

Abstract
Plasmacytoid dendritic cells (pDCs) express Toll like receptors (TLRs) that modulate the immune response by production of type I interferons. Here, we report that sphingosine kinase 1 (SphK1) which produces the bioactive sphingolipid metabolite, sphingosine 1-phosphate (S1P), plays a critical role in the pDC functions and interferon production. Although dispensable for the pDC development, SphK1 is essential for the pDC activation and production of type I IFN and pro-inflammatory cytokines stimulated by TLR7/9 ligands. SphK1 interacts with TLRs and specific inhibition or deletion of SphK1 in pDCs mitigates uptake of CpG oligonucleotide ligands by TLR9 ligand. In the pristane-induced murine lupus model, pharmacological inhibition of SphK1 or its genetic deletion markedly decreased the IFN signature, pDC activation, and glomerulonephritis. Moreover, increases in the SphK1 expression and S1P levels were observed in human lupus patients. Taken together, our results indicate a pivotal regulatory role for the SphK1/S1P axis in maintaining the balance between immunosurveillance and immunopathology and suggest that specific SphK1 inhibitors might be a new therapeutic avenue for the treatment of type I IFN-linked autoimmune disorders.
Keywords: auto-immunity, interferon, plasmacytoid dendritic cells, sphingosine 1-Phosphate, sphingosine kinase, systemic lupus erythematosus
1 |. INTRODUCTION
The innate immune system maintains a robust immunosurveillance which includes an extensive array of sentinel cells that continuously monitor the surroundings for any invading pathogens. Of the varied immune cells that are capable of antigen recognition and the subsequent activation of the adaptive arm of the immune system, the plasmacytoid dendritic cells (pDCs) represent a unique class in itself.1,2 The “immature” pDCs with plasmacytoid morphology are specialized in antigen capture and type I Interferon production, wherein they differentiate to “mature” dendritic cells capable of antigen presentation.1,2 This unique cell type thus serves as a link between the innate and adaptive arms of the immune response. Though low in frequency in comparison to other hematopoietic cells, pDCs are the highest known producers of type I interferon in response to a viral attack.3,4 Hence, they are considered to be professional interferon producing cells.
Plasmacytoid dendritic cells are equipped with artillery for defending against a pathogenic invasion, especially of the viral genre. Toll like receptors (TLRs) constitute the most extensively studied pattern recognition receptors that have been purposed to recognize evolutionarily conserved pathogen associated molecular signatures.4,5 Of the large repertoire of TLRs, TLR3, 7, and 9 are strategically localized in the endosomal compartments where they recognize the ssRNA or dsDNA of invading viruses. The location of the endosomal TLRs also ensures that self-nucleic acids are not recognized and thereby lead to a state of autoimmunity.6 TLRs have a bipartite role in protecting against viruses and also regulation of autoimmunity. This requires an intricate regulatory balance to ensure proper functioning of the immune system w ithout an unduly heightened response.
In addition to their roles in host defense, pDCs have the ability to recognize self-nucleic acids, auto-antibodies, and immune complexes which can result in the exacerbated interferon secretion.1 This elicits a condition of autoimmunity with a typical interferon signature. Systemic lupus erythematosus (SLE) represents a classical example of such an autoimmune disease characterized by multi-organ involvement, heterogeneous clinical manifestations, and an unpredictable course marked by flares and remission.7,8 Currently, there is no cure for this disease but only management of symptoms. The importance of pDCs in the early stages of the disease has been identified,9,10 where an early depletion of pDCs alleviates autoimmunity10 and affects the maturation and distribution of pDCs in a mouse model of SLE prior to the clinical onset of the disease.11
The pleiotropic bioactive sphingolipid, sphingosine-1-phosphate (S1P), a ligand of 5 specific G protein-coupled receptors (GPCR), known as S1PR1–5, regulates a myriad of physiological and pathological processes, including lymphocyte egress,12 migration of dendritic cells,13 inflammation,14 cancer,15 and arthritis.16 Recent studies have suggested that S1PR1 activation in pDCs inhibits IFN-auto amplification by induced degradation of the IFN-alpha receptor 1 and suppression of STAT1 signaling.17 Moreover, S1PR4 signaling limits the IFN-alpha production by inhibiting internalization of the human pDC-specific inhibitory receptor, Ig-like transcript 7.18 However, not much is still known of the role of sphingosine kinases (SphK1 and SphK2), the enzymes that produce S1P,14,15 in the regulation of type 1 IFN responses and SLE, although SphK1 has been recently suggested to play a role in anti-viral responses.19–21 Therefore, it was of interest to examine the role of the SphK1/S1P axis in TLR-mediated innate immune functions of pDCs. Here, we report previously unknown functions of SphK1 in pDC-mediated immunological defense and immunopathology. Pharmacological inhibition or genetic deletion of SphK1 resulted in impaired production of type I IFN and other pro-inflammatory cytokines and amelioration of autoimmunity in a mouse model of SLE, thus providing an additional layer of regulation in TLR7/9-dependent production of IFN by pDCs.
2 |. MATERIALS AND METHODS
2.1 |. Cell culture and mice
HEK293 cells were purchased from the American Type Culture Collection (Manassas, VA). 293XL hTLR cells stably expressing either HA tagged TLR7 or TLR9 were purchased from Invivogen (San Diego, CA, USA). Cell lines were maintained in DMEM medium supplemented with 10% fetal bovine serum and 1X penicillin/streptomycin. Plasmacytoid dendritic cell line CAL-1 was maintained as described previously.22 Briefly, cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, NY, USA) supplemented with 10% fetal bovine serum, 1X penicillin streptomycin, and 1 mM glutamine.
C57Bl/6 mice (JAX stock # 005304, Jackson Laboratories (Bar Harbor, ME, USA) and SphK1 knockout mice and were backcrossed for at least seven generations (SphK1−/−, stock # 019095, Jackson Laboratories). About 6 to 8-weeks-old mice were housed in individually ventilated cages and fed with standard rodent chow and water ad libitum and followed a 12 hour light/dark cycle. All animal experiments were performed with prior approval from the Institutional Animal Ethics Committee (IAEC) of RGCB and followed the rules and regulations mandated by Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India.
2.2 |. In vivo experiments
For TLR ligand studies, mice were pretreated with the SphK1 inhibitor SK1-I (10 mg/kg, BML-E1411–0025, Enzo) by i.p injections 60 minutes prior to tail vein injections of R848 (2 μg/mouse, Tlrl-r848, Invivogen). After 2 hours, mice were euthanized by CO2 asphyxiation and blood collected by cardiac puncture. Serum was used for cytokine measurements.
For lupus studies, mice received a single i.p injection of 500 μL pristane as described previously.23 Experiments were carried out in 4 groups––untreated normal, pristane alone, pristane together with SK1-I (10 mg/kg i.p, twice a week, either in simultaneous or therapeutic mode) prior to onset of disease markers. Blood was collected regularly through the tail vein and monitored for changes in disease markers and when detected, treatment was initiated for the SK1-I therapeutic group at the same dose as the SK1-I simultaneous group. After 2 months, mice in all four groups were euthanized by CO2 asphyxiation and organs and blood collected.
2.3 |. Bone marrow isolation and generation of Flt3L-pDCs
Bone marrow isolation was carried out as described previously.24 Briefly, mice were euthanized by CO2 asphyxiation and femurs dissected out carefully, keeping the head of the bone intact and without any breaks. The bones were kept in 1X PBS and transported to a laminar hood where the rest of the isolation procedure was done under aseptic conditions. The bone marrows were flushed out into sterile centrifuge tubes and centrifuged at 1200 rpm for 5 minutes at room temperature. Pellets were washed with twice with PBS containing 1% serum. Red blood cells (RBCs) were lysed by incubating cell pellets with 1 mL of RBC lysis solution (R7757, Sigma-Aldrich, St. Louis, MO, USA) for 3 minutes at room temperature. 10 mL of 1X PBS were then added followed by centrifugation at 750 g for 10 minutes at room temperature. Pellets were then re-suspended in RPMI and cells counted. 1 × 106/mL of cells were plated per well in a 24-well plate in RPMI media containing 10% serum, 1X penicillin/streptomycin, 2 mM glutamine, 10 mM Hepes, 5 μM β-mercaptoethanol, and 100 ng/mL of Flt3L (murine Flt3-ligand, Peprotech, Rehovot, Israel). The plate was left undisturbed for 5 days when half of the old media was removed and replenished with new media containing all the components. After a period of 8 days, cells were collected and sorted by fluorescence-activated cell sorting (FACS) for pDC populations. Cells were collected and re-suspended in 100 μL of wash buffer and stained with CD11C-FITC and PDCA1-APC antibodies (Biolegend). Double positive cells that represent the pDC population were sorted and collected with a BDFACS Aria II flow cytometer. The cells were counted and plated (1×105cells) in RPMI medium containing 2% serum and 1X antibiotic. Cells were then treated with CpG-ODN2216 (3 μM), R848 (10 μg/mL), murine cytomegalovirus (MCMV) Smith strain (VR-1399, ATCC) (MOI = 1) for 18 hours (for Enzyme linked immunosorbent assay [ELISA]) or 3 hours (for RNA isolation).
2.4 |. Preparation of splenocytes
To isolate splenocytes, spleens were collected from mice after euthanasia. This was followed by macerating the spleen in 1X PBS, and centrifugation at 2000 rpm for 3 minutes at room temperature. RBCs were lysed by incubating with RBC lysis solution for 3 minutes. The cells were washed, counted, and plated for subsequent experiments. For pDC activation assays, splenocytes were incubated with R848 for 24 hours, with or without 30 minutes SK1-I pretreatment. Cells were then stained with PE-CD80 and PE-Cy7-CD86 antibodies (Biolegend) and expression was analyzed by flow cytometry. For ligand uptake studies, splenocytes were treated with 50 nM CpG-ODN FITC (Tlrl-2216f, Invivogen) and analyzed by flow cytometry.
2.5 |. RNA isolation
RNA from cultured cells and peripheral blood mononuclear cells (PBMCs) was isolated with the RNeasy Micro kit (74004, Qiagen) and from mouse blood with RNeasy Protect animal blood kit (73224, Qiagen) following the manufacturer’s instructions. An intermediate DNase incubation was included to eliminate genomic DNA contamination. RNA was reverse transcribed to cDNA using the High Capacity cDNA reverse transcription kit (4368814, Applied Biosystems). Gene expression studies were performed with Power SYBR green PCR master mix (4367659, Applied Biosystems) and the respective primers. Fold changes were calculated by the 2(-ΔΔCt) method, after normalizing with glyceralde-hyde-3-phosphate dehydrogenase (GAPDH) or β-actin. The primer sequences used for QPCR are given below. β-actin (h)-(F) 5’-CCAGCTCACCATGGATGATG-3’,(R)5’-AT GCCGGAGCCGTTGTC-3’; IFNβ (h)-(F) 5’-CTTTGCT ATTTTCAGACAAGATTCA-3’;(R) 5’-GCCAGGAGGTTCTCAACAAT-3’; IL-6 (h)-(F) 5’-AAATTCGGTACATCCT CGACGGCA-3’, (R)5’-GTGCCTCTTTGCTGCTTTCACA CA-3’;TNF-α (h)-(F)5’-CACCACTTCGAAACCTGGGA-3’, (R)5’-CACTTCACTGTGCAGGCCAC-3’; IFN-α (h)-(F)5’TCCTGCTTGAAGGACAGACA-3’,(R)5’-TTTCAGCCTTTTGGAACTGG-3’; CCL5 (h)-(F)5’ GGAGTATTTCTACACCAGTGGCA-3’, (R)5’-TGACAAAGCTTGCCC TTGTTC-3’; ISG15 (h)-(F)5’-AGATCACCCAGAAGATCGGC-3’,(R)5’-GTTCGTCGCATTTGTCCACC-3’; IFIT-1(h)-(F)5’-GGCTTTGCTACAAGGCACAAA-3’, 5’AGGTCTAGATGAGCCACCTCAA-3’; CXCL-10 (h)-(F) 5’-CTAAGTGGCATTCAAGGAGTACCT-3’, (R)5’ACGTGGACAAAATTGGCTTGC-3’; CXCL-1(h)-(F)5’-ACACTCAAGAATGGGCGGAAA-3’, (R)5’CAGTTGGATTTGTCACTGTTCAGC-3’; NF-κB1(h)-(F)5’-TTTGGGAAGGCCTGAACAAATG-3’, (R)5’-AATCCTCTCTGTTTAGGTTGCTCT-3’; IL-8 (h)-(F)5’-CACTGCGCCAACACAGAAATTA-3’, (R)5’-TTCTCAGCCCTCTTCAAAAACTTC-3’; CCL-4(h)-(F)5’-ACCGCCTGCTGCTTTTCTTA-3’, (R)5’-GATTCACTGGGATCAGCACAGA-3’; CXCL-9 (h)-(F)5’-GTTC TGATTGGAGTGCAAGGAAC-3’, (R)5’-AGGGCTTGGG GCAAATTGTT-3’; IFIT-2 (h)-(F)5’-AGAAAGCTGATGAGGCCAATGA-3’, (R)5’-AGCAGTTGTTTCGCTACAG GA-3’; ISG-20(h)-(F)5’-AGGCACTGAAAGAGGACATGAG-3’, (R)5’GTTCTGGATGCTCTTGTGTAGGA-3’; SphK1(h)-(F)5’-AGCTTCCTTGAACCATTATGCTG-3’, (R)5’ AGGTCTTCATTGGTGACCTGCT-3’; SLC15A4 (h)-(F) 5’-ATCCCTCTGAAGGACAAACTGG-3’, (R)5’-TCCAAAATTCCTGCAGCAAAGG-3’; Granulin (h)-(F)5’-CTCCTGCATCTTTACCGTCTCA-3’, (R) 5’-CGGAGTTGTTACCTGATCTTTGGA-3’; PRAT4A (h)-(F) 5’-TGTTGCTGTGGAGCTGAAGT-3’, (R)5’-GGGGATCTGAAATGGACTTGGT-3’; S1PR1 (h)-(F)5’ TATCATCGTCCGGCATTACA-3’,(R)5’-GAACACCACCGAGGTAGTT-3’; S1PR2 (h)-(F)5’-TGGCCGCCTCCGATCT-3’, (R)5’-GAGAGCAAGGTATTGGCTACGAA-3’; S1PR3 (h)-(F)5’-TCCCTTTGAAATGAATGTTCCTGG-3’, (R)5’-CAGTTGCCATCACTTGGCATT-3’; S1PR4 (h)-(F)5’-AGTCTTGCGTGTGGATGGTG-3’, (R)5’-ACACAGCTTCCTGTACC CCA-3’; S1PR5 (h)-(F)5’-TTGACGTTCTGCTTGGGAACA-3’, (R)5’-ATCTCTGTCGTTTGTCACTGGAA-3’; GA PDH (m)-(F)5’-TGCCCCCATGTTTGTGATG-3’, (R)5’-TGTGGTCATGAGCCCTTCC-3’; U1A (m)-(F)5’-CGGGGA AGATAGTTGTGTGTCT-3’,(R)5’AGGGACTTCTTGAGCTCATCCT-3’; Sca-1(m)-(F)5’-ACTGTGTGCAGAAAGAGCTCAG-3’, (R)5’-TGCCTCCTGAGTAACACAGACT-3’; IFN-β (m)-(F)5’-AGCTCCAAGAAAGGACGAACA-3’, (R)5’-TGGATGGCAAAGGCAGTGTA-3’; Mx-1(m)-(F)5’-GCAGACGGAATATTGGGAGA-3’, (R)5’-GAGCCTCATCCAGCCTTAAA-3’; ICAM-1(m)-(F)5’-TGAAAGATGAGCTCGAGAGTGG-3’, (R)5’-CGGAAACGAATACACGG TGATG-3’; BAFF (m)-(F)5’-TTCTTCATCTACAGCCAGGTTCT-3’, (R)5’-AGCCGAGTAGCAGGAATTGT-3’; Oas-3(m)-(F)5’-TGATAAGGATTGCCAAGGGAGG-3’, (R)5’-TCACCAAAGCTCTGGAAGCA-3’; Siglec-H (m)-(F)5’-TGTGCATGTGACAGACCTCA-3’, (R)5’-GCTGACATCCAGGAAAAGATGG-3’, E2–2 (m)-(F) 5’-AAACCGAGCCAGGTGCATAA-3’, (R)5’-CACAAACCTTCAATGGCCACA-3’; SpiB (m)-(F)5’-CCACACTTAAGCTGTTTGTACCC-3’, (R)5’-TGAACAGTTTGGGAGTGGCT-3’; TL R7(m)-(F)5’-CTCAGTGGGTGTTTTCGATGTG-3’, (R)5’-CACATGGGCCTCTGGGATATTT-3’; TLR9 (m)-(F)5’-ACTTACTGTTGGAGGTGCAGAC-3’,(R)5’-AAAGGCCAAAGCAGTCCCAA-3’; S1PR1 (m)-(F)5’-ATTAGCAGGCGTGGCTTACA-3’, (R)5’-AAACATACTCCCTTCCCGCA-3’; S1PR2 (m)-(F)5’-GCCTTCATCATCATCTTGTGCTG-3’, (R)5’-AGGTACATTGCTGAGTGGAACT-3’, S1PR3 (m)-(F)5’-TTCATCGGCAACTTGGCTCT-3’, (R)5’ TGTTGGAGACAGACTGAACGTC-3’; S1PR4 (m)-(F)5’-CATCTTTGGTTCTAATGTCTGGGC-3’; (R)5’-TGAGAG GATTAATGGCTGAGTTGA-3’; S1PR5 (m)-(F)5’-TGCTATTACTGGATGTCGCGT-3’, (R)5’-TTCAGCAGCGAG TTAGCCAT-3’.
2.6 |. Western blotting
After treatments, cells were collected in protein lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% NP40, 50 mM EDTA, 50 mM NaF, and 10 μM Na2VO3 with protease inhibitors. After removing the cytoplasmic fractions, nuclear pellets were suspended in nuclear extraction buffer (20 mM Hepes, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, and protease inhibitors). For SDS-PAGE, bis-acrylamide gels of required percentage were used and proteins were transferred to nitrocellulose membranes. The membranes were blocked with 5% skim milk and antibodies were added and incubated overnight at 4°C. HRP conjugated secondary antibodies were added and immunopositive bands visualized by chemiluminescence.25 The source of antibodies as follows. p-ERK (9106), p-JNK (9251) and p-IKK (2697) all from Cell Signaling Technology; p-SphK1 (SP1641, ECM Biosciences); SphK1 (ab46719), and His (ab18184) from Abcam; ERK (sc93), JNK (sc474), p65 (sc109); IKKα/β (sc7607), β-tubulin (sc9104), Lamin (sc20681), IRF3 (sc9082), and IRF7(sc9083) from Santacruz biotechnology. HA (AKR-006; Cell biolabs.Inc); V5 (R-96025; Invitrogen).
2.7 |. Enzyme linked immunosorbent assay
ELISAs were carried out with supernatants from cell cultures as well as with mouse serum according to the suppliers’ instructions. Human IFNα and β kits were procured from R&D Systems and mouse IFNα and β kits were from ebiosciences and Biolegend, respectively.
2.8 |. Ni-NTA affinity purification
Ni-NTA affinity beads were used to purify 6X-histidine tagged SphK1. Briefly, 50 μL of Ni-NTA agarose beads (30210, Qiagen) were suspended in IP buffer containing a protease inhibitor, then centrifuged at 1000 rpm for 1 minute at 4°C, and washed twice with IP buffer. Protein lysates (350 μg) were added to the washed beads and then mixed by rotation for 4 hours. After two washes, the pellets were resuspended in SDS gel loading buffer, boiled for 5 minutes, and centrifuged. Supernatants were separated by SDS-PAGE and His-tagged proteins visualized with the indicated antibodies.
2.9 |. Pull downs with anti-HA beads
TLR7 and TLR9 were tagged with HA in the 293 XL hTLR7-HA and 293 XL hTLR9-HA pDCs. Briefly, HA-tagged protein complexes were pulled down with anti-HA beads as follows: 50 μL of were extensively washed anti-HA-agarose beads (A2095, Sigma) were added to 400 μg of protein lysates and incubated overnight at 4°C in a rotary mixer. The beads were washed four times with IP buffer containing a protease inhibitor. SDS sample buffer was added to the beads, boiled for 5 minutes, and proteins separated by SDS-PAGE. After transfer to nitrocellulose, HA tagged and associated proteins were detected with the indicated antibodies.
2.10 |. Immunofluorescence
CAL-1 cells were collected onto glass slides by cytospin in a Thermo CytoSpin4 Cytocentrifuge for 3 minutes at 1000 rpm. The slides were then air dried for 10 minutes at room temperature. For experiments with HEK293XL hTLR9-HA, cells were cultured on polylysine-coated coverslips. The cells were fixed with 4% paraformaldehyde, and then washed three times with 1X PBS. Cells were permeabilized with 0.1% Triton X-100 and blocked with 3% bovine serum albumin (BSA) in PBS. Cover slips were then incubated with the corresponding primary antibodies overnight at 4°C. After 1X PBS washes, cover slips were incubated with the appropriate fluorophore-conjugated secondary antibodies, and nuclei were stained with Hoechst 33342 (1 μg/mL). The cover slips were mounted on slides with glycerol and examined with a Nikon A1R Laser scanning confocal microscope with 60X oil immersion objective.
2.11 |. Renal IgG deposition to assess glomerulonephritis
Renal immune complex deposition was analyzed as described previously.24 Briefly, tissue cryosections from snap frozen kidneys were fixed in ice cold acetone followed by 1X PBS wash. Slides were blocked with 3% BSA and stained with FITC goat anti-mouse IgG antibody (Biolegend, San Diego, USA) prepared in 3% BSA and incubated at 4°C, overnight in a moist dark chamber. Slides were mounted with glycerol and analyzed by scanning confocal microscopy.
2.12 |. Measurements of CpG-FITC uptake
For flow cytometry measurements, Flt3L-pDCs cultured for 8 days were treated with 50 nM of CpG-ODN2216-FITC as indicated. In some experiments, pDCs were pre-treated with SK1-I for 30 minutes and then stimulated with CpG-ODNFITC. After fixation with 4% paraformaldehyde, pDCs were stained with APC-PDCA1, and then analyzed by flow cytometry for FITC uptake. Gating was set to eliminate debris and for APC positive cells, which represent the pDC population that has taken up CpG-FITC.
For immunofluorescence uptake assays, pDCs were pretreated with SK1-I for 30 minutes prior to incubation with CpG-FITC for 20 minutes. After staining with Hoechst 33342, slides were mounted with glycerol and analyzed by scanning confocal microscopy.
2.13 |. Autoantigen arrays
Auto-antibodies in serum from pristane-treated mice were measured by the University of Texas Southwestern Medical Center Genomics and Microarray Core facility with Autoantigen Microarray Panel I (https://microarray.swmed.edu/products/product/autoantigen-microarray-panel-i/). Each panel has the capacity to analyze 16 samples including two internal controls. The IgG auto-antibodies in the samples (n = 2 for wild-type untreated mice and n = 4 for other groups) were determined as signals at 532 nm. The Tiff images thus generated were analyzed with Genepix Pro 6.0 software.26 The average signal to noise ratios was normalized to a PBS negative control and the data presented as a heat map.
2.14 |. Clinical lupus samples and pDC isolation
Peripheral blood was collected by venipuncture from 10 healthy human subjects and 22 SLE patients after an IRB (Institute Review Board) and human ethical committee approved the study protocol (RGCB—IHEC/01/2017/01 and HEC 04/18/2018/MCT). Patients attending Nephrology department, Government Medical College, Thiruvananthapuram, who met the 2012 Systemic Lupus International Collaborating Clinics (SLICC) classification criteria for SLE were selected.24 The disease activity was assessed using SLE Disease Activity Index (SLEDAI) score. Healthy adults (female, age group 22–32) who did not have any acute or chronic illnesses with absent clinical symptoms and signs of SLE were selected as control group. For measuring the S1P levels, serum samples (10 healthy controls and 9 SLE samples) were commercially obtained from Discovery Life Sciences (Los Osos, USA).
PBMCs were isolated from the buffy coats obtained by Ficol gradient centrifugation with Histopaque. For isolation of pDCs (human ethical committee approval no. RGCBIHEC/1/2019/14), the PBMCs from human blood were further subjected to magnetic sorting with Plasmacytoid Dendritic Cell Isolation Kit II, human (130-097-415, Miltenyi Biotec, San Diego CA, USA). The pDCs were obtained by a negative labeling technique whereby the pDCs remained unlabeled and the labeled non-pDCs were separated out magnetically using LD columns (130-042-901, Miltenyi Biotec). The pDCs after magnetic sorting were labeled with anti-human CD123-FITC (Biolegend) and APC-BDCA-2-human (Miltenyi Biotec) and sorted by flow cytometry. The collected pDCs were then counted and plated in RPMI-1640 media containing 2% fetal bovine serum and 1X antibiotic (105 cells/100 μL). After stimulation with TLR ligands as described above, pCDs were collected for gene expression analyses. For determination of SphK1 protein expression, pDCs were lysed, and the proteins analyzed by immunoblotting.
2.15 |. Quantification of sphingolipids
Sphingolipids in pristane-treated mice and in clinical lupus samples were quantified by liquid chromatography, electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS, 6500 QTRAP, ABI) at the Virginia Commonwealth University Lipidomics Facility as described.27
2.16 |. Docking studies
Bioinformatics approaches to model the protein-protein interactions between SphK1 and TLRs used three different docking software packages: ZDOCK, ClusPro, and GRAMM-X. The best docked structures were visualized in Discovery Studio and the inter-molecular H-bonds were also depicted. SphK1 was docked with SK1-I using the automated docking platform, AUTODOCK 4.2.
2.17 |. Statistical analyses
The data are from experiments conducted three or more times and are expressed as mean ± SD Statistical analyses (GraphPad Prism 7; GraphPad software, CA, USA) between two groups were performed with unpaired two-tailed Student’s t test. Wilcoxon paired t test was used where indicated. The significance between multiple groups was analyzed by ANOVA followed by post hoc Tukey test. P value <.05 was considered statistically significant.
3 |. RESULTS
3.1 |. Inhibition or deletion of SphK1 diminishes TLR7/9-mediated interferon production in pDCs
pDCs are specialized in the secretion of IFNs in response to viral invasion. TLR9 is activated by both type A (ODN2216) and B (ODN2006) unmethylated CpG oligodeoxynucleotide (ODN) classes whereas TLR7 is stimulated by ssRNA and the synthetic ligand Resiquimod, R848. As an initial approach to delineate the importance of SphK1 in pDCs, we used SK1-I, a specific competitive inhibitor of SphK128 that does not decrease SphK1 level.29 Pharmacological inhibition of SphK1 with SK1-I decreased the induction of mRNAs of both IFN subtypes in CAL-1 cells, a human pDC cell line22 induced by stimulation of TLR7/9 (Figure 1A,B). The secretion of IFN-β from TLR-stimulated CAL-1 cells determined by ELISA was also markedly reduced by SK1-I treatment (Figure 1C). Consistent with this, TLR7/9-induced production of pro-inflammatory cytokines (TNF and IL-6) was greatly suppressed by inhibition of SphK1 with SK1-I (Supplemental Figure S1A). Similarly, when SphK1 expression was transiently downregulated in HEK293 cells stably over expressing TLR9 followed by stimulation with CpGODN 2216, levels of both IFN-β and IL-6 were significantly decreased (Supplemental Figure S1B). As inhibition of production of the pro-growth S1P by SK1-I may induce apoptosis,28,30 it was important to examine its effect on survival of pDCs as assessed by MTT assay and Annexin V staining (Supplemental Figure S1C,D). However, SK1-I treatment did not affect survival or induce apoptosis of pDCs indicasting that SK1-I-induced cell death was not responsible for the decreased IFN response.
FIGURE 1.
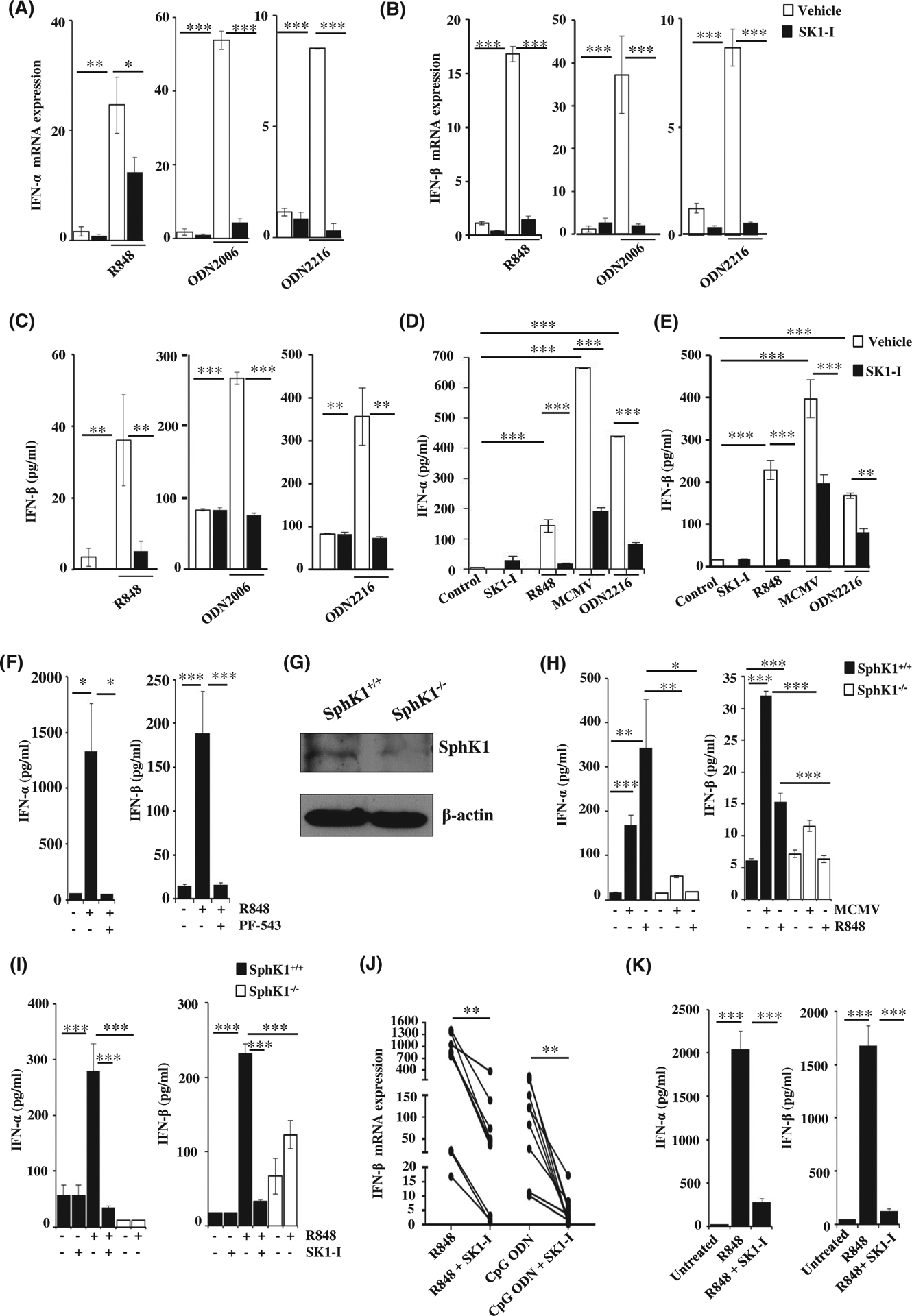
SphK1 is essential for TLR7/9-mediated type I interferon response. A-C, CAL-1 cells pre-treated with SK1-I (10 μM) or vehicle followed by stimulation with R848 (10 μg/mL), CpG-ODN 2006 (1 μM), CpG-ODN 2216 (3 μM) for 3 h (A, B) or 18 h (C) as indicated. IFN-α (A) and IFN-β mRNA (B) measured by qPCR and the mRNA levels normalized to β-actin. IFN-β easured by ELISA (C). Data are mean ± SD of three independent experiments. D-E, Flt3L-derived mouse pDCs pre-treated with SK1-I or vehicle and then stimulated with the indicated ligands. Levels of IFN-α (D) and IFN-β (E) measured by ELISA. Data are representative of four independent experiments. F, Flt3L-derived mouse pDCs treated with PF543 (10 μM), followed by R848 for 18 h. Levels of IFN-α and β in cell supernatant determined by ELISA. G, Expression of SphK1 determined in Flt3L-pDCs derived from SphK1+/+ and SphK1−/− mice by immunoblotting (H) Flt3L-derived mouse pDCs isolated from SphK1+/+ and SphK1−/− mice and treated with R848 (10 μg/mL) and MCMV (MOI = 1) and levels of IFN-α and IFN-β measured by ELISA. Data are mean ± S.D from four independent experiments. I, SphK1+/+ and SphK1−/− mice (n = 4/group) intraperitoneally injected with R848 (2 μg). One group of SphK1+/+ mice (n = 4) pre-treated with SK1-I (10 mg/kg) for 1 h before challenging with R848. IFN-α and IFN-β in serum measured by ELISA. J, PBMCs isolated from healthy human subjects (n = 10) stimulated with R848 (10 μg/mL) and CpG-ODN (1 μM) in the presence and absence of SK1-I IFN-β mRNA measured by qPCR and normalized to β-actin. K, Human pDCs isolated from peripheral blood of healthy human subjects treated with R848 either with or without SK1-I pretreatment. ELISA analyses of IFN-α and β in cell supernatants after 18 h of ligand treatment. Statistical significance was determined by ANOVA followed by post hoc Tukey test. (A-F, H, I, K), Wilcoxon paired t test (J). *P < .05, **P < .005, ***P < .001
Having observed the effect of SphK1 in vitro on cultured pDCs, we extended our findings to the ex vivo model of Flt3L-induced pDCs, as this cytokine promotes the differentiation of hematopoietic cells into the pDC lineage.31 pDCs differentiated from mouse bone marrow cells by culturing in the presence of Flt-3L were sorted (CD11C+/PDCA-1+) and then stimulated with various ligands, including mouse cytomegalovirus (MCMV) that stimulates TLR9. In the presence of SK1-I, murine pDCs failed to produce both IFN subtypes compared to vehicle treated pDCs (Figure 1D,E). These findings were further confirmed with another potent and specific SphK1 inhibitor, PF54332 (Figures 1F and S1E,F).
To further validate the role of SphK1 in type I IFN production, pDCs were generated from SphK1+/+ and SphK1−/−mice and protein expression determined by western blotting (Figure 1G). Importantly, production of IFN-α and IFN-β induced by stimulation of TLR7/9 in Flt-3L-derived pDCs devoid of SphK1 was drastically reduced compared to wild-type pDCs (Figure 1H). Furthermore, as expected, intraperitoneal injections of the potent TLR7 agonist R848 elicited a dramatic increase of type 1 IFNs detected in the serum. In sharp contrast, production of both IFNs was greatly attenuated in SphK1 knockout mice or by administration of SK1-I to wild-type mice (Figure 1I).
Similarly, when SphK1 was inhibited in peripheral blood mononuclear cells (PBMC) isolated from healthy human volunteers, IFN-β mRNA levels were significantly decreased following stimulation with TLR7/9 ligands (Figure 1J). Expression of SphK1 was confirmed in pDCs purified from human PBMCs with magnetic beads, both at the mRNA and protein level (Figure S2A). Stimulation of these pDCs with TLR7/9 ligands led to the production of IFN-α and IFN-β and both were markedly reduced by SK1-I treatment (Figures 1K and S2B). Hence, pharmacological and genetic deletion of SphK1 leads to impaired production of IFNs induced by TLR activation.
3.2 |. SphK1 reciprocally regulates TLR7/9 activation and is a determinant of pDC activation
The next question we asked was whether SphK1 is a determinant of pDC homeostasis. To this end, pDC populations in spleens from SphK1−/− mice were compared to those from WT mice. There were no significant differences in the numbers of these cells indicating that SphK1 deficiency did not alter pDC development (Figure 2A). Exposure to infection triggers the expression of various co-stimula-tory molecules such as CD80 and CD86 that aid pDCs in stimulating and activating T cells.33 The role of SphK1 in regulating this unique property of pDCs that links the innate and adaptive arms of the immune response was next examined. Flow cytometric analysis of pDCs showed that treatment with TLR ligands resulted in an increased expression of the pDC activation markers CD80 and CD86 (Figure 2B,C). Prior treatment with SK1-I markedly diminished activation of pDCs in response to TLR stimulation. Similarly, R848 failed to increase levels of CD80 and CD86 in SphK1 null pDCs (Figure 2B,C), supporting the importance of SphK1 in activation of pDCs in response to TLR triggering. Taken together, these results indicate that SphK1 is dispensable for pDC development; however, it is essential for pDC activation and subsequent production of type I IFN and pro-inflammatory cytokines.
FIGURE 2.
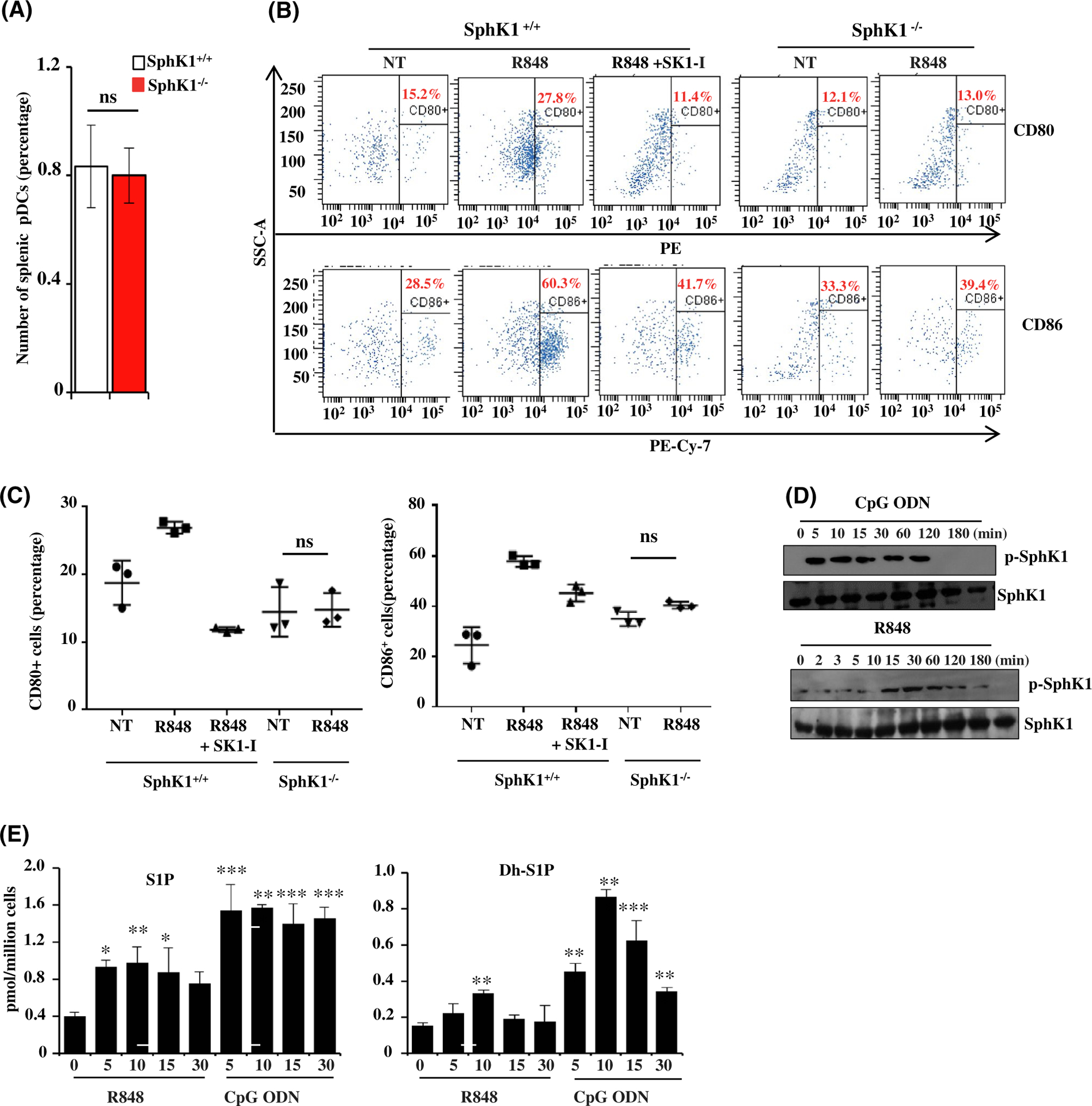
Involvement of SphK1 in pDC activation. A, Numbers of splenic pDCs isolated from wild-type and SphK1−/− mice determined by flow cytometry. Data are mean ± SD of four independent experiments. B, Activation status (CD80-PE/CD86-PECy7) of splenic pDCs from SphK1+/+ and SphK1−/− mice determined by flow cytometry after TLR7 stimulation. Where indicated, cells were pretreated for 30 min with SK1-I prior to R848 treatment. C, Quantification of flow cytometry data in Figure 2B. D, CAL-1 cells treated with CpG-ODN (1 μM) or R848 (10 μg/mL) for the indicated times. Proteins separated by SDS-PAGE and immunoblotted with the indicated antibodies. Blots are representative of three independent experiments. E, CAL-1 cells treated with R848 (10 μg/mL) or CpG-ODN (1 μM) for the indicated times (in min) and the level of S1P and dihydro-S1P (DH-S1P) in the cell culture supernatant determined by LC-ESI-MS/MS. Statistical significance was determined by Student’s t test (A) or ANOVA followed by post hoc Tukey test (C, E). n.s—P > .05, *P < .05, **P < .005, ***P < .001
Robust effects on TLR-mediated responses by SphK1 led us to ask whether SphK1 is activated in response to TLR stimulation. As previously shown, SphK1 is activated by numerous agonists including cytokines and growth factors by phosphorylation at Ser225.34 This aids in its translocation to the plasma membrane where it phosphorylates its substrate sphingosine and produces S1P.35 Indeed, stimulation of CAL-1 cells with TLR7/9 ligands rapidly increased phosphorylation of SphK1 within 5 minutes, as determined by immunoblotting with a phospho-specific SphK1 antibody (Figure 2D). Consistent with these effects, CpG-ODN rapidly increased S1P within 5 minutes, which remained elevated for 30 minutes (Figure 2E). Similarly, there was also a rapid increase in dihydro-S1P (Figure 2E). These results suggest that stimulation of TLR7/9 leads to activation of SphK1 and production of S1P that contribute to pDC activation and subsequent type 1 IFN production.
3.3 |. SphK1 regulates nuclear import of IRF3, IRF7, and p65 and determines IRF3- and NF-κB-dependent gene expression
Viral infections induce elevated production of type I IFN; this is mediated by the IRF family of transcription factors.36 IRF3 and IRF7 are considered to be the “master regulators” of type I interferon expression. IRF3 is important in early events, remains latent in the cytoplasm, and moves into the nucleus after it is phosphorylated in response to a pathogenic invasion.37 IRF7, which has a predominant role toward the later phase of the immune response, is constitutively expressed by pDCs,38 which equips them with the ability to rapidly produce IFN. IRF7, like IRF3, is transported to the nucleus in response to TLR stimulation. Immunofluorescence analysis was used to investigate the effects of SphK1 inhibition by SK1-I on TLR-stimulated nuclear import of both of these transcription factors. Treatment with SK1-I prevented their nuclear transport (Figure 3A). Moreover, pretreatment with SK1-I significantly decreased the expression of a panel of IRF3-dependent genes (Figure 3B). The NF-κB transcription machinery represents another pathway that acts as a direct effector of interferon responses during TLR stimulation.39 Inhibition of SphK1 likewise interfered with the NF-κB pathway as demonstrated by the diminished activation of the IKK complex (Figure 3C), a decline in the nuclear import of the p65 transcription factor (Supplemental Figure S3A,B), and reduced expression of NF-κB -dependent genes (Figure 3D) when treated with a TLR ligand.
FIGURE 3.
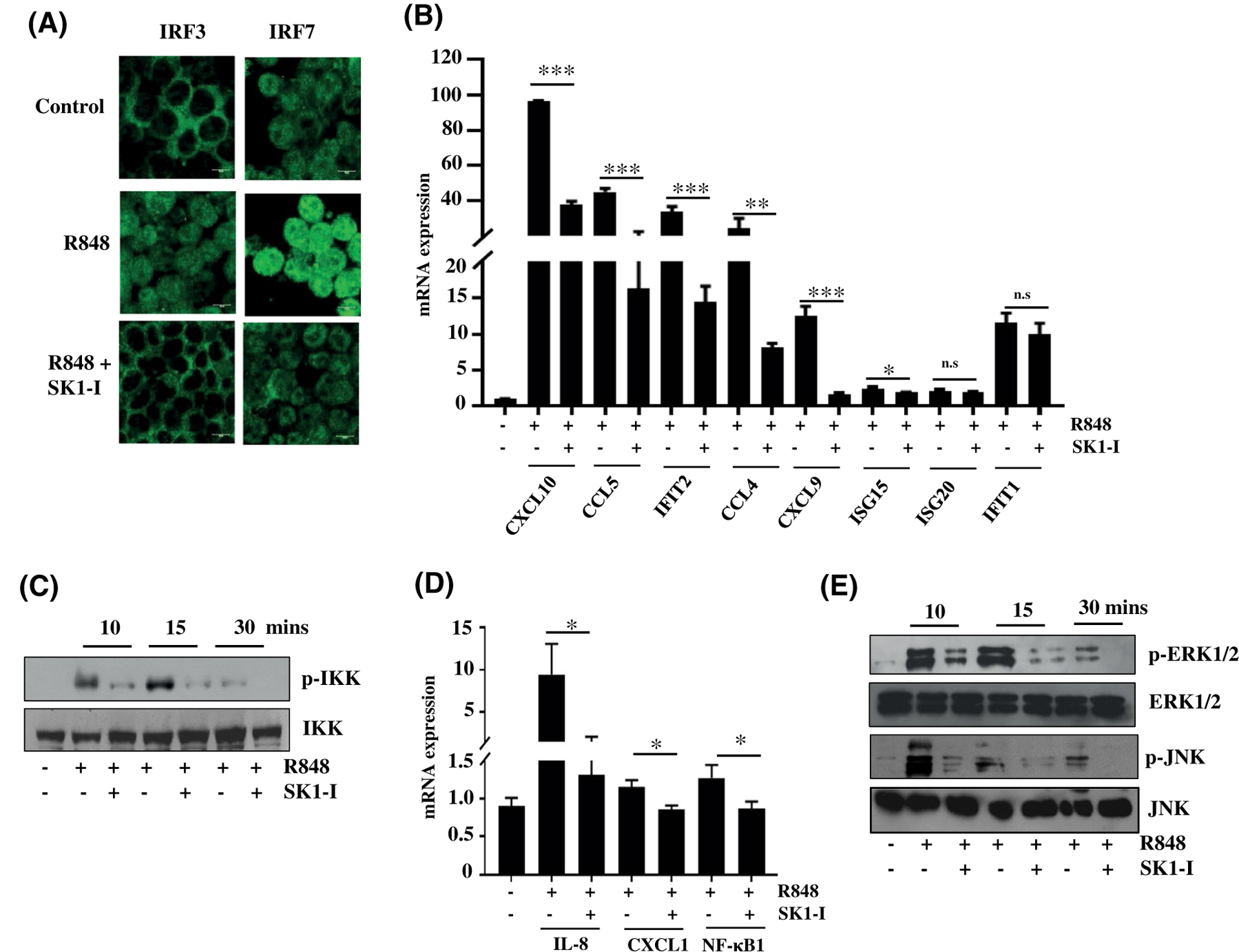
SphK1 regulates nuclear import of IRF3, IRF7, and NF-κB and downstream MAPK signaling. A-F, CAL-1 cells treated with R848 (10 μg/mL) in the presence or absence of SK1-I and (A) nuclear import of IRF3 and IRF7 was analyzed by immunofluorescence microscopy after staining with antibody against IRF3 and IRF7. Images are representative of five independent experiments. Scale bar: 10 μm. B, IRF3-dependent gene expression determined by qPCR. mRNA levels of the indicated genes after normalization to β-actin. Data are mean ± SD from three independent experiments. C, Proteins separated and immunoblotted with the indicated antibodies after treating the cells with TLR ligand for the indicated time points following SK1-I pretreatment. Similar results were obtained in two independent experiments. D, NF-κB-dependent gene expression was determined by qPCR. mRNA levels of the indicated genes normalized with β-actin. E, Proteins were separated and immunoblotted with the indicated antibodies after treating cells as in Figure 3C. Similar results were obtained in three independent experiments. Statistical significance for difference between two groups for each indicated gene was determined by Student’s t-test (B, D), *P < .05, **P < .005, ***P < .001
Mitogen activated protein kinases, including ERK1/2 and JNK1/2 signalling pathways, have also been implicated in IFN production in response to TLR activation.40,41 Inhibition of SphK1 attenuated activation of these signalling pathways during TLR stimulation (Figures 3E and S3C). In sum, SphK1 is a key molecule that regulates the direct transducers of viral PAMP sensing and responses.
3.4 |. TLR7/9 and SphK1 form a complex
TLR3, TLR7/8, and TLR9 are endosome localized TLRs2; however, pDCs express TLR7 and TLR 9 only, and do not express TLR8.42 Induction of IFN, TNF-α, or IL-6 could not be detected in CAL-1 cells stimulated with the TLR3 ligand poly (I:C), even after prolonged incubation (data not shown). This is likely due to expression of TLR7/9 by CAL-1 cells as reported previously,43 but not TLR3. As previous studies have suggested that SphK1 is also localized to endosomes,44–46 it was of interest to examine whether TLR7/9 co-localized or interacted with SphK1. Confocal fluorescence microscopy revealed a high co-localization of SphK1 and TLR9 (Figure 4A). In agreement, co-immunoprecipitation revealed that SphK1 associated with TLR7 (Figure 4B) and TLR9 (Figure 4C), even in the absence of ligand stimulation or by treatment with SK1-I. These observations were further supported by bioinformatics tools and molecular modelling using ZDOCK (Figure 4D,E), ClusPro (Figure S4A), and GRAMM-X (Figure S4B) servers that predict the interaction between TLR7/9 and SphK1. The interacting regions identified by the different molecular modeling approaches were similar and the H-bond forming regions are highlighted in each structure. The docking studies also suggest that SK1-I and TLRs interact with SphK1 on different sites (Figure S4C) and might explain why SK1-I did not affect constitutive complex formation between SphK1 and TLRs.
FIGURE 4.
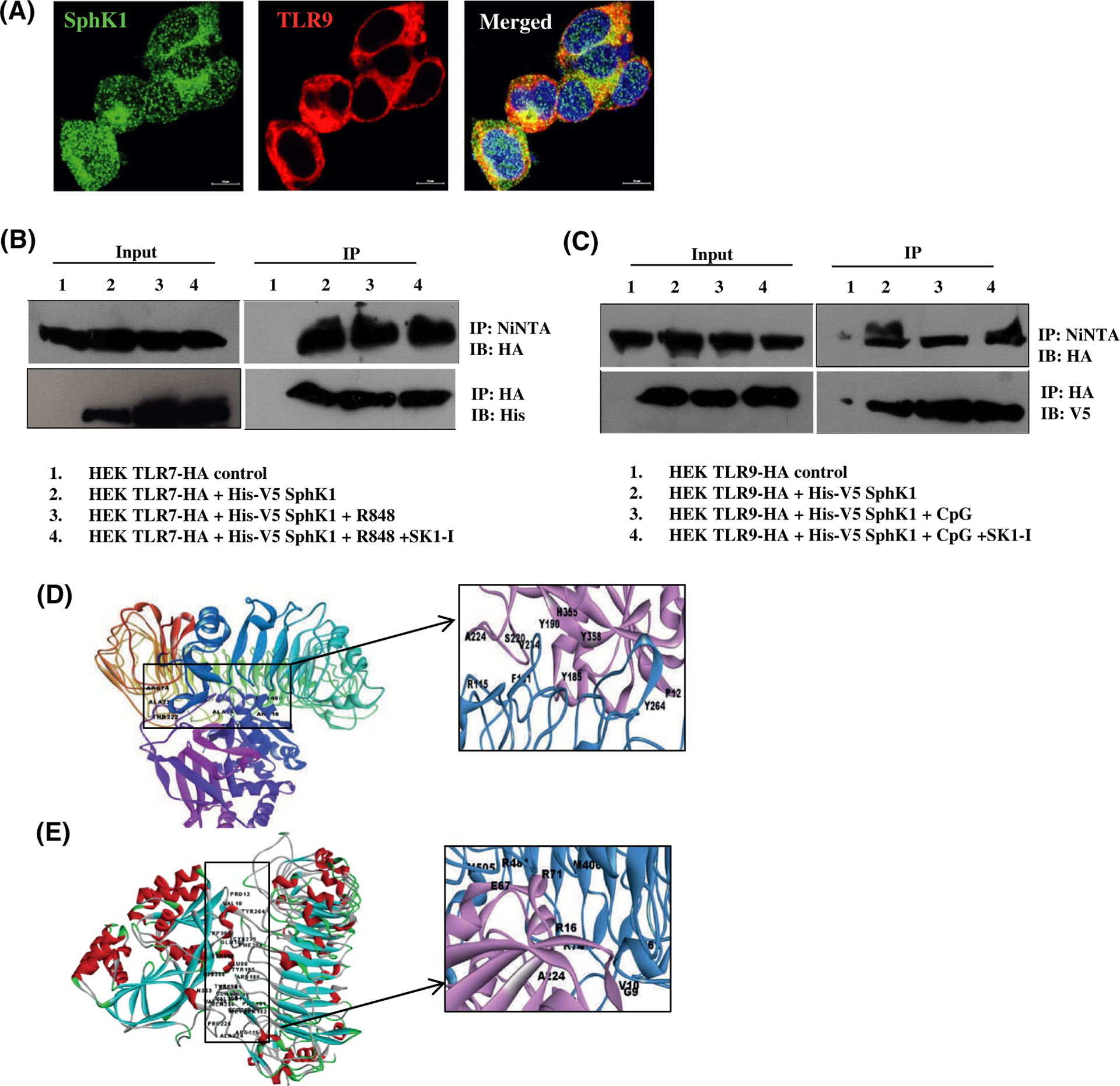
SphK1 interacts with TLR7 and TLR9. A, HEK293 cells stably expressing HA-TLR9 transiently transfected with His-V5-SphK1. Representative images of immunofluorescence microscopy after staining with antibodies against TLR9 and SphK1. Images are representative of three independent experiments. Scale bar: 10 μm (B, C) HEK293 cells stably expressing HA-TLR7 (B) or HA-TLR9 (C) transiently transfected with His-V5-SphK1 and treated with R848, CpG-ODN or SK1-I as indicated. SphK1, TLR7, and TLR9 pulled down with Ni-NTA or HA affinity purification beads, separated by SDS-PAGE, and immunoblotted as indicated. Input exposed differently (D-E) Docked complexes of TLR9 and SphK1 (D) and TLR7 and SphK1 (E) obtained from ZDOCK server. The H-bond forming residues are marked in the inset (TLR9, TLR7- blue, SphK1- pink)
3.5 |. Inhibition or deletion of SphK1 impairs CpG-ODN uptake
Since SphK1 had a robust influence on TLR-mediated innate immune responses and SphK1 and TLR interact, it was of interest to examine the involvement of SphK1 in early steps of endosomal TLR7/9 activation, namely uptake of the ligand, followed by intracellular trafficking to endosomes. Pre-treatment of CAL-1 cells or HEK293 cells stably expressing TLR9 with SK1-I significantly decreased uptake of FITC tagged CpG-ODN (Figure 5A and data not shown). Similarly, SK1-I treatment decreased CpG-ODN uptake by splenic pDCs from 18.9% to 1.1% (Figure 5B). Moreover, uptake of CpG-ODN was totally ablated in SphK1−/− splenic pDCs (0.7%) (Figure 5B), suggesting that both pharmacological inhibition and genetic deletion of SphK1 mitigated CpG-ODN internalization by pDCs. Nevertheless, as was reported previously,18 S1P had no significant effect on intracellular accumulation of FITC tagged CpG-ODN as determined by flow cytometric analysis (Figure 5C). Similar to previous reports,17,18 expression of S1PR1 and S1PR4 were reduced in pDCs after TLR stimulation and no effects were observed on expression of S1PR2, S1PR3, or S1PR5 (Figure S5A,B).
FIGURE 5.
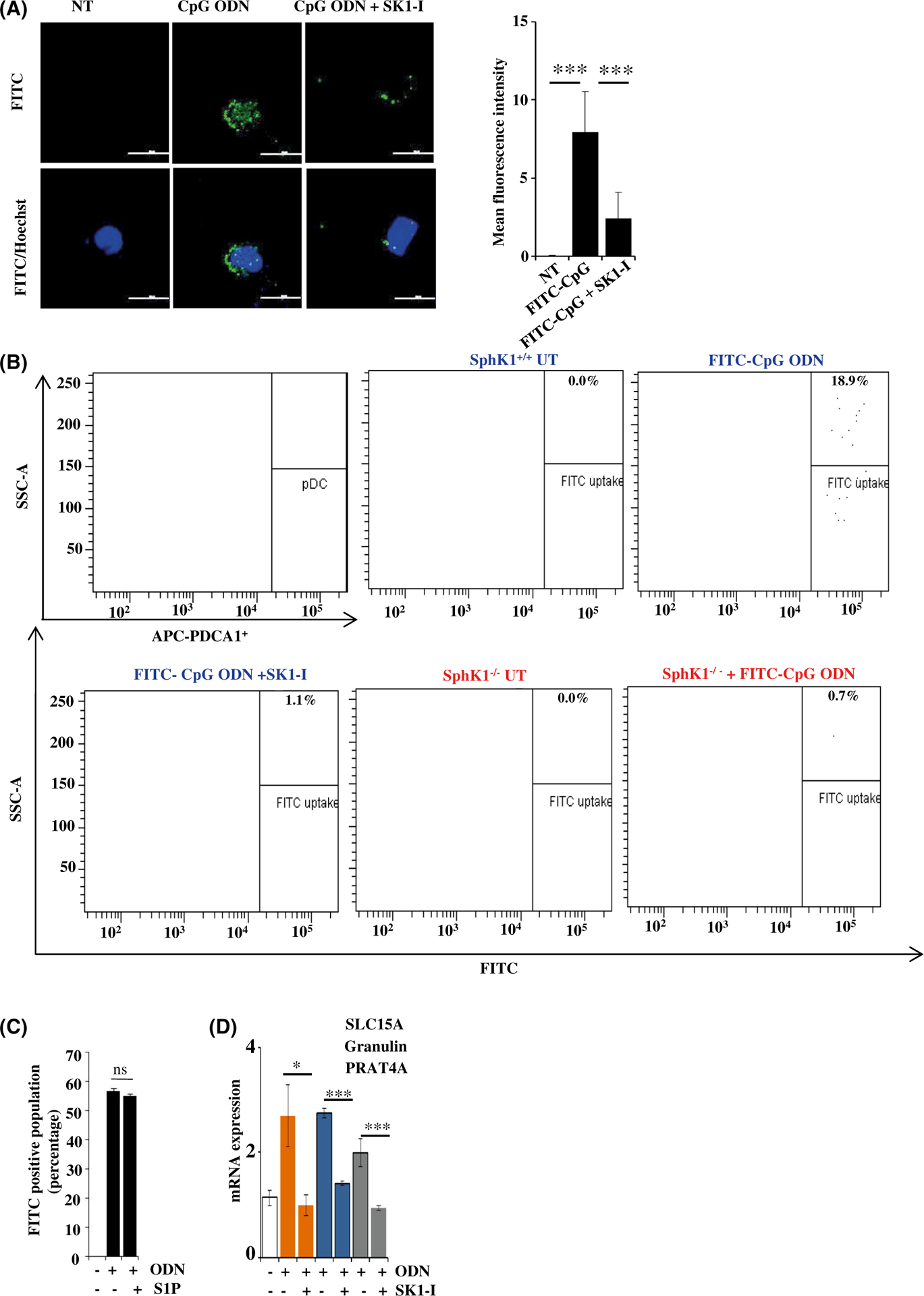
SphK1 regulates uptake of CpG-ODN by dendritic cells. A, CAL-1 cells pretreated with SK1-I (10 μM) for 30 min followed by stimulation without or with FITC-CpG-ODN (50 nM) for 20 min. CpG uptake was analyzed by immunofluorescence microscopy. Nuclei counter stained with Hoechst 33342.Images representative of five independent experiments. Scale bar: 20 μm. Graphical representation of FITC-CpG ODN uptake by Image J software. B, Splenocytes isolated from SphK1+/+ and SphK1−/−mice (n = 4/group) and gated for splenic pDCs by staining with APC-PDCA1. Cells were treated as in Figure 5A. CpG uptake by splenic pDCs was analyzed by flow cytometry as indicated by FITC fluorescence. C, Uptake of FITC-CpG-ODN analyzed by flow cytometry and data expressed as percentage of FITC-positive cells. Data are mean ± SD from three independent experiments. D, CAL-1 cells were treated with CpG-ODN as in Figure 1A. mRNA levels of SLC15A, Granulin, PRAT4A determined by qPCR and normalized to β-actin. Data are mean ± SD of five independent experiments. Statistical significance was determined by Student’s t test to compare between two experimental groups (D) and ANOVA (A,C), post hoc Tukey test, *P < .05, **P < .005, ***P < .001
We next sought to examine the involvement of different cofactors and accessory molecules that are involved in CpG-ODN uptake and endosomal delivery. These included granulin, a cofactor that binds to CpG-ODN and helps in its delivery to endosomes47 and also has the potential to bind to TLR9; SLC15A, a transporter protein involved in TLR9 and endosomal functions48; and PRAT4A, a chaperone that is needed for proper trafficking of TLR9.49 SK1-I pretreatment decreased the expression of all these genes induced by CpGODN (Figure 5D), supporting the notion that SphK1 may be critical in CpG-ODN uptake and processing.
3.6 |. SphK1 promotes susceptibility to experimental SLE in a pDC-dependent manner
The profound influence of SphK1 on IFN production and subsequent pro-inflammatory cytokines led us to investigate the role of the multifaceted SphK1 in SLE, a prototypic autoimmune disorder that is characterized by a heightened production of type I IFNs.7–10 Since pDCs and the secreted IFNs play a major role in the initiation of SLE,10,11 we decided to investigate the importance of SphK1 in the early stages when the disease is initiated rather than in a full-fledged stage with characteristic flares and remission. A chemically induced murine lupus model by administration of the hydrocarbon oil TMPD (2, 6, 10, 14-tetramethylpentadecane, also known as pristane) was used50,51 (Figure 6A). Pristane-induced lupus simulates the disease parameters that are observed in humans and is an appropriate model where type I IFN plays a central role in its development.50,51 As expected, administration of pristane induced the onset of lupus as shown by the large increase in IFN-β and by the expression of disease-associated markers such as U1a and Sca-152 (Figure 6B). The increases of these markers were mitigated in mice treated with the SphK1 inhibitor SK1-I (10 mg/kg, twice weekly) simultaneously with pristane injections. Intriguingly, treatment with SK1-I, even after induction of early disease-associated markers (therapeutic mode), also significantly reduced IFN-β, U1a, and Sca-1 after 2 months. Pristane failed to induce these disease markers in SphK1−/− mice (Figure 6B). Similarly, deposition of immune complexes in the kidney composed of IgG auto-antibodies, a hallmark of glomerulonephritis,53 was drastically reduced in mice treated simultaneously or therapeutically with SK1-I as well as in the SphK1 knockout mice (Figures 6C and S6A). Although pristane treatment reduced the level of pDCs in the peritoneum, their activation status was greatly increased (Figure 6D). Simultaneous or therapeutic treatment with SK1-I as well as deletion of SphK1 significantly reduced pDC activation markers CD80 and CD86 in pristane treated mice (Figure 6D). Similar results were observed in splenic pDCs (Figure S6B), where the expression of CD80/86 was decreased by genetic and pharmacological inhibition of SphK1. Moreover, the expression pattern of known markers of pDC function in blood, Siglec-H, E2–2, and Spi-B (1), which were increased several fold by pristane, were greatly reduced by SK1-I treatment and in SphK1 null mice (Figure 7A).Taken together, these results suggest a pivotal role for SphK1 in determining initiation of lupus by modulating the functions of pDCs.
FIGURE 6.
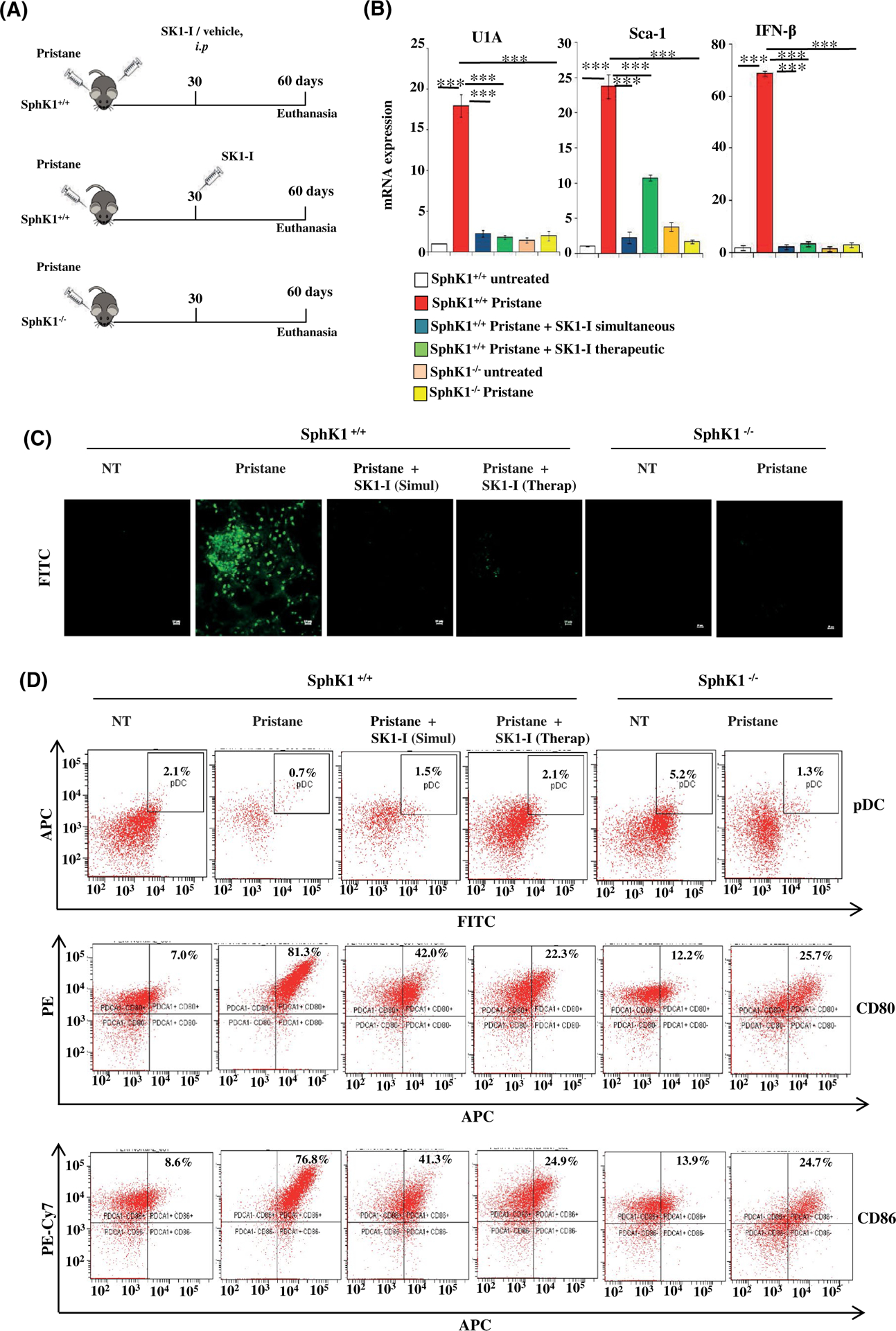
Inhibition or deletion of SphK1 reduces disease parameters in pDC-dependent murine SLE. A, Schematic experimental diagram for pristane-induced SLE model. Wild-type or SphK1−/− mice were injected with pristane (500 μL/mouse, intraperitoneal). Where indicated, mice were treated with SK1-I (10 mg/kg, i.p) twice a week starting at day 0 (simultaneous) or after 30 days (therapeutic). Mice were euthanized after 60 days (n = 5/group). B, mRNA levels of the indicated genes in blood samples from mice were measured by qPCR and normalized with GAPDH. Data are mean ± SD (n = 5/group). C, Immunofluorescence microscopy of mouse kidney cryosections stained with FITC-labeled anti-IgG antibody to analyze glomerulonephritis. Representative images from three independent experiments. Scale bar: 10 μm. D, Peritoneal cells isolated from the indicated mice groups, pDC percentage (FITC-CD11C+/APC-PDCA-1+) and activation status (PE-CD80, PE-Cy7-CD86) determined by flow cytometric analysis. Statistical significance determined by ANOVA with post hoc Tukey test, *P < .05, **P < .005, ***P < .001
FIGURE 7.
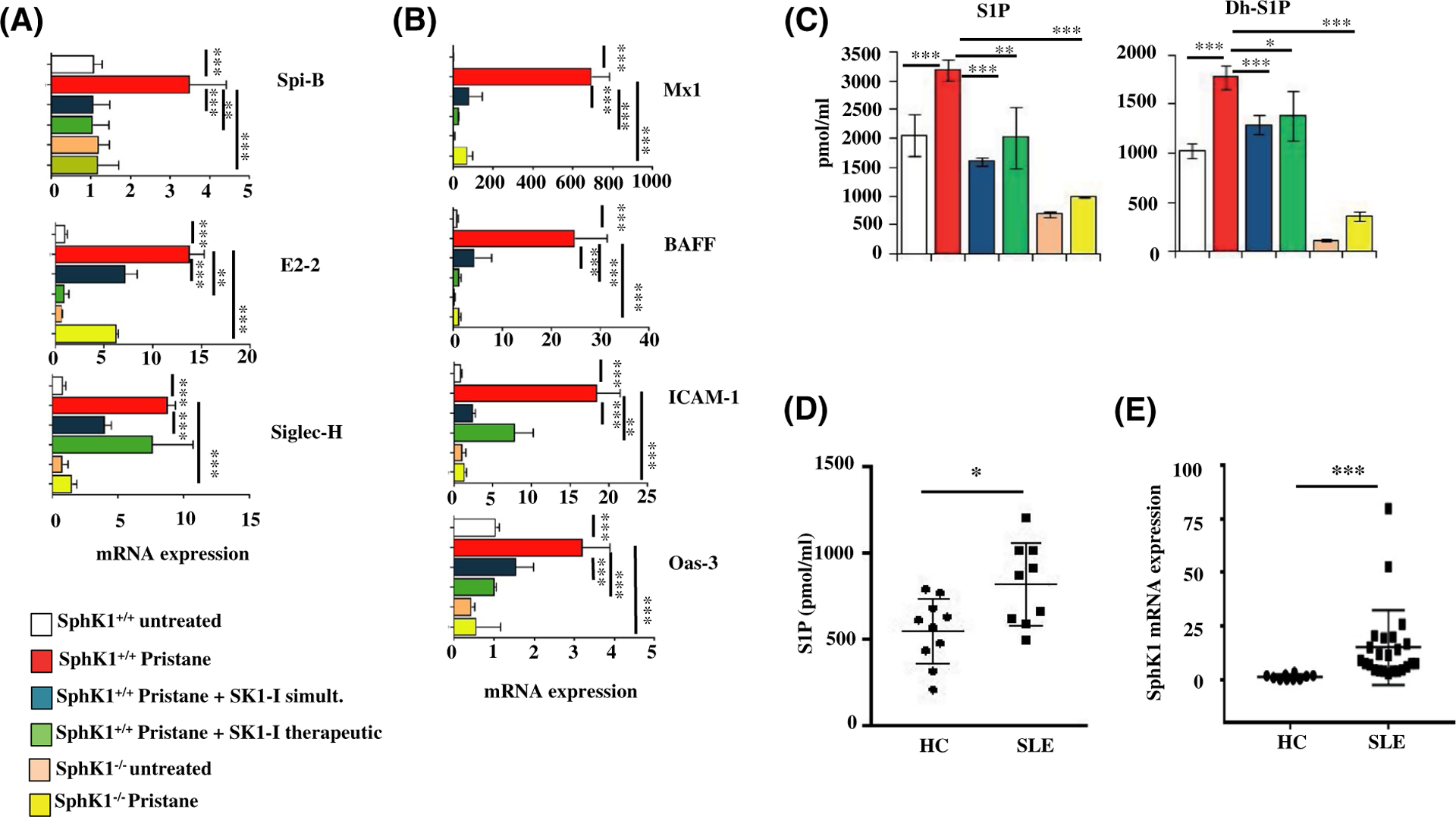
Inhibition or deletion of SphK1 mitigates IFN gene signature in a mouse model of lupus, and SphK1 is elevated in human lupus patients. A-D, Mice were treated as described in Figure 6A. A, mRNA levels of Spi-B, E2–2, and Siglec-H in blood determined by qPCR and normalized to GAPDH. Data are mean ± SD of three independent experiments. B, mRNA levels of genes induced by interferon in blood determined by qPCR and normalized to GAPDH. Data are mean ± SD of three independent experiments. C, Levels of S1P and dihydro-S1P in serum of experimental mice measured by LC-ESI-MS/MS. D, Levels of S1P in serum from healthy human controls (HC) (n = 10) and lupus patients (SLE) (n = 9) determined by LC-ESI-MS/MS. E, PBMCs isolated from healthy controls (n = 10) and lupus patients (n = 22). SphK1 mRNA levels were determined by qPCR and normalized to β-actin. Whisker plots show mean ± SD Statistical significance determined by ANOVA, post hoc Tukey test (A-C), Student’s t test (D) or Welch’s t test (E).*P < .05, **P < .005, ***P < .001
3.7 |. SphK1 regulates the pristane-driven type I IFN signature
Lupus is characterized by an elevated “interferon signature” of increased expression of IFN-stimulated genes (ISGs). Decreased expression of ISGs, namely Mx1, Oas3, BAFF, and ICAM-1 were observed in SphK1 deleted mice and after treatment of wild-type mice with SK1-I, compared to mice treated with pristane alone (Figure 7B). However changes in autoantibodies were not apparent in these early time points (Figure S6C) in agreement with a previous report.10 Consistent with the pivotal role of SphK1 in SLE, increased levels of S1P and dihydro-S1P were observed in mice treated with pristane and were significantly attenuated by SK1-I treatment (Figure 7C). Moreover, pristane failed to increase levels of these phosphorylated sphingoid bases in SphK1 null mice (Figure 7C). Importantly, S1P levels were also significantly higher in SLE patients (Figure 7D). Consistent with previous studies54 and SphK1 expression was upregulated in PBMCs from human SLE patients (classified per SLEDAI criteria) compared to those from healthy controls (Figure 7E).
4 |. DISCUSSION
When TLRs on pDCs are engaged by viruses and other microbial components they produce copious amounts of type I IFNs that stimulate adaptive immune cells. pDCs, due to their extensive potential for production of Type I IFNs, act as integral links between the innate and adaptive arms of the immune machinery.1,5 Immune responses are carefully regulated to maintain the delicate balance between immune surveillance and immunopathology. We have uncovered the decisive role played by SphK1, one of the isoenzymes that produce S1P, in determining this equilibrium (Figure 8). SphK1 regulates activation of pDCs and production of Type I IFNs and pro-inflammatory cytokines. Several lines of evidence support this conclusion. First, specific inhibition or deletion of SphK1 diminishes TLR7/9-mediated INF production in pDCs. We found that SphK1, though dispensable for pDC development, is essential for pDC activation. This is critical as pDCs are causal effectors in the pathogenesis of autoimmune disorders, including lupus,10,55 and have been linked to disease progression in animal models of lupus. Second, SphK1 regulates nuclear import of IRF3, IRF7, and p65 and and consequently IRF3 and NF-κB-dependent gene expression that is controlled by these transcription factors that play important roles in production of Type I IFNs and pro-inflammatory cytokines. Third, there is a reciprocal regulation between TLR7/9 and SphK1 where TLR7/9 ligands activate SphK1 leading to increased cellular levels of S1P. TLRs, in turn, depend on SphK1 for activation and responses as inhibition of SphK1 affects TLR stimulation as evidenced by the diminished immunological outcome. Fourth in the lupus murine model, pharmacological inhibition of SphK1 or its genetic deletion prevented pristane-induced S1P elevation and markedly decreased pDC activation, IFN signature, and glomerulonephritis. Finally, in human lupus patients, SphK1 expression and S1P levels are significantly increased.
FIGURE 8.
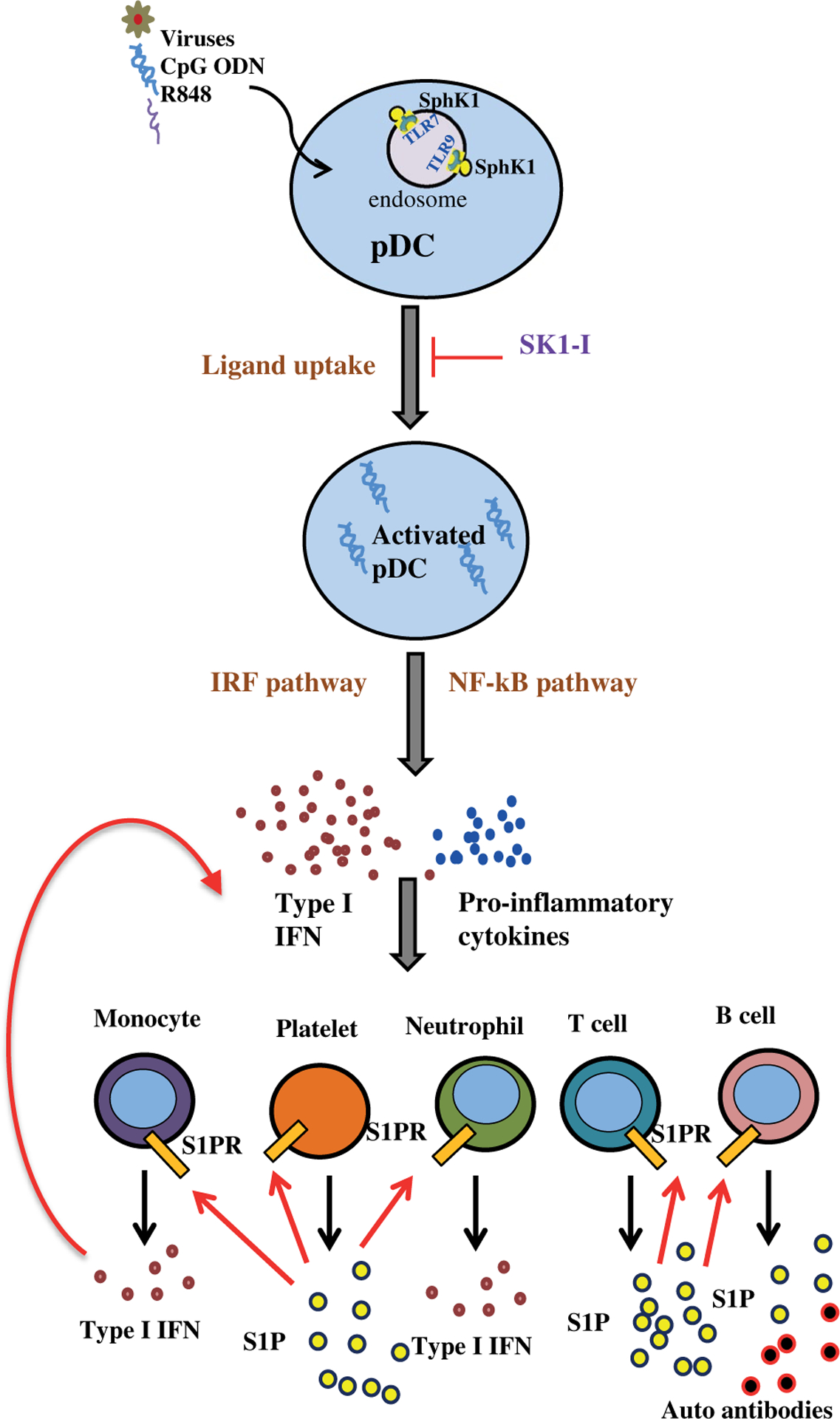
A model illustrating the role of SphK1 in type I interferon-dependent innate and autoimmune activation. In systemic lupus erythematosus, activation of pDCs by TLR7/9 ligands leads to massive IFN production and also activation of sphingosine kinase 1. Intracellular SphK1 is important for efficient uptake of TLR7/9 ligands and trafficking to endosomes. pDCs are involved in the initial stages of the lupus and as disease advances, IFN and production of pro-inflammatory cytokines and chemokines lead to further recruitment and activation of other immune cells, such as monocytes, neutrophills, T and B cells, as well as platelets. These cells secrete S1P into the circulation, which further amplifies their immune functions by binding to its receptors on these cells but not on pDCs. Thus, our work suggests that SphK1 is at the crossroads of immunosurveillance and immuno pathology and upregulation of the SphK1/S1P axis results in a state of autoimmunity with an exacerbated production of interferon
Interestingly, specific SphK1 inhibition with SK1-I or its genetic deletion in pDCs significantly reduced CpG-ODN uptake and thus interfered with their intracellular trafficking to endosomes where TLR7/9 activation occurs. However In agreement with a previous report, exogenous S1P did not alter CpG-ODN uptake, TLR9 trafficking, or pDC maturation.18 Moreover, consistent with this report, we also observed that stimulation of pDCs with CpG reduced expression of S1PR1 and S1PR4.18 Thus, although previous studies showed that binding of S1P to these receptors on pDCs inhibited IFN-α production,17,18 it seems unlikely that activation of SphK1 in pDCs produces sufficient S1P for secretion and autocrine activation of S1PR1 and S1PR4 on pDCs. Hence, our work describes an unexpected function of SphK1 in pDCs and the intracellular conversion of sphingosine to S1P in regulation of TLR7/9 uptake that is independent of the extracellular actions of S1P as a ligand of S1PRs. This is consistent with recent studies describing a new role for SphK1 and the conversion of sphingosine to S1P in endocytic trafficking independent of S1PRs.45,46,56 SphK1 is recruited to nascent endosomes and knockdown of SphKs results in endocytic recycling defects.45 Similarly, treating cells with sphingosine or a sphingosine-based SphK1 inhibitor resulted in delayed endocytic maturation and the accumulation of dilated dysfunctional late endosomes,46,56 further supporting a role for SphK1 and sphingosine phosphorylation in endocytic membrane trafficking. Unfortunately, there is a lack of technology with sufficient resolution to directly follow dynamic changes in endogenous sphingosine and S1P during membrane trafficking and thus the intracellular role of these bioactive sphingolipids and their intracellular targets are unknown. Furthermore, SphK1 interacts with TLR7/9 and co-localizes with TLR9 at endosomes. Because there are reports that endosomal TLRs transiently exhibit plasma membrane translocation,57 we cannot exclude the possibility that SphK1 also interacts with TLR7/9 at the plasma membrane. As the first steps of endosomal TLR7/9 activation are the recognition and uptake of the ligand, followed by intracellular trafficking to early endosomes, it will be important in the future to understand how interaction between TLR7/9 and SphK1 and activation of SphK1 and subsequent changes in intracellular levels of sphingosine or S1P or both can affect early events in viral response. Altogether, our data suggest that in pDCs stimulated with TLR7/9 ligands, intracellular SphK1 plays an important role in regulating IFN expression.
Despite its chemical nature of induction, the pristane-induced SLE mouse model recapitulates many of the hallmarks of human SLE, including immune-complex glomerulonephritis and loss of kidney function.58,59 We found that knockout of SphK1 significantly reduced these symptoms in pristane-treated mice. Moreover, not only simultaneous treatment with the specific SphK1 inhibitor SK1-I, but more importantly, therapeutic treatment with SK1-I thirty days after initiation of the disorder, greatly reduced known markers of pDC function in blood, as well as glomerulonephritis, and mitigated increased expression of IFN-stimulated genes. Although the role of monocyte recruitment in pristane-induced IFN and auto-antibody production by activation of TLR7 is well-known,60 in agreement with our study, several previous reports have also highlighted the important roles of pDCs in early stages of lupus and that depletion of pDCs ameliorates autoimmunity in a lupus model.9–11,61 Therefore, it has been suggested that pDCs are initiators of the disease. Though low in number in comparison to other hematopoietic cells, pDCs are the first cell type to respond and are the highest known producers of Type I IFN in response to a viral attack. This IFN then activates a wide array of cells including NK cells, monocytes, myeloid DCs, and T cells, to release pro-inflammatory cytokines and S1P into the circulation. It is well known that increased S1P in circulation is pro-inflammatory by stimulating production and secretion of many pro-inflammatory cytokines and chemokines (TNF, IL-6, etc) that further amplifies immune functions. Based on our findings, S1P in the circulation likely does not have a significant effect on decreasing IFN via the S1PRs in pDCs,17,18 but is rather a potent pro-inflammatory mediator.14 Indeed administration of FTY720, which is phosphorylated in vivo and then acts as a functional antagonist of S1PR1, ameliorates systemic lupus manifestations in several mouse models.62–64
The IFN pathway has been recognized as a therapeutic target in SLE. Because in a phase II clinical trial, patients with a high IFN gene signature were more responsive to a monoclonal antibody targeting the IFNα receptor,65 it is possible that SK1-I may provide therapeutic benefit to specific SLE patient populations with distinct IFN profiles. Given the efficacy demonstrated by SK1-I in a preclinical animal model, and the elevation of SphK1 and serum S1P in murine models of SLE and in human patients, our data suggests that SK1-I may warrant clinical evaluation as a potential treatment for SLE.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Drs. M. Radhakrishna Pillai, T. R. Santhoshkumar, and Ruby John Anto for sharing reagents, Eugene Y. Kim for technical discussions and Jeremy Allegood for LC-MS analysis. Abgenex, India for providing TLR7 and TLR9 antibodies. Expressing our gratitude to the personnel of FACS core facility and Bio-imaging facility for their excellent technical assistance and Animal research facility for maintaining the mouse colony. SM and MBL acknowledge senior research fellowships from the Indian Council of Medical Research and University Grant Commission, respectively. SM acknowledges doctoral advisory committee for their valuable suggestions. The work is supported US National Institute of Health grant R01GM043880 (to S.S), a fast track grant from Department of Science and Technology, Government of India (No. SR/FT/LS-159/2012) and grand-in-aid scheme of Council for Scientific and Industrial Research (No.37 (1720)/18/EMR-II) and in part by faculty start-up grant and DBT-Ramalingaswami fellowship (No. BT/RLF/Reentry/38/2011) to KBH
Abbreviations:
- APC
allophycocyanin
- BSA
bovine serum albumin
- FACS
fluorescence-activated cell sorting
- FITC
fluorescein isothiocyanate
- Flt3L
FMS-like tyrosine kinase 3 ligand
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IFIT
interferon induced protein with tetratricopeptide repeats
- LC-ESI-MS/MS
liquid chromatography–electrospray ionization–tandem mass spectrometry
- MCMV
murine cytomegalovirus
- PBMC
peripheral blood mononuclear cell
- PRAT4A
protein associated with toll-like receptor 4A
- RBC
red blood cell
- RPMI
Roswell Park Memorial Institute
- Sca-1
stem cell antigen 1
- Siglec H
sialic acid–binding Ig-like lectin H
- SLC15A4
solute carrier family 15 member 4
- STAT1
signal transducer and activator of transcription 1
- U1a
U1 small nuclear ribonucleoprotein A
Footnotes
CONFLICT OF INTEREST
Dr Spiegel is a co-inventor on patent number US 8,372,888 B2 titled Sphingosine kinase type 1 inhibitors, compositions, and processes for using same. The other authors declare no competing interests.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. 2015;15:471–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alculumbre S, Raieli S, Hoffmann C, Chelbi R, Danlos FX, Soumelis V. Plasmacytoid pre-dendritic cells (pDC): from molecular pathways to function and disease association. Semin Cell Dev Biol. 2019;86:24–35. [DOI] [PubMed] [Google Scholar]
- 3.Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–1837. [DOI] [PubMed] [Google Scholar]
- 4.Asselin-Paturel C, Boonstra A, Dalod M, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. 2001;2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. [DOI] [PubMed] [Google Scholar]
- 6.Theofilopoulos AN. TLRs and IFNs: critical pieces of the autoimmunity puzzle. J Clin Invest. 2012;122:3464–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet. 2014;384:1878–1888. [DOI] [PubMed] [Google Scholar]
- 8.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–2121. [DOI] [PubMed] [Google Scholar]
- 9.Sisirak V, Ganguly D, Lewis KL, et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med. 2014;211:1969–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rowland SL, Riggs JM, Gilfillan S, et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med. 2014;211:1977–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott JL, Wirth JR, EuDaly JG, Gilkeson GS, Cunningham MA. Plasmacytoid dendritic cell distribution and maturation are altered in lupus prone mice prior to the onset of clinical disease. Clin Immunol. 2017;175:109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. [DOI] [PubMed] [Google Scholar]
- 13.Czeloth N, Bernhardt G, Hofmann F, Genth H, Forster R. Sphingosine-1-phosphate mediates migration of mature dendritic cells. J Immunol. 2005;175:2960–2967. [DOI] [PubMed] [Google Scholar]
- 14.Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12:688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai WQ, Irwan AW, Goh HH, et al. Anti-inflammatory effects of sphingosine kinase modulation in inflammatory arthritis. J Immunol. 2008;181:8010–8017. [DOI] [PubMed] [Google Scholar]
- 17.Teijaro JR, Studer S, Leaf N, et al. S1PR1-mediated IFNAR1 degradation modulates plasmacytoid dendritic cell inter-feron-alpha autoamplification. Proc Natl Acad Sci U S A. 2016;113:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dillmann C, Ringel C, Ringleb J, et al. S1PR4 signaling attenuates ILT 7 internalization to limit IFN-alpha production by human plasmacytoid dendritic cells. J Immunol. 2016;196:1579–1590. [DOI] [PubMed] [Google Scholar]
- 19.Aloia AL, Calvert JK, Clarke JN, et al. Investigation of sphingosine kinase 1 in interferon responses during dengue virus infection. Clin Transl Immunology. 2017;6:e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vijayan M, Seo YJ, Pritzl CJ, Squires SA, Alexander S, Hahm B. Sphingosine kinase 1 regulates measles virus replication. Virology. 2014;450–451:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seo YJ, Blake C, Alexander S, Hahm B. Sphingosine 1-phosphate-metabolizing enzymes control influenza virus propagation and viral cytopathogenicity. J Virol. 2010;84:8124–8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maeda T, Murata K, Fukushima T, et al. A novel plasmacytoid dendritic cell line, CAL-1, established from a patient with blastic natural killer cell lymphoma. Int J Hematol. 2005;81:148–154. [DOI] [PubMed] [Google Scholar]
- 23.Satoh M, Reeves WH. Induction of lupus-associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. J Exp Med. 1994;180:2341–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohammed S, Vineetha NS, James S, et al. Examination of the role of sphingosine kinase 2 in a murine model of systemic lupus erythematosus. FASEB J. 2019;33:7061–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lankadasari MB, Aparna JS, Mohammed S, et al. Targeting S1PR1/STAT3 loop abrogates desmoplasia and chemosensitizes pancreatic cancer to gemcitabine. Theranostics. 2018;8:3824–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li QZ, Xie C, Wu T, et al. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115:3428–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newton J, Hait NC, Maceyka M, et al. FTY720/fingolimod increases NPC1 and NPC2 expression and reduces cholesterol and sphingolipid accumulation in Niemann-Pick type C mutant fibro-blasts. FASEB J. 2017;31:1719–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paugh SW, Paugh BS, Rahmani M, et al. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112:1382–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lima S, Takabe K, Newton J, et al. TP53 is required for BECN1-and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy. 2018;14:942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapitonov D, Allegood JC, Mitchell C, et al. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009;69:6915–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brasel K, De Smedt T, Smith JL, Maliszewski CR. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood. 2000;96:3029–3039. [PubMed] [Google Scholar]
- 32.Schnute ME, McReynolds MD, Kasten T, et al. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem J. 2012;444:79–88. [DOI] [PubMed] [Google Scholar]
- 33.Khan N, Gowthaman U, Pahari S, Agrewala JN. Manipulation of costimulatory molecules by intracellular pathogens: veni, vidi, vici!!. PLoS Pathog. 2012;8:e1002676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pitson SM, Moretti PA, Zebol JR, et al. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003;22:5491–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol. 2013;78:105–116. [DOI] [PubMed] [Google Scholar]
- 37.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. [DOI] [PubMed] [Google Scholar]
- 38.Honda K, Yanai H, Negishi H, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. [DOI] [PubMed] [Google Scholar]
- 39.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. [DOI] [PubMed] [Google Scholar]
- 40.Takauji R, Iho S, Takatsuka H, et al. CpG-DNA-induced IFN-alpha production involves p38 MAPK-dependent STAT1 phosphorylation in human plasmacytoid dendritic cell precursors. J Leukoc Biol. 2002;72:1011–1019. [PubMed] [Google Scholar]
- 41.Janovec V, Aouar B, Font-Haro A, et al. The MEK1/2-ERK pathway inhibits type I IFN production in plasmacytoid dendritic cells. Front Immunol. 2018;9:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hornung V, Rothenfusser S, Britsch S, et al. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–4537. [DOI] [PubMed] [Google Scholar]
- 43.Steinhagen F, Rodriguez LG, Tross D, Tewary P, Bode C, Klinman DM. IRF5 and IRF8 modulate the CAL-1 human plasmacytoid dendritic cell line response following TLR9 ligation. Eur J Immunol. 2016;46:647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayashi S, Okada T, Igarashi N, Fujita T, Jahangeer S, Nakamura S. Identification and characterization of RPK118, a novel sphingosine kinase-1-binding protein. J Biol Chem. 2002;277:33319–33324. [DOI] [PubMed] [Google Scholar]
- 45.Shen H, Giordano F, Wu Y, et al. Coupling between endocytosis and sphingosine kinase 1 recruitment. Nat Cell Biol. 2014;16:652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lima S, Milstien S, Spiegel S. Sphingosine and sphingosine kinase 1 involvement in endocytic membrane trafficking. J Biol Chem. 2017;292:3074–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park B, Buti L, Lee S, et al. Granulin is a soluble cofactor for toll-like receptor 9 signaling. Immunity. 2011;34:505–513. [DOI] [PubMed] [Google Scholar]
- 48.Sasawatari S, Okamura T, Kasumi E, et al. The solute carrier family 15A4 regulates TLR9 and NOD1 functions in the innate immune system and promotes colitis in mice. Gastroenterology. 2011;140:1513–1525. [DOI] [PubMed] [Google Scholar]
- 49.Takahashi K, Shibata T, Akashi-Takamura S, et al. A protein associated with Toll-like receptor (TLR) 4 (PRAT4A) is required for TLR-dependent immune responses. J Exp Med. 2007;204:2963–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kienhofer D, Hahn J, Stoof J, et al. Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI. Insight 2017;2:e92920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Freitas EC, de Oliveira MS, Monticielo OA. Pristane-induced lupus: considerations on this experimental model. Clin Rheumatol. 2017;36:2403–2414. [DOI] [PubMed] [Google Scholar]
- 52.Xu Y, Zhuang H, Han S, et al. Mechanisms of tumor necrosis factor alpha antagonist-induced lupus in a murine model. Arthritis Rheumatol. 2015;67:225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyer-Schwesinger C, Dehde S, Klug P, et al. Nephrotic syndrome and subepithelial deposits in a mouse model of immune-mediated anti-podocyte glomerulonephritis. J Immunol. 2011;187:3218–3229. [DOI] [PubMed] [Google Scholar]
- 54.Watson L, Tullus K, Marks SD, Holt RC, Pilkington C, Beresford MW. Increased serum concentration of sphingosine-1-phosphate in juvenile-onset systemic lupus erythematosus. J Clin Immunol. 2012;32:1019–1025. [DOI] [PubMed] [Google Scholar]
- 55.Liao X, Li S, Settlage RE, et al. Cutting edge: plasmacytoid dendritic cells in late-stage lupus mice defective in producing IFN-alpha. J Immunol. 2015;195:4578–4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Young MM, Takahashi Y, Fox TE, Yun JK, Kester M, Wang HG. Sphingosine kinase 1 cooperates with autophagy to maintain endocytic membrane trafficking. Cell Rep. 2016;17:1532–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guerrier T, Pochard P, Lahiri A, Youinou P, Pers JO, Jamin C. TLR9 expressed on plasma membrane acts as a negative regulator of human B cell response. J Autoimmun. 2014;51:23–29. [DOI] [PubMed] [Google Scholar]
- 58.Zhuang H, Szeto C, Han S, Yang L, Reeves WH. Animal models of interferon signature positive lupus. Front Immunol. 2015;6:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30:455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee PY, Kumagai Y, Li Y, et al. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008;205:2995–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou Z, Ma J, Xiao C, et al. Phenotypic and functional alterations of pDCs in lupus-prone mice. Sci Rep. 2016;6:20373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okazaki H, Hirata D, Kamimura T, et al. Effects of FTY720 in MRL-lpr/lpr mice: therapeutic potential in systemic lupus erythematosus. J Rheumatol. 2002;29:707–716. [PubMed] [Google Scholar]
- 63.Ando S, Amano H, Amano E, et al. FTY720 exerts a survival advantage through the prevention of end-stage glomerular inflammation in lupus-prone BXSB mice. Biochem Biophys Res Commun. 2010;394:804–810. [DOI] [PubMed] [Google Scholar]
- 64.Sui M, Zhou J, Xie R, et al. The sphingosine-1-phosphate receptor agonist FTY720 prevents the development of anti-glomerular basement membrane glomerulonephritis. Mol Biol Rep. 2012;39:389–397. [DOI] [PubMed] [Google Scholar]
- 65.Furie R, Khamashta M, Merrill JT, et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. 2017;69:376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.