

Abstract
Objective
Fulminant type 1 diabetes (FT1D) is a type of type 1 diabetes, which is characterized by rapid onset of disease and severe metabolic disorders. We intend to screen for crucial genes and potential molecular mechanisms in FT1D in this study.
Method
We downloaded GSE44314, which includes six healthy controls and five patients with FT1D, from the GEO database. Identification of differentially expressed genes (DEGs) was performed by NetworkAnalyst. The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of DEGs were screened by an online tool—Database for Annotation, Visualization, and Integration Discovery (DAVID). Protein-protein interaction (PPI) network and hub genes among DEGs were analyzed by NetworkAnalyst. And we also use NetworkAnalyst to find out the microRNAs (miRNAs) and transcription factors (TFs) which regulate the expression of DEGs.
Result
We identified 130 DEGs (60 upregulated and 70 downregulated DEGs) between healthy controls and FT1D patients. GO analysis results revealed that DEGs were mostly enriched in generation of precursor metabolites and energy, neurohypophyseal hormone activity, and mitochondrial inner membrane. KEGG pathway analysis demonstrated that DEGs were mostly involved in nonalcoholic fatty liver disease. Results indicated that NCOA1, SRF, ERBB3, EST1, TOP1, UBE2S, INO80, COX7C, ITGAV, and COX6C were the top hub genes in the PPI network. Furthermore, we recognized that LDLR, POTEM, IFNAR2, BAZ2A, and SRF were the top hub genes in the miRNA-target gene network, and SRF, TSPAN4, CD59, ETS1, and SLC25A25 were the top hub genes in the TF-target gene network.
Conclusion
Our study pinpoints key genes and pathways associated with FT1D by a sequence of bioinformatics analysis on DEGs. These identified genes and pathways provide more detailed molecular mechanisms of FT1D and may provide novel therapeutic targets.
1. Introduction
Fulminant type 1 diabetes (FT1D) is a novel type of type 1 diabetes (T1DM) raised by Imagawa et al. in 2000 [1], which is featured by abrupt disease onset, no C-peptide secretion, negative islet-related autoantibodies, and elevated pancreatic enzymes. At first, FT1D was identified as idiopathic T1DM because patients with FT1D lack autoimmune markers such as protein tyrosine phosphatase antibody or glutamic acid decarboxylase autoantibody. Over the past 20 years, the understanding of FT1D has increased. And a sequence of studies indicated that the immunity has a role in the occurrence and development of FT1D, which convinced that FT1D is possibly an autoimmune disease [2–4].
There are studies that reported that genetic and environmental factors take part in the initiation and progression of FT1D. Numbers of studies indicated that CTLA-4, HLA-B, and HLA DR-DQ are related with FT1D [5–7]. Many studies advocate that in FT1D, immune response against viral infection in islets caused the β cell destruction [8–10]. Numerous virus infections were covered in FT1D patients, including coxsackievirus, enterovirus, and human cytomegalovirus [11–13]. Genes such as lymphocyte cytosolic protein 1, melanoma differentiation-associated protein 5, DEAD box helicase 5, and C-X-C motif chemokine 10, which take part in the virus infection, have been proved to be associated with the pathogenesis of FT1D [3, 11, 14]. To further reveal the mechanism of FT1D, a microarray data numbered GSE44314 was deposited by Nakata et al., and it has reported that NKG2E-CD94 were significantly reduced in FT1D, indicating that the reduced expression of NK activating receptor gene and low proportion of NK cells are probably involved in the progression of FT1D [15]. However, there are no studies that had reported the possible regulatory mechanisms of transcription factors (TFs) and microRNAs (miRNAs) related to the development of FT1D.
In our study, we reanalyzed the dataset of GSE44314 by the method of bioinformatics, which includes screening differentially expressed genes (DEGs), functional enrichment analysis, protein-protein interaction (PPI) analysis, and the regulatory TFs/miRNAs related to DEG prediction. Through these analyses, we expect to determine novel insights for the knowledge of FT1D and provide more detailed molecular mechanisms underlying the development of FT1D.
2. Materials and Methods
2.1. Microarray Data
We downloaded the gene expression profile data of GSE44314 from the Gene Expression Omnibus (GEO) database in the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/geo/). The microarray data was based on the platform of GPL6480 (Agilent-014850 Whole Human Genome Microarray 4x44K G4112F). The datasets available in this analysis were uploaded by Nakata et al. [15], which include 11 samples, containing 6 healthy controls and 5 patients with FT1D.
2.2. Identification of Differentially Expressed Genes
NetworkAnalyst [16, 17] (https://www.networkanalyst.ca), a website for integrative statistical and visualizing tool, was used to determine the DEGs between healthy controls and FT1D patients. The cutoff of the P value was adjusted to 0.05, and ∣log fold change | (∣log FC∣) > 0.585 for the DEG discrimination, using the false discovery rate (FDR) found on the Benjamini-Hochberg program and moderated t-test based on the Limma algorithm.
2.3. Functional and Pathway Enrichment Analysis
We used an online tool named DAVID [18] (https://david.ncifcrf.gov/) in conducting the Gene Ontology (GO) term [19] and Kyoto Encyclopedia of Genes and Genomes (KEGG) [20] pathway enrichment analyses of DEGs, with the thresholds of count ≥ 2 and P value < 0.05.
2.4. Protein-Protein Interaction (PPI) Network Analysis and Hub Gene Searching
Based on the analyzed DEGs, NetworkAnalyst [21] was used to perform the PPI Network identification with a hypergeometric algorithm, and P < 0.05 was identified as having statistically significant differences. Besides, we used NetworkAnalyst to recognize the most significant modules of hub genes using the “module explorer tool,” found on the random walk-dependent Walktrap algorithm.
2.5. Prediction of Target Gene-MicroRNA Network
The gene expression was affected by microRNAs in a disease condition through posttranscriptional control. In the present study, the online tool NetworkAnalyst [17] was used to search the miRNAs associated with DEGs, which integrates microRNA databases miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/php/download.php) [22] and TarBase (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index) [23].
2.6. Prediction of Target Gene-Transcription Factor Network
The gene expression was influenced by TFs in a disease condition by transcriptional control. In our study, NetworkAnalyst [17] was used for recognizing the TFs associated with DEGs, which combines TF database JASPAR (http://jaspar.genereg.net/) [24].
3. Results
3.1. Identification of Differentially Expressed Genes in Fulminant Type 1 Diabetes
We identified 130 DEGs in FT1D patients compared to healthy controls in total, including 60 upregulated genes and 70 downregulated genes (Supplementary Table 1). We draw a volcano plot of the DEGs (Figure 1) and a hierarchical clustering heat map of DEGs (Figure 2). It turned out that these DEGs were well distinguished between the FT1D group and the healthy control group. NK2 homeobox 3 (NKX2-3) and Ring finger protein 182 (RNF182) were, respectively, identified as the most significantly upregulated and downregulated genes in FT1D patients.
Figure 1.
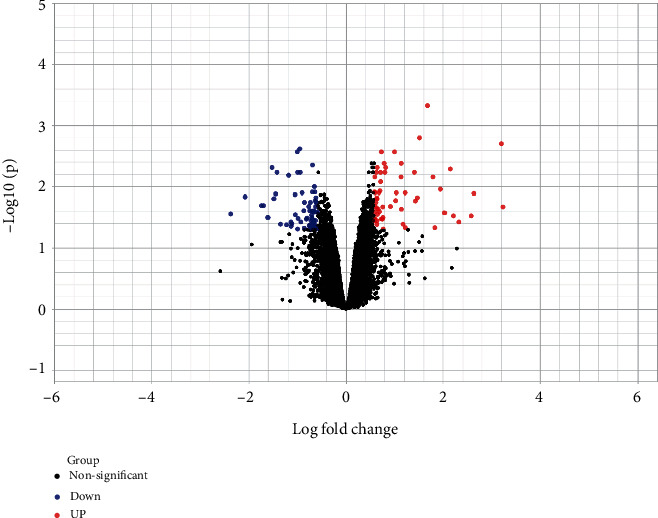
Volcano plot of differentially expressed genes. Genes with a significant change of more than 1.5-fold were selected.
Figure 2.
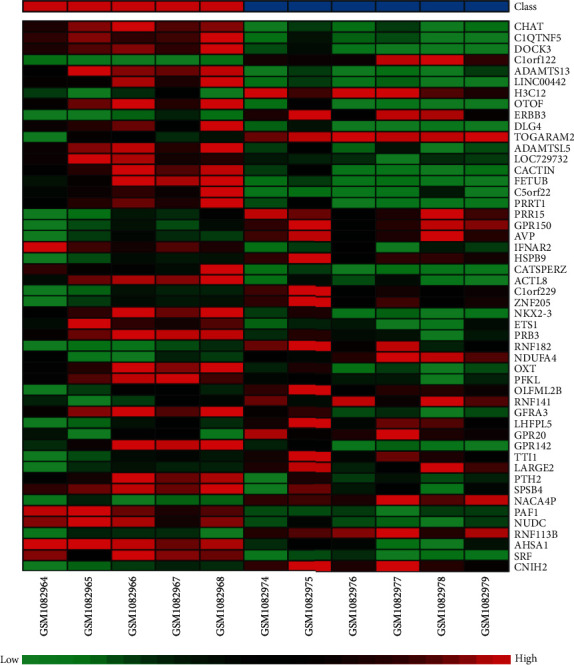
Heat map of differentially expressed genes. The abscissa represents different samples, and the ordinate represents different genes. The red boxes indicate upregulated genes, and the green boxes indicate downregulated genes.
3.2. Functional Enrichment Analysis
We recognized 21 Gene Ontology terms (Table 1) and 5 KEGG pathways (Table 2) when analyzed with DAVID. The DEGs were mainly focused on the generation of precursor metabolites and energy, hydrogen ion transmembrane transport, and mitochondrial electron transport, cytochrome c to oxygen by biological process (BP) analysis. For the cellular component (CC) group, mitochondrial inner membrane, extracellular space, and cell junction were the enriched terms. Molecular function (MF) analysis showed that the DEGs were remarkably focused on neurohypophyseal hormone activity, cytochrome c oxidase activity, and neuregulin binding. Moreover, the KEGG pathway analysis indicated that the DEGs were significantly involved in nonalcoholic fatty liver disease, Huntington's disease, Alzheimer's disease, and Parkinson's disease as well as oxidative phosphorylation.
Table 1.
The results of Gene Ontology (GO) of DEGs ranked by P value.
| Term | Count | P value | Genes |
|---|---|---|---|
| GO-BPs | |||
| Generation of precursor metabolites and energy | 4 | 0.004 | AVP, UQCR11, COX7C, COX6C |
| Hydrogen ion transmembrane transport | 4 | 0.006 | NDUFA4, UQCR11, COX7C, COX6C |
| Mitochondrial electron transport, cytochrome c to oxygen | 3 | 0.006 | NDUFA4, COX7C, COX6C |
| Extrinsic apoptotic signaling pathway in the absence of ligand | 3 | 0.017 | MOAP1, ERBB3, ITGAV |
| Positive regulation of female receptivity | 2 | 0.018 | NCOA1, OXT |
| Positive regulation of gene expression | 6 | 0.02 | AMH, ATF4, AVP, LDLR, ERBB3, GPER1 |
| Maternal aggressive behavior | 2 | 0.024 | AVP, OXT |
| Hyperosmotic salinity response | 2 | 0.029 | AVP, OXT |
| Cellular response to lipopolysaccharide | 4 | 0.03 | TNFRSF1B, ADAMTS13, PAF1, CACTIN |
| Social behavior | 3 | 0.033 | AVP, OXT, DLG4 |
| Positive regulation of apoptotic process | 6 | 0.034 | MOAP1, ATF4, NCOA1, ARHGEF6, GPER1, PDCD1 |
| Male mating behavior | 2 | 0.035 | NCOA1, OXT |
| Positive regulation of uterine smooth muscle contraction | 2 | 0.041 | OXT, GPER1 |
| Drinking behavior | 2 | 0.041 | HTR1B, OXT |
| Positive regulation of cytosolic calcium ion concentration | 4 | 0.046 | AVP, OXT, DLG4, GPER1 |
| GO-MFs | |||
| Neurohypophyseal hormone activity | 2 | 0.011 | AVP, OXT |
| Cytochrome c oxidase activity | 3 | 0.013 | NDUFA4, COX7C, COX6C |
| Neuregulin binding | 2 | 0.028 | ERBB3, ITGAV |
| GO-CCs | |||
| Mitochondrial inner membrane | 8 | 0.014 | NDUFA4, UQCR11, SLC25A25, COX7C, ROMO1, MRPL30, NDUFB1, COX6C |
| Extracellular space | 14 | 0.046 | INA, AVP, CXCL5, ERBB3, ADAMTS13, OXT, FETUB, AMH, IFNAR2, C1QTNF5, CLEC3B, CD59, SEMA4D, PRSS33 |
| Cell junction | 7 | 0.05 | CNIH2, OTOF, PRRT1, DLG4, PAF1, GPER1, GPR142 |
Table 2.
The results of Kyoto Encyclopedia of Genes and Genomes (KEGG) of DEGs ranked by P value.
| Term | Count | P value | Genes |
|---|---|---|---|
| Nonalcoholic fatty liver disease (NAFLD) | 6 | 0.0017 | NDUFA4, ATF4, UQCR11, COX7C, NDUFB1, COX6C |
| Huntington's disease | 6 | 0.0048 | NDUFA4, UQCR11, DLG4, COX7C, NDUFB1, COX6C |
| Oxidative phosphorylation | 5 | 0.0071 | NDUFA4, UQCR11, COX7C, NDUFB1, COX6C |
| Parkinson's disease | 5 | 0.0089 | NDUFA4, UQCR11, COX7C, NDUFB1, COX6C |
| Alzheimer's disease | 5 | 0.0158 | NDUFA4, UQCR11, COX7C, NDUFB1, COX6C |
3.3. PPI Network and Hub Gene Identification
There were 363 nodes and 409 edges in the PPI network (Figure 3). In this PPI network, sixteen genes with degrees > 10 were found as key genes (Table 3). The node size is influenced by the fold change between FT1D patients and healthy controls, and the red or orange color nodes indicate that they have a higher score. The core of the whole PPI network was the most key genes in this cluster, including NCOA1, SRF, ERBB3, ETS1, TOP1, UBE2S, INO80, COX7C, ITGAV, COX6C, ATF4, PAF1, YARS, TTI1, UBC, EEF1B2, and AHSA1. Thence, the seventeen genes were recognized as the hub genes.
Figure 3.

Protein-protein interaction network of the differentially expressed genes. Red and orange nodes stand for hub genes.
Table 3.
Sixteen genes with degrees < 10 in the protein-protein interaction network of the differentially expressed genes.
| Gene | Regulation | Degree | Betweenness | Expression |
|---|---|---|---|---|
| ETS1 | Up | 26 | 15103.54 | 1.145 |
| AHSA1 | Up | 11 | 3565 | 0.82 |
| TOP1 | Up | 23 | 10312.37 | 0.764 |
| NCOA1 | Up | 34 | 9967.16 | 0.752 |
| PAF1 | Up | 18 | 5908.12 | 0.732 |
| SRF | Up | 31 | 22498.06 | 0.647 |
| YARS | Up | 15 | 4237.33 | 0.644 |
| INO80 | Up | 22 | 7030.5 | 0.606 |
| ITGAV | Down | 20 | 9973.24 | -0.603 |
| ATF4 | Down | 18 | 11878.52 | -0.705 |
| COX6C | Down | 20 | 3314.17 | -0.759 |
| COX7C | Down | 22 | 4037.83 | -0.801 |
| EEF1B2 | Down | 11 | 11109 | -0.817 |
| UBE2S | Down | 23 | 7532.83 | -0.858 |
| TTI1 | Down | 14 | 11460.5 | -1.226 |
| ERBB3 | Down | 29 | 19037.55 | -1.422 |
3.4. miRNA-DEG and TF-DEG Regulating Network Analysis
The miRNAs and TFs for DEGs are displayed in Figures 4 and 5, respectively. The top five targeted genes regulated by miRNA are shown in Supplementary Table 2. It turned out that 167 miRNAs regulate LDLR, 124 miRNAs regulate POTEM, 109 miRNAs regulate IFNAR2, 107 miRNAs regulate BAZ2A, and 92 miRNAs regulate SRF. The top five targeted genes regulated by TFs are shown in Supplementary Table 3. It turned out that 25 TFs regulate SRF, 18 TFs regulate TSPAN4, 16 TFs regulate CD59, 16 TFs regulate ETS1, and 15 TFs regulate SLC25A25.
Figure 4.

Target gene-miRNA regulatory network. Red and orange nodes stand for differentially expressed genes; blue diamonds stand for miRNA.
Figure 5.
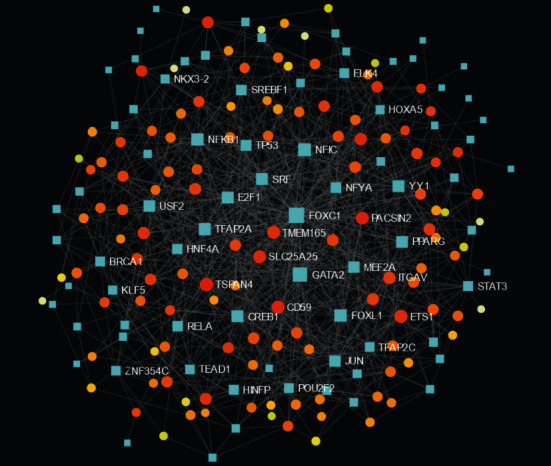
Transcription factor-target DEG regulatory network. Orange and yellow nodes stand for differentially expressed genes; blue diamonds stand for transcription factors.
4. Discussion
FT1D is a disease with a state of insulin dependency due to the rapid destruction of almost all pancreatic β cells, which causes the radical onset of ketoacidosis in a few days after the appearance of hyperglycemic symptoms [25–27]. It has been reported that most of the patients with FT1D are found in East Asia, but recently, Western countries also reported this disease [8, 28, 29]. FT1D makes up about 20% of abrupt-onset T1DM cases in Japan [8]. It is important to understand the molecular mechanisms of FT1D. We downloaded and analyzed a dataset (GSE44314) that contains five FT1D patients and six healthy controls from the GEO database. We identified 130 DEGs in total, including 60 upregulated DEGs and 70 downregulated DEGs. Among the 130 DEGs, we noticed that programmed cell death-1 (PD-1) was downregulated in FT1D patients. PD-1 is a critical member of the B7-CD28 family and is one of the important costimulatory molecules [30]. PD-1 can regulate the T cell response and keep maintaining peripheral tolerance by delivering critical inhibitory signals [30]. Inhibiting the PD-1 pathway would bring about excessive T cell proliferation, failure of tolerance, and autoimmune activation [31]. Therefore, PD-1 has gained popularity in the treatment of several advanced cancers [32, 33]. Studies have proved that treatment with PD-1 inhibitors can cause FT1D [34–36]. And the termination of anti-PD1 antibody therapy may preserve inherent insulin secretion capacity in “anti-PD1 antibody-induced” FT1D [37]. It seems that PD-1 should be upregulated in FT1D, which is totally opposite to our result. Various researchers have identified that cellular immunity, especially T cell, played a crucial role in β cell destruction in FT1D [38–40]. However, a Japanese study that compares PD-1 expression in peripheral CD4+ T cells between type 1A diabetes (classical type 1 diabetes), FT1D, and healthy controls found that there is no difference between FT1D and healthy controls in PD-1 expression and that there is lower PD-1 expression in CD4+ T cells in patients with type 1A diabetes [41]. Different studies have different conclusions in PD-1 expression in FT1D, which need further studies to confer this question and explore how PD-1 take part in the occurrence and progression of FT1D. Among the increased DEGs, NK2 homeobox 3 (NKX2-3) is the most upregulated gene in FT1D, and an animal study has indicated that NKX2-3 is related to T1DM [42], but further study is needed to figure out how NKX2-3 acts in FT1D.
In the current study, the most significant GO BP term for DEGs is generation of precursor metabolites. UQCR11, COX7C, and COX6C are the new biomarkers for the progression of FT1D. The most significant GO MF term for DEGs is neurohypophyseal hormone activity. Arginine vasopressin (AVP) and oxytocin are associated with type 2 diabetes but are new biomarkers for the progression of FT1D. The most significant GO CC term for DEGs is mitochondrial inner membrane. NDUFA4, SLC25A25, ROMO1, MRPL30, and NDUFB1 are novel biomarkers for the development of FT1D. Nonalcoholic fatty liver disease is the most significant KEGG pathway for DEGs. Activation of activating transcription factor 4 (ATF4) contributes to diabetic hepatotoxicity by ER stress [43]. Besides, ATF4 is a transcription factor implicated in β cell survival and susceptibility to stress [44]. ATF4 is a new biomarker for the progression of FT1D. Parkinson's disease, Alzheimer's disease, and Huntington's disease also are significant KEGG pathways for DEGs. Diabetes mellitus (DM) adversely affects multiple organ systems, including the brain [45]. These evidences suggest that FT1D may also lead to neurodegenerative diseases and adversely affect cognition. Discs large MAGUK scaffold protein 4 (DLG4) is related to neurological disorders and type 2 diabetes [46–48]; DLG4 is a new biomarker for the progression of FT1D.
In the present study, NCOA1, SRF, ERBB3, ETS1, TOP1, UBE2S, INO80, COX7C, ITGAV, and COX6C were recognized as top 10 hub genes in the PPI network. A genome-wide meta-analysis study confirmed that nuclear receptor coactivator 1 (NCOA1) is a T1DM susceptibility gene [49]. An animal study suggests that serum response factor (SRF) is decreased in diabetic nephropathy compared to healthy controls [50]. Many studies confirmed that ERBB3 was the most important T1DM association locus in the non-HLA gene [51–53]. ETS proto-oncogene 1 (EST1) was found associated with T1DM in the NOD mouse and then confirmed in human population [54–56]. Tissues derived from the T1DM animals show that DNA topoisomerase I (TOP1) activity and enzyme protein level decreased, whereas the enzyme mRNA level was not altered, which demonstrates that TOP1 activity is regulated by high glucose levels and may lead to the pathogenesis of diabetic complications [57]. Ubiquitin-conjugating enzyme E2 (UBE2S) takes part in T1DM by enhancing M2 macrophage polarization [58]. Jin et al. compared integrin subunit alpha V (ITGAV) expression between diabetic nephropathy and normal human kidney and found that ITGAV is higher in diabetic nephropathy [59]. Although there are evidences that the hub genes are contacted with T1DM, they are novel biomarkers for the development of FT1D.
LDLR, POTEM, IFNAR2, BAZ2A, and SRF were identified as top five targeted genes in the miRNA-target gene regulatory network. Low-density lipoprotein receptor (LDLR) is increased in a NOD mouse compared with a nondiabetic mouse [60]. A study in Ins2(Akita)Ldlr−/− mice revealed that lack of LDLR will accelerate atherosclerosis in T1DM animals [61]. When lacking the r type II interferon receptor (IFNAR2), diabetes happened only in female NOD mice [62]. POTEM and BAZ2A are novel biomarkers for the development of FT1D. SRF, TSPAN4, CD59, ETS1, and SLC25A25 were identified as top five targeted genes in the TF-DEG regulatory network. Due to the lack of complement regulatory protein CD59, the development of diabetes-induced atherosclerosis in mice is accelerated [63]. Besides, CD59 is reduced in diabetic subjects compared with healthy controls [64]. Tetraspanin 4 (TSPAN4) is a new biomarker for the progression of FT1D.
We noticed that there are two bioinformatics analysis of type 1 diabetes, and there are some the same conclusions between our study and theirs [65, 66]. Fang et al. reported that programmed cell death ligand 1 (PD-L1) was upregulated in the new-onset T1DM samples [66]. This is identical with our result. PD-1/PD-L1 is a negative modulatory signaling pathway for activation of T cell. The upregulated PD-L1 and downregulated PD-l cause the same result, which are the inactivation of T cell and the progression of immune tolerance, which play a protective role in the pathogenesis of T1DM. Liu et al. found that HLA-DQA1 and HLA-DRB4 might be targets for the treatment of T1D, and IL8 is likely to be a new marker for the diagnosis of T1D [65]. These results indicated that T1DM is an autoimmune disease, which is in accordance with our result.
5. Conclusions
Our data provide a comprehensive bioinformatics analysis of DEGs to search molecular mechanisms related to the progression of FT1D. We found a set of useful genes for future research into the molecular mechanisms of FT1D progression, while further molecular biological experiments are needed to confirm the effect of these DEGs in the progression of FT1D.
Acknowledgments
The authors thank Nakata and his colleges for depositing the dataset of GSE44314. This study is supported by grants from the National Natural Science Foundation of China (Grant Numbers 81770843 and 81974107).
Contributor Information
Wen Kong, Email: wenly-kong@163.com.
Lu-lu Chen, Email: cheria_chen@126.com.
Data Availability
The data used to support the findings of this study are included within the article.
Conflicts of Interest
The authors declare that there are no conflicts of interests associated with the manuscript.
Supplementary Materials
Supplementary Table 1: the list of all differentially expressed genes. Supplementary Table 2: the top five targeted genes regulated by miRNA. Supplementary Table 3: the top five targeted genes regulated by transcription factor.
References
- 1.Imagawa A., Hanafusa T., Miyagawa J., Matsuzawa Y. A novel subtype of type 1 diabetes mellitus characterized by a rapid onset and an absence of diabetes-related antibodies. The New England Journal of Medicine. 2000;342(5):301–307. doi: 10.1056/NEJM200002033420501. [DOI] [PubMed] [Google Scholar]
- 2.Luo S., Ma X., Li X., Xie Z., Zhou Z. Fulminant type 1 diabetes: a comprehensive review of an autoimmune condition. Diabetes/Metabolism Research and Reviews. 2020;36, article e3317 doi: 10.1002/dmrr.3317. [DOI] [PubMed] [Google Scholar]
- 3.Hosokawa Y., Hanafusa T., Imagawa A. Pathogenesis of fulminant type 1 diabetes: genes, viruses and the immune mechanism, and usefulness of patient-derived induced pluripotent stem cells for future research. Journal of Diabetes Investigation. 2019;10(5):1158–1164. doi: 10.1111/jdi.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu L., Zeng L., Sang D., Lu Z., Shen J. Recent findings on fulminant type 1 diabetes. Diabetes/Metabolism Research and Reviews. 2018;34(1) doi: 10.1002/dmrr.2928. [DOI] [PubMed] [Google Scholar]
- 5.Imagawa A., Hanafusa T., Uchigata Y., et al. Different contribution of class II HLA in fulminant and typical autoimmune type 1 diabetes mellitus. Diabetologia. 2005;48(2):294–300. doi: 10.1007/s00125-004-1626-x. [DOI] [PubMed] [Google Scholar]
- 6.Kawasaki E., Imagawa A., Makino H., et al. Differences in the contribution of the CTLA4 gene to susceptibility to fulminant and type 1A diabetes in Japanese patients. Diabetes Care. 2008;31(8):1608–1610. doi: 10.2337/dc08-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawabata Y., on behalf of the Committee on Type 1 Diabetes, Society J. D., et al. Differential association of HLA with three subtypes of type 1 diabetes: fulminant, slowly progressive and acute-onset. Diabetologia. 2009;52(12):2513–2521. doi: 10.1007/s00125-009-1539-9. [DOI] [PubMed] [Google Scholar]
- 8.Imagawa A., Hanafusa T., Uchigata Y., et al. Fulminant type 1 diabetes: a nationwide survey in Japan. Diabetes Care. 2003;26(8):2345–2352. doi: 10.2337/diacare.26.8.2345. [DOI] [PubMed] [Google Scholar]
- 9.Imagawa A., Hanafusa T., Makino H., Miyagawa J. I., Juto P. High titres of IgA antibodies to enterovirus in fulminant type-1 diabetes. Diabetologia. 2005;48(2):290–293. doi: 10.1007/s00125-004-1624-z. [DOI] [PubMed] [Google Scholar]
- 10.Shibasaki S., Imagawa A., Tauriainen S., et al. Expression of toll-like receptors in the pancreas of recent-onset fulminant type 1 diabetes. Endocrine Journal. 2010;57(3):211–219. doi: 10.1507/endocrj.K09E-291. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka S., Nishida Y., Aida K., et al. Enterovirus infection, CXC chemokine ligand 10 (CXCL10), and CXCR3 circuit: a mechanism of accelerated beta-cell failure in fulminant type 1 diabetes. Diabetes. 2009;58(10):2285–2291. doi: 10.2337/db09-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohara N., Kaneko M., Nishibori T., et al. Fulminant type 1 diabetes mellitus associated with Coxsackie virus type A2 infection: a case report and literature review. Internal Medicine. 2016;55(6):643–646. doi: 10.2169/internalmedicine.55.5292. [DOI] [PubMed] [Google Scholar]
- 13.Yoneda S., Imagawa A., Fukui K., et al. A histological study of fulminant type 1 diabetes mellitus related to human cytomegalovirus reactivation. The Journal of Clinical Endocrinology and Metabolism. 2017;102(7):2394–2400. doi: 10.1210/jc.2016-4029. [DOI] [PubMed] [Google Scholar]
- 14.Aida K., Nishida Y., Tanaka S., et al. RIG-I- and MDA5-initiated innate immunity linked with adaptive immunity accelerates beta-cell death in fulminant type 1 diabetes. Diabetes. 2011;60(3):884–889. doi: 10.2337/db10-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakata S., Imagawa A., Miyata Y., et al. Low gene expression levels of activating receptors of natural killer cells (NKG2E and CD94) in patients with fulminant type 1 diabetes. Immunology Letters. 2013;156(1-2):149–155. doi: 10.1016/j.imlet.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 16.Xia J., Fjell C. D., Mayer M. L., Pena O. M., Wishart D. S., Hancock R. E. INMEX--a web-based tool for integrative meta-analysis of expression data. Nucleic Acids Research. 2013;41(W1):W63–W70. doi: 10.1093/nar/gkt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia J., Gill E. E., Hancock R. E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nature Protocols. 2015;10(6):823–844. doi: 10.1038/nprot.2015.052. [DOI] [PubMed] [Google Scholar]
- 18.Da W. H., Sherman B. T., Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 19.Ashburner M., Ball C. A., Blake J. A., et al. Gene ontology: tool for the unification of biology. Nature Genetics. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanehisa M., Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia J., Benner M. J., Hancock R. E. NetworkAnalyst--integrative approaches for protein-protein interaction network analysis and visual exploration. Nucleic Acids Research. 2014;42(W1):W167–W174. doi: 10.1093/nar/gku443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou C. H., Shrestha S., Yang C. D., et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Research. 2018;46(D1):D296–D302. doi: 10.1093/nar/gkx1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vlachos I. S., Paraskevopoulou M. D., Karagkouni D., et al. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Research. 2015;43(D1):D153–D159. doi: 10.1093/nar/gku1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan A., Fornes O., Stigliani A., et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Research. 2018;46(D1):p. D1284. doi: 10.1093/nar/gkx1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanafusa T., Imagawa A. Fulminant type 1 diabetes: a novel clinical entity requiring special attention by all medical practitioners. Nature Clinical Practice, Endocrinology & Metabolism. 2007;3(1):36–45. doi: 10.1038/ncpendmet0351. [DOI] [PubMed] [Google Scholar]
- 26.Imagawa A., Hanafusa T. Fulminant type 1 diabetes--an important subtype in East Asia. Diabetes/Metabolism Research and Reviews. 2011;27(8):959–964. doi: 10.1002/dmrr.1236. [DOI] [PubMed] [Google Scholar]
- 27.Imagawa A., Hanafusa T., Awata T., et al. Report of the Committee of the Japan Diabetes Society on the research of fulminant and acute-onset type 1 diabetes mellitus: new diagnostic criteria of fulminant type 1 diabetes mellitus (2012) Journal of Diabetes Investigation. 2012;3(6):536–539. doi: 10.1111/jdi.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung T. S., Chung S. I., Kim M. A., et al. A Korean patient with fulminant autoantibody-negative type 1 diabetes. Diabetes Care. 2004;27(12):3023–3024. doi: 10.2337/diacare.27.12.3023. [DOI] [PubMed] [Google Scholar]
- 29.Moreau C., Drui D., Arnault-Ouary G., Charbonnel B., Chaillous L., Cariou B. Fulminant type 1 diabetes in Caucasians: a report of three cases. Diabetes & Metabolism. 2008;34(5):529–532. doi: 10.1016/j.diabet.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Keir M. E., Latchman Y. E., Freeman G. J., Sharpe A. H. Programmed death-1 (PD-1):PD-ligand 1 interactions inhibit TCR-mediated positive selection of thymocytes. Journal of Immunology. 2005;175(11):7372–7379. doi: 10.4049/jimmunol.175.11.7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lazar-Molnar E., Chen B., Sweeney K. A., et al. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(30):13402–13407. doi: 10.1073/pnas.1007394107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohaegbulam K. C., Assal A., Lazar-Molnar E., Yao Y., Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends in Molecular Medicine. 2015;21(1):24–33. doi: 10.1016/j.molmed.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balar A. V., Weber J. S. PD-1 and PD-L1 antibodies in cancer: current status and future directions. Cancer Immunology, Immunotherapy : CII. 2017;66(5):551–564. doi: 10.1007/s00262-017-1954-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marchand L., Thivolet A., Dalle S., et al. Diabetes mellitus induced by PD-1 and PD-L1 inhibitors: description of pancreatic endocrine and exocrine phenotype. Acta Diabetologica. 2019;56(4):441–448. doi: 10.1007/s00592-018-1234-8. [DOI] [PubMed] [Google Scholar]
- 35.Okamoto M., Okamoto M., Gotoh K., et al. Fulminant type 1 diabetes mellitus with anti-programmed cell death-1 therapy. Journal of Diabetes Investigation. 2016;7(6):915–918. doi: 10.1111/jdi.12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Araujo M., Ligeiro D., Costa L., et al. A case of fulminant type 1 diabetes following anti-PD1 immunotherapy in a genetically susceptible patient. Immunotherapy. 2017;9(7):531–535. doi: 10.2217/imt-2017-0020. [DOI] [PubMed] [Google Scholar]
- 37.Sakai G., Saito D., Nakajima R., et al. Intrinsic insulin secretion capacity might be preserved by discontinuing anti-programmed cell death protein 1 antibody treatment in 'anti-programmed cell death protein 1 antibody-induced' fulminant type 1 diabetes. Journal of Diabetes Investigation. 2018;9(2):448–449. doi: 10.1111/jdi.12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimada A., Morimoto J., Kodama K., et al. T-cell-mediated autoimmunity may be involved in fulminant type 1 diabetes. Diabetes Care. 2002;25(3):635–636. doi: 10.2337/diacare.25.3.635. [DOI] [PubMed] [Google Scholar]
- 39.Shimada A., Oikawa Y., Shigihara T., Senda T., Kodama K. A case of fulminant type 1 diabetes with strong evidence of autoimmunity. Diabetes Care. 2002;25(8):1482–1483. doi: 10.2337/diacare.25.8.1482. [DOI] [PubMed] [Google Scholar]
- 40.Aoki K., Taniyama M., Nagayama C., Oikawa Y., Shimada A. T cell immunity to glutamic acid decarboxylase in fulminant type 1 diabetes without significant elevation of serum amylase. Annals of the New York Academy of Sciences. 2006;1079(1):181–185. doi: 10.1196/annals.1375.028. [DOI] [PubMed] [Google Scholar]
- 41.Fujisawa R., Haseda F., Tsutsumi C., et al. Low programmed cell death-1 (PD-1) expression in peripheral CD4(+) T cells in Japanese patients with autoimmune type 1 diabetes. Clinical and Experimental Immunology. 2015;180(3):452–457. doi: 10.1111/cei.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiss H., Bleich A., Hedrich H. J., et al. Genetic analysis of the LEW.1AR1-iddm rat: an animal model for spontaneous diabetes mellitus. Mammalian Genome. 2005;16(6):432–441. doi: 10.1007/s00335-004-3022-8. [DOI] [PubMed] [Google Scholar]
- 43.Pandey V. K., Mathur A., Khan M. F., Kakkar P. Activation of PERK-eIF2α-ATF4 pathway contributes to diabetic hepatotoxicity: attenuation of ER stress by Morin. Cellular Signalling. 2019;59:41–52. doi: 10.1016/j.cellsig.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 44.Juliana C. A., Yang J., Cannon C. E., Good A. L., Haemmerle M. W., Stoffers D. A. A PDX1-ATF transcriptional complex governs β cell survival during stress. Molecular Metabolism. 2018;17:39–48. doi: 10.1016/j.molmet.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J., Liang L., Zhan L., et al. ZiBuPiYin recipe protects db/db mice from diabetes-associated cognitive decline through improving multiple pathological changes. PLoS One. 2014;9(3, article e91680) doi: 10.1371/journal.pone.0091680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim G. H., Park E. C., Yun S. H., et al. Proteomic and bioinformatic analysis of membrane proteome in type 2 diabetic mouse liver. Proteomics. 2013;13(7):1164–1179. doi: 10.1002/pmic.201200210. [DOI] [PubMed] [Google Scholar]
- 47.Matsunaga Y., Negishi T., Hatakeyama A., Kawagoe Y., Sawano E., Tashiro T. Impairment of synaptic development in the hippocampus of diabetic Goto-Kakizaki rats. International Journal of Developmental Neuroscience. 2016;53(1):58–67. doi: 10.1016/j.ijdevneu.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 48.Tucsek Z., Noa Valcarcel-Ares M., Tarantini S., et al. Hypertension-induced synapse loss and impairment in synaptic plasticity in the mouse hippocampus mimics the aging phenotype: implications for the pathogenesis of vascular cognitive impairment. GeroScience. 2017;39(4):385–406. doi: 10.1007/s11357-017-9981-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bradfield J. P., Qu H. Q., Wang K., et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genetics. 2011;7(9, article e1002293) doi: 10.1371/journal.pgen.1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kostic S., Williams B., Ksouri S., et al. Changes in snail and SRF expression in the kidneys of diabetic rats during ageing. Acta Histochemica. 2020;122(1):p. 151460. doi: 10.1016/j.acthis.2019.151460. [DOI] [PubMed] [Google Scholar]
- 51.Sun C., Wei H., Chen X., et al. ERBB3-rs2292239 as primary type 1 diabetes association locus among non-HLA genes in Chinese. Meta Gene. 2016;9:120–123. doi: 10.1016/j.mgene.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frohnert B. I., Laimighofer M., Krumsiek J., et al. Prediction of type 1 diabetes using a genetic risk model in the Diabetes Autoimmunity Study in the Young. Pediatric Diabetes. 2018;19(2):277–283. doi: 10.1111/pedi.12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang D., Pan G. The association between rs2292239 Polymorphism in ERBB3 Gene and type 1 diabetes: a meta-analysis. BioMed Research International. 2019;2019:7. doi: 10.1155/2019/7689642.7689642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prochazka M., Serreze D. V., Worthen S. M., Leiter E. H. Genetic control of diabetogenesis in NOD/Lt mice. Development and analysis of congenic stocks. Diabetes. 1989;38(11):1446–1455. doi: 10.2337/diab.38.11.1446. [DOI] [PubMed] [Google Scholar]
- 55.Aparicio J. M., Wakisaka A., Takada A., Matsuura N., Yoshiki T. Non-HLA genetic factors and insulin dependent diabetes mellitus in the Japanese: TCRA, TCRB and TCRG, INS, THY1, CD3D and ETS1. Disease Markers. 1990;8(5):294. [PubMed] [Google Scholar]
- 56.Aparicio J. M. HLA and non-HLA genetic factors in Japanese IDDM, [Hokkaido igaku zasshi] The Hokkaido Journal of Medical Science. 1991;66(6):780–793. [PubMed] [Google Scholar]
- 57.Levi I., Segev Y., Priel E. Type 1 diabetes affects topoisomerase I activity and GlcNAcylation in rat organs: kidney, liver and pancreas. Glycobiology. 2012;22(5):704–713. doi: 10.1093/glycob/cws008. [DOI] [PubMed] [Google Scholar]
- 58.Wang F., Sun F., Luo J., et al. Loss of ubiquitin-conjugating enzyme E2 (Ubc9) in macrophages exacerbates multiple low-dose streptozotocin-induced diabetes by attenuating M2 macrophage polarization. Cell Death & Disease. 2019;10(12):p. 892. doi: 10.1038/s41419-019-2130-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jin D. K., Fish A. J., Wayner E. A., et al. Distribution of integrin subunits in human diabetic kidneys. Journal of the American Society of Nephrology: JASN. 1996;7(12):2636–2645. doi: 10.1681/ASN.V7122636. [DOI] [PubMed] [Google Scholar]
- 60.Aaron-Brooks L. M., Sasaki T., Vickman R. E., et al. Hyperglycemia and T cell infiltration are associated with stromal and epithelial prostatic hyperplasia in the nonobese diabetic mouse. The Prostate. 2019;79(9):980–993. doi: 10.1002/pros.23809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou C., Pridgen B., King N., Xu J., Breslow J. L. Hyperglycemic Ins2AkitaLdlr(-)/(-) mice show severely elevated lipid levels and increased atherosclerosis: a model of type 1 diabetic macrovascular disease. Journal of Lipid Research. 2011;52(8):1483–1493. doi: 10.1194/jlr.M014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carrero J. A., Benshoff N. D., Nalley K., Unanue E. R. Type I and II interferon receptors differentially regulate type 1 diabetes susceptibility in male versus female NOD mice. Diabetes. 2018;67(9):1830–1835. doi: 10.2337/db18-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu F., Sahoo R., Ge X., et al. Deficiency of the complement regulatory protein CD59 accelerates the development of diabetes-induced atherosclerosis in mice. Journal of Diabetes and its Complications. 2017;31(2):311–317. doi: 10.1016/j.jdiacomp.2016.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soggiu A., Piras C., Bonizzi L., Hussein H. A., Pisanu S., Roncada P. A discovery-phase urine proteomics investigation in type 1 diabetes. Acta Diabetologica. 2012;49(6):453–464. doi: 10.1007/s00592-012-0407-0. [DOI] [PubMed] [Google Scholar]
- 65.Liu H., Xu R., Liu X., Sun R., Wang Q. Bioinformatics analysis of gene expression in peripheral blood mononuclear cells from children with type 1 diabetes in 3 periods. Experimental and Clinical Endocrinology & Diabetes. 2014;122(8):477–483. doi: 10.1055/s-0034-1372599. [DOI] [PubMed] [Google Scholar]
- 66.Fang C., Huang Y., Pei Y., et al. Genome-wide gene expression profiling reveals that CD274 is up-regulated new-onset type 1 diabetes mellitus. Acta Diabetologica. 2017;54(8):757–767. doi: 10.1007/s00592-017-1005-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: the list of all differentially expressed genes. Supplementary Table 2: the top five targeted genes regulated by miRNA. Supplementary Table 3: the top five targeted genes regulated by transcription factor.
Data Availability Statement
The data used to support the findings of this study are included within the article.