

Abstract
Neurotoxic side effects of traditional systemic chemotherapy are abundantly described. The introduction of newly developed biologic therapeutics and cellular immune effector therapies has expanded the spectrum of neurotoxicity. Multifocal necrotizing leukoencephalopathy (MNL) is a pathologic condition of unknown etiology that has been observed in patients after prolonged critical illness. We observed a case of MNL in a patient treated with extensive multimodal therapy including chimeric antigen receptor T cells. A month before death, MRI demonstrated signs of inflammation and developing edema in brainstem structures. At autopsy the abnormal MRI regions showed a wave-like loss of microglia with hemorrhagic MNL in regions closest to the brain surface. These findings reiterate the susceptibility of white matter to antineoplastic therapy and suggest new mechanisms of neurotoxicity when traditional chemotherapy is combined with biologic or cellular effector therapy.
Keywords: Chemotherapy, Chimeric antigen receptor T-cell (CAR-T) therapy, Inotuzumab-ozogamicin, Multifocal necrotizing leukoencephalopathy (MNL), Neurotoxicity
INTRODUCTION
Multifocal necrotizing leukoencephalopathy (MNL) is pathologic condition affecting patients with prolonged critical illnesses in the terminal stages of life. The signature white matter lesion presents in the basis pontis. Histologically, the white matter shows discrete foci of white matter vacuolation, myelin loss, axonal damage, and relatively minimal glial reaction (1–3). The majority of affected patients have impaired immune function in the context of chronic illness, classically in the form of HIV/AIDS or systemic chemotherapy for malignancy. The clinical presentation of this process is not well characterized and the diagnosis is made on postmortem examination.
Although many traditional chemotherapeutic agents have well described patterns of neurotoxicity, the neuropathologic effects of newly developed biologic and immune effector cellular therapy is far less established. Chimeric antigen receptor T-cell (CAR-T) therapy for large B-cell lymphoma and pediatric acute lymphoblastic leukemia utilizes patient T cells transduced with a chimeric anti-CD19 antibody fragment/intracellular CD3 signaling domains to target B cells for non-MHC dependent T-cell directed killing (4). Initial studies were complicated by a high frequency of unexpected neurologic side effects, including tremor, aphasia, seizure, and fatal cerebral edema (5–8). Although the clinical presentation of CAR-T neurotoxicity in the acute setting is now well established, the mechanisms of toxicity or possible long-term effects are not understood.
There are only 3 published detailed neuropathologic autopsy studies of patients treated with CAR-T. The first by Gust et al (9) examined 2 patients who experienced severe clinical neurotoxicity and died in the acute window after CAR-T administration (3–13 days after administration). Neuropathologic analysis revealed evidence of severe blood-brain barrier disruption in the form of red blood cell extravasation, intravascular fibrin thrombi, and fibrinoid vascular necrosis with much of the damage occurring in the brainstem. One of the 2 patients showed significant CAR-T infiltration into the CNS and CSF. These pathologic findings were associated with severe systemic cytokine response. The authors proposed that in a subset of patients, the peripheral cytokine release triggered by CAR-T administration results in endothelial activation and stress with subsequent blood-brain barrier disruption and injury. Torre et al (10) published a similar study of a patient who died of acute cerebral edema 5 days after CAR-T administration. This study further demonstrated acute vascular injury, showing perivascular edema, fibrin deposition, and acute glial injury. This patient did not show significant parenchymal T-cell infiltrate and CAR-T cells were not demonstrated in the CNS. These findings further supported the cytokine based acute vascular injury model. Finally, Gust et al (11) published a case report of a pediatric patient who died of disease recurrence 3 years after CAR-T therapy. This study demonstrated periventricular Chaslin gliosis, evidence of prior vascular injury, and suggestion of neuronal loss.
Inotuzumab-ozogamicin is another recent successful biologic treatment for refractory B-cell malignancies, primarily acute lymphoblastic leukemia. It consists of a humanized monoclonal anti-CD22 antibody conjugated to ozogamicin, a potent pan-cellular toxin. CD22 is a member of the immunoglobin superfamily of transmembrane proteins, and is involved in inhibition of B-cell receptor signaling (12). It is expressed predominantly on B cells. Upon binding CD22, the antibody-toxin conjugate is rapidly endocytosed, allowing ozogamicin to exert its cytotoxic effects via DNA strand scission (13). The primary toxicity associated with this treatment is bone marrow suppression and hepatic toxicity (13), and no significant neurotoxicity has been associated with this treatment to date.
Here, we present a case of a variant of MNL with predominantly superficial brainstem distribution associated with selective microglial toxicity in a patient previously treated with traditional chemotherapy, CAR-T therapy, and Inotuzumab-ozogamicin.
Case Presentation
The patient was a 59-year-old woman who was diagnosed with diffuse large B-cell lymphoma 2 years previously. At initial presentation, she had extensive bone marrow and CNS meningeal involvement. She was treated with Rituximab with hyper-fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (R-hyper-CVAD) and intrathecal methotrexate and cytarabine with complete response. Part B of the last cycle of hyper-CVAD treatment was discontinued due to lower extremity motor weakness, not due to the lymphoma, and from which she experienced substantial improvement by withholding the treatment. She remained disease free for 14 months when she experienced her first relapse, detected by flow cytometry and confirmed by bone marrow biopsy. Salvage therapy was started with dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin (DA-EPOCH) with minimal response.
Given the refractoriness of the lymphoma to standard salvage chemotherapy, the clinical team pursued CAR-T therapy. She received lymphodepletion chemotherapy with fludarabine and cyclophosphamide and a subsequent single dose of Axicabtagene ciloleucel CAR-T. Her course was complicated by grade 4 cytokine release syndrome and grade 4 neurotoxicity, according to the American Society for Transplantation and Cellular Therapy consensus grading, and treated with Tocilizumab and steroids (14). Her neurotoxicity was initially characterized by tremor and word finding difficulties eventually progressing to unresponsiveness not requiring intubation on days 3–6 after CAR-T administration. Her symptoms began to resolve on day 7 postadministration, eventually returning to her neurologic baseline. Her next bone marrow biopsy showed complete response.
Unfortunately, the patient experienced another relapse roughly 6 months later. She received additional treatment with hyper-CVAD and Inotuzumab-ozogamicin with minimal response. She was switched to EPOCH and received additional intrathecal methotrexate and cytarabine. A month after her relapse during the salvage treatment she began to experience high fever and progressive altered mental status, characterized by confusion, aphasia, and obtundation. An extensive metabolic, infectious, and endocrine workup failed to reveal a specific etiology. Inflammatory markers were significantly elevated (C-Reactive Protein 10–15 mg/dl, sedimentation rate >145). MRI ∼1 month prior to death demonstrated diffuse failure of CSF signal suppression on the T2 FLAIR sequences (not shown), which was concerning for an inflammatory or infectious process. CSF sampling revealed elevated protein (993 mg/dl) and only a single white blood cell. In addition, symmetric edema within the brainstem, medial cerebellar hemispheres, and portions of the deep gray nuclei developed over a course of 5 days (Fig. 1A–C). There was no evidence of restricted diffusion to suggest an acute infarct or infection. No hemorrhage was evident on blood sensitive sequences.
FIGURE 1.
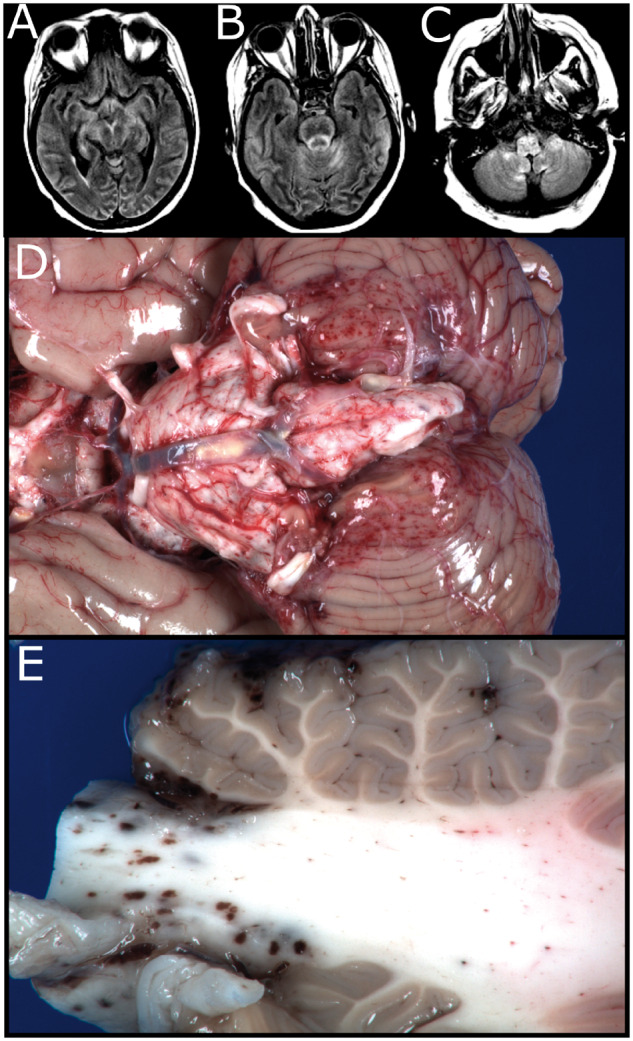
Imaging and gross findings. Selected axial T2 FLAIR images, ∼1 month prior to death. (A) Development of edema involving the thalami, lentiform nuclei, and mesial temporal lobe structures. (B, C) Development of symmetric bilateral edema involving the mesial temporal lobe structures and throughout the brainstem and medial cerebellar hemispheres. (D) Gross image of the unfixed brain showing superficial petechial hemorrhages over the brainstem, cerebellum, and basilar supratentorial structures. (E) Cut surface of the cerebellar peduncle showing extent of petechial hemorrhages involving predominantly white matter and focally involving the cerebellar folia.
Despite therapy with empiric antibiotics, pulse dose steroids, and intravenous immunoglobin, the patient’s mental status continued to deteriorate and she passed away 1 month after the onset of her cognitive decline. The family requested a brain-only autopsy.
Pathologic Findings
The fresh brain weighed 1030 g. External examination demonstrated petechial hemorrhages over the bilateral cerebellar tonsils, bilateral anterior-superior cerebellar hemispheres, bilateral cerebral peduncles, optic chiasm, optic tracts, and mamillary bodies (Fig. 1D). There were no cingulate, uncal, or cerebellar tonsillar herniations. On sectioning of the fresh brain, additional petechial hemorrhages were found on the superficial aspects of the medulla, pons, midbrain, posterior thalamus, and cerebellar peduncles (Fig. 1E). The cerebral hemispheres were grossly unremarkable and no gross infarctions or masses were identified.
Microscopic examination revealed numerous acute lesions corresponding to the grossly observed petechial hemorrhages. There was severe vacuolation of the involved parenchyma, which was predominantly but not exclusively white matter, with many lesions showing freshly extravasated RBCs (Fig. 2A). The lesions were otherwise hypocellular on hematoxylin and eosin (H&E) stain without a significant inflammatory component and with frequent axonal spheroids (Fig. 2B). The lesions penetrated to a depth of 1–3 mm, and were nearly circumferential in areas such as the cervical cord and medulla. Luxol fast blue/periodic acid-Schiff stains demonstrated severe loss of myelin in the lesions without signs of active demyelination, (Fig. 2C). No B cells and only rare T cells were present on CD20 and CD3 immunostains, respectively (data not shown). Astrocytes were present at the periphery of the lesions and showed moderate activation and some cellular process beading (Figs. 2D and 3A, B). Neurofilament and amyloid precursor protein stains showed partially preserved neurofilament but severe axonal damage within the vacuolated lesions (Fig. 2E). CD31 stains showed decreased endothelial cells in and surrounding the lesions and staining for membrane attack complex demonstrated strong staining in the remaining vessels in the lesions, with some labeling in endothelia beyond perimeter of lesion (Fig. 2F, G). Demyelination and axonopathic changes were confirmed by electron microscopy (Fig. 3D, E). Osmiophilic deposits, compatible with complement deposition, were seen on opposed endothelial processes and basement membrane (Fig. 4F). TUNEL staining was positive in endothelial cells and possible residual microglia within the lesions (Supplementary Fig. S1).
FIGURE 2.

Routine histology and immunohistochemistry. (A) Low-power hematoxylin and eosin-stained formalin-fixed paraffin-embedded section of cerebellar peduncle showing areas of vacuolation and acute hemorrhage (scale bar: 1 mm). Areas of cerebellar gray matter were also focally involved (not shown). (B) Higher-power view of region of interest showing numerous axonal spheroids and minimal cellular reaction (scale bar: 300 µm). (C) Luxol fast blue/periodic acid-Schiff stain shows loss of myelin without evidence of active demyelination (scale bar: 1 mm). (D) GFAP stain shows loss of astrocytes in the lesion with moderate activation of the surrounding astrocytes (scale bar: 1 mm). (E) Amyloid precursor protein stain highlights the axonal damage (scale bar: 1 mm). (F) Membrane attack complex highlights vessels in and at the periphery of the lesion (scale bar: 1 mm). (G) CD31 shows widespread paucity of vessels in region of the peripheral lesion, with relative preservation in the central white matter (scale bar: 1 mm). (H) Iba1 stain shows near total loss of microglia in the peripheral lesion and surrounding parenchyma, in comparison to the relative normal density in the central white matter (scale bar: 1 mm).
FIGURE 3.

In situ hybridization and electron microscopy. (A) In situ hybridization for GFAP of formalin-fixed paraffin-embedded embedded medulla demonstrating expected perivascular distribution and loss of expression in the vacuolated lesion (scale bar: 500 µm). (B) In situ hybridization demonstrates YKL expression in activated astrocytes (scale bar: 500 µm). (C) In situ hybridization for CSF1R fails to highlight any residual microglia (scale bar: 500 µm). (D) Thick section of plastic embedded brainstem stained with toluidine blue. Upper right corner shows lesion edge with preserved myelinated fibers the remaining tissue shows severe vacuolation with some residual cellular elements and vessels at bottom. (E) Low-power transmission electron micrograph from edge of lesion demonstrating axons (yellow) with retained surrounding compact myelin. Dilated axonal profiles with and without retained subcellular organelles are interspersed amongst the myelinated fibers. A single blood vessel courses from top to bottom on the right side. The endothelium is colored green, whereas the lumen is colored blue (scale bar: 5 µm). (F) Higher-power transmission electron micrograph of blood vessel showing electron dense junction between apposed endothelial processes (between arrows) and electron dense deposits along endothelial basement membrane (arrowheads) consistent with complement deposition (scale bar: 1 µm). (G) Rare perivascular microglial (red) present at the lesional edge shows cytoplasmic vacuolation with electron dense bodies (scale bar: 1 µm).
FIGURE 4.
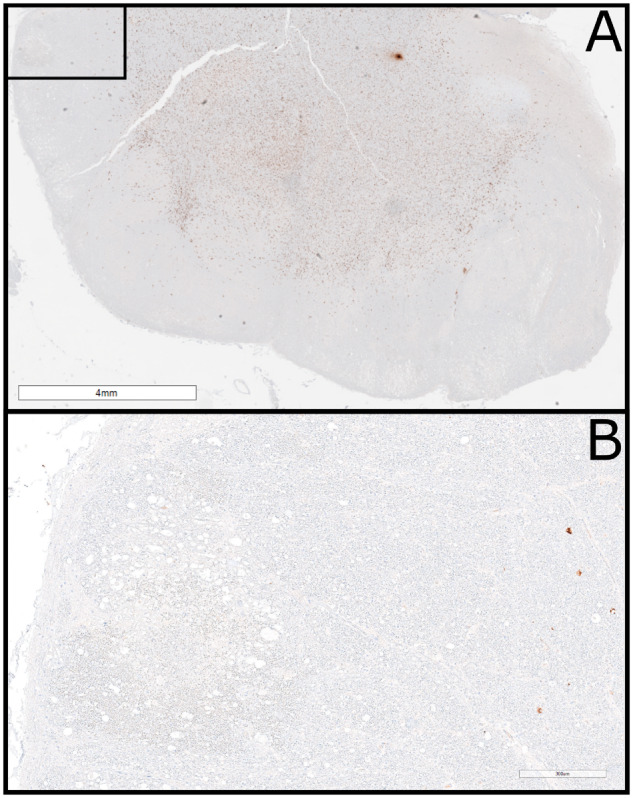
Extensive loss of microglia. (A) Formalin-fixed paraffin-embedded section of medulla immunostained for Iba1 showing extensive loss of microglia in a superficial circumferential pattern (scale bar: 4 mm). (B) Higher-power view of the inset in Figure 3A showing microglia loss in the medulla with microglial loss extending much deeper than the vacuolated white matter lesions (scale bar: 300 µm).
The most striking finding was an almost total loss of microglia on the superficial aspects of the brain regions with MNL (Figs. 2H and 4A, B). The loss of microglia extended to a depth of 6 mm, often significantly deeper than the acute necrotizing lesions. It was predominantly present in white matter and involved areas distant from the acute lesions but was occasionally noted in adjacent gray matter (cerebellar cortex). Loss of microglia was confirmed by additional immunostains for CD163, P2RY12, and CD68 (not shown) as well as by in situ hybridization for CSF1R (Fig. 3C). Electron microscopy demonstrated loss of microglia within the lesions, and only rare small vessels at the perimeter of the lesion were surrounded by microglia with vacuolated cytoplasm and electron dense subcellular structures (Fig. 3G).
Additional pathologic changes included pallor and rarefication of the deep white matter and a few scattered microscopic subacute to remote infarctions. There was no morphologic, immunohistochemical, or flow cytometric evidence of residual lymphoma in the CNS. Molecular studies performed by Kite pharmaceuticals found no persistence of CAR-T cells in the brain.
DISCUSSION
In addition to protecting us from infectious agents, our immune system is critical to staunching emergent neoplasia. Therefore, it is not surprising that immunosuppression is associated with both novel infectious agents and malignancies. Neuropathologists are familiar with opportunistic infections and neoplasms that have characterized immunosuppression with classical protocols employing antimetabolites and steroids; however, great strides have been made in the advent of novel immunological therapies to treat cancer, and with these advances novel forms of neurotoxicity have emerged. Recognizing these diseases and their mechanism of action provide new challenges for the neuropathologist. The speed of development of the new immunological therapies accents the importance of learning quickly from these cases as they emerge.
Traditionally, the study of new diseases is aided by utilizing animal models. However, in the case of precision medicine where new immunological therapies are tailored to the individual patient, this classic approach may not be feasible. Therapies are so targeted that neurotoxicity may be species specific, or possibly even patient specific. Analysis of individual human cases is complicated by variations in therapeutic protocols and time of death relative to presentation of neurotoxicity. This is particularly true in the case of CAR-T neurotoxicity where current neuropathologic reports to date are limited to either the periods of hyperacute cerebral edema or remote from treatment when only nonspecific chronic changes are present.
This case demonstrated a unique pattern of neurologic injury associated with extensive chemotherapeutic, biologic, and immune effector cellular therapy for diffuse large B-cell lymphoma. The imaging studies performed a month prior to the patient’s death at the onset of her cognitive decline demonstrated symmetric edema throughout the brainstem and deep gray nuclei. After excluding infection, these findings in this clinical setting are most compatible with a toxic etiology. These imaging findings could correlate with the microglial toxicity or vasculopathy, which are favored to be more chronic than the acute MNL type lesions that developed in the days before death and were not imaged.
Histologically, the individual acute lesions resembled those described in MNL, but with an atypical distribution involving predominantly superficial brainstem structures. These acute lesions were characterized by hemorrhage, axonal injury, myelin loss, endothelial injury, and complement deposition. Interestingly, these lesions are histologically somewhat similar to those described in the acute window of CAR-T therapy by Gust et al (9), but no evidence of residual CAR-T cells was found on molecular analysis by Kite pharmaceuticals in our case. In addition, our case also showed extensive specific loss of microglia in the superficial surfaces of the posterior fossa, a phenomenon that has not been previously reported. The relationship between the leukoencephalopathy and the microglial loss is not clear, with the microglial injury extending beyond and involving areas not affected by the leukoencephalopathy.
The superficial pattern of injury strongly suggests a topical toxic insult. The patient did receive standard intrathecal methotrexate and cytarabine in the month before and in the initial stages of her cognitive decline. Intrathecal methotrexate and cytarabine are known to cause disseminated leukoencephalopathy in a subset of exposed patients, especially those receiving concomitant radiation therapy (15); however, lesions in these traditionally treated patients are characterized by abundant macrophage infiltration in the acute stages and gliosis in the chronic stages, neither of which were seen in our patient. The other standard chemotherapeutic agents she received also have the potential for significant neurotoxicity, most notably fludarabine (16, 17), but they do not show this pattern of injury. Some of the pathologic features share similarities with osmotic demyelination syndromes; however, the patient’s sodium was extensively monitored throughout her course and remained close to the normal range in the 6 months prior to her death (130–153 mMol/l), without rapid shifts or corrections.
There are several possible explanations for the unusual pattern of injury seen in our patient, involving possible unexpected interactions of the multimodal therapies to which she was exposed. One possibility involves a subacute vascular injury caused by the CAR-T therapy. Acute brain vascular injury secondary to CAR-T in lymphoma has been well established, but little is known about the subacute or long-term vascular effects. Similar to the acute vascular injury, a subacute vascular injury initiated by the systemic cytokine release may have predisposed the patient to a severe and abnormal pattern of injury in response to the subsequent intrathecal chemotherapy. The idea of a predisposing vascular injury is supported by the imaging studies she received a month prior to her death, showing T2 abnormalities in a distribution similar to her eventual acute necrotizing injury. In addition, the lesions in this patient showed significant acute complement deposition and a more chronic loss of vasculature.
The selective toxicity to microglia is more difficult to explain and has not been previously reported in response to standard chemotherapy or CAR-T therapy. The superficial distribution suggests a process related to an intrathecal toxin, but the distribution of loss is similar to the MR abnormalities seen 1 month before death and more extensive and chronic than the acute necrotizing lesions. One intriguing possibility relates to the use of Inotuzumab-ozogomacin in this patient. Although CD22 is predominantly expressed on B cells, recent studies in mice have demonstrated microglial expression of CD22, particularly in aged and activated microglia (18). Microglial activation in response to intrathecal chemotherapy could result in microglial expression of CD22, and susceptibility to subsequent Inotuzumab treatment. This could explain the specificity and distribution of microglial loss in this patient. We attempted to demonstrate CD22 expression in microglia in this patient through immunohistochemistry and in situ hybridization, but these studies were negative (data not shown). Previous in vitro studies have also demonstrated neuronal expression of CD22 in inflammatory states, raising an alternate pathway of possible neuronal toxicity that was not observed in this case (19). Future studies will need to address the possibility of human microglial and/or neuronal CD22 expression, and what effect this may have on current and future CD22 directed therapies.
In conclusion, we have shown a novel pattern of MNL associated with extensive loss of microglia. As the introduction of new treatment modalities for cancer accelerates, extensive neuropathologic monitoring for unexpected adverse neuropathologic reactions or interactions is warranted.
Supplementary Material
The banking and processing of the brain was supported by the University of Pittsburgh Brain Institute and National Institute on Aging (NIA) P50 AG005133.
The authors have no duality or conflicts of interest to declare.
REFERENCES
- 1. Anders KH, Becker PS, Holden JK, et al. Multifocal necrotizing leukoencephalopathy with pontine predilection in immunosuppressed patients: A clinicopathologic review of 16 cases. Hum Pathol 1993;24:897–904 [DOI] [PubMed] [Google Scholar]
- 2. Premji S, Kang L, Rojiani MV, et al. Multifocal necrotizing leukoencephalopathy: Expanding the clinicopathologic spectrum. J Neuropathol Exp Neurol 2019;78:340–7 [DOI] [PubMed] [Google Scholar]
- 3. Rubinstein LJ, Herman MM, Long TF, et al. Disseminated necrotizing leukoencephalopathy: A complication of treated central nervous system leukemia and lymphoma. Cancer 1975;35:291–305 [DOI] [PubMed] [Google Scholar]
- 4. June CH, Sadelain M.. Chimeric antigen receptor therapy. N Engl J Med 2018;379:64–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karschnia P, Jordan JT, Forst DA, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood 2019;133:2212–21 [DOI] [PubMed] [Google Scholar]
- 6. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.JCAR015. JCAR015 in ALL: A Root-Cause Investigation. Cancer Discov 2018;8:4–5 [DOI] [PubMed] [Google Scholar]
- 9. Gust J, Hay KA, Hanafi LA, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov 2017;7:1404–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Torre M, Solomon IH, Sutherland CL, et al. Neuropathology of a case with fatal car t-cell-associated cerebral edema. J Neuropathol Exp Neurol 2018;77:877–82 [DOI] [PubMed] [Google Scholar]
- 11. Gust J, Finney OC, Li D, et al. Glial injury in neurotoxicity after pediatric CD19-directed chimeric antigen receptor T cell therapy. Ann Neurol 2019;86:42–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walker JA, Smith KG.. CD22: An inhibitory enigma. Immunology 2008;123:314–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yurkiewicz IR, Muffly L, Liedtke M.. Inotuzumab ozogamicin: A CD22 mAb-drug conjugate for adult relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Drug Des Devel Ther 2018;12:2293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant 2019;25:625–38 [DOI] [PubMed] [Google Scholar]
- 15. Kim JY, Kim ST, Nam DH, et al. Leukoencephalopathy and disseminated necrotizing leukoencephalopathy following intrathecal methotrexate chemotherapy and radiation therapy for central nerve system lymphoma or leukemia. J Korean Neurosurg Soc 2011;50:304–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beitinjaneh A, McKinney AM, Cao Q, et al. Toxic leukoencephalopathy following fludarabine-associated hematopoietic cell transplantation. Biol Blood Marrow Transplant 2011;17:300–8 [DOI] [PubMed] [Google Scholar]
- 17. Spriggs DR, Stopa E, Mayer RJ, et al. Fludarabine phosphate (NSC 312878) infusions for the treatment of acute leukemia: Phase I and neuropathological study. Cancer Res 1986;46:5953–8 [PubMed] [Google Scholar]
- 18. Pluvinage JV, Haney MS, Smith BAH, et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 2019;568:187–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mott RT, Ait-Ghezala G, Town T, et al. Neuronal expression of CD22: Novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia 2004;46:369–79 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.