

Summary
Diabetic peripheral neuropathy (DPN) is a common diabetic complication and has yet no efficient medication. Here, we report that antispasmodic drug drofenine (Dfe) blocks Kv2.1 and ameliorates DPN-like pathology in diabetic mice. The underlying mechanisms are investigated against the DPN mice with in vivo Kv2.1 knockdown through adeno associated virus AAV9-Kv2.1-RNAi. Streptozotocin (STZ) induced type 1 or db/db type 2 diabetic mice with DPN exhibited a high level of Kv2.1 protein in dorsal root ganglion (DRG) tissue and a suppressed neurite outgrowth in DRG neuron. Dfe promoted neurite outgrowth by inhibiting Kv2.1 channel and/or Kv2.1 mRNA and protein expression level. Moreover, it suppressed inflammation by repressing IκBα/NF-κB signaling, inhibited apoptosis by regulating Kv2.1-mediated Bcl-2 family proteins and Caspase-3 and ameliorated mitochondrial dysfunction through Kv2.1/CaMKKβ/AMPK/PGC1α pathway. Our work supports that Kv2.1 inhibition is a promisingly therapeutic strategy for DPN and highlights the potential of Dfe in treating this disease.
Subject Areas: Human Metabolism, Immunology
Graphical Abstract
Highlights
-
•
Antispasmodic drug drofenine (Dfe) ameliorates DPN-like pathology in diabetic mice
-
•
Dfe inhibits Kv2.1 channel and/or Kv2.1 mRNA and protein expression level
-
•
Dfe represses inflammation, apoptosis, and mitochondrial dysfunction in DPN mice
-
•
Kv2.1 inhibition is a therapeutic tactic and Dfe shows therapeutic potential for DPN
Human Metabolism; Immunology
Introduction
Diabetic peripheral neuropathy (DPN) is a microvascular complication of diabetes afflicting more than two-thirds of diabetic patients (Liu et al., 2017; Hur et al., 2015; Vincent et al., 2011). Clinical characteristics of DPN include pain, paresthesia, and sensory loss, which are the risk factors for burns, injuries, and foot ulceration (Tesfaye et al., 2012; Bönhof et al., 2019). DPN severely affects the life quality of diabetic patients (Tesfaye et al., 2012; Boulton et al., 2005), but there is yet no efficient treatment against this disease.
DPN is a complicated disease with complex pathological mechanisms. For example, mitochondrial dysfunction is closely linked to the occurrence and development of DPN, and it occurs in sensory neurons and contributes to distal axonopathy in animal models of diabetic neuropathy (Roy Chowdhury et al., 2012). In addition, apoptosis is also associated with DPN, as exemplified by the finding that apoptosis is increased in acutely dissociated DRG neuron from 3- to 6-week-old diabetic rats (Srinivasan et al., 2000). Besides, inflammation is severely implicated in DPN, as indicated by the published reports that patients with DPN have high levels of TNF-α and IL-6 in plasma and rodents with DPN show increased levels of these two proinflammatory cytokines in sciatic nerve (Nguyen et al., 2012; Wilson, 2011; Duksal et al., 2016). Currently, dozens of clinical medications are available, including antioxidant alfa lipoic acid, aldose reductase inhibitor epalrestat, neurotrophic agent mecobalamin, and vasoactive drug alprostadil, but they only can alleviate the symptoms of DPN (Zhao et al., 2018; Hotta et al., 2006; Jiang et al., 2015, 2016). Thus, it is full of challenges to design new generation of anti-DPN drugs based on new therapeutic strategies.
Voltage-gated potassium (Kv) channels participate in varied physiological events, such as neuronal discharge pattern, synaptic integration, and neurotransmitter release (Yang et al., 2018; Romer et al., 2016; Wei et al., 2018). In structure, Kv channels are tetramers consisting of six transmembrane helixes and one pore subunit. Abnormal structure and dysfunction of Kv channel are tightly linked to many kinds of diseases, including neurodegenerative diseases (Chao et al., 2017; Frazzini et al., 2016). Kv channel family is divided into 12 subfamilies, including Kv1-12. Among them, Kv2 family mainly has two members, Kv2.1 and Kv2.2 (Johnson et al., 2018). Kv2.1 is enriched in varied tissues and organs such as brain tissue, peripheral tissue, retina, heart, and pancreas (Frolov et al., 2014; Jacobson et al., 2007; MacDonald, 2002). It has been also reported recently that Kv2.1 is also expressed in dorsal root ganglion (DRG) tissue (Tsantoulas et al., 2014) together with other Kv channels, including Kv1.1, Kv1.2, Kv1.4, Kv2.2, Kv3.4, Kv4.2, Kv4.3 (Cao et al., 2010). Kv2.2 is mainly distributed in brain tissue and participates in the periodic physiological processes of sleep and wakefulness (Hermanstyne et al., 2013).
Kv2.1 channel is involved in multiple functions of neuron, including cell discharge, classical delayed rectifier potassium current encoding, and neuronal action potential repolarization (Pathak et al., 2016). Abnormal sensory symptoms of DPN include pain and hypoalgesia in late stage of DPN (Liu et al., 2017), and Kv2.1 channel may function potently as an excitatory brake that can be compromised under chronic pain conditions (Cao et al., 2010). Demyelination is one of the mainly pathophysiological features of DPN, which can be relieved by protecting and promoting the neurite outgrowth of sensory neuron (Calcutt et al., 2017). Recently, it has been reported that Kv2.1 is essential for the excitability of neuron and functions of afferent myelination (Jensen et al., 2017). Abnormal Kv2.1 regulation may cause outflow of potassium ions resulting in apoptotic reaction cascades and further mitochondrial dysfunction and inflammation reaction (Ramirez et al., 2016; Pal et al., 2003). Thus, it is suggested that Kv2.1 channel be associated with demyelination, apoptosis, mitochondrial dysfunction, and inflammation, further addressing the mediation of Kv2.1 channel in the pathological process of DPN.
In the current work, we reported that antispasmodic drug drofenine (Dfe, drofenine hydrochloride was here used for better solubility. Figure 1A) as a Kv2.1 inhibitor efficiently ameliorated DPN-like pathology of diabetic mice. The mechanisms underlying the Kv2.1 inhibition-mediated amelioration have been intensively investigated by in vivo Kv2.1 knockdown assay with adeno associated virus AAV9-Kv2.1-RNAi in diabetic mice with DPN. Our work has highly supported that Kv2.1 inhibition is a promising therapeutic strategy for DPN and shed light on the potential of Dfe in the treatment of this disease.
Figure 1.

Dfe Was an Inhibitor of Kv2.1 Channel
SP6616 or GxTx-1E (GE) was used as a positive control.
(A) Chemical structure of Dfe.
(B) CHO-Kv2.1 cell was incubated with SP6616 (20 μM) or GxTx-1E (GE, 100 nM) and membrane potential dye for 30 min, and signal was recorded. Data were shown as area under the curve (AUC). N = 6. SP6616 versus DMSO by Student's t test: ∗∗∗p < 0.001. GE versus DMSO by Student's t test: ∗∗∗p < 0.001.
(C) CHO-Kv2.1 cell was incubated with SP6616 (20 μM) or Dfe (5, 10, 20 μM) and membrane potential dye for 30 min, and signal was recorded. Data were shown as area under the curve (AUC). N = 3. SP6616 versus DMSO by Student's t test: ∗p < 0.05. Dfe (5, 10, 20 μM) versus DMSO by one-way ANOVA with Dunnett's post hoc test: F (3, 8) = 29.5. ∗∗p < 0.01; ∗∗∗p < 0.001.
(D) Membrane potential assay in CHO cell was conducted using the similar procedure to that in CHO-Kv2.1 cell. N = 6. SP6616 versus DMSO by Student's t test: ns. Dfe (5, 10, 20 μM) versus DMSO by one-way ANOVA with Dunnett's post hoc test: F (3, 20) = 0.2102. ns.
(E) Effect of Dfe (5, 10, 20 μM) on cell viability in CHO-Kv2.1 cell was detected by MTT assay. N = 6. Dfe (5, 10, 20 μM) versus DMSO by one-way ANOVA with Dunnett's post hoc test: F (3, 20) = 0.9416. ns.
(F) The whole-cell patch-clamp technique was used to detect the inhibitory effect of different concentrations of Dfe on Kv2.1 current in different cells, and the current curve of CHO-Kv2.1 cell was recorded.
(G) The electrophysiological data of multiple concentrations of Dfe on the same cell. N = 5. Dfe (1, 3, 10 μM) versus DMSO by one-way ANOVA with Dunnett's post hoc test: F (3, 16) = 41.48. ∗∗∗p < 0.001.
(H) The 50% inhibitory concentration (IC50) of Dfe at 9.53 μM was detected by whole-cell patch-clamp technique. N = 4–9. All data were presented as means ± SEM.
Results
Dfe Inhibits the Kv2.1 Channel
Dfe inhibited membrane potential in CHO-Kv2.1 cell: Kv2.1 inhibitor candidate was assayed by evaluating its inhibition against the membrane potential in CHO-Kv2.1 cell according to our previously published approach (Zhou et al., 2016). As indicated in Figures 1B and 1C, the known Kv2.1 inhibitors GxTx-1E (GE, 100 nM) (Lee et al., 2010) and SP6616 (20 μM) (Zhou et al., 2016) as well as Dfe could inhibit the membrane potential in CHO-Kv2.1 cell, F (3, 8) = 29.5. Furthermore, Dfe could neither inhibit the membrane potential in normal CHO cell (Figure 1D) (F (3, 20) = 0.2102) nor affect the cell viability of CHO-Kv2.1 cell (Figure 1E) (F (3, 20) = 0.9416).
Thus, all data revealed that Dfe might inhibit the Kv2.1 channel (Zhou et al., 2016).
Patch-clamp assay confirmed the inhibition of Dfe against Kv2.1 channel: We performed whole-cell patch-clamp on Kv2.1 channel stably expressed in CHO cells. As indicated in Figure 1H, Dfe blocked the channel with an IC50 of 9.53 μM. Shown in Figures 1F and 1G were the example traces of Dfe in patch-camp electrophysiological recordings at 1, 3, and 10 μM (F (3, 16) = 41.48).
Dfe exhibits selectivity for Kv2.1 over Kv2.2: Since Kv2.1 is similar in sequence to Kv2.2 (Johnson et al., 2018; IUPHAR Ion Channel Database, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=547&familyId=81&familyType=IC), we tested Dfe on the Kv2.2 channel by measuring membrane potential in CHO cells transfected with the pcDNA3.1a-Kv2.2 plasmid (Zhou et al., 2016) (Figure S1C). In whole-cell patch clamp, Dfe exhibited minimal inhibition of Kv2.2 at 1, 3, and 10 μM (Figures S1A and S1B) (F (3, 32) = 12.64). Taken together, Dfe is a weak (micromolar potency) inhibitor of Kv2.1 with selectivity over Kv2.2. However, it modulates butyrylcholinesterase and TRPV3 (Bodur et al., 2001; Deering-Rice et al., 2014) at the same low micromolar concentrations as that it blocks Kv2.1, indicating that it is not a selective Kv2.1 inhibitor. Actually, further studies are needed to determine if Dfe inhibits other Kv channels expressed in DRG neurons (Cao et al., 2010).
Dfe Treatment Suppressed Protein and mRNA Expressions of Kv2.1 in the DRG Tissue of DPN Mice
Next, we investigated the protein level of Kv2.1 channel in DRG tissue from DPN mice by western blot assay. As indicated in Figures 2A and 2B, Kv2.1 level in DRG tissue from either type 1 (STZ) or type 2 (db/db) DPN mice was obviously increased compared with that from normal mice (Control: age-matched C57BL/6 mice as a control in STZ mice-related assay; db/m: age-matched heterozygotes mice with nonpenetrant genotype as a control in db/db mice-related assay; the same hereinafter). Notably, the DRG tissue from the Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) exhibited lower level of Kv2.1 protein compared with that from the vehicle-treated DPN mice (STZ, db/db) (F (2, 6) = 162.2 for STZ mice, 13.31 for db/db mice).
Figure 2.

Dfe Reduced Kv2.1 Expression in DPN Mice
(A) Western blot results of Kv2.1 protein expression in control mice (Control, age-matched C57BL/6 mice as control in the related assay for STZ-induced type 1 diabetic mice with DPN; db/m, age-matched heterozygotes mice with nonpenetrant genotype as control in the related assay for db/db type 2 diabetic mice with DPN; the same hereinafter), DPN mice (STZ, type 1 diabetic mice with DPN; db/db, type 2 diabetic mice with DPN), and Dfe (10, 20 mg/kg)-treated DPN mice (STZ + Dfe, db/db + Dfe). T-ERK served as the loading control. N = 3.
(B) Quantitative analyses for (A). STZ versus Control by Student's t test: ∗∗p < 0.01. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 162.2. ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 13.31. ∗∗p < 0.01.
(C and D) Representative micrographs stained by Kv2.1 (brown) and quantitative analysis results based on Image (J) Scale bar: 25 μm. N = 3. STZ versus Control by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 183.7. ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 79.87. ∗∗p < 0.01. ∗∗∗p < 0.001.
(E and F) The qPCR result of Kv2.1 mRNA level in (E) control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (F) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe). N = 4–8. STZ versus Control by Student's t test: ∗p < 0.05. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 18) = 7.103. ∗p < 0.05, ∗∗p < 0.01; db/db versus db/m by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 13) = 15.26. ∗p < 0.05. ∗∗∗p < 0.001.
(G) Example traces of whole-cell currents in DPN mice were recorded by the whole-cell patch clamp. N = 19–26.
(H) Whole-cell currents changes of DRG neuron from DPN mice under different voltages and the current value at the highest voltage +50 mv was recorded by the whole-cell patch clamp. N = 19–26. db/db versus db/m by Student's t test: ∗∗p < 0.01. Dfe (20 mg/kg) versus db/db by Student's t test: ∗∗p < 0.01. ∗∗∗p < 0.001. All data were presented as means ± SEM.
In addition, immunohistochemistry (IHC)-based assay was also applied to investigate Kv2.1 expression in the DRG tissue from DPN mice by micrographs stained with Kv2.1 protein (brown) (Figure 2C), and the quantitation was indicated in Figure 2D by ImageJ. Similarly, the results also demonstrated the high level of Kv2.1 protein in the DRG tissue from either type of DPN mice (STZ, db/db) and Dfe treatment efficiently suppressed Kv2.1 protein level in DPN mice (Figures 2C and 2D) (F (2, 6) = 183.7 for STZ mice, 79.87 for db/db mice).
Moreover, qPCR assay results (Figures 2E and 2F, Figure S7) demonstrated that mRNA level of Kv2.1 was elevated in the DRG tissue of DPN mice compared with that of control mice and Dfe treatment obviously decreased the mRNA level of Kv2.1 (Figures 2E and 2F) (F (2, 18) = 7.103 for STZ mice, F (2, 13) = 15.26 for db/db mice).
Finally, we investigated the effect of Dfe on potassium flow in the DRG neuron from DPN mice by whole-cell patch clamp-related assay. As indicated in Figures 2G and 2H, the level of the total voltage-gated potassium current was upregulated in the DRG neuron from DPN mice compared with that from normal mice, and Dfe treatment efficiently suppressed such a level in DPN mice. These results are consistent with the above-mentioned western blot, IHC, and qPCR results.
Taken together, Dfe treatment suppressed protein and mRNA expression of Kv2.1 in the DRG tissue of DPN mice. The mechanism underlying this suppression remains to be determined but may not be related to Dfe's block of Kv2.1 current. Future studies are needed to determine whether Dfe treatment suppresses the expression of other Kv channels reported in DRG tissue. It must be noted here that, in a study of diabetic rats, the mRNA level of Kv2.1 was unchanged in DRGs, whereas mRNA levels of Kv1.4, Kv3.4, Kv4.2, and Kv4.3 were decreased (Cao et al., 2010). The reason for the difference between their results and ours is not clear.
Dfe Treatment Promoted Neurite Outgrowth of DRG Neurons in DPN Mice by Modulating Kv2.1 Channel
Dfe treatment promoted the neurite outgrowth of DRG neuron in DPN mice: By considering that neurodegeneration is highly related to the pathology of DPN (Jia et al., 2016), we detected the potential effect of Dfe treatment on the neurite outgrowth of DRG neuron in DPN mice.
As indicated in Figures 3A and 3B, the total neurite outgrowth of DRG neuron from either type of DPN mice (STZ, db/db) was repressed compared with that from the control mice (Control, db/m). Obviously, the total neurite outgrowth of DRG neuron was promoted from the Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) compared with that from the vehicle-treated DPN mice (STZ, db/db) (Figures 3C and 3D) (F (2, 6) = 197.8 for STZ mice, 215 for db/db mice). These results thus implied that Dfe treatment promoted the neurite outgrowth of DRG neuron in DPN mice.
Figure 3.

Dfe Treatment Promoted Neurite Outgrowth of DRG Neuron in DPN Mice by Inhibiting Kv2.1 Channel
(A and B) β-Tubulin III-immunostained sensory neurons from: (A) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (B) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe). Scale bar: 100 μm.
(C and D) Quantitative analyses of total neurite outgrowth for (A, B). N = 3. STZ versus Control by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 197.8. ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 215. ∗∗∗p < 0.001.
(E) β-Tubulin III immunofluorescence analyses in DRG neuron from Control, AAV9-NC-injected STZ mice (STZ + AAV9-NC), AAV9-Kv2.1-RNAi-injected STZ (STZ + AAV9-Kv2.1) and Dfe (10, 20 mg/kg)-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe). Scale bar: 100 μm.
(F) Quantitative analyses of total neurite outgrowth for (E). N = 3. STZ + AAV9-NC versus Control by Student's t test: ∗∗p < 0.01. STZ + AAV9-Kv2.1 versus STZ + AAV9-NC by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ + AAV9-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 0.06414. ns. All data were presented as means ± SEM.
Dfe treatment promoted the neurite outgrowth of DRG neuron in DPN mice by inhibiting Kv2.1 channel: Next, to verify that the above-mentioned Dfe-mediated promotion on the neurite growth of DRG neuron was via Kv2.1 inhibiting, in vivo Kv2.1 knockdown assay was carried out by injection of adeno associated virus AAV9-Kv2.1-RNAi into STZ mice. As indicated in Figures 3E and 3F, the neurite outgrowth of DRG neuron was enhanced from AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) compared with that from AAV9-negative control (NC) injected STZ mice (STZ + AAV9-NC). Notably, no significant difference was determined in neurite outgrowth of DRG neuron between Dfe-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe) and vehicle-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) (F (2, 6) = 0.06414).
Given that Dfe was also ever reported to be M1 receptor antagonist (Kunysz et al., 1988), butyrylcholinesterase blocker (Bodur et al., 2001), and TRPV3 inhibitor (Deering-Rice et al., 2014), we also performed related assays to inspect whether or not the above-mentioned Dfe-mediated promotion on neurite outgrowth of DRG neuron was also through any of these three targets. As indicated in Figures S4A–S4G, none of M1 inhibitor Pirenzepine, TRPV3 si-RNA, and BCHE si-RNA could block the capability of Dfe in promoting the neurite growth of DRG neuron. These results thereby demonstrated that Dfe promoted the neurite growth of DRG neuron independent of any of these three targets.
Therefore, all results demonstrated that Dfe treatment enhanced neurite outgrowth of DRG neuron in DPN mice by inhibiting Kv2.1 channel.
Dfe Treatment Ameliorated Nerve Conduction Deficiency and Hypoalgesia of DPN Mice by Inhibiting Kv2.1 Channel
Next, the behavioral indicators (tactile allodynia and thermal sensitivity) and motor nerve conduction velocity of STZ and db/db mice were detected. As shown in Figure 4A and 4B, orange wide arrows above axis indicated Dfe injection, orange triangle below axis indicated behavioral tests (tactile allodynia and thermal sensitivity), orange pentacles indicated motor nerve conduction velocity (MNCV). The following neurological changes were found in DPN mice (STZ and db/db): nerve conduction deficiency, hypoalgesia, impairment in motor nerve conduction velocity (MNCV) (Figures 4C and 4D; F (2, 33) = 9.92 for STZ mice, 20.87 for db/db mice), enhanced sensitivity to mechanical stimuli (Figures 4E and 4F; F (2, 33) = 130.2 for STZ mice, 70.06 for db/db mice), and augmented thermal response (Figures 4G and 4H; F (2, 33) = 6.211 for STZ mice, 13.3 for db/db mice). Dfe treatment (10, 20 mg/kg) attenuated these pathological features in DPN mice (Figures 4C–4H).
Figure 4.

Dfe Treatment Ameliorated Nerve Conduction Deficiency and Hypoalgesia in DPN Mice by Inhibiting Kv2.1 Channel
(A and B) Experimental schedule of STZ and db/db mice. Orange wide arrows above axis indicated Dfe injection, orange triangle below axis indicated behavioral tests (tactile allodynia and thermal sensitivity), orange pentacles indicated motor nerve conduction velocity (MNCV).
(C and D) MNCV was assayed to detect nerve function of (C) STZ mice and (D) db/db mice. N = 12. STZ versus Control by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 33) = 9.92. ##p < 0.01. ###p < 0.001; db/db versus db/m by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 33) = 20.87. ###p < 0.001.
(E and F) Tactile allodynia test was used to evaluate neurological function of (E) STZ mice and (F) db/db mice. N = 12. STZ versus Control by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 33) = 130.2. ###p < 0.001; db/db versus db/m by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 33) = 70.06. ###p < 0.001.
(G and H) radial heat plate test was performed to evaluate neurological function of (G) STZ mice and (H) db/db mice. N = 12. STZ versus Control by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 33) = 6.211. ##p < 0.01; db/db versus db/m by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 33) = 13.3. ###p < 0.001.
(I–K) (I) MNCV assay, (J) tactile allodynia test, and (K) radial heat plate test were carried out in Control, AAV9-NC or AAV9-Kv2.1-RNAi-injected STZ (STZ + AAV9-NC, STZ + AAV9-Kv2.1) and Dfe (10, 20 mg/kg)-treated AAV9-Kv2.1-RNAi-injected STZ (STZ + AAV9-Kv2.1 + Dfe) mice. N = 12. In MNCV assay, STZ + AAV9-NC versus Control by Student's t test: ∗∗∗p < 0.001, STZ + AAV9-Kv2.1 versus STZ + AAV9-NC by Student's t test: ###p < 0.001, Dfe (10, 20 mg/kg) versus STZ + AAV9-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 33) = 0.3592. ns; In tactile allodynia test, STZ + AAV9-NC versus Control by Student's t test: ∗∗∗p < 0.001, STZ + AAV9-Kv2.1 versus STZ + AAV9-NC by Student's t test: ###p < 0.001, Dfe (10, 20 mg/kg) versus STZ + AAV9-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 33) = 0.6631. ns; In radial heat plate test, STZ + AAV9-NC versus Control by Student's t test: ∗∗∗p < 0.001, STZ + AAV9-Kv2.1 versus STZ + AAV9-NC by Student's t test: ###p < 0.001, Dfe (10, 20 mg/kg) versus STZ + AAV9-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 33) = 1.486. ns. All data were presented as means ± SEM.
Next, AAV9-Kv2.1-RNAi-based in vivo Kv2.1 knockdown assay against STZ mice was carried out to verify that the above-mentioned Dfe-mediated improvement on DPN-like pathology of DPN mice was through inhibiting Kv2.1 channel. As expected, the results in Figures 4I–4K demonstrated that AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) exhibited improvements on MNCV, sensitivity to mechanical stimuli, and thermal response compared with AAV9-NC-injected STZ mice (STZ + AAV9-NC). Notably, no significant difference was determined in any of the potential improvements on the mentioned pathological behaviors between Dfe-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe) and vehicle-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) (F (2, 33) = 0.3592 for MNCV, F (2, 33) = 0.6631 for tactile allodynia test, F (2, 33) = 1.486 for thermal sensitivity measurement).
Moreover, Dfe was also applied in the assay against normal (non-DPN) animals by performing the same behavioral tests as DPN mice (motor nerve conduction velocity test, tactile allodynia test, and thermal sensitivity measurement). As shown in Figure S11, Dfe treatment for 4 weeks rendered no effects on normal animals in terms of any of these behavioral activities.
Therefore, all data verified that Dfe treatment ameliorated nerve conduction deficiency and hypoalgesia of DPN mice by inhibiting Kv2.1 channel.
Dfe Treatment Improved Mitochondrial Dysfunction of DRG Neuron in DPN Mice through Kv2.1/CaMKKβ/AMPK/PGC-1α Pathway
Given that mitochondrial dysfunction is tightly linked to diabetic neuropathy and the lesion of peripheral sensory neuron (Chowdhury et al., 2013), we investigated the potential of Dfe treatment in ameliorating mitochondrial dysfunction of DRG neuron in DPN mice.
Dfe treatment improved mitochondrial dysfunction of DRG neuron in DPN mice by inhibiting Kv2.1 channel: In the assay, oxygen consumption rate (OCR) was investigated to evaluate mitochondrial respiratory function of DRG neuron from DPN mice. To our expect, Dfe treatment improved ATP production, maximal respiration, basal respiration, and spare respiratory capacity of DRG neuron from either STZ (Figures 5A and 5B) or db/db (Figures 5C and 5D) mice. Notably, no significant difference was found in the improvement on OCR of DRG neuron between Dfe-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe) and vehicle-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) (Figures 5E and 5F).
Figure 5.

Dfe Improved Mitochondrial Dysfunction of DRG Neuron in DPN Mice through Kv2.1/CaMKKβ/AMPK/PGC-1α Pathway
(A and C) Oxygen consumption rate (OCR) was measured in DRG neuron from (A) Control, STZ or Dfe (20 mg/kg)-treated STZ mice (STZ + Dfe 20 mg/kg) and (C) db/m, db/db or Dfe (20 mg/kg)-treated db/db mice (db/db + Dfe 20 mg/kg) using XF96 analyzer. N = 4–7. STZ + Dfe (20 mg/kg) versus STZ by Student's t test: #p < 0.05; db/db + Dfe (20 mg/kg) versus db/db by Student's t test: ##p < 0.01.
(B and D) ATP production, basal respiration, spare respiratory capacity, and maximal respiration were measured in DRG neuron from (B) Control, STZ or Dfe (20 mg/kg)-treated STZ mice (STZ + Dfe 20 mg/kg) and (D) db/m, db/db or Dfe (20 mg/kg)-treated db/db mice (db/db + Dfe 20 mg/kg). N = 3. For ATP production, STZ versus Control by Student's t test: ∗∗p < 0.01, STZ + Dfe (20 mg/kg) versus STZ by Student's t test: ∗∗p < 0.01; db/db versus db/m by Student's t test: ∗∗p < 0.01, db/db + Dfe (20 mg/kg) versus db/db by Student's t test: ∗p < 0.05. For basal respiration, STZ versus Control by Student's t test: ∗p < 0.05, STZ + Dfe (20 mg/kg) versus STZ by Student's t test: ∗∗∗p < 0.01; db/db versus db/m by Student's t test: ∗p < 0.05, db/db + Dfe (20 mg/kg) versus db/db by Student's t test: ∗p < 0.05. For spare respiratory capacity, STZ versus Control by Student's t test: ns, STZ + Dfe (20 mg/kg) versus STZ by Student's t test: ∗p < 0.05; db/db versus db/m by Student's t test: ∗p < 0.05, db/db + Dfe (20 mg/kg) versus db/db by Student's t test: ns. For maximal respiration, STZ versus Control by Student's t test: ∗p < 0.05, STZ + Dfe (20 mg/kg) versus STZ by Student's t test: ∗∗∗p < 0.01; db/db versus db/m by Student's t test: ∗p < 0.05, db/db + Dfe (20 mg/kg) versus db/db by Student's t test: ∗p < 0.05.
(E) OCR data in DRG neuron from Control, AAV9-NC or AAV9-Kv2.1-RNAi-injected STZ (STZ + AAV9-NC, STZ + AAV9-Kv2.1) and Dfe (20 mg/kg)-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + 20 mg/kg) were obtained. N = 5–7. STZ + AAV9-Kv2.1 + Dfe versus STZ + AAV9-Kv2.1 by Student's t test: ns.
(F) ATP production, basal respiration, spare respiratory capacity, and maximal respiration were measured in DRG neuron from Control, AAV9-NC or AAV9-Kv2.1-RNAi-injected STZ (STZ + AAV9-NC, STZ + AAV9-Kv2.1) and Dfe (10, 20 mg/kg)-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe). N = 3. For ATP production, STZ + AAV9-NC versus Control by Student's t test: ∗∗p < 0.01, STZ + AAV9-Kv2.1 versus STZ + AAV9-NC by Student's t test: ∗p < 0.05. Dfe (20 mg/kg) versus STZ + AAV9-Kv2.1 by Student's t test: ns; For basal respiration, STZ + AAV9-NC versus Control by Student's t test: ∗∗p < 0.01, STZ + AAV9-Kv2.1 versus STZ + AAV9-NC by Student's t test: ∗p < 0.05. Dfe (20 mg/kg) versus STZ + AAV9-Kv2.1 by Student's t test: ns; For spare respiratory capacity, STZ + AAV9-NC versus Control by Student's t test: ∗p < 0.05, STZ + AAV9-Kv2.1 versus STZ + AAV9-NC by Student's t test: ∗p < 0.05. Dfe (20 mg/kg) versus STZ + AAV9-Kv2.1 by Student's t test: ns; For maximal respiration, STZ + AAV9-NC versus Control by Student's t test: ∗∗p < 0.01, STZ + AAV9-Kv2.1 versus STZ + AAV9-NC by Student's t test: ∗∗p < 0.01. Dfe (20 mg/kg) versus STZ + AAV9-Kv2.1 by Student's t test: ns.
(G and H) Mitochondrial membrane potential levels in DRG neuron from (G) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (H) db/m, db/db and Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe). N = 3. STZ versus Control by Student's t test: ∗p < 0.05. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 40.94. ∗∗p < 0.01, ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗p < 0.05. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 7.388. ∗p < 0.05.
(I and J) Western blot results of CaMKKβ, AMPK, p-AMPK, PGC-1α and p-ACC in DRG homogenates from (I) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (J) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe). T-ERK was used as an internal control. N = 3.
(K and L) Quantitative analyses for (I) and (J). p-AMPK (STZ versus Control by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 91.83. ##p < 0.01, ###p < 0.001; db/db versus db/m by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 56.34. ##p < 0.01, ###p < 0.001), AMPK (STZ versus Control by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 3.235. ns; db/db versus db/m by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 1.179. ns), CaMKKβ (STZ versus Control by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 22.41. ##p < 0.01; db/db versus db/m by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 10.58. #p < 0.05), PGC1-α (STZ versus Control by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 79.92. ##p < 0.01, ###p < 0.001; db/db versus db/m by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 20.91. ##p < 0.01), p-ACC (STZ versus Control by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 3.615. ##p < 0.01; db/db versus db/m by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 4.675. #p < 0.05).
(M) Western blot results of CaMKKβ, AMPK, p-AMPK, PGC-1α, and p-ACC in DRG homogenates from Control, AAV9-NC or AAV9-Kv2.1-RNAi-injected STZ (STZ + AAV9-NC, STZ + AAV9-Kv2.1), and Dfe (10, 20 mg/kg)-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe). T-ERK was used as an internal control. DRG tissue isolated from three mice per group was mixed and homogenized.
(N) Quantitative analysis for (M). p-AMPK (STZ + AAV-NC versus Control by Student's t test: ∗∗∗p < 0.001. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ##p < 0.01. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 3.97. ns), AMPK (STZ + AAV-NC versus Control by Student's t test: ns. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ns. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 1.121. ns), CaMKKβ (STZ + AAV-NC versus Control by Student's t test: ∗∗p < 0.01. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ###p < 0.001. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 3.787. ns), PGC1-α (STZ + AAV-NC versus Control by Student's t test: ∗∗p < 0.01. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: #p < 0.05. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 0.4457. ns), p-ACC (STZ + AAV-NC versus Control by Student's t test: ∗∗p < 0.01. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ##p < 0.01. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 10.19. &p < 0.05). All data were presented as means ± SEM.
By considering that downregulation of mitochondrial membrane potential (MMP) features mitochondrial apoptosis (Srinivasan et al., 2000), we also investigated the potential amelioration of Dfe treatment on MMP of DRG neuron in DPN mice by commercial MMP kit. As indicated in Figures 5G and 5H, MMP level of DRG neuron from either type of DPN mice (STZ, db/db) was expectedly reduced compared with that from normal mice (Control, db/m) but reversed by Dfe treatment as comparing the corresponding results from the Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) with those from the vehicle-treated DPN mice (STZ, db/db) (F (2, 6) = 40.94 for STZ mice, 7.388 for db/db mice). Obviously, these results implied that Dfe protected mitochondria from apoptosis in DRG neuron of DPN mice.
Together, all results demonstrated that Dfe treatment improved mitochondrial dysfunction of DRG neuron in DPN mice by inhibiting Kv2.1 channel.
Dfe treatment improved mitochondrial dysfunction through Kv2.1/CaMKKβ/AMPK/PGC-1α pathway: Given the pivotal role of AMPK/PGC-1α signaling in mitochondrial energy homeostasis (Calcutt et al., 2017; Herzig et al., 2018; Hashem et al., 2017), we detected the potential of Dfe treatment in regulating AMPK/PGC-1α signaling in DRG tissue of DPN mice by western blot. The results demonstrated that the levels of phosphorylated AMPK and PGC-1α were decreased from DPN mice (STZ, db/db) compared with those from control mice (Control, db/m) but increased from the Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) compared with the vehicle-treated DPN mice (STZ, db/db) (Figures 5I–5L) (for p-AMPK, F (2, 6) = 91.83 in STZ mice, 56.34 in db/db mice; for PGC1-α, F (2, 6) = 79.92 in STZ mice, 20.91 in db/db mice; for AMPK, F (2, 6) = 3.235 in STZ mice, 1.179 in db/db mice; for p-ACC, F (2, 6) = 3.615 in STZ mice, 4.675 in db/db mice).
It was noticed that AMPK can be phosphorylated then activated by Ca2+/CaM-dependent protein kinase Came β (CaMKKβ) or the tumor suppressor LKB1 (Shaw et al., 2005). Since western blot results revealed that Dfe rendered no effects on LKB1 as comparing the LKB1 protein level in DRG tissue from Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) with that from vehicle-treated DPN mice (STZ, db/db) (Figure S2) (F (2, 6) = 0.6136 for STZ mice, 0.3575 for db/db mice), we here focused on the study on CaMKKβ in response to Dfe-mediated Kv2.1 regulation. To our expect, the results in Figures 5I–5L demonstrated that Dfe treatment promoted the level of CaMKKβ protein in DRG tissue from either type of DPN mice (F (2, 6) = 22.41 for STZ mice, 10.58 for db/db mice).
Moreover, western blot results (Figures 5M and 5N) indicated that the DRG tissue from AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) exhibited increased levels of CaMKKβ, p-AMPK, and PGC-1α proteins compared with that from AAV9-NC-injected STZ mice (STZ + AAV9-NC) and no significant difference was found in the regulation of any of these three proteins (CaMKKβ, p-AMPK, and PGC-1α) in DRG tissue between Dfe-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe) and vehicle-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) (F (2, 6) = 3.97 for p-AMPK, F (2, 6) = 1.121 for AMPK, F (2, 6) = 3.787 for CaMKKβ, F (2, 6) = 0.4457 for PGC-1α, F (2, 6) = 10.19 for p-ACC).
Thus, all findings indicated that Dfe treatment improved mitochondrial dysfunction by regulating Kv2.1/CaMKKβ/AMPK/PGC-1α signaling. Additional studies will have to be undertaken to elucidate the molecular mechanisms coupling Kv2.1 to CaMKKβ/AMPK/PGC-1α signaling and to determine if Dfe-mediated suppression of Kv2.1 expression or blockade of Kv2.1 current by Dfe underlies this modulatory effect.
Dfe Treatment Alleviated Apoptosis of DRG Neuron in DPN Mice Involving Regulation of Bcl-2 Family Proteins and Caspase-3 by Inhibiting Kv2.1 Channel
Dfe treatment alleviated apoptosis of DRG neuron in DPN mice: As apoptosis was also tightly linked to DPN (Srinivasan et al., 2000), we detected the potential of Dfe treatment in preventing against apoptosis of DRG neuron in DPN mice (STZ, db/db). In the assay, images of double immune-fluorescent staining were detected to inspect the Annexin V-FITC and PI-labeled early and late apoptosis of DRG neuron from mice.
As indicated in Figures 6A–6C, the number of early or late apoptotic cells of DRG neuron from DPN mice (STZ, db/db) was increased compared with that from normal mice (Control, db/m) and the apoptotic response was markedly decreased nearly to normal level in DRG neuron from Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) compared with that from vehicle-treated DPN mice (STZ, db/db) (F (2, 6) = 27.24 for STZ mice, 14.77 for db/db mice).
Figure 6.

Dfe Treatment Alleviated Apoptosis of DRG Neuron in DPN Mice Involving Regulation of Bcl-2 Family Proteins and Caspase-3 by Inhibiting Kv2.1 Channel
(A and B) Representative images of double immune-fluorescent staining showing Annexin V-FITC and PI labeled early and late stage of apoptosis of DRG neuron from (A) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (B) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe). Hoechst was used to mark nuclei. Scale bar: 250 μm.
(C) Quantitative analyses for (A, B). N = 3. STZ versus Control by Student's t test: ∗∗p < 0.01. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 27.24. ∗p < 0.05, ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗p < 0.01. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 14.77. ∗∗p < 0.01.
(D and E) The paraffin sections of DRG tissue from (D) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (E) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe) were fluorescently stained for IHC assay. Neurons were located by NeuN and apoptotic cells were labeled by tunnel. Scale bar: 25 μm.
(F) Quantitative analyses for (D, E). N = 3. STZ versus Control by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 54.09. ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 41.48. ∗∗p < 0.01, ∗∗∗p < 0.001.
(G and I) Western blot results demonstrated that Kv2.1 inhibitor Dfe reduced apoptosis in DRG neuron from (G) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (I) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe) by regulating apoptosis-related proteins. T-ERK was used as loading control. N = 3.
(H and J) Quantitative analyses for (G, I). Bcl-xl (STZ versus Control by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 14.33. ##p < 0.01; db/db versus db/m by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 4.535. #p < 0.05), Bcl-2 (STZ versus Control by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 4.205. #p < 0.05; db/db versus db/m by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 15.61. #p < 0.05, ##p < 0.01), Bax (STZ versus Control by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 11.06. #p < 0.05. ##p < 0.01; db/db versus db/m by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 8.733. #p < 0.05), p-Bad (STZ versus Control by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 5.134. #p < 0.05; db/db versus db/m by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 5.357. #p < 0.05), Cleaved-caspase-3 (STZ versus Control by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 4.543. #p < 0.05; db/db versus db/m by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 20.9. ##p < 0.01).
(K and L) Western blot assays in DRG tissue from Control, AAV9-NC or AAV9-Kv2.1-RNAi-injected STZ (STZ + AAV9-NC, STZ + AAV9-Kv2.1) and Dfe (10, 20 mg/kg)-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe). DRG tissue from three mice per group was mixed and homogenized. Bcl-xl (STZ + AAV-NC versus Control by Student's t test: ∗∗p < 0.01. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ###p < 0.001. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 5.186. ns), Bcl-2 (STZ + AAV-NC versus Control by Student's t test: ∗∗∗p < 0.001. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ##p < 0.01. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 6.773. ns. #p < 0.05), Bax (STZ + AAV-NC versus Control by Student's t test: ∗p < 0.05. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ##p < 0.01. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 2.492. ns), p-Bad (STZ + AAV-NC versus Control by Student's t test: ∗p < 0.05. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ns. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 5.101. &p < 0.05, ns), Cleaved caspase-3 (STZ + AAV-NC versus Control by Student's t test: ∗∗p < 0.01. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ##p < 0.01. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 4.272. &p < 0.05, ns). All data were presented as means ± SEM.
In addition, we also investigated the prevention of Dfe treatment against the apoptosis of DRG neuron from DPN mice by IHC assay. Paraffin sections of DRG tissue were used, and DRG neuron was located by NeuN antibody and apoptotic cells were labeled by tunnel. As shown in Figures 6D–6F, the level of green fluorescence marked by tunnel (apoptotic cells) in DRG neuron was increased from DPN mice (STZ, db/db) compared with that from control mice (Control, db/m), and reduced from the Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) compared with the vehicle-treated DPN mice (STZ, db/db) (F (2, 6) = 54.09 for STZ mice, 41.48 for db/db mice).
Dfe treatment alleviated apoptosis of DRG neuron in DPN mice involving regulation of Bcl-2 family proteins and Caspase-3 by inhibiting Kv2.1 channel: Next, we investigated the potential regulation of Dfe against Bcl-2 family proteins (Bcl-xl, Bcl-2, p-Bad, and Bax) and Caspase-3 in response to the Dfe-mediated suppression against DRG neuron apoptosis in DPN mice by western blot assay.
As shown in Figures 6G–6J, the levels of anti-apoptotic (Bcl-xl, Bcl-2 and p-Bad) and pro-apoptotic (Bax and cleaved Caspase-3) proteins in DRG tissue were, respectively, decreased and increased from DPN mice (STZ, db/db) compared with those from control mice (Control, db/m), indicative of the apoptosis of DRG neuron in DPN mice. Obviously, the levels of these anti-apoptotic and pro-apoptotic proteins of DRG tissue were, respectively, increased and decreased from the Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) compared with those from the vehicle-treated DPN mice (STZ, db/db) (for Bcl-xl, F (2, 6) = 14.33 in STZ mice, 4.535 in db/db mice; for Bcl-2, F (2, 6) = 4.205 in STZ mice, 15.61 in db/db mice; for Bax, F (2, 6) = 11.06 in STZ mice, 8.733 in db/db mice; for p-Bad, F (2, 6) = 5.134 in STZ mice, 5.357 in db/db mice; for cleaved Caspase-3, F (2, 6) = 4.543 in STZ mice, 20.9 in db/db mice). All results demonstrated that Dfe treatment alleviated apoptosis of DRG neuron in DPN mice involving regulation of Bcl-2 family proteins and Caspase-3.
As indicated in Figures 6K and 6L by western blot, DRG tissue from AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) exhibited, respectively, increased and decreased levels of the above-mentioned anti-apoptotic and pro-apoptotic proteins compared with those from AAV9-NC-injected STZ mice (STZ + AAV9-NC). However, no significant difference was determined in the levels of any anti-apoptotic or pro-apoptotic proteins in DRG tissue between Dfe-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe) and vehicle-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) (F (2, 6) = 5.186 for Bcl-xl, 6.773 for Bcl-2, 2.492 for Bax, 5.101 for p-Bad, and 4.272 for cleaved Caspase-3).
Therefore, all data demonstrated that Dfe treatment alleviated apoptosis of DRG neurons from DPN mice involving regulation of Bcl-2 family proteins and Caspase-3 by inhibiting the Kv2.1 channel. It must be noted here that Kv2.1 has been identified as the channel responsible for the pro-apoptotic K+ current increase in cortical, hippocampal, and cerebellar granule neurons; increased phosphorylation of Kv2.1, enhanced plasma membrane delivery of Kv2.1, and larger Kv2.1 K + currents produce an intracellular environment that enables DNA fragmentation, caspase activation, and apoptosis (Redman et al., 2007; Yao et al., 2009; Shah et al., 2014). Kv2.1 may play a similar pro-apoptotic role in DRG neurons, and Dfe by suppressing Kv2.1 expression or inhibiting Kv2.1 current may protect DRGs from apoptosis.
Dfe Treatment Reduced Inflammation in DPN Mice Involving IκBα/NF-κB Signaling by Inhibiting Kv2.1 Channel
Dfe treatment suppressed proinflammatory cytokines in DPN mice by inhibiting Kv2.1 channel: Given the close linkage of inflammation to DPN, we inspected the potential anti-inflammation effect of Dfe treatment on DPN mice by ELISA assay.
As indicated in Figures 7A and 7B, the levels of proinflammatory factors (IL-6, IL-1β, and TNF-α) were increased in the serum of DPN mice (STZ, db/db) compared with those of control mice (Control, db/m) but repressed in the serum of Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) compared with those of vehicle-treated DPN mice (STZ, db/db) (for IL-6, F (2, 6) = 55.44 in STZ mice, 1.884 in db/db mice; for IL-1β, F (2, 6) = 13.92 in STZ mice, 23.84 in db/db mice; for TNF-α, F (2, 6) = 25.92 in STZ mice, 45.48 in db/db mice). As indicated in Figure 7C, the levels of the proinflammatory cytokines (IL-6, IL-1β, and TNF-α) were all decreased in the serum of AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) compared with those of AAV9-NC-injected STZ mice (STZ + AAV9-NC). All these results thus demonstrated that either Kv2.1 inhibition or Dfe treatment could alleviate proinflammatory cytokines in DPN mice.
Figure 7.

Dfe Treatment Reduced Inflammation in DPN Mice by Inhibiting Kv2.1 Channel
(A and B) ELISA analysis of proinflammatory cytokines IL-6, IL-1β, and TNF-α in the serum from (A) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (B) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe). N = 3. IL-6 (STZ versus Control by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 55.44. ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 1.884. ∗∗p < 0.01), IL-1β (STZ versus Control by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 13.92. ∗p < 0.05, ∗∗p < 0.01; db/db versus db/m by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 23.84. ∗∗p < 0.01, ∗∗∗p < 0.001), TNF-α (STZ versus Control by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 25.92. ∗∗p < 0.01, ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 45.48. ∗∗p < 0.01, ∗∗∗p < 0.001).
(C) ELISA analysis of proinflammatory cytokines IL-6, IL-1β, and TNF-α in the serum from Control, AAV9-NC or AAV9-Kv2.1-RNAi-injected STZ (STZ + AAV9-NC, STZ + AAV9-Kv2.1) and Dfe (10, 20 mg/kg)-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe). N = 3. IL-6 (STZ + AAV-NC versus Control by Student's t test: ∗∗p < 0.01. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ∗p < 0.05. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 7.015. ns), IL-1β (STZ + AAV-NC versus Control by Student's t test: ∗∗p < 0.01. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ∗p < 0.05. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 2.671. ns), TNF-α (STZ + AAV-NC versus Control by Student's t test: ∗∗∗p < 0.001. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ∗∗p < 0.01. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 2.023. ns).
(D and E) Paraffin sections of DRG tissue from (D) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (E) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe) for NF-κB immunostaining. Scale bar: 25 μm.
(F and G) Quantitative analyses for (D, E). N = 3. STZ versus Control by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 16.15. ∗p < 0.05, ∗∗p < 0.01; db/db versus db/m by Student's t test: ∗∗p < 0.01. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 42.06. ∗∗p < 0.01, ∗∗∗p < 0.001).
(H and I) Paraffin sections of DRG tissue from Control, AAV9-NC or AAV9-Kv2.1-RNAi-injected STZ (STZ + AAV9-NC, STZ + AAV9-Kv2.1) and Dfe (10, 20 mg/kg)-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe) for NF-κB immunostaining and quantitative analysis. N = 3. STZ + AAV-NC versus Control by Student's t test: ∗∗∗p < 0.001. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 0.3936. ns.
(J and K) Western blot results indicating the expressions of iNOS, TNF-α, phosphorylated IκBα and IκBα in DRG tissue from (J) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (K) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe). N = 3.
(L and M) Quantitative analyses for (J, K). iNOS (STZ versus Control by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 9.918. ∗p < 0.05; db/db versus db/m by Student's t test: ns, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 12.43. ns. ∗∗p < 0.01), p-IκBα (STZ versus Control by Student's t test: ns, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 3.739. ∗p < 0.05; db/db versus db/m by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 15.79. ∗∗p < 0.01), TNF-α (STZ versus Control by Student's t test: ∗p < 0.05, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 11.13. ns, ∗∗p < 0.01; db/db versus db/m by Student's t test: ns, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 25.83. ns, ∗∗∗p < 0.001).
(N and O) Western blot assays in DRG tissue from Control, AAV9-NC or AAV9-Kv2.1-RNAi-injected STZ (STZ + AAV9-NC, STZ + AAV9-Kv2.1) and Dfe (10, 20 mg/kg)-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe). DRG tissue from three mice per group were mixed and homogenized. iNOS (STZ + AAV-NC versus Control by Student's t test: ∗∗∗p < 0.001. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ∗p < 0.05. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 1.224. ns), p-IκBα (STZ + AAV-NC versus Control by Student's t test: ∗∗∗p < 0.001. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 3.089. ns), TNF-α (STZ + AAV-NC versus Control by Student's t test: ∗∗∗p < 0.001. STZ + AAV-Kv2.1 versus STZ + AAV-NC by Student's t test: ∗∗p < 0.01. Dfe (10, 20 mg/kg) versus STZ + AAV-Kv2.1 by one-way ANOVA with Dunnett's post hoc test: F (2, 6) = 0.2739. ns). All data were presented as means ± SEM.
Notably, no significant difference was determined in the levels of any proinflammatory cytokines of the serum between Dfe-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe) and vehicle-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) (F (2, 6) = 7.015 for IL-6, 2.671 for IL-1β, and 2.023 for TNF-α).
Together, all results demonstrated that Dfe treatment suppressed proinflammatory cytokines in DPN mice by inhibiting Kv2.1 channel.
Dfe treatment repressed NF-κB nuclear translocation by inhibiting Kv2.1 channel: NF-κB is a widely influential transcriptional regulator in chronic inflammatory process and, NF-κB dimer can be activated by various post-translational modifications to translocate into the nucleus, which is thought to be indicative of inflammation (Liu et al., 2018; Tak et al., 2001). With these facts, we inspected the potential regulation of Dfe treatment against NF-κB nuclear translocation in DRG neuron from DPN mice by immunostaining assay.
As shown in Figures 7D–7G, the number of NF-κB translocated into nuclei of DRG neuron was increased from DPN mice (STZ, db/db) compared with that from normal mice (Control, db/m) and decreased from the Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) compared with that from the vehicle-treated DPN mice (STZ, db/db) (F (2, 6) = 16.15 for STZ mice, 42.06 for db/db mice). Notably, the number of NF-κB translocated into the nuclei of DRG neuron from AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) was slightly decreased compared with that from AAV9-NC-injected STZ mice (STZ + AAV9-NC) (Figures 7H and 7I), and no significant difference was found in this number between Dfe-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe) and vehicle-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) (Figures 7H and 7I) (F (2, 6) = 0.3936).
Therefore, all results indicated that Dfe treatment repressed NF-κB nuclear translocation by inhibiting Kv2.1 channel.
Dfe treatment suppressed inflammatory factors of DRG tissue in DPN mice by inhibiting Kv2.1 channel: Given that IκBα phosphorylation displays IκBα degradation and activates NF-κB dimer release into nucleus (Kumar et al., 2012), we detected the potential of Dfe treatment in regulating key inflammatory factors iNOS, TNF-α, and p-IκBα in DRG tissue of DPN mice by western blot assay.
As indicated in Figures 7J–7M, the protein levels of iNOS, TNF-α, and p-IκBα in DRG tissue were increased from DPN mice (STZ, db/db) compared with those from normal mice (Control, db/m) and decreased from Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) compared with vehicle-treated DPN mice (STZ, db/db) (for iNOS, F (2, 6) = 9.918 in STZ mice, 12.43 in db/db mice; for p-IκBα, F (2, 6) = 3.739 in STZ mice, 15.79 in db/db mice; for TNF-α, F (2, 6) = 11.13 in STZ mice, 25.83 in db/db mice). Thus, these results demonstrated that Dfe treatment suppressed inflammatory factors of DRG tissue in DPN mice.
Notably, we also determined that the protein levels of the above-mentioned inflammatory factors were repressed in DRG tissue from AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) compared with those from AAV9-NC-injected STZ mice (STZ + AAV9-NC), and no significant difference was found in any levels of these inflammatory factors of DRG neuron between Dfe-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1 + Dfe) and vehicle-treated AAV9-Kv2.1-RNAi-injected STZ mice (STZ + AAV9-Kv2.1) (Figures 7N and 7O) (F (2, 6) = 1.224 for iNOS, 3.089 for p-IκBα, and 0.2739 for TNF-α).
Together, all results implied that Dfe treatment suppressed inflammatory factors of DRG tissue in DPN mice by inhibiting Kv2.1 channel.
Dfe Treatment Improved Neurovascular Impairment in DPN Mice
Dfe treatment improved peripheral tissue perfusion in DPN mice: Considering that neurovascular impairment is one of the leading causes for DPN (Nguyen et al., 2012; Van et al., 2013; Demiot et al., 2006), we inspected the potential amelioration of Dfe on neurovascular impairment in DPN mice. In the assay, Laser Speckle Contrast Imaging system was applied. As indicated in Figures 8A–8F, Dfe treatment increased the blood flow velocity and blood perfusion area of sciatic nerve and footpad in DPN mice (for blood flow of footpad, F (2, 9) = 28.2 in STZ mice, 12.95 in db/db mice; for blood flow of sciatic nerve, F (2, 9) = 34.78 in STZ mice, 33.6 in db/db mice; for blood perfusion of footpad, F (2, 9) = 14 in STZ mice, 37.14 in db/db mice; for blood perfusion of sciatic nerve, F (2, 9) = 73.09 in STZ mice, 24 in db/db mice).
Figure 8.

Dfe Treatment Improved Neurovascular Dysfunction in DPN Mice
(A and D) Representative images of blood flow velocity and blood perfusion unite of foot pad and sciatic nerve in (A) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (D) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe). N = 4.
(B, C, E, and F) Quantitative analyses of blood flow and blood perfusion of foot pad and sciatic nerve for (A, D). For blood flow of foot pad, STZ versus Control by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 9) = 28.2. ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗p < 0.01, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 9) = 12.95. ∗∗p < 0.01. For blood perfusion of foot pad, STZ versus Control by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 9) = 14. ∗p < 0.05, ∗∗p < 0.01; db/db versus db/m by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 9) = 37.14. ∗p < 0.05, ∗∗∗p < 0.001. For blood flow of sciatic nerve, STZ versus Control by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 9) = 34.78. ns, ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 9) = 33.6. ns, ∗∗∗p < 0.001. For blood perfusion of sciatic nerve, STZ versus Control by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 9) = 73.09. ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗∗p < 0.001, Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 9) = 24. ∗p < 0.05, ∗∗∗p < 0.001.
(G and H) Representative sections of mice paw skin for PGP9.5 immunostaining (white arrows) in (G) Control, STZ or Dfe (10, 20 mg/kg)-treated STZ mice (STZ + Dfe) and (H) db/m, db/db or Dfe (10, 20 mg/kg)-treated db/db mice (db/db + Dfe). Scale bar: 100 μm. N = 4.
(I and J) Quantitative analysis results for (G, H). STZ versus Control by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus STZ by one-way ANOVA with Dunnett's post hoc test: F (2, 9) = 34.68. ns, ∗∗∗p < 0.001; db/db versus db/m by Student's t test: ∗∗∗p < 0.001. Dfe (10, 20 mg/kg) versus db/db by one-way ANOVA with Dunnett's post hoc test: F (2, 9) = 27.84. ns, ∗∗∗p < 0.001. All data were presented as means ± SEM.
Dfe treatment improved intraepidermal nerve fiber density in DPN mice: Neurovascular function is closely related to peripheral nerves and intradermal nerve fibers (IENFs) innervating dermis and epidermis, whereas skin biopsy-based IENF density measurement has been widely used in clinical testing of peripheral nerve function (Cameron et al., 2001; Lauria et al., 2010). With these facts, immunostaining assay was performed to examine the density of PGP9.5-positive IENF. As indicated in Figure 8, Dfe-treated DPN mice (STZ + Dfe, db/db + Dfe) exhibited increased density of PGP9.5-positive IENF in the plantar skin tissue compared with vehicle-treated DPN mice (STZ, db/db) (F (2, 9) = 34.68 for STZ mice, and 27.84 for db/db mice). Therefore, these results demonstrated that Dfe treatment improved intraepidermal nerve fiber density in DPN mice.
Discussion
DPN is a long-term complication of diabetes with complicated pathogenesis, and there has yet been no efficient treatment against this disease. It is actually full of challenges to develop effective medication to treat DPN by new target and strategy. In the present work, we determined that inhibition of Kv2.1 channel is an effective therapeutic strategy for DPN and Dfe as an inhibitor of Kv2.1 is expected to find its valuable application in new generation of anti-DPN drug design. Dfe was found to efficiently alleviate DPN-like pathology by counteracting multiple risk factors including mitochondrial dysfunction, apoptosis, and inflammation in DPN mice. Additionally, Dfe is currently a clinically drug, its efficient amelioration of DPN-like pathology in the current work may provide valuable references for further anti-DPN drug discovery.
Mitochondrial dysfunction and apoptosis are mainly pathogenetic features of DPN (Srinivasan et al., 2000), and improvement of mitochondrial dysfunction is believed to be a promising strategy for DPN (Roy Chowdhury et al., 2012). Kv2.1 overexpression in β cell was reported to enhance cell response to mitochondrial dysfunction and endoplasmic reticulum stress thus promoting cell apoptosis (Kim et al., 2012; Pal et al., 2003). Here, we found that Dfe as a Kv2.1 inhibitor was capable of improving mitochondrial bioenergetics profiles in DRG neuron. Consistent with the previous report that AMPK activation in sensory neuron was related to the transient elevation of intracellular Ca2+ concentration (Calcutt et al., 2017), we also determined that Dfe stimulated extracellular calcium influx in DRG neuron (Figure S5) and activated Ca2+-dependent kinase CaMKKβ activity in DPN mice (Figure 5). We found that Dfe improved mitochondrial dysfunction of DRG neuron by Kv2.1/CaMKKβ/AMPK/PGC-1α signaling. Bcl-2 family proteins are central regulators of apoptosis with either anti-apoptotic or pro-apoptotic function (Chan et al., 2004), and Caspase-3 is indispensable for apoptotic chromatin condensation and DNA fragmentation (Porter et al., 1999). Here, we determined that Dfe alleviated apoptosis of DRG neuron involving regulation of Bcl-2 family proteins and Caspase-3 by inhibiting Kv2.1 channel. All these findings have further strengthened the potential of Kv2.1 inhibition as a therapeutic strategy for DPN.
Notably, neurovascular function damage is tightly associated with inflammation in DPN, and inflammation reduction promotes the recovery of vascular function (Liu et al., 2017; Nguyen et al., 2012). Here, we determined that Kv2.1 inhibition improved peripheral blood perfusion and alleviated the loss of intraepithelial nerve fibers. It is suggested that the neurovascular function improvement should be ascribed to the Dfe-mediated anti-inflammatory effect through Kv2.1 inhibition.
Although the detailed mechanism underlying the beneficial effect of Dfe on DPN-like pathology relating to potassium flux could not be well presented, we tentatively suggested that Dfe repressed Kv2.1 expression on the surface of cell membrane causing the opening of potassium channels and reducing of potassium conductance. As indicated in Figure S9, Dfe reduced the post-polarization potential and shortened the latency of DRG neuron action potential release, thus facilitating action potential release. As a result, the level of opening of potassium ion channels and the difference between the resting potential and the threshold potential decreased, resulting in an increase in the conduction velocity of the action potential. In addition, neuronal excitability changes led to an improvement in DPN-like pathology including neuropathic pain. Moreover, by considering that Kv2 downregulation augments firing by limiting the Kv2 inhibitory effect on spike frequency and Kv2 blocker against DRG neuron promotes myelinated neuron hyperexcitability (Tsantoulas et al., 2014), we here tentatively proposed that the beneficial effect of Dfe on DPN-like pathology in our work might be also related to the Dfe-mediated regulation of myelinated neuron hyperexcitability.
It was noticed that the densities of total Kv, IA, and IK currents were reduced in medium and large-diameter DRG neurons in DPN rats (Cao et al., 2010) and K+ channel expression was also reduced in sensory neurons of other models of chronic peripheral pain at early pathological stage (Tsantoulas et al., 2014; Tsantoulas et al., 2012; Tsantoulas et al., 2014; Ishikawa et al., 1999; Uchida et al., 2010; Ritter et al., 2012; Chien et al., 2007; Cao et al., 2010). In contrast, our current work mainly involved the study on the late stage of DPN mice with evident hypersensitivity, demonstrating different result concerning Kv2.1 expression. Thus, our finding that Dfe could effectively improve the late stage of the DPN-like pathology might highlight its potential in preventing the occurrence of diabetic foot.
Finally, in the current work, apart from Kv2 (Kv2.1 and Kv2.2), we also assayed the mRNA levels of other Kv channels Kv1.1, Kv1.2, Kv1.4, Kv3.4, Kv4.2, and Kv4.3 in DRG tissue from DPN mice (Figures S7K–S7P), and the results indicated that the mRNA levels of the tested Kv channels were all upregulated in DRG tissue of type 1 diabetic mice but there were no obvious changes of mRNA levels for the other tested Kv genes except for the upregulations of Kv3.4 and Kv4.2 in DRG tissue of type 2 diabetic mice compared with those of Control and db/m mice. These results thus revealed the DPN-like pathology difference in Kv channel expression between type 1 and type 2 DPN model mice.
Based on the literature that elevated expression of channel protein may lead to the amplification of whole-cell current, we speculated that the decrease of Kv2.1 expression could reduce the whole-cell current (Leung et al., 2005; Wang et al., 2014). Thus, we tentatively hypothesized that the functional consequences caused by Kv2.1 current inhibition were in response to the functional consequences caused by the decrease of Kv2.1 channel protein expression.
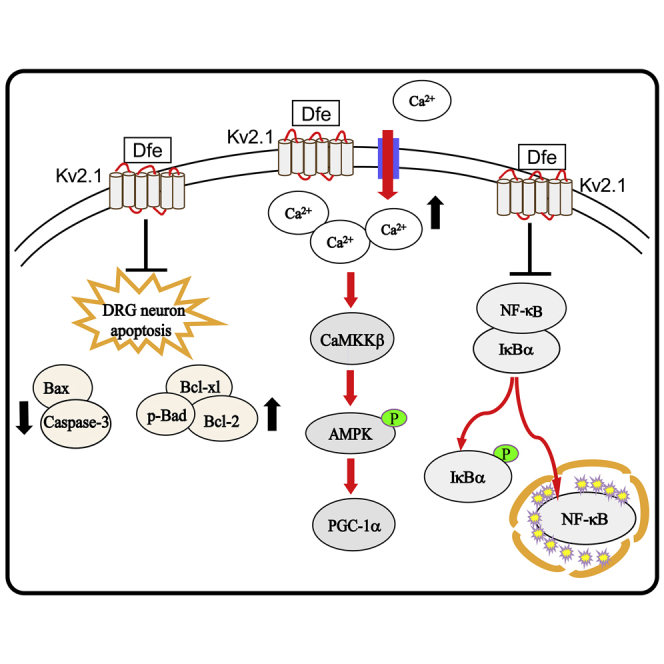
In summary, we reported that antispasmodic drug Dfe as a Kv2.1 inhibitor ameliorated DPN-like pathology and the underlying mechanisms have been investigated by assay against the DPN mice with in vivo Kv2.1 knockdown by injection of adeno associated virus AAV9-Kv2.1-RNAi. As summarized in the Graphical Abstract, Dfe ameliorated mitochondrial dysfunction through the Kv2.1/CaMKKβ/AMPK/PGC-1α pathway, suppressed inflammation by IκBα/NF-κB signaling, and alleviated apoptosis by regulating Bcl-2 family proteins and Caspase-3.
Limitations of the Study
In our study, we determined that Dfe as a Kv2.1 inhibitor efficiently ameliorated DPN-like pathology and the underlying mechanism has been intensively investigated. As Dfe is a clinical drug for treating spasmolysis, the structure of Dfe needs to be rationally modified and optimized for developing Dfe-based anti-DPN reagents with high specificity and low side effects.
Resource Availability
Lead Contact
Further information and requests should be directed to and will be fulfilled by the Lead Contact, Xu Shen (xshen@njucm.edu.cn).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplemental Information. Original data have been deposited to Mendeley Data: [https://doi.org/10.17632/gmxth7wsh8.1]
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” China (Number:2018ZX09711002), the National Natural Science Foundation of China for Young Scientists of China (81703806), Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX18_1600), the Open Project Program of Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica (No. JKLPSE201801), the Project of the Priority Academic Program Development of Jiangsu Higher Education Institutions of Jiangsu Higher Education Institutions (PAPD), Priority Academic Program Development of Jiangsu Higher Education Institutions (Integration of Chinese and Western Medicine) and Innovative Research Team of Six Talent Peaks Project in Jiangsu Province (TD-SWYY-013).
Author Contributions
Xiaoju Xu and X.S. designed the study. X.S. reviewed the manuscript. Xiaoju Xu and X.Z. performed the animal experiments. Y.H. conducted the patch clamp experiment. Xiaoju Xu analyzed and interpreted data. Xiaoju Xu and Xu Xu wrote the manuscript. Xiaoju Xu, X.S., Y.Li., and J.W. are the guarantors of this work and, as such, have full access to all data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors approved the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101617.
Contributor Information
Yang Li, Email: liyang@simm.ac.cn.
Jiaying Wang, Email: wangjy@njucm.edu.cn.
Xu Shen, Email: xshen@njucm.edu.cn.
Supplemental Information
References
- Bodur E., Cokuğraş A.N., Tezcan E.F. Inhibition effects of benactyzine and drofenine on human serum butyrylcholinesterase. Arch. Biochem. Biophys. 2001;386:25–29. doi: 10.1006/abbi.2000.2188. [DOI] [PubMed] [Google Scholar]
- Bönhof G.J., Herder C., Strom A., Papanas N., Roden M., Ziegler D. Emerging biomarkers, tools, and treatments for diabetic polyneuropathy. Endocr. Rev. 2019;40:153–192. doi: 10.1210/er.2018-00107. [DOI] [PubMed] [Google Scholar]
- Boulton A.J.M., Vinik A.I., Arezzo J.C., Bril V., Feldman E.L., Freeman R., Malik R.A., Maser R.E., Sosenko J.M., Ziegler D. Diabetic neuropathies: a statement by the american diabetes association. Diabetes Care. 2005;28:956–962. doi: 10.2337/diacare.28.4.956. [DOI] [PubMed] [Google Scholar]
- Calcutt N.A., Smith D.R., Frizzi K., Sabbir M.G., Chowdhury S.K., Mixcoatlzecuatl T., Saleh A., Muttalib N., Van P.R., Ochoa J. Selective antagonism of muscarinic receptors is neuroprotective in peripheral neuropathy. J. Clin. Invest. 2017;127:608–622. doi: 10.1172/JCI88321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron N.E., Eaton S.E.M., Cotter M.A., Tesfaye S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia. 2001;44:1973–1988. doi: 10.1007/s001250100001. [DOI] [PubMed] [Google Scholar]
- Cao X.H., Byun H.S., Chen S.R., Cai Y.Q., Pan H.L. Reduction in voltage-gated k+ channel activity in primary sensory neuron in painful diabetic neuropathy: role of brain-derived neurotrophic factor. J. Neurochem. 2010;114:1460–1475. doi: 10.1111/j.1471-4159.2010.06863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S.L., Yu V.C. Proteins of the bcl-2 family in apoptosis signalling: from mechanistic insights to therapeutic opportunities. Clin. Exp. Pharmacol. Physiol. 2004;31:119–128. doi: 10.1111/j.1440-1681.2004.03975.x. [DOI] [PubMed] [Google Scholar]
- Chao R.Y., Cheng C.H., Wu S.N., Chen P.C. Defective trafficking of kv2.1 channels in mptp-induced nigrostriatal degeneration. J. Neurochem. 2017;144:483–497. doi: 10.1111/jnc.14282. [DOI] [PubMed] [Google Scholar]
- Chien L.Y., Cheng J.K., Chu D., Cheng C.F., Tsaur M.L. Reduced expression of A-type potassium channels in primary sensory neurons induces mechanical hypersensitivity. J. Neurosci. 2007;27:9855–9865. doi: 10.1523/JNEUROSCI.0604-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S.K., Smith D.R., Fernyhough P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol. Dis. 2013;51:56–65. doi: 10.1016/j.nbd.2012.03.016. [DOI] [PubMed] [Google Scholar]
- Demiot C., Tartas M., Fromy B., Abraham P., Saumet J.L., Sigaudo-Roussel D. Aldose reductase pathway inhibition improved vascular and c-fiber functions, allowing for pressure-induced vasodilation restoration during severe diabetic neuropathy. Diabetes. 2006;55:1478–1483. doi: 10.2337/db05-1433. [DOI] [PubMed] [Google Scholar]
- Deering-Rice C.E., Mitchell V.K., Romero E.G., Abdel Aziz M.H., Ryskamp D.A., Križaj D., Gopal V.R., Reilly C.A. Drofenine: a 2-APB analogue with greater selectivity for human TRPV3. Pharmacol. Res. Perspect. 2014;2:e00062. doi: 10.1002/prp2.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duksal T., Tiftikcioglu B.I., Bilgin S., Kose S., Zorlu Y. Role of inflammation in sensory neuropathy in prediabetes or diabetes. Acta Neurol. Scand. 2016;133:384–390. doi: 10.1111/ane.12474. [DOI] [PubMed] [Google Scholar]
- Frazzini V., Guarnieri S., Bomba M., Navarra R., Morabito C., Mariggiò M.A., Mariggi, Sensi S.L. Altered Kv2.1 functioning promotes increased excitability in hippocampal neurons of an Alzheimer's disease mouse model. Cell Death Dis. 2016;7:e2100. doi: 10.1038/cddis.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov R.V., Singh S. Celecoxib and ion channels: a story of unexpected discoveries. Eur. J. Pharmacol. 2014;730:61–71. doi: 10.1016/j.ejphar.2014.02.032. [DOI] [PubMed] [Google Scholar]
- Hashem R.M., Rashed L.A., Hassanin K.M.A., Hetta M.H., Ahmed A.O. Effect of 6-gingerol on AMPK-NF-κB axis in high fat diet fed rats. Biomed. Pharmacother. 2017;88:293–301. doi: 10.1016/j.biopha.2017.01.035. [DOI] [PubMed] [Google Scholar]
- Hermanstyne T.O., Kalpana S., Wei L.W., Hoffman G.E., Meredith A.L., Mong J.A., Misonou H. Kv2.2: a novel molecular target to study the role of basal forebrain gabaergic neuron in the sleep-wake cycle. Sleep. 2013;36:1839–1848. doi: 10.5665/sleep.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta N., Akanuma Y., Kawamori R., Matsuoka K., Oka Y., Shichiri M., Toyota T., Nakashima M., Yoshimura I., Sakamoto N. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy. Diabetes Care. 2006;29:1538. doi: 10.2337/dc05-2370. [DOI] [PubMed] [Google Scholar]
- Hur J., Dauch J.R., Hinder L.M., Hayes J.M., Backus C., Pennathur S., Kretzler M., Brosius F.C., III, Feldman E.L. The metabolic syndrome and microvascular complications in a murine model of type 2 diabetes. Diabetes. 2015;64:3294–3304. doi: 10.2337/db15-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K., Tanaka M., Black J.A., Waxman S.G. Changes in expression of voltage-gated potassium channels in dorsal root ganglion neurons following axotomy. Muscle Nerve. 1999;22:502–507. doi: 10.1002/(sici)1097-4598(199904)22:4<502::aid-mus12>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Jacobson D.A., Kuznetsov A., Lopez J.P., Kash S., Philipson L. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab. 2007;6:229–235. doi: 10.1016/j.cmet.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen C.S., Watanabe S., Stas J.I., Klaphaak J., Yamane A., Schmitt N., Olesen S.P., Trimmer J.S., Rasmussen H.B., Misonou H. Trafficking of kv2.1 channels to the axon initial segment by a novel non-conventional secretory pathway. J. Neurosci. 2017;37:11523–11536. doi: 10.1523/JNEUROSCI.3510-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L., Wang L., Chopp M., Zhang Y., Szalad A., Zhang Z.G. MicroRNA 146a locally mediates distal axonal growth of dorsal root ganglia neuron under high glucose and sildenafil conditions. Neuroscience. 2016;329:43–53. doi: 10.1016/j.neuroscience.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D.Q., Li M.X., Ma Y.J., Wang Y., Wang Y. Efficacy and safety of prostaglandin e1 plus lipoic acid combination therapy versus monotherapy for patients with diabetic peripheral neuropathy. J. Clin. Neurosci. 2016;27:8–16. doi: 10.1016/j.jocn.2015.07.028. [DOI] [PubMed] [Google Scholar]
- Jiang D.Q., Li M.X., Wang Y., Wang Y. Effects of prostaglandin e1 plus methylcobalamin alone and in combination with lipoic acid on nerve conduction velocity in patients with diabetic peripheral neuropathy: a meta-analysis. Neurosci. Lett. 2015;594:23–29. doi: 10.1016/j.neulet.2015.03.037. [DOI] [PubMed] [Google Scholar]
- Johnson B.T., Ashley L., Michael K., Emily M., James T. The kv2.1 potassium channel forms endoplasmic reticulum/plasma membrane junctions via interaction with vap-a and vap-b. Biophys. J. 2018;114:295a. [Google Scholar]
- Kim S.J., Widenmaier S.B., Choi W.S., Nian C., Ao Z., Warnock G., Mclntosh C.H. Pancreatic beta-cell prosurvival effects of the incretin hormones involve post-translational modification of Kv2.1 delayed rectifier channels. Cell Death Differ. 2012;19:333–344. doi: 10.1038/cdd.2011.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Negi G., Sharma S.S. Suppression of NF-κB and NF-κB regulated oxidative stress and neuroinflammation by BAY 11-7082 (IκB phosphorylation inhibitor) in experimental diabetic neuropathy. Biochimie. 2012;94:1158–1165. doi: 10.1016/j.biochi.2012.01.023. [DOI] [PubMed] [Google Scholar]
- Kunysz E.L., Michel A.D., Whiting R.L. Functional and direct binding studies using subtype selective muscarinic receptor antagonists. Br. J. Pharmacol. 1988;93:491–500. doi: 10.1111/j.1476-5381.1988.tb10303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria G., Hsieh S.T., Johansson O., Kennedy W.R., Leger J.M., Mellgren S.I., Nolano M., Merkies I.S., Polydefkis M., Smith A.G. European federation of neurological societies/peripheral nerve society guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the European Fe-deration of neurological societies and the peripheral nerve society. Eur. J. Neurol. 2010;17:903–E49. doi: 10.1111/j.1468-1331.2010.03023.x. [DOI] [PubMed] [Google Scholar]
- Lee S., Milescu M., Jung H.H., Lee J.Y., Bae C.H., Lee C.W., Kim H.H., Swartz K.J., Kim J.I. Solution structure of GXTX-1E, a high-affinity tarantula toxin interacting with voltage sensors in kv2.1 potassium channels. Biochemistry. 2010;49:5134–5142. doi: 10.1021/bi100246u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung Y.M., Kang Y., Xia F., Sheu L., Gao X., Xie H., Tsushima R.G., Gaisano H.Y. Open form of syntaxin-1A is a more potent inhibitor than wild-type syntaxin-1A of Kv2.1 channels. Biochem. J. 2005;387:195–202. doi: 10.1042/BJ20041625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.W., Chang S.J. Moderate exercise suppresses NF-κB signaling and activates the SIRT1-AMPK-PGC1alpha Axis to attenuate muscle loss in diabetic db/db mice. Front. Physiol. 2018;9:636. doi: 10.3389/fphys.2018.00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.S., Fan B., Szalad A., Jia L., Wang L., Wang X., Pan W., Zhang L., Zhang R., Hu J. Microrna-146a mimics reduce the peripheral neuropathy in type II diabetic mice. Diabetes. 2017;66:3111–3121. doi: 10.2337/db16-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald P.E. Inhibition of Kv2.1 voltage-dependent K+ channels in pancreatic beta-cell enhances glucose-dependent insulin secretion. J. Biol. Chem. 2002;277:44938–44945. doi: 10.1074/jbc.M205532200. [DOI] [PubMed] [Google Scholar]
- Nguyen D.V., Shaw L.C., Grant M.B. Inflammation in the pathogenesis of microvascular complications in diabetes. Front. Endocrinol. 2012;3:170. doi: 10.3389/fendo.2012.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S., Hartnett K.A., Nerbonne J.M., Levitan E.S., Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J. Neurosci. 2003;23:4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak D., Guan D., Foehring R.C. Roles of specific Kv channel types in repolarization of the action potential in genetically identified subclasses of pyramidal neuron in mouse neocortex. J. Neurophysiol. 2016;115:2317–2329. doi: 10.1152/jn.01028.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter A.G., JaÈnicke R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Ramírez A., Vázquez-Sánchez A.Y., Carrión-Robalino N., Javier C. Ion channels and oxidative stress as a potential link for the diagnosis or treatment of liver diseases. Oxid. Med. Cell. Longev. 2016;2016:3928714. doi: 10.1155/2016/3928714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman P.T., He K., Hartnett K.A., Jefferson B.S., Hu L., Rosenberg P.A., Levitan E.S., Aizenman E. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc. Natl. Acad. Sci. 2007;104:3568–3573. doi: 10.1073/pnas.0610159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter D.M., Ho C., O’Leary M.E., Covarrubias M. Modulation of Kv3.4 channel N-type inactivation by protein kinase C shapes the action potential in dorsal root ganglion neurons. J. Physiol. 2012;590:145–161. doi: 10.1113/jphysiol.2011.218560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer S.H., Deardorff A.S., Fyffe R.E. Activity-dependent redistribution of Kv2.1 ion channels on rat spinal motoneuron. Physiol. Rep. 2016;4:e13039. doi: 10.14814/phy2.13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy Chowdhury S.K., Smith D.R., Saleh A., Schapansky J., Marquez A., Gomes S., Akude E., Morrow D., Calcutt N.A., Fernyhough P. Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain. 2012;135:1751–1766. doi: 10.1093/brain/aws097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N.H., Aizenman E. Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl. Stroke. Res. 2014;5:38–58. doi: 10.1007/s12975-013-0297-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw R.J. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S., Stevens M., Wiley J.W. Diabetic peripheral neuropathy: evidence for apoptosis and associated mitochondrial dysfunction. Diabetes. 2000;49:1932–1938. doi: 10.2337/diabetes.49.11.1932. [DOI] [PubMed] [Google Scholar]
- Tak P.P., Firestein G.S. NF-κB: a key role in inflammatory diseases. J. Clin. Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfaye S., Selvarajah D. Advances in the epidemiology, pathogenesis and management of diabetic peripheral neuropathy. Diabetes Metab. Res. Rev. 2012;28:8–14. doi: 10.1002/dmrr.2239. [DOI] [PubMed] [Google Scholar]
- Tsantoulas C., Zhu L., Shaifta Y., Grist J., Ward J.P.T., Raouf R., Michael G.J., Mcmahon S.B. Sensory neuron downregulation of the Kv9.1 potassium channel subunit mediates neuropathic pain following nerve injury. J. Neurosci. 2012;32:17502–17513. doi: 10.1523/JNEUROSCI.3561-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsantoulas C., Zhu L., Yip P., Grist J., Mcmahon S.B. Kv2 dysfunction after peripheral axotomy enhances sensory neuron responsiveness to sustained input. Exp. Neurol. 2014;251:115–126. doi: 10.1016/j.expneurol.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida H., Ma L., Ueda H. Epigenetic gene silencing underlies C-Fiber dysfunctions in neuropathic pain. J. Neurosci. 2010;30:4806–4814. doi: 10.1523/JNEUROSCI.5541-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van D.P.S., Cotter M.A., Bravenboer B., Cameron N.E. Pathogenesis of diabetic neuropathy: focus on neurovascular mechanisms. Eur. J. Pharmacol. 2013;719:180–186. doi: 10.1016/j.ejphar.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Vincent A.M., Callaghan B.C., Smith A.L., Feldman E.L. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat. Rev. Neurol. 2011;7:573–583. doi: 10.1038/nrneurol.2011.137. [DOI] [PubMed] [Google Scholar]
- Wang C.Y., Huang A.Q., Zhou M.H., Mei Y.A. GDF15 regulates Kv2.1-mediated outward K+ current through the Akt/mTOR signalling pathway in rat cerebellar granule cells. Biochem. J. 2014;460:35–47. doi: 10.1042/BJ20140155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y., Shin M.R., Sesti F. Oxidation of KCNB1 channels in the human brain and in mouse model of Alzheimer's disease. Cell Death Dis. 2018;9:820. doi: 10.1038/s41419-018-0886-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson N.M.W.D. Inflammatory mediators in diabetic neuropathy. J. Diabetes Metab. 2011;S5:004. [Google Scholar]
- Yang C.T., Lu G.L., Hsu S.F., Macdonald I., Chiou L.C., Hung S.Y., Chen Y.H. Paeonol promotes hippocampal synaptic transmission: the role of the kv2.1 potassium channel. Eur. J. Pharmacol. 2018;827:227–237. doi: 10.1016/j.ejphar.2018.03.020. [DOI] [PubMed] [Google Scholar]
- Yao H., Zhou K., Yan D., Li M., Wang Y. The Kv2.1 channels mediate neuronal apoptosis induced by excitotoxicity. J. Neurochem. 2009;108:909–919. doi: 10.1111/j.1471-4159.2008.05834.x. [DOI] [PubMed] [Google Scholar]
- Zhao M., Chen J.Y., Chu Y.D., Zhu Y.B., Bu S.Z. Efficacy of epalrestat plus α-lipoic acid combination therapy versus monotherapy in patients with diabetic peripheral neuropathy: a meta-analysis of 20 randomized controlled trials. Neural Regen. Res. 2018;13:1087. doi: 10.4103/1673-5374.233453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.T., Quan L.L., Chen L.P., Du T., Sun K.X., Zhang J.C., Yu L., Li Y., Wan P., Chen L.L. Sp6616 as a new kv2.1 channel inhibitor efficiently promotes β-cell survival involving both PKC/Erk1/2 and CaM/PI3K/Akt signaling pathways. Cell Death Dis. 2016;7:e2216. doi: 10.1038/cddis.2016.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplemental Information. Original data have been deposited to Mendeley Data: [https://doi.org/10.17632/gmxth7wsh8.1]