including the three spacers that partially match the ΦNM1γ6PAM genome (Supplementary Data File 1). The presence of these sequences, resulting from spacer acquisition from ΦNM4γ4 DNA following cleavage of this phage by Cas9 loaded with the spc174 crRNA guide, and which are not required to provide immunity against this ΦNM4γ4, demonstrates that these spacers indeed can be used for the targeting of ΦNM1γ6PAM. Altogether, the results presented in Figure 6 demonstrate that primed spacer acquisition occurs during the type II-A CRISPR-Cas immune response, enabling the containment of seed escaper phages. More importantly, priming within already-immune cells allows a pre-emptive strike against future infection by escaper phages and/or related viruses.
DISCUSSION
Our results demonstrate that the presence of pre-existing spacers enhances further spacer acquisition during the type II-A CRISPR-Cas immune response through a mechanism that requires the cleavage of the target DNA and that results in the capture of sequences in the vicinity of the cut site. This phenomenon was observed for both an heterologous S. pyogenes SF370 and the native CRISPR3 locus of S. thermophilus DGCC7710, suggesting that it is a common feature of type 2 CRISPR systems.
As a result of this mechanism, the abundance of the newly acquired spacers is determined by the efficiency of cleavage mediated by the preexisting priming spacer. Spacers with fully functional targets lead to the highest level of spacer acquisition, spacers that target a protospacer with seed mutations promote an intermediate level of priming and those that target a site with a non-functional PAM do not enhance spacer acquisition. We believe that the strict correlation between target cleavage and primed spacer acquisition is a result of the mechanism behind type II-A spacer capture, which displays a marked preference for sequences near free double-stranded DNA (dsDNA) ends (Modell et al., 2017). In this model, the dsDNA ends generated by Cas9 cleavage become substrates for spacer acquisition. The previously reported physical association between Cas9, Csn2 and the Cas1-Cas2 integrase of the type II-A systems (Heler et al., 2015; Wilkinson et al., 2019), could provide an efficient avenue for the recruitment of the spacer acquisition machinery to the free dsDNA ends generated by Cas9. A previous study showed that when the dsDNA end is either the cos site injected by a phage or the product of DNA restriction, processing by the AddAB nuclease extends the region of spacer acquisition to the first chi site capable of inhibit AddAB (Modell et al., 2017), presumably by generating degradation products with new dsDNA ends for the spacer acquisition machinery. In AddAB mutants, however, the region of acquisition is narrowed and centered around the dsDNA end, lacking the expansion to the chi sites. Interestingly, our data shows an acquisition hotspot concentrated at the cleavage site, not limited by chi sites, similar to the spacer acquisition pattern in AddAB mutants (Modell et al., 2017). This suggests that the dsDNA ends produced by Cas9 cannot be processed by AddAB, due to either the tight binding of Cas9 to its cleavage products (Garneau et al., 2010; Sternberg et al., 2014) or the expression of an AddAB inhibitor by ΦNM4γ4 (Bobay et al., 2013). Alternatively, as a result of the close association between the acquisition machinery and Cas9, the Csn2/Cas1-Cas2 complex could have preferential access to the dsDNA ends generated after cleavage and prevent the loading of AddAB to the ends.
Although conceptually similar, our results show important differences in the mechanism of primed spacer acquisition carried out by type I and type II CRISPR-Cas systems. In these systems, the crRNA-guided Cascade complex requires both seed and PAM sequences to recognize a protospacer sequence and recruit the Cas3 nuclease to destroy the target genome by nicking one DNA strand at different length intervals (Dillard et al., 2018; Loeff et al., 2018; Mulepati and Bailey, 2013; Sinkunas et al., 2011). The lack of robust priming against perfect targets has been explained by two hypotheses in the context of this targeting mechanism. Some studies support the idea that, upon interaction with a mutated, but not with a perfect, protospacer, the Cascade complex adopts a “priming” conformation that enables the recruitment of Cas1/Cas2 to initiate spacer acquisition from the target genome (Blosser et al., 2015; Redding et al., 2015). Other work has shown that target cleavage is required for type I priming, but perfect protospacers lead to the rapid degradation of the target genome and prevent Cas1/Cas2 from extracting additional spacers from it (Musharova et al., 2019; Semenova et al., 2016). Indeed, only when an anti-CRISPR Cascade inhibitor is over-expressed in hosts to impair target destruction, high levels of “interference-driven acquisition” can be detected (Staals et al., 2016). Mutated protospacers, on the other hand, lead to the recruitment by Cascade of both Cas3 and Cas1/Cas2, which promote slow degradation of the target genome and primed spacer acquisition, respectively (Musharova et al., 2019; Semenova et al., 2016; Staals et al., 2016). Moreover, in type I-F systems that encode a Cas2-Cas3 fusion protein, Cas1 recruitment to the mutated target site, an essential step for primed acquisition, inhibits the nuclease activity (within Cas3) of the fusion (Rollins et al., 2017), supporting the idea that slow target degradation is important for primed spacer acquisition. In contrast to the results reported for type I priming, where acquisition mediated by targeted protospacers can only be detected when target degradation is artificially decreased, we observed the highest levels of type II primed spacer acquisition in the presence of a perfect protospacer that can be rapidly cleaved. We believe that this difference could be related to the lack of processive degradation after target cleavage by Cas9 compared with Cas3 (Sternberg et al., 2014); i.e. the rapid degradation of DNA containing a perfect target for Cas3 prevents the acquisition of new spacers from sequences that surround the target site. Alternatively, even if the Cas1/Cas2 complex interacts with both the Cas3 and Cas9 nucleases, the integration substrates (also known as pre-spacers) could be more immediately accessible to the integrase complex in the vicinity of the target site after Cas9 cleavage versus Cas3 nicking.
Another difference is the influence of the protospacer location in the orientation of the new spacers. Whereas during type I priming the acquired spacers match the same strand as the priming protospacer (Datsenko et al., 2012; Staals et al., 2016; Swarts et al., 2012), this is not the case for the spacers acquired through type II primed spacer acquisition. This dissimilitude could be related to the disparities in DNA cleavage between Cas3 and Cas9. The recognition of a mutated protospacer by Cascade leads to the recruitment of Cas3 and the Cas1/Cas2 integrase to the target DNA. This priming complex moves unidirectionally along one of the DNA strands (Dillard et al., 2018; Redding et al., 2015), a process that could cause the observed polarity of spacer acquisition during type I priming. In contrast, Cas9 remains bound to the cleaved DNA ends (Sternberg et al., 2014); i.e. the lack of strand-specific or directional movement of this nuclease prevents any influence on the extraction of spacers by Cas1/Cas2.
Primed spacer acquisition has clear advantages for the bacterial population: it increases the diversity of spacer sequences to prevent the raise of escaper phages (van Houte et al., 2016) and to generate new immunity against related viruses. In type I systems, the presence of a target mutation would trigger additional spacer acquisition from other regions of the escaper or related phage to neutralize these threats. In type II systems, however, given the strong priming effect of fully targeting spacers, spacer diversity is generated not only after, but also before the cell is infected with an escaper or a related phage. This is a unique feature of type II primed spacer acquisition that allows “preventive” immunity against future threats that could overcome the defense provided by the initial spacer acquired. We believe that cleavage-mediated spacer acquisition is an integral part of the type II-A CRISPR-Cas immune response that immediately follows the acquisition of the first spacers by naïve cells (an argument supported by the data in Figure 6A) and leads to a more rapid increase in the diversity and abundance of spacers in the population that limits the rise of escapers. Phage escapers, however, are only one of the many threats that bacteria face in their natural habitats. How cleavage-mediated spacer acquisition impacts the dynamics of the virus-host co-evolution in ecological contexts remains largely unknown. In Figure 6F we explored conditions in which the host is exposed to two phages and showed that this mechanism can immunize the host against both infections if the viral genomes are related. In more complex environments, the number of co-existing phages, their genetic divergence and their relative frequencies will most likely affect the benefits of type II priming. Finally, since the strength of the immunity provided by type II spacers decreases as their positions within the CRISPR array upon new integration events (McGinn and Marraffini, 2016), the accelerated acquisition mediated by Cas9 cleavage will also “downgrade” the previously acquired spacers. How this potential cost of priming affects the overall type II CRISPR-Cas immune response is not known.
Finally, the wide use of Cas9 as a tool for genome engineering of the human genome (Doudna and Charpentier, 2014) have revealed the existence of “off-target” sites. These are not simply mutated targets, but sequences with random and imperfect complementarity to the crRNA guide of Cas9 that can nevertheless be recognized and cleaved by the nuclease at low rates (Fu et al., 2013; Hsu et al., 2013). Limiting Cas9 off-target effects represents a fundamental obstacle for the continued improvement of CRISPR-Cas9 biotechnology. According to our results, these off-target cleavage events should lead to primed spacer acquisition, and therefore it is interesting to speculate that this “defect” of Cas9 targeting has been selected through evolution to boost the otherwise naïve type II CRISPR-Cas immune response and to achieve immunization against phages for which a priori there are no partially matching pre-existing spacers.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Luciano A. Marraffini (marraffini@rockefeller.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and growth conditions
Cultivation of S. aureus RN4220 (Kreiswirth et al., 1983) was carried out in brain-heart infusion (BHI) broth medium or tryptic soy broth (TSB) at 37°C with shaking. Wherever applicable, media were supplemented with chloramphenicol at 10 μg/ml and or erythromycin at 10 μg/ml to ensure pC194-derived and pE194-derived plasmid maintenance respectively. Cultivation of S. thermophilus DG7710 (Horvath et al., 2008) was carried out in M17 broth supplemented with 10% lactose (LM17) at 42 °C without shaking. Wherever applicable, media were supplemented with 200 μg/ml spectinomycin. Strains and plasmids used in this study are listed in Supplementary Data File 2.
METHOD DETAILS
Plasmid Construction
All cloning was performed with electrocompetent S. aureus RN4220 cells as described previously (Goldberg et al., 2014). The sequences and oligonucleotides used in this study are listed in Supplementary Data File 2. To construct pPN86, pE194 was amplified with oPN283 and oPN284, pGG32 (Goldberg et al., 2014) was amplified with oPN285 and oPN286, and a two-piece Gibson assembly was preformed. To construct pPN249, pC194 was amplified with oPN548 and oPN549, φNM4γ4 genomic DNA was amplified with oPN550 and oPN551, and a two-piece Gibson assembly was preformed. pPN250 was then constructed by amplifying pPN249 with AV186 and oPN565 as well as AV187 and oPN564 and a two-piece Gibson assembly was preformed. pPN23 was constructed by amplifying pJM62 (Modell et al., 2017) with H237 and H238 (Heler et al., 2015) and preforming a one-piece Gibson assembly. BsaI cloning was used to make pPN183 by inserting spacer174 (annealed primers RH486-RH487) into the BsaI site of pPN23. pPN77 was constructed by amplifying pJM62 with JM110 and JM115 (McGinn and Marraffini, 2016) and preforming a one piece Gibson assembly. pPN91 then was constructed in a two-piece Gibson by amplifying pPN77 with JM90 and oPN280 as well as JM91 and oPN279 and a two-piece Gibson assembly was preformed. BsaI cloning was used to make pPN174 and pPN256 by inserting spacer174 (annealed primers RH486-RH487) and spacer256 (annealed oPN91-oPN92) into the BsaI site of pPN91. The E220A mutation was introduced into pPN174 to generate pPN285 by amplifying pPN174 with PS285 and H293 as well as PS284 and H294 and then preforming a two-piece Gibson assembly as described previously (Heler et al., 2015). pPN136 was constructed by amplying pGG32 (Heler et al., 2015) with JM91 and oGG140 and JM90 and JM115 and preforming a two piece Gibson assembly (McGinn and Marraffini, 2016). pPN290 was constructed by replacing the wild-type Cas9 with the nuclease-dead form of Cas9 (D10A, H840A mutations) by amplying pPN91 with H294 and H295 (Heler et al., 2015), amplyfing pDB182 (Bikard et al., 2014) with H293 and H296 (Heler et al., 2015), and preforming a two-piece Gibson assembly. pPN294 was then made by inserting spacer174 (annealed primers RH486-RH487) into the BsaI site of pPN290. pPN92 was constructed by amplifying pJM62 (Modell et al., 2017) with JM90 and oPN280, amplifying pPN77 with JM91 and oPN279, and preforming a two piece Gibson assembly (McGinn and Marraffini, 2016). BsaI cloning was used to make pPN182 by inserting spacer174 (annealed primers RH486-RH487) into the BsaI site of pPN92.
Strain Construction
To create the S. thermophilus strain JAV25 we employed the method described previously (Varble et al., 2019). Briefly, deletions were made by transforming PCR amplicons with 2-kb homology arms flanking an spectinomycin resistance cassette into wild-type strains. The sequences and oligonucleotides used in this study are listed in Supplementary Data File 2. To delete CRISPR1, DG7710 genomic DNA was amplified with AV664 and AV665 as well as AV666-AV667 (Varble et al., 2019) and pLZ12spec55 was amplified with AV672 and AV673 (Varble et al., 2019) followed by a thee-piece Gibson assembly to create the final PCR amplicon for transformation.
Isolation of JAV25 CRISPR BIMs
BIMs derived from JAV25 (CRISPR3-naïve in the text) were isolated using a previously described method (Hynes et al., 2017) with minor modification. Overnight cultures of JAV25 were infected with Φ2972 at an MOI of 0.1, and mixed with with LM17 media soft agar supplemented with 10 mM CaCl2 and then plated LM17 agar plates supplements on 10 mM CaCl2. Plates were incubated at 42 °C overnight after drying at room temperature from 30 minutes. The resulting colonies were then screened for spacer acquisition by TopTaq PCR amplification (Qiagen) with the primers oPN737 with oPN738 and three of these colonies containing a single, unique spacer targeting Φ2972 were saved for subsequent study (CRISPR3α, CRISPR3β, and CRISPR3γ).
Plaque formation assay in S. aureus
Serial dilutions of phage stock were prepared in triplicate, spotted on fresh top agar lawns of RN4220 in BHI agar supplemented with appropriate antibiotic and 5 mM CaCl2. Plates were incubated at 37 °C overnight after drying at room temperature from 30 minutes.
Plaque formation assay in S. thermophilus
Plaque formation of was measured as described previously (Hynes et al., 2017) with minor variations. Briefly, overnight cultures were launched from single colonies, infected with serials dilutions of phage stock Φ2972 were prepared in triplicate, and mixed with with LM17 media soft agar supplemented with 10 mM CaCl2 and then plated LM17 agar plates supplements on 10 mM CaCl2. Plates were incubated at 42 °C overnight after drying at room temperature from 30 minutes.
Quantifying CRISPR phage escapers
Thirty escaper plaques were isolated from soft agar lawns of RN4220 harboring a type II-A CRISPR plasmid targeting φNM4γ4 in BHI soft agar supplemented with the appropriate antibiotic, 5 mM CaCl2, and resuspended in 20 μL BHI. 2 μL of the phage mixture was added to 30 μL of Colony Lysis Buffer (Pyenson et al., 2017) and boiled at 98 °C degrees for 10 minutes. 1 μL of the resulting phage lysate was then used as template for PCR amplification. For escapers of CRISPR174, oPN246 and oPN247 (all oligonucleotides used in this study are listed in Supplementary Data File 2) were used to amplify a portion of φNM4γ4 amidase gene (gp68) (Bae et al., 2006). PCR products were purified with the QIAquick PCR Purification Kit (Qiagen) and submitted for Sanger sequencing.
Isolation of spontaneous CRISPR escaper phage
φNM4γ4PAM, φNM4γ4Seed, and φNM1γ6PAM were isolated from spontaneous escaper plaques following infection of φNM4γ4 on soft agar lawns of S. aureus RN4220 cells carrying pPN174 that encodes a type II-A CRISPR system targeting the amidase gene (gp68) of φNM4γ4 (CRISPR174) (Fig. 2A). PCR and Sanger sequencing of the resulting PCR amplicons confirmed escape mutation in the PAM and seed of the target of CRISPR174 (Fig. S2).
Phage construction
We used a variation of a previously described method (Lemay et al., 2017). φNM4γ4256rev was created by propagating φNM4γ4 liquid culture of cells harboring pPN250 which contains the protospacer and PAM of CRISPR256 flipped onto the opposite strand as well as the surrounding upstream and downstream homology regions for recombination with the φNM4γ4 genome. Recombinant φNM4γ4256rev plaques were isolated on a soft agar lawn of RN4220 pGG14, which encodes a type III-A CRISPR-Cas system targeting the sequence present in φNM4γ4 and not in φNM4γ4256rev. Subsequent PCR and Sanger sequencing of the resulting PCR amplicons confirmed the flipped target.
Spacer Acquisition in S. aureus Liquid Culture
Overnight cultures were launched from single colonies and diluted to an optical density at 600 nm (OD600) of ~0.1 in BHI, 5 mM CaCl2, and the appropriate antibiotics. After 1 hour and ten minutes, optical density (OD600) was measured for each culture, and each sample was normalized to an equal cell density. Cultures were then infected with φNM4γ4, φNM4γ4PAM, φNM4γ4Seed, φNM4γ4256rev, or mock infected at a multiplicity of infection (MOI) of 100 for thirty minutes prior pelleting infected cells, removing the supernatant, and flash freezing the pellets in liquid nitrogen. Frozen pellets were kept at −80°C until ready for DNA extraction. For 24 hour spacer acquisition assay (Figs. 5 and S5), three cultures of pWJ40 prepared as above and each normalized to an OD600 of ~0.5 prior to infection with φNM4γ4 at a MOI of 1 for 24 hours. Infected cultures were pelleted, the supernatant was removed, and immediately prepared for DNA extraction.
Spacer Acquisition in S. thermophilus Liquid Culture
Overnight cultures were launched from single colonies and diluted to an optical density at 600 nm (OD600) of ~0.1 in LM17 with 10 mM CaCl2. After 1 hour and 30 minutes of outgrowth, the optical density (OD600) was measured for each culture, and each sample was normalized to an equal cell density. Cultures were then infected with Φ2972 or mock infected at a multiplicity of infection (MOI) of 10 for thirty minutes prior pelleting infected cells, removing the supernatant, and flash freezing the pellets in liquid nitrogen. Frozen pellets were kept at −80°C until ready for DNA extraction.
PCR amplification of expanded CRISPR loci for high-throughput sequencing in S. aureus
CRISPR plasmids were isolated from RN4220 cells with modified QIAprep Spin Miniprep Kit protocol (Qiagen) as previously described (Modell et al., 2017). We used 200 ng (log phase) of plasmid as input for encrichment PCR of the CRISPR locus with Phusion DNA Polymerase (Thermo) with the following primer mix: oPN287 and cocktail containing an equal mixture of oPN288, oPN289, and oPN290. For 24 hour infection assay, H188 and JM257, JM248 and JM258, and JM249 and JM259 were used to amplify the CRISPR for the three cultures respectively. To differentiate between samples during multiplexed high-throughput sequencing, variants of oPN287 containing randomized 5 nucleotides (NNNNN) followed by 3-6 nucleotide barcode at 5’ end. Amplicons corresponding to the size of expanded CRISPR arrays were gel purified allowing for the removal of unexpanded CRISPR arrays. Purified amplicons were then prepared for sequencing with the TrueSeq Nano DNA Library Prep protocol (Illumina), using a final concentration of 1.36x Sample Purification Beads (Ilumina) following end repair for further size selection, followed by high-throughput sequencing with the MiSeq platform.
PCR amplification of expanded CRISPR loci for high-throughput sequencing in S. thermophilus
Genomic DNA was isolated from DG7710 cells by first treating with modified Wizard Genomic DNA Purification protocol for gram-positive bacteria (Promega): bacterial cell pellets were resuspended in 0.5 M EDTA, pH 8.0 supplemented with 200 μg ml−1 lysozyme (Sigma) and incubated at 37 °C for 25 min prior to pelleting and removing the supernatant. The standard Wizard protocol for gram-positives was then followed as described by the manufacturer (Promega). We used 200 ng (log phase) of plasmid as input for PCR of the CRISPR locus with Phusion DNA Polymerase (Thermo) with the following primer mix: oPN737 with oPN738, oPN757, oPN758, or oPN759 depending on whether JAV25, JAV25-BIM01, JAV25-BIM02, or JAV25-BIM03 was used. To differentiate between samples during multiplexed high-throughput sequencing, variants of oPN737 containing randomized 5 nucleotides (NNNNN) followed by 3-6 nucleotide barcode at 5’ end. Amplicons corresponding to the size of expanded CRISPR arrays were gel purified allowing for the removal of unexpanded CRISPR arrays. Purified amplicons were then prepared for sequencing with the TrueSeq Nano DNA Library Prep protocol (Illumina), using a final concentration of 1.36x Sample Purification Beads (Ilumina) following end repair for further size selection, followed by high-throughput sequencing with the MiSeq platform.
Spacer acquisition in soft agar
Overnight cultures were launched from single colonies and diluted to an optical density at 600 nm (OD600) of ~0.1 in BHI and the appropriate antibiotics. After 1 hour and ten minutes, optical density (OD600) was measured for each culture, and each sample was normalized to an equal cell density. Cultures were then concentrated to a final density of OD600 ~125, infected at with φNM4γ4, φNM4γ4PAM or φNM4γ4Seed at MOI 2.5, mixed with soft BHI soft agar containing 5 mM CaCl2 and then plated BHI agar with the appropriate antibiotics. In order to quantify initial number of cells infected, prior to infection an aliquot of each culture was serially diluted and plated on BHI plates. Following overnight incubation at 37 °C, soft agar plates were photographed and the number of surviving bacteriophage-immune mutant colonies (BIMs) were quantified with ImageJ32. To assay for spacer acquisition, individual colonies were picked and lysed with lysis buffer containing 50 ng/μl lysostaphin (Ambi) as described before (Heler et al., 2015). Following centrifugation (21,000g), the supernatant of each sample was used a template for TopTaq PCR amplification (Qiagen) with the primers oPN479 and oPN292. The resultant PCR amplicons were then analyzed on 2% agarose gels and the percent of BIMs containing expanded arrays (CRISPR BIMs) was determined (Fig. S3). The spacer acquisition rate for each infection was then calculated based on the percent of CRISPR BIMs divided by the initial number of cells infected (Figs. 2C and S3).
In vitro CRISPR-Cas9 cleavage assay
174CRISPR RNA and tracrRNA (IDT cat# 1072532) were purchased from IDT and annealed to form crRNA174:tracrRNA RNA duplexes according Alt-R CRISPR-Cas9: In vitro cleavage of target DNA with ribonucleoprotein complex protocol (IDT). All RNA oligonucleotides used in this study are listed in Supplementary Data File 2.Target DNA substrates were generated by PCR of purified φNM4γ4 genomic DNA with Phusion DNA Polymerase (Thermo) using oPN144 and oPN562 and then purified by gel extraction. Cleavage assays were preformed similarly as described previously (Jinek et al., 2012). Briefly, crRNA:tracrRNA and Cas9 (NEB #MO386) were allowed form RNP complexes at room temperature for 10 minutes and then diluted to a final concentration of 6.25, 12.5, 25, 50 and 100 nM following to the addition of target DNA. All reactions were incubated at 37°C for 5 minutes before the digestion with proteinase K (NEB #P8107) to stop the reactions and liberate target DNA and then stored at −80°C until ready for further analyzed. Samples were visualized on 2% agarose gel with SYBR Gold Nucleic Acid Gel Stain (Invitrogen #S11494) and the abundance of cleavage products quantified by automated electrophoresis and imaging using a Tapestation 4200 (Agilent).
In vivo CRISPR-Cas9 cleavage of viral DNA
To observe CRISPR-Cas9 cleavage of anti-viral targets, overnight culture of RN4220 cells carrying the pC194 (CRISPR-), pPN136 (SR), or pPN174 (CRISPR174) were diluted to an OD600 of ~0.1 with the appropriate antibiotic and 5 mM CaCl2. After 1 hour and ten minutes of growth, optical density (OD600) was measured for each culture, and each sample was normalized to equal cell density. Cultures were then infected at MOI 5 with φNM4γ4, φNM4γ4PAM or φNM4γ4Seed for 20 minutes prior to centrifugation and flash-freezing of cell pellets. All samples were stored at −80°C until ready for genomic DNA (gDNA) purification using the DNeasy Blood and Soft Tissue protocol for Gram-Positive organisms (Qiagen). In order assay for viral gDNA cleavage we preformed modified Anchor PCR by utilizing components of the 5’ RACE System for Rapid Amplification of cDNA Ends Version 2.0 kit (Invitrogen Life Sciences). In brief, 200 ng of purified genomic DNA was incubated with dCTP at final concentration of 200 μM for 2.5 minutes at 94°C degrees to denature dsDNA. The reactions were chilled on ice for 1 minute prior to the addition of recombinant terminal deoxynucleotidyl transferase (TdT) (Invitrogen Life Sciences). Incubation at 37°C for 10 minutes allows for the addition dC-homopolymeric tail to the 3’ ends of dsDNA ends generated by Cas9-mediated cleavage and creates abridged anchor primer (AAP) binding site (Invitrogen Life Sciences). TdT is then heat inactivated at 65°C for 10 minutes. dC-tailed DNA was then amplified with Taq DNA polymerase (Invitrogen Life Sciences) using the AAP and oPN656 upstream of CRISPR174 protospacer. Finally amplicons were visualized on 2% agarose gel and amplicons were purified by gel extraction for Zero Blunt TOPO PCR cloning (Invitrogen Life Sciences).
Bacterial infection growth curves
Viral infections were preformed in a microplate reader as previously described (Goldberg et al., 2014; McGinn and Marraffini, 2016) with minor alterations. Overnight cultures were launched in triplicate from single colonies and diluted to an OD600 of ~0.1 with the appropriate antibiotic and 5 mM CaCl2. After 1 hour and ten minutes of growth, optical density (OD600) was measured for each culture, and each sample was normalized to equal cell density (OD600 ~ 0.4) and loaded into a 96-well plate (Cellstar, 655180). Individual cultures were then infected with φNM4γ4, φNM4γ4PAM or φNM4γ4Seed at an MOI 1. For infections at high MOIs (Fig. 5D), cells were prepared as above but infected at an MOI 100 in bulk culture (5 ml) and the infection were allowed to proceed in the shaking incubator for 30 minutes before an aliquiot of each culture was then loaded into a 96-well plate. Optical density measurements were then taken every 10 minutes for 24 hours in a microplate reader (TECAN Infinite 200 PRO) to generate growth curves. At the end of 24 hours, an aliquot of each culture was lysed in colony lysis buffer contains 50 ng/μl lysostaphin (Ambi) as described before (Heler et al., 2015) and the supernatant was used as template in PCR to assay for spacer acquisition using oPN479 and oPN292 or oPN635 and oPN292. The resulting amplicons were then analyzed on a 2% agarose gel.
Bacterial co-infection growth curve assays
As before, overnight cultures were launched, diluted to an OD600 of ~0.025, and then normalized to equal cell density (OD600 ~ 0.1) prior to infection with φNM4γ4 at a MOI 10. The infections were allowed to proceed in the shaking incubator for 30 minutes before the optical density (OD600) was re-measured. Cultures were then loaded into a 96-well plate and infected with φNM1γ6PAM at MOI 1. Optical density measurements were then taken every 10 minutes for 24 hours in a microplate reader (TECAN Infinite 200 PRO) to generate growth curves. At the end of 24 hours, an aliquot of each culture was lysed in colony lysis buffer contains 50 ng/μl lysostaphin as described before (Heler et al., 2015) and the supernatant was used as template in PCR to assay for spacer acquisition using oPN635 and oPN292. The resulting amplicons were then analyzed on a 2% agarose gel.
QUANTIFICATION AND STASTICAL ANALYSIS
High Throughput Sequencing Data Analysis
Spacers were extracted from raw MiSeq FASTQ files and aligned to reference phage genomes using Python. Spacers that aligned perfectly to each reference genome were assigned to genomic position at the 5’ end of the alignment and reads were aggregated based on each unique spacer sequence. In order to avoid bias introduced into the data set due to duplication of the CRISPR array, reads containing expanded CRISPR arrays comprising of the original priming spacer (spc174, spc256, spc300, spc303, spc305, spcα, spcβ , or spcγ) were discarded. Following aggregation, the flanking protospacer adjacent motif (PAM) and strand for each spacer was determined based on the reference genome. To account sequencing bias introduced as result of enrichment PCR, all spacer read counts were normalized as described previously (Modell et al., 2017). When conventional spacers primers were used to amplify CRISPR loci this normalization step was skipped. Each unique spacer was then sorted into 1-kb bins and each bin was divided by the number of 5’-NGG-3’ sequences CRISPR loci derived from S. pyogenes SF370 or 5’-NGGNG-3’ for CRISPR loci derived from S. thermophilus DGCC7710 CRISPR3 within each bin in order to account for observation that spacer acquisition occurs primarily from sequences immediately upstream of PAMs (Heler et al., 2015). Reads per million (RPM) were calculated as RPMtot or RPMphage as desired previously (Modell et al., 2017).
When assessing expanded CRISPR arrays with two anti-viral spacers (Figs. 5A and S5A-E), both spacers were extracted pooled from three liquid cultures, aligned to φNM4γ4 genome, and the genomic position, PAM, and strand were determined for each spacer as before. Each anti-viral spacer pair was then grouped into 1-kb bins based on the distance between their respective positions in the φNM4γ4 genome. The proportion of anti-viral spacer pairs in each bin was then calculated by dividing by the total number of pairs. To examine groups of doubly expanded arrays that all arise from primed acquisition against the same viral target within φNM4γ4 (Figs. S5A-E), CRISPR loci were sorted into groups based on the first acquired spacer in the array and the distance between each anti-viral spacer was calculated.
Growth Curve Analysis
For growth curves in Figure 5 and S6, error bars represent the standard error of four biological replicates
In vitro CRISPR-Cas9 Cleavage Assay Analysis
For the cleavage assay in Figure 2, error bars represent the standard error of 3 biological replicates.
Efficiency of Plaquing Assay Analysis
For EOP assays in Figure 2, 3, and 5, error bars represent the standard error of 3 biological replicates.
DATA AND CODE AVAILABILITY
All the datasets generated during this study are all available and the accession number is SRA:SUB6246845.
Supplementary Material
Supplemantary Data File S1. Next generation sequencing data obtained in this study. Related to all figures.
Supplemantary Data File S2. Oligonucleotides, spacers and strains used in this study. Related to STAR methods.
ACKNOWLEDGEMENTS.
We would like to thank the Rockefeller University Genomics Resource Center for assistance with next generation sequencing experiments. P.N. is supported by an NIH Medical Scientist Training Program grant (T32GM07739). L.A.M. is supported by a Burroughs Wellcome Fund PATH Award, and a NIH Director’s Pioneer Award (DP1GM128184). L.A.M. is an investigator of the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS. L.A.M. is a cofounder and Scientific Advisory Board member of Intellia Therapeutics, and a co-founder of Eligo Biosciences.
REFERENCES
- Bae T, Baba T, Hiramatsu K, and Schneewind O (2006). Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol Microbiol 62, 1035–1047. [DOI] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Bikard D, Euler CW, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X, Fischetti VA, and Marraffini LA (2014). Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol 32, 1146–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, and Marraffini LA (2013). Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41, 7429–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blosser TR, Loeff L, Westra ER, Vlot M, Kunne T, Sobota M, Dekker C, Brouns SJ, and Joo C (2015). Two Distinct DNA Binding Modes Guide Dual Roles of a CRISPR-Cas Protein Complex. Mol Cell 58, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobay LM, Touchon M, and Rocha EP (2013). Manipulating or superseding host recombination functions: a dilemma that shapes phage evolvability. PLoS Genet 9, e1003825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Sorokin A, and Ehrlich SD (2005). Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561. [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carte J, Christopher RT, Smith JT, Olson S, Barrangou R, Moineau S, Glover CV 3rd, Graveley BR, Terns RM, and Terns MP (2014). The three major types of CRISPR-Cas systems function independently in CRISPR RNA biogenesis in Streptococcus thermophilus. Mol Microbiol 93, 98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabas H, Lion S, Nicot A, Meaden S, van Houte S, Moineau S, Wahl LM, Westra ER, and Gandon S (2018). Evolutionary emergence of infectious diseases in heterogeneous host populations. PLoS Biol 16, e2006738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, and Semenova E (2012). Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun 3, 945. [DOI] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, and Charpentier E (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, and Moineau S (2008). Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190, 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillard KE, Brown MW, Johnson NV, Xiao Y, Dolan A, Hernandez E, Dahlhauser SD, Kim Y, Myler LR, Anslyn EV, et al. (2018). Assembly and Translocation of a CRISPR-Cas Primed Acquisition Complex. Cell 175, 934–946 e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, and Charpentier E (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096. [DOI] [PubMed] [Google Scholar]
- Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lecrivain AL, Bzdrenga J, Koonin EV, and Charpentier E (2014). Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res 42, 2577–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, and Sander JD (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, and Moineau S (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. [DOI] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, and Siksnys V (2012). Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109, E2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg GW, Jiang W, Bikard D, and Marraffini LA (2014). Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature 514, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale C, Kleppe K, Terns RM, and Terns MP (2008). Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA 14, 2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, and Marraffini LA (2015). Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature 519, 199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heler R, Wright AV, Vucelja M, Doudna JA, and Marraffini LA (2019). Spacer Acquisition Rates Determine the Immunological Diversity of the Type II CRISPR-Cas Immune Response. Cell Host Microbe 25, 242–249 e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Romero DA, Coute-Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, and Barrangou R (2008). Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol 190, 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes AP, Lemay ML, Trudel L, Deveau H, Frenette M, Tremblay DM, and Moineau S (2017). Detecting natural adaptation of the Streptococcus thermophilus CRISPR-Cas systems in research and classroom settings. Nat Protoc 12, 547–565. [DOI] [PubMed] [Google Scholar]
- Jiang F, Taylor DW, Chen JS, Kornfeld JE, Zhou K, Thompson AJ, Nogales E, and Doudna JA (2016). Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jore MM, Lundgren M, van Duijn E, Bultema JB, Westra ER, Waghmare SP, Wiedenheft B, Pul U, Wurm R, Wagner R, et al. (2011). Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat Struct Mol Biol 18, 529–536. [DOI] [PubMed] [Google Scholar]
- Kazlauskiene M, Tamulaitis G, Kostiuk G, Venclovas C, and Siksnys V (2016). Spatiotemporal Control of Type III-A CRISPR-Cas Immunity: Coupling DNA Degradation with the Target RNA Recognition. Mol Cell 62, 295–306. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, and Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, and Novick RP (1983). The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–712. [DOI] [PubMed] [Google Scholar]
- Lemay ML, Tremblay DM, and Moineau S (2017). Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth Biol 6, 1351–1358. [DOI] [PubMed] [Google Scholar]
- Loeff L, Brouns SJJ, and Joo C (2018). Repetitive DNA Reeling by the Cascade-Cas3 Complex in Nucleotide Unwinding Steps. Mol Cell 70, 385–394 e383. [DOI] [PubMed] [Google Scholar]
- Marraffini LA, and Sontheimer EJ (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinn J, and Marraffini LA (2016). CRISPR-Cas systems optimize their immune response by specifying the site of spacer integration. Mol Cell 64, 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell JW, Jiang W, and Marraffini LA (2017). CRISPR-Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature 544, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulepati S, and Bailey S (2013). In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J Biol Chem 288, 22184–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musharova O, Sitnik V, Vlot M, Savitskaya E, Datsenko KA, Krivoy A, Fedorov I, Semenova E, Brouns SJJ, and Severinov K (2019). Systematic analysis of Type I-E Escherichia coli CRISPR-Cas PAM sequences ability to promote interference and primed adaptation. Mol Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson TJ, Jackson SA, Croft BI, Staals RHJ, Fineran PC, and Brown CM (2018). Bioinformatic evidence of widespread priming in type I and II CRISPR-Cas systems. RNA Biol, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Espino D, Morovic W, Sun CL, Thomas BC, Ueda K, Stahl B, Barrangou R, and Banfield JF (2013). Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nature communications 4, 1430. [DOI] [PubMed] [Google Scholar]
- Pyenson NC, Gayvert K, Varble A, Elemento O, and Marraffini LA (2017). Broad Targeting Specificity during Bacterial Type III CRISPR-Cas Immunity Constrains Viral Escape. Cell Host Microbe 22, 343–353 e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, and Lim WA (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding S, Sternberg SH, Marshall M, Gibb B, Bhat P, Guegler CK, Wiedenheft B, Doudna JA, and Greene EC (2015). Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System. Cell 163, 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins MF, Chowdhury S, Carter J, Golden SM, Wilkinson RA, Bondy-Denomy J, Lander GC, and Wiedenheft B (2017). Cas1 and the Csy complex are opposing regulators of Cas2/3 nuclease activity. Proc Natl Acad Sci U S A 114, E5113–E5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, and Marraffini LA (2015). Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell 161, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E, Savitskaya E, Musharova O, Strotskaya A, Vorontsova D, Datsenko KA, Logacheva MD, and Severinov K (2016). Highly efficient primed spacer acquisition from targets destroyed by the Escherichia coli type I-E CRISPR-Cas interfering complex. Proc Natl Acad Sci U S A 113, 7626–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkunas T, Gasiunas G, Fremaux C, Barrangou R, Horvath P, and Siksnys V (2011). Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J 30, 1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staals RH, Jackson SA, Biswas A, Brouns SJ, Brown CM, and Fineran PC (2016). Interference-driven spacer acquisition is dominant over naive and primed adaptation in a native CRISPR-Cas system. Nat Commun 7, 12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, and Doudna JA (2014). DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarts DC, Mosterd C, van Passel MW, and Brouns SJ (2012). CRISPR interference directs strand specific spacer acquisition. PLoS One 7, e35888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Houte S, Ekroth AK, Broniewski JM, Chabas H, Ashby B, Bondy-Denomy J, Gandon S, Boots M, Paterson S, Buckling A, et al. (2016). The diversity-generating benefits of a prokaryotic adaptive immune system. Nature 532, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varble A, Meaden S, Barrangou R, Westra ER, and Marraffini LA (2019). Recombination between phages and CRISPR-cas loci facilitates horizontal gene transfer in staphylococci. Nat Microbiol 4, 956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chesne MT, Terns RM, and Terns MP (2015a). Sequences spanning the leader-repeat junction mediate CRISPR adaptation to phage in Streptococcus thermophilus. Nucleic Acids Res 43, 1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Terns RM, and Terns MP (2015b). Cas9 function and host genome sampling in Type II-A CRISPR-Cas adaptation. Genes Dev 29, 356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, van Erp PB, Kunne T, Wong SP, Staals RH, Seegers CL, Bollen S, Jore MM, Semenova E, Severinov K, et al. (2012). CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell 46, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, van Houte S, Oyesiku-Blakemore S, Makin B, Broniewski JM, Best A, Bondy-Denomy J, Davidson A, Boots M, and Buckling A (2015). Parasite Exposure Drives Selective Evolution of Constitutive versus Inducible Defense. Curr Biol 25, 1043–1049. [DOI] [PubMed] [Google Scholar]
- Wigley DB (2013). Bacterial DNA repair: recent insights into the mechanism of RecBCD, AddAB and AdnAB. Nat Rev Microbiol 11, 9–13. [DOI] [PubMed] [Google Scholar]
- Wilkinson M, Drabavicius G, Silanskas A, Gasiunas G, Siksnys V, and Wigley DB (2019). Structure of the DNA-Bound Spacer Capture Complex of a Type II CRISPR-Cas System. Mol Cell 75, 90–101 e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AV, and Doudna JA (2016). Protecting genome integrity during CRISPR immune adaptation. Nat Struct Mol Biol 23, 876–883. [DOI] [PubMed] [Google Scholar]
Methods references
- Bae T, Baba T, Hiramatsu K, and Schneewind O (2006). Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol Microbiol 62, 1035–1047. [DOI] [PubMed] [Google Scholar]
- Bikard D, Euler CW, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X, Fischetti VA, and Marraffini LA (2014). Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol 32, 1146–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg GW, Jiang W, Bikard D, and Marraffini LA (2014). Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature 514, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, and Marraffini LA (2015). Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature 519, 199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Romero DA, Coute-Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, and Barrangou R (2008). Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol 190, 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes AP, Lemay ML, Trudel L, Deveau H, Frenette M, Tremblay DM, and Moineau S (2017). Detecting natural adaptation of the Streptococcus thermophilus CRISPR-Cas systems in research and classroom settings. Nat Protoc 12, 547–565. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiswirth BN, Lofdahl S, Betley MJ, O’Reilly M, Schlievert PM, Bergdoll MS, and Novick RP (1983). The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–712. [DOI] [PubMed] [Google Scholar]
- Lemay ML, Tremblay DM, and Moineau S (2017). Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth Biol 6, 1351–1358. [DOI] [PubMed] [Google Scholar]
- McGinn J, and Marraffini LA (2016). CRISPR-Cas systems optimize their immune response by specifying the site of spacer integration. Mol Cell 64, 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell JW, Jiang W, and Marraffini LA (2017). CRISPR-Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature 544, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyenson NC, Gayvert K, Varble A, Elemento O, and Marraffini LA (2017). Broad Targeting Specificity during Bacterial Type III CRISPR-Cas Immunity Constrains Viral Escape. Cell Host Microbe 22, 343–353 e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varble A, Meaden S, Barrangou R, Westra ER, and Marraffini LA (2019). Recombination between phages and CRISPR-cas loci facilitates horizontal gene transfer in staphylococci. Nat Microbiol 4, 956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemantary Data File S1. Next generation sequencing data obtained in this study. Related to all figures.
Supplemantary Data File S2. Oligonucleotides, spacers and strains used in this study. Related to STAR methods.
Data Availability Statement
All the datasets generated during this study are all available and the accession number is SRA:SUB6246845.