

Abstract
Background
HbH disease results from dysfunction of three, less commonly two, α-globin genes through various combinations of deletion and non-deletion mutations. Characterization of the mutations and the underlying genotypes is fundamental for proper screening and prevention of thalassaemia in any region. The aim of this study was to explore the genetic arrangements of HbH disease and to correlate the genotypes with the clinical phenotypes.
Methods
A total of 44 HbH disease patients were enrolled in this study. They were clinically and haematologically assessed. The patients were tested for 21 common α-globin gene mutations using multiplex PCR and reverse hybridization. According to the genotype, the patients were categorized into two separate sub-groups, deletion and non-deletion types HbH disease.
Results
Within the studied HbH disease patients, eight different α-globin gene mutations were detected in nine different genetic arrangements. The --MED and -α3.7 deletions were the two most frequently encountered mutations (37.5 and 35.2% respectively). Patients with deletion genotypes constituted 70.4%. The most common detected genotype was --MED/−α3.7 (59.1%), followed by αpoly-A1α/αpoly-A1α (13.6%). For the first time, coinheritance of two relatively mild mutations (−α3.7/ααAdana) was unpredictably detected in a 1.5 year-old child with Hb of 7.1 g/dL.
Conclusion
The HbH disease patients’ clinical characteristics were variable with no ample difference between the deletion and non-deletion types. These results can be of benefit for the screening and management of thalassaemia in this region.
Keywords: HbH disease, Genotype-phenotype correlation, Alpha-thalassaemia, Erbil, Iraq
Background
α-thalassaemia is said to be the most common monogenic disorder worldwide. The genetic defects in the α-globin genes, which are mostly gene deletions, result in the absence or reduced production of α-globin chain in the hemoglobin tetramer [1]. Numerous mutations, deletion and non-deletion types, have been so far identified in the α-globin genes; some causes dysfunction of one α-globin gene, denoted as α+ mutations; others resulting in deletion or deactivation of two α-globin genes, denoted as α0 alleles [2]. The clinical manifestations of these genetic defects vary. In heterozygous state, these mutations are usually of little clinical significance. However, the coinheritance of these mutations may result in a variety of clinical and haematological phenotypes. The most severe phenotype is hydrops fetalis, where all four α-globin genes are missing (−−/−−). Hemoglobin H (HbH) disease is a moderate clinical form of α-thalassaemia in which three of the four α-globin genes are affected (−−/−α). The unstable HbH can precipitate in the red cells causing hemolysis and jaundice [3–5]. Less commonly, HbH disease may result from two point mutations in the α-globin genes [6–8]. Based on the nature of the genetic defects, two types of HbH disease exist. The first, and commonest, form is called deletion HbH disease that results from a large deletion mutation, which deactivates both α-globin genes on one chromosome such as --MED, −-SEA, −-Thai, or --FIL combined with a single-gene deletion like -α3.7 or -α4.2 on the other chromosome. The second type is the non-deletion form of HbH disease, which ensues when a non-deletion mutations in either the α1 or α2 globin genes on one chromosome combines with another non-deletion mutation, or with a deletion of both α-globin genes on the other chromosome [9, 10].
α-thalassaemia mutations, which are apparently not common in this geographical location, have not been studied before, nor HbH disease genotypes were explored. This study aimed to characterize the α-globin gene mutations in this cohort of patients, discover the mutation combinations resulting in HbH disease, and correlate the disease genotypes with clinical and haematological phenotypes.
Methods
Over 6 years, starting from February 2014, 44 cases of HbH disease were recruited to this study at Erbil Thalassaemia Care Centre, Erbil, Northern Iraq. Most of the enrolled patients were referred to the aforementioned centre from other hospitals and/or private clinics with the suspicion of having a hemoglobinopathy. All enrolled patients were interviewed and physically examined. Demographic data and disease history, specifically transfusion history, were obtained. Informed written consent was obtained from the patients. In the case of minors (less than 16 years), the guardian’s consent was obtained before obtaining their assent. The study was approved by the Research Ethics Committee of Hawler Medical University, Erbil, Iraq. The diagnosis of HbH disease was made following performing routine complete blood count, using automated haematology analyzer (Swelab, Spagna, Sweden), and peripheral blood smear; HbH inclusion test, using 1% brilliant cresyl blue staining of the red cells and incubation at 37 °C for 1 h; and measuring the HbH fraction using high performance liquid chromatography (HPLC), with the VARIANT™ HPLC system on fresh blood samples (Bio-Rad Laboratories, Hercules, CA, US) [11]. Serum ferritin level was measured with Cobas E411 analyzer, Roche®, Germany.
The molecular analysis started with DNA extraction using Gentra Puregene blood kit (Qiagen, Germantown, MD, USA). α-globin genotyping was then performed using multiplex PCR and reverse hybridization assay according to the manufacturer’s instructions (Alpha-globin StripAssay; ViennaLab Diagnostics, Vienna, Austria), covering the following 21 mutations: two single gene deletions (−α3.7, −α4.2), five double gene deletions [−-MED, −-SEA, −-THAI, −-FIL, −α20.5 kb], the αααanti-3.7 gene triplication, two point mutations on the α1 gene [codon 14 (TGG > TAG); codon 59 (GGC > GAC) (Hb Adana)], and 11 mutations on the α2 gene [initiation codon ATG > ACG; codon 19 (GCG > GC-), IVS-I(− 5 nt) (−TGAGG); codon 59 (GGC > GAC); codon 125 (CTG > CCG) (Hb Quong Sze); Hb Constant Spring (HbCS) codon 142, Term → Gln, TAA > CAA; Hb Icaria, codon 142, Term→ Lys, TAA > AAA; Hb Pakse´, codon 142, Term→ Tyr, TAA > TAT, Hb Koya Dora, codon 142, Term→ Ser, TAA > TCA; polyadenylation signal site, poly-A1 (AATAAA>AATAAG); and poly-A2 (AATAAA>AATGAA) [12]. Based on the nature of the α-globin genetic defects, and for the sake of comparing patients’ phenotypic characteristics, we categorized the patients into two distinct genotypic groups: deletion and non-deletion HbH disease subgroups. In the latter subgroup, we included patients with both non-deletion/non-deletion and the deletion/non-deletion arrangements together.
Statistical analysis was performed using Mircosoft® Excel, Professional Edition, 2010. Data was described in number and percentage. Quantitative data were described using mean, standard deviation median, and range. Chi-square test was used to compare categorical data. Mann-Whitney test was used to compare two sets of numerical data. Correlations between two quantitative variables were assessed using Spearman’s coefficient. Significance was considered at P < 0.05.
Results
Over 6 years, 44 HbH disease patients, 19 males and 25 females, were recruited to this study. Their age ranged from 1 to 68 years, with a median of 19 years. The median Hb was 9.15 g/dL, ranging from 6.4 to 12.7. Table 1 shows some demographic and clinical parameters of the patients.
Table 1.
Demographic and clinical characteristics of patients with HbH disease
| Parameters | |
|---|---|
| Age, y (median, range) | 19 (1–68) |
| Gender | |
| Male | 19 (43.2%) |
| Female | 25 (56.8%) |
| Hb, g/dL (median, range) | 9.15 (6.4–12.7) |
| HbH, % (median, range) | 7.35 (1–25.7) |
| Ferritin, ng/mL (median, range) | 442 (11–2950) |
| Splenectomized | 5 (11.4%) |
| Transfusion status | |
| Never transfused | 23 (52.3%) |
| Occasional (≤1 unit per year) | 17 (38.6%) |
| Irregular (2 to 4 units per year) | 4 (9.1%) |
Serum ferritin showed a positive correlation with age (r = 0.761) (Fig. 1). Splenectomized patients were 5 (11.4%), we found their mean age was noticeably higher compared to the non-splenectomized (34.6 versus 20.5 years respectively, p = 0.05). A significant relation (p < 0.001) was found between patients’ age and blood transfusion. There were 23 (52.3%) patients who never received a transfusion; their mean age was 10.5 years versus 35.4 years in the transfused patients. The Hb and ferritin levels were not different in the splenectomized and non-splenectomized patients (p-values 0.061 and 0.138, respectively). We also found significantly higher ferritin levels among transfused patients (p < 0.001).
Fig. 1.
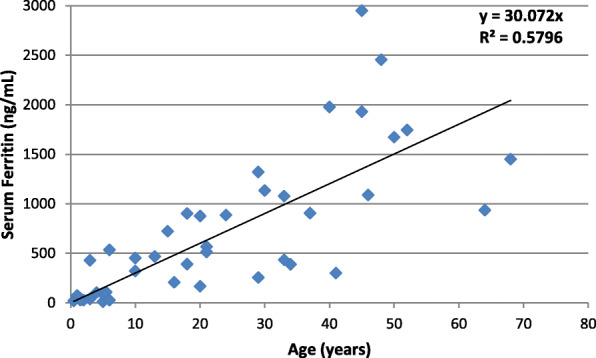
Correlation of serum ferritin with the age of HbH disease patients
The molecular characterization detected eight different α-globin mutations within the studied group. As illustrated in Table 2, −-MED double-gene deletion was the most common mutation, which was observed in 33 (37.5%) alleles, followed by mutations of -α3.7 (35.2%), α2poly-A1 (15.9%), −α4.2 (3.4%), in addition to four other mutations which collectively comprised 8% of all mutated alleles. Totally, deletion mutations constituted 78.4% and non-deletion mutations 21.6% of all mutated alleles.
Table 2.
Mutation frequency in HbH disease patients
| Mutation | Type | Frequency | % |
|---|---|---|---|
| --MED | Deletion | 33 | 37.5 |
| -α3.7 | Deletion | 31 | 35.2 |
| α2poly-A1 (AATAAA>AATAAG) | Point mutation | 14 | 15.9 |
| -α4.2 | Deletion | 3 | 3.4 |
| -α20.5 | Deletion | 2 | 2.3 |
| Codon 59 (GGC > GAC) (Hb Adana) | Point mutation | 2 | 2.3 |
| α2 IVS-I(−5 nt) (−TGAGG) | Splicing site 5 nt. Deletion | 2 | 2.3 |
| α2poly-A2 (AATAAA>AATGAA) | Point mutation | 1 | 1.1 |
| Total | 88 | 100 |
The mutated alleles were found in nine different genetic arrangements. The most frequent genotype was --MED/−α3.7 arrangement, which was detected in 26 (59.1%) patients. The second most common genotype was homozygosity of αPoly-A1α, which was observed in 6 (13.6%) patients. Deletion genotypes we encountered in 31 patients (70.45%), while non-deletion genotypes were observed in 13 cases (29.54%). The α-globin genotypes and their relative frequencies are illustrated in Table 3.
Table 3.
HbH disease genotypes
| Genotypes | Frequency | % |
|---|---|---|
| --MED/−α3.7 | 26 | 59.1 |
| Αpoly-A1α/αpoly-A1α | 6 | 13.6 |
| --MED/−α4.2 | 3 | 6.8 |
| -α20.5/−α3.7 | 2 | 4.5 |
| --MED/αivs1(−5 nt)α | 2 | 4.5 |
| -α3.7/αpoly-A1α | 2 | 4.5 |
| --MED/αpoly-A2α | 1 | 2.3 |
| --MED/ααAdana | 1 | 2.3 |
| -α3.7/ααAdana | 1 | 2.3 |
| Total | 44 | 100 |
Some relevant clinical and haematological parameters within the deletion and non-deletion HbH disease subgroups are illustrated in Table 4. The median values of the red cell count and Hb were higher in patients with deletion HbH disease; however, the differences were not statistically significant. All other haematological and clinical parameters did not show significant differences between the two groups.
Table 4.
Clinical and haematological parameters in the deletion and non-deletion types HbH Disease
| Parameters | Deletion (31 cases) |
Non-Deletion (13 cases) |
p-value |
|---|---|---|---|
| RBC count, (median, range) | 5.05 (3.5–6.61) | 4.51 (2.96–6.74) | 0.069 |
| Hb, g/dL (median, range) | 9.2 (8.0–11.5) | 8.3 (6.4–12.7) | 0.086 |
| MCV, fl (median, range) | 61.3 (52.2–68.7) | 62.6 (52.5–81.9) | 0.309 |
| MCH, pg (median, range) | 18.0 (16.1–21.9) | 18.9 (16.1–28) | 0.241 |
| RDW, % (median, range) | 25.6 (16.9–30.7) | 25.0 (18.7–36.3) | 0.816 |
| HbH, % (median, range) | 7.5 (1–15) | 7.2 (5.7–25.7) | 0.125 |
| Ferritin, ng/mL (median, range) | 467 (11–2950) | 389 (29–1726) | 0.403 |
| Splenectomized | 4 (12.9%) | 1 (7.7) | 0.20 |
| Transfusion | |||
| Never transfused | 16 (51.6%) | 7 (53.8%) | 0.57 |
| Occasional (≤1 unit per year) | 13 (41.9%) | 4 (30.8%) | |
| Irregular (2 to 4 units per year) | 2 (6.5%) | 2 (15.4%) | |
Discussion
HbH disease is considered the most symptomatic form of α-thalassaemias that is compatible with life. The disease, however, has a heterogeneous clinical and haematological phenotype ranging from very mild phenotypes to more severe forms requiring regular or irregular blood transfusion. The severity of the disease is assumed to be related to the degree of α:β globin chain imbalance, which is principally determined by the nature of the underlying α-globin mutations [1, 10]. However, variations in the clinical phenotypes have been observed even in the presence of similar genotype, which is probably due to genetic and environmental modifiers [13].
Erbil province in northern Iraq covers an area of 15,075 km2 with a population of about two million people. Majority of the population are Kurds; the minorities of Turkmen, Assyrians, and Arabs comprise about 5% of its total population. The province is bordered by Turkey from the north-east and Iran from the east. We know much about β-thalassaemia in northern Iraq. Many studies have described the spectrum of β-globin gene mutations, the carrier rate, and the genotypic mechanisms in both thalassaemia major and thalassaemia intermedia [14–19]. Scrutinizing α-thalassaemia is not as easy as for β-thalassaemia because of the lower carrier rate in our region, and the difficulty in confirming diagnosis of α-thalassaemia carriers via the routine Hb-electrophoresis. The carrier rate of α-thalassaemia in Iraq is estimated at < 1%, but no study has yet rectified the exact carrier rate in this region. So far, a couple of small studies by Al-Allawi et al. have described the spectrum of α-globin gene mutations among carriers in the neighboring provinces of Dohuk and Sulaimaniyah [20, 21]. Results of the current study, which is the first to tackle a relatively large cohort of patients with HbH disease in this region, will be complementary to improve the existing national thalassemia carrier screening and genetic counseling program that was established in 2009, and will be helpful for prenatal diagnosis.
The clinical and haematological features of the enrolled patients showed obvious heterogeneity. Substantial variation was noticed in the patients’ age, Hb level, and transfusion requirement. Many clinical parameters strongly related to patients’ age, primarily the ferritin level and transfusion requirement. It is widely reported that the clinical severity of thalassaemias and other hemoglobinopathies is extremely age dependent [22].
In the current study, we have identified eight different α-globin gene mutations, four of which are deletion mutations (−-MED, −α3.7, −α4.2, and -α20.5) and the remaining four are non-deletion mutations (α2 poly-A1, Hb Adana, α2 IVS1 (− 5 nt), and α2 poly-A2). Comparable to the results of earlier reports by Al-Allawi et al. [20, 21], we found the spectrum of α-globin gene defects restricted to a relatively limited number of mutations. However, this finding is unlike to what have been observed in Iran, Turkey, Sardinia, and Cyprus, where the mutations are more heterogeneous [23, 24]. Within our HbH disease patients, the --MED double-gene deletion was the most commonly encountered mutation (33 alleles) followed by the worldwide prevalent -α3.7 single gene deletion (31 alleles). Similar findings have been reported by the vast majority of studies done in the Mediterranean and Middle East regions [25]. The α2 poly-A1 non-deletion mutation, which is known to be prevalent in the Arabian Peninsula, was encountered in 14 alleles and was the most frequently encountered non-deletion mutation. Homozygosity for the latter mutation is the main genotype causing HbH disease in Saudi Arabia, UAE, Kuwait, and Bahrain [26–29]. None of the south-east Asian mutations were observed in this cohort. A previous study has reported the --FIL double-gene deletion in one β-thalassaemia intermedia case who was homozygous for IVS-II-1 (G > A). The same deletion was also observed in Hatay Province of Turkey [18, 25].
We, in the current cohort, observed nine different genotypes causing HbH disease, most of which were deletion genotypes (70.45%, 31 patients). The --MED/−α3.7 arrangement was the most frequently encountered genotype as in the neighboring areas of Turkey and Iran. High prevalence of deletion type HbH disease have been reported in the Mediterranean area, Turkey, Iran, Greece, and Sardinia, (80, 50.5, 56.1, and 86% respectively) [10, 26, 30, 31]. We detected 19 alleles carrying non-deletion mutations of which 17 involved the α2 gene. Mutations of the α2-globin gene are associated with more severe phenotype because more than two-third of the α-globin chain synthesis is controlled by the dominant α2 globin gene transcription [32]. Hb Adana was the only detected α1 globin gene mutation in our patients; it was observed in compound heterozygosity with --MED deletion in one patient and with -α3.7 deletion in another. We could not find any previous study reporting the latter combination (−α3.7/ααAdana) to have a HbH equivalent phenotype; though, we surprisingly found it in a 1.5 year-old boy with Hb of 7.1 g/dL. Coinheritance of -α3.7 deletion, with many other point mutations in the α2 globin gene, are reported in many studies [33]. In our cohort, coinheritance of two point mutations was detected in six patients (13.6%) only, who all were homozygous for α2 poly-A1 mutation. Non-deletion genotypes are reported to be the predominant HbH disease genotypes in the Arabia area, Jordan, and northern Iran [23, 27, 34, 35].
It has been previously documented that patients with deletion/deletion HbH disease genotype are of least severe phenotype, followed by deletion/non-deletion arrangement, and the non-deletion/non-deletion genotype which is supposed to be the most severe [6, 30]. In our cohort, we did not find any significant difference in the clinical and haematological parameters, which we studied in both deletion and non-deletion HbH disease subgroups. The median Hb and red cell count were notably lower in the non-deletion HbH subgroup, however not to a statistically significant level. The remaining red cell parameters, the ferritin level, the HbH fraction, splenectomy, and blood transfusion status did not show any difference in the two subgroups.
Conclusion
This study showed that our HbH disease patients had variable clinical and haematological phenotypes ranging from very mild cases not requiring transfusion to more severe others needed irregular transfusions. The spectrum of α-globin gene mutations and then the underlying genotypic arrangements were not many. The majority of our HbH disease patients were found to have deletion genotypes; however, their clinical phenotypes did not differ much from those having non-deletion genotypes. Knowing all these will definitely have a positive impact on thalassaemia diagnosis and management guidelines in this region.
Acknowledgements
The authors very much appreciate the assistance and support provided by Erbil Thalassemia Care Centre and the Lab Division of PAR Hospital.
Abbreviations
- HbH
Hemoglobin-H
- PCR
Polymerase chain reaction
- HPLC
High performance liquid chromatography
- Hb
Hemoglobin
- RBCs
Red blood cells
- MCV
Mean cell volume
- MCH
Mean cell hemoglobin
- RDW
Red cell distribution width
Authors’ contributions
RPS contributed in conceiving and designing the study, molecular testing, analyzing the data, and editing and finalizing the manuscript. AKY and MDA contributed in conceptualization and designing the study and took part in writing the initial draft of the manuscript. RKP contributed in conceptualization and designing the study, data collection, and the initial draft of the manuscript. The author(s) read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
The dataset used for the current study is available in the Mendeley datasets repository (DOI: 10.17632/bjgf3kcpgk.1). https://data.mendeley.com/datasets/bjgf3kcpgk/1
Ethics approval and consent to participate
The study protocol was reviewed and approved by the Research Ethics Committee at Hawler Medical University. Each participant provided written informed consent before participation in the study. In the case of minors, the guardian’s written consent was obtained before obtaining their assent. The anonymity of participants and the confidentiality of collected information were explained for the participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Rawand P. Shamoon, Email: rawand.shamoon@hmu.edu.krd
Ahmed K. Yassin, Email: ahmed.khudair@hmu.edu.krd
Ranan K. Polus, Email: ranan.kardagh@hmu.edu.krd
Mohamad D. Ali, Email: mohamed.daherali@gmail.com
References
- 1.Farashi S, Harteveld CL. Molecular basis of α-thalassemia. Blood Cells Mol Dis. 2018;70:43–53. doi: 10.1016/j.bcmd.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Harteveld CL, Higgs DR. α-thalassaemia. Orphanet J Rare Dis. 2010;5:13. doi: 10.1186/1750-1172-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vichinsky EP. Clinical manifestations of α-thalassemia. Cold Spring Harbor Perspect Med. 2013;3:a011742. doi: 10.1101/cshperspect.a011742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chui DH. α-Thalassemia: Hb H disease and Hb Barts hydrops fetalis. Ann N Y Acad Sci. 2005;1054:25–32. doi: 10.1196/annals.1345.004. [DOI] [PubMed] [Google Scholar]
- 5.Traivaree C, Boonyawat B, Monsereenusorn C, Rujkijyanont P, Photia A. Clinical and molecular genetic features of Hb H and AE Bart’s diseases in central Thai children. Appl Clin Genet. 2018;11:23. doi: 10.2147/TACG.S161152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003;101:791–800. doi: 10.1182/blood-2002-07-1975. [DOI] [PubMed] [Google Scholar]
- 7.Lal A, Goldrich ML, Haines DA, Azimi M, Singer ST, Vichinsky EP. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011;364:2069–2070. doi: 10.1056/NEJMoa1010174. [DOI] [PubMed] [Google Scholar]
- 8.Quadri MI, Islam SA, Nasserullah Z. Hemoglobin H disease in the eastern region of Saudi Arabia. Saudi Med J. 2000;21:666–671. [PubMed] [Google Scholar]
- 9.Lin P-C, Chang T-T, Liao Y-M, Tsai S-P, Chen Y-C, Hsu W-Y, et al. Clinical features and genotypes of patients with hemoglobin H disease in Taiwan. Lab Med. 2019;50:168–173. doi: 10.1093/labmed/lmy043. [DOI] [PubMed] [Google Scholar]
- 10.Valaei A, Karimipoor M, Kordafshari A, Zeinali S. Molecular basis of α-thalassemia in Iran. Iran Biomed J. 2018;22:6–14. doi: 10.22034/ibj.22.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabath DE. Molecular diagnosis of thalassemias and hemoglobinopathies: an ACLPS critical review. Am J Clin Path. 2017;148:6–15. doi: 10.1093/ajcp/aqx047. [DOI] [PubMed] [Google Scholar]
- 12.Puehringer H, Najmabadi H, Law H-Y, Krugluger W, Viprakasit V, Pissard S, et al. Validation of a reverse-hybridization StripAssay for the simultaneous analysis of common α-thalassemia point mutations and deletions. Clin Chem Lab Med. 2007;45:605–610. doi: 10.1515/CCLM.2007.125. [DOI] [PubMed] [Google Scholar]
- 13.Piel FB, Weatherall DJ. The α-thalassemias. N Engl J Med. 2014;371:1908–1916. doi: 10.1056/NEJMra1404415. [DOI] [PubMed] [Google Scholar]
- 14.Al-Allawi N, Al-Dousky A. Frequency of haemoglobinopathies at premarital health screening in Dohuk, Iraq: implications for a regional prevention programme. East Mediterr Health J. 2010;16:381–385. doi: 10.26719/2010.16.4.381. [DOI] [PubMed] [Google Scholar]
- 15.Jalal SD, Al-Allawi NA, Bayat N, Imanian H, Najmabadi H, Faraj A. β-Thalassemia mutations in the Kurdish population of northeastern Iraq. Hemoglobin. 2010;34:469–476. doi: 10.3109/01676830.2010.513591. [DOI] [PubMed] [Google Scholar]
- 16.Al-Allawi NA, Hassan K, Sheikha AK, Nerweiy FF, Dawood RS, Jubrael J. Mol Biol Int. 2010;2010. https://www.ncbi.nlm.nih.gov/pmc/issues/202494/. 10.4061/2010/479282. [DOI] [PMC free article] [PubMed]
- 17.Al-Allawi NA, Jubrael JM, Hughson M. Molecular characterization of β-thalassemia in the Dohuk region of Iraq. Hemoglobin. 2006;30:479–486. doi: 10.1080/03630260600868097. [DOI] [PubMed] [Google Scholar]
- 18.Shamoon RP, Al-Allawi NA, Cappellini MD, Di Pierro E, Brancaleoni V, Granata F. Molecular basis of β-thalassemia intermedia in Erbil province of Iraqi Kurdistan. Hemoglobin. 2015;39:178–183. doi: 10.3109/03630269.2015.1032415. [DOI] [PubMed] [Google Scholar]
- 19.Polus RK. Prevalence of hemoglobinopathies among marrying couples in Erbil province of Iraq. Iraq J Hemat. 2017;6:90–93. doi: 10.4103/ijh.ijh_27_17. [DOI] [Google Scholar]
- 20.Al-Allawi NA, Badi AI, Imanian H, Nikzat N, Jubrael JM, Najmabadi H. Molecular characterization of α-thalassemia in the Dohuk region of Iraq. Hemoglobin. 2009;33:37–44. doi: 10.1080/03630260802626053. [DOI] [PubMed] [Google Scholar]
- 21.Al-Allawi NA, Jalal SD, Rasheed NS, Bayat N, Imanian H, Najmabadi H, Faraj A. The spectrum of α-thalassemia mutations in the Kurdish population of northeastern Iraq. Hemoglobin. 2013;37:56–64. doi: 10.3109/03630269.2012.749490. [DOI] [PubMed] [Google Scholar]
- 22.Origa R. β-Thalassemia. Genet Med. 2018;19:609–619. doi: 10.1038/gim.2016.173. [DOI] [PubMed] [Google Scholar]
- 23.Ebrahimkhani S, Azarkeivan A, Bayat N, Houry-Parvin M, Jalil-Nejad S, Zand S, et al. Genotype-phenotype correlation in Iranian patients with Hb H disease. Hemoglobin. 2011;35:40–46. doi: 10.3109/03630269.2010.546314. [DOI] [PubMed] [Google Scholar]
- 24.Origa R, Paglietti ME, Sollaino MC, Desogus MF, Barella S, Loi D, et al. Complexity of the alpha-globin genotypes identified with thalassemia screening in Sardinia. Blood Cells Mol Dis. 2014;52:46–49. doi: 10.1016/j.bcmd.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 25.Celik MM, Gunesacar R, Oktay G, Duran GG, Kaya H. Spectrum of α-thalassemia mutations including first observation of--FIL deletion in Hatay Province, Turkey. Blood Cells Mol Dis. 2013;51:27–30. doi: 10.1016/j.bcmd.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 26.Dehbozorgian J, Moghadam M, Daryanoush S, Haghpanah S, Imani fard J, Aramesh A, et al. Distribution of alpha-thalassemia mutations in Iranian population. Hematology. 2015;20:359–362. doi: 10.1179/1607845414Y.0000000227. [DOI] [PubMed] [Google Scholar]
- 27.Baysal E. α-Thalassemia syndromes in the United Arab Emirates. Hemoglobin. 2011;35:574–580. doi: 10.3109/03630269.2011.634698. [DOI] [PubMed] [Google Scholar]
- 28.Laosombat V, Viprakasit V, Chotsampancharoen T, Wongchanchailert M, Khodchawan S, Chinchang W, et al. Clinical features and molecular analysis in Thai patients with HbH disease. Ann Hematol. 2009;88:1185–1192. doi: 10.1007/s00277-009-0743-5. [DOI] [PubMed] [Google Scholar]
- 29.Jassim N, Al-Arrayed S, Gerard N, Al-Mukharraq H, Al-Ajami A, Ducrocoq R, et al. Krishnamoorthy R. molecular basis of α-thalassemia in Bahrain. Bahrain Med Bull. 2001;23:3–7. [Google Scholar]
- 30.Kanavakis E, Papassotiriou I, Karagiorga M, Vrettou C, Metaxotou-Mavrommati A, Stamoulakatou A, Kattamis C, Traeger-Synodinos J. Phenotypic and molecular diversity of haemoglobin H disease: a Greek experience. Br J Haematol. 2000;111:915–923. [PubMed] [Google Scholar]
- 31.Origa R, Sollaino M, Giagu N, Barella S, Campus S, Mandas C, et al. Clinical and molecular analysis of haemoglobin H disease in Sardinia: haematological, obstetric and cardiac aspects in patients with different genotypes. Br J Haematol. 2007;136:326–332. doi: 10.1111/j.1365-2141.2006.06423.x. [DOI] [PubMed] [Google Scholar]
- 32.Galanello R, Cao A. Alpha-thalassemia. Genet Med. 2011;13:83–88. doi: 10.1097/GIM.0b013e3181fcb468. [DOI] [PubMed] [Google Scholar]
- 33.Farashi S, Bayat N, Vakili S, Faramarzi Garous N, Ashki M, Imanian H, et al. Point mutations which should not be overlooked in Hb H disease. Expert Rev Hematol. 2016;9:107–113. doi: 10.1586/17474086.2016.1107470. [DOI] [PubMed] [Google Scholar]
- 34.Hellani A, Fadel E, El-Sadadi S, El-Sweilam H, El-Dawood A, Abu-Amero KK. Molecular spectrum of α-thalassemia mutations in microcytic hypochromic anemia patients from Saudi Arabia. Genet Test Mol Biomarkers. 2009;13:219–221. doi: 10.1089/gtmb.2008.0123. [DOI] [PubMed] [Google Scholar]
- 35.Mw A-G. Subtypes of alpha thalassemia diagnosed at a medical center in Jordan. TAF Prev Med Bull. 2008;7:373. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset used for the current study is available in the Mendeley datasets repository (DOI: 10.17632/bjgf3kcpgk.1). https://data.mendeley.com/datasets/bjgf3kcpgk/1