

Abstract
Background
Duchenne muscular dystrophy (DMD) is a severe neuromuscular disorder and is one of the most common muscular dystrophies. There are currently few effective therapies to treat the disease, although many small-molecule approaches are being pursued. Certain histone deacetylase inhibitors (HDACi) have been shown to ameliorate DMD phenotypes in mouse and zebrafish animal models. The HDACi givinostat has shown promise for DMD in clinical trials. However, beyond a small group of HDACi, other classes of epigenetic small molecules have not been broadly and systematically studied for their benefits for DMD.
Methods
We used an established animal model for DMD, the zebrafish dmd mutant strain sapje. A commercially available library of epigenetic small molecules was used to treat embryonic-larval stages of dmd mutant zebrafish. We used a quantitative muscle birefringence assay in order to assess and compare the effects of small-molecule treatments on dmd mutant zebrafish skeletal muscle structure.
Results
We performed a novel chemical-combination screen of a library of epigenetic compounds using the zebrafish dmd model. We identified candidate pools of epigenetic compounds that improve skeletal muscle structure in dmd mutant zebrafish. We then identified a specific combination of two HDACi compounds, oxamflatin and salermide, that ameliorated dmd mutant zebrafish skeletal muscle degeneration. We validated the effects of oxamflatin and salermide on dmd mutant zebrafish in an independent laboratory. Furthermore, we showed that the combination of oxamflatin and salermide caused increased levels of histone H4 acetylation in zebrafish larvae.
Conclusions
Our results provide novel, effective methods for performing a combination of small-molecule screen in zebrafish. Our results also add to the growing evidence that epigenetic small molecules may be promising candidates for treating DMD.
Keywords: Chemical screen, Duchenne muscular dystrophy, Epigenetic small molecules, HDAC inhibitors, Zebrafish
Background
Duchenne muscular dystrophy (DMD) is a severe neuromuscular disorder caused by mutations in the X-linked DMD gene, which encodes the protein dystrophin [1, 2]. Dystrophin is a key component of the dystrophin-associated protein complex (DAPC), a multi-protein assembly localized at the sarcolemma of muscle cells [3, 4]. Dystrophin and the DAPC serve a structural role in muscle cells, functioning as a stabilizing link between the cytoskeleton and extracellular matrix during muscle fiber contraction [5–7]. Dystrophin and the DAPC also regulate signaling pathways such as nitric oxide production and Ca2+ entry [6–9]. Loss of dystrophin and DAPC function makes muscle cell membranes susceptible to contraction-induced damage and leads to progressive calcium dysregulation, satellite cell dysfunction, inflammation, fibrosis, and necrosis [7, 10–13].
DMD is the most common type of muscular dystrophy, affecting approximately 1 in 3500-5000 male births [14, 15]. It first presents as motor difficulties in early childhood and progresses rapidly, leaving most affected boys in need of a wheelchair in their early teens and in need of respiratory aid in their 20s [16]. The disease is usually fatal in the third or fourth decade due to respiratory or cardiovascular failure.
Treatment options for DMD are still quite limited. The current standard of care is corticosteroid treatment, which delays the progression of muscle dysfunction but has serious side effects [16–18]. Besides corticosteroids, two other drugs are approved for a subset of DMD patients: eteplirsen, an exon-skipping oligonucleotide drug for patients with DMD exon 51 mutations, has received accelerated FDA approval in the USA, and ataluren, a stop codon read-through drug, has conditional approval in Europe for patients with DMD nonsense mutations [16, 19]. DMD gene therapy and gene editing approaches are very promising but face many challenges [20–23]. Many small-molecule approaches are being identified that could benefit DMD by modulating different pathological mechanisms downstream of the dystrophin mutation [7, 16, 19, 24]. A current view in the DMD field is that a combination of therapies targeting different pathological mechanisms may ultimately be most beneficial for patients [19, 24].
Histone deacetylase inhibitors (HDACi) are one class of small molecules that have shown promise in mouse and zebrafish DMD animal models and in DMD clinical trials (ClinicalTrials.gov NCT02851797, NCT03373968) [25–28]. HDACi are examples of “epigenetic small molecules,” compounds that target chromatin modifications and transcriptional regulators. Histone acetylation, which is often linked with open chromatin and active transcription, is one example of an epigenetic modification and is the target of many HDACi [29]. There are four different classes of histone deacetylase (HDAC) proteins, with different functions and tissue expression patterns, and different HDACi often selectively inhibit specific HDACs [30]. Studies in the mdx mouse model of DMD have identified epigenetic mechanisms involved in the pathogenesis of DMD, including constitutive activation of HDAC2 and dysregulation of certain histone modifications, leading to downstream transcriptional perturbations [31–34]. Dystrophin and the DAPC can regulate chromatin signaling through nitric oxide inhibition of HDAC2 [25, 32]. HDACi that have been shown to ameliorate pathology in the mdx mouse are the pan-HDACi Trichostatin A (TSA), valproic acid, phenylbutyrate, SAHA, and givinostat (ITF 2357), and the class I HDACi MS-275 [25, 28, 35–37]. TSA also ameliorates the zebrafish dmd mutant [27, 38], and givinostat has shown promise in DMD clinical trials [26]. However, beyond these HDACi, other classes of epigenetic small molecules have not been broadly and systematically tested in DMD models for their potential benefits for DMD.
Zebrafish are an outstanding animal model for DMD [39–43]. A zebrafish dmd mutant strain, dmdta222a, also known as sapje, has a nonsense mutation in exon 4 and is a dystrophin-deficient model of DMD [39, 41, 44]. The zebrafish dmdta222a mutation is autosomal recessive, and, following a Mendelian ratio, approximately 25% of the offspring from a heterozygous dmd+/− cross exhibit degenerative muscle lesions by about 3 days post-fertilization. dmd mutant zebrafish exhibit many aspects of human DMD pathology; in particular, skeletal muscle fibrosis and inflammation, including infiltration of mononuclear cells [40, 41].
dmd mutant zebrafish offer several advantages for screening and evaluating small molecule therapies [42, 43]. Zebrafish eggs can be rapidly produced in large numbers, and the resulting embryos readily absorb small molecules. Because skeletal muscle lesions can be observed within 3-4 days of development, dmd mutant zebrafish are amenable to rapid and high-throughput screening. An exceptional range of approaches are available for assessing treatment outcomes in dmd mutant zebrafish, including assays of skeletal muscle structure and function as well as survival [42, 43, 45, 46]. In particular, muscle structure can easily be observed in zebrafish larvae using polarized light birefringence techniques [44, 45, 47]. Large-scale chemical screens have highlighted the potential of dmd mutant zebrafish for identifying new therapeutic compounds and targets as well as for understanding the molecular mechanisms behind DMD [45, 48, 49]. These large-scale screens, which applied either pools of eight chemicals or individual chemicals to dmd mutant zebrafish, have tested over 4000 compounds and identified 25 positive hits [43]. In addition, some small molecules, such as TSA, have been shown to ameliorate both dmd mutant zebrafish and mdx mice [27, 45, 50–52]. Because of this strong conservation, insight from zebrafish can inform our understanding of human DMD disease, while also taking advantage of the utility of high-throughput analysis.
Here, we performed a pilot screen of the commercially available Cayman Chemical Epigenetics Screening Library to identify additional epigenetic small molecules that could improve the dmd mutant zebrafish muscle phenotype. We identified candidate pools of epigenetic compounds that improve skeletal muscle structure in dmd mutant zebrafish. We then identified a specific combination of two HDACi compounds, oxamflatin and salermide, that ameliorated dmd mutant zebrafish skeletal muscle degeneration. We validated the effects of oxamflatin and salermide on dmd mutant zebrafish in an independent laboratory. Furthermore, we showed that the combination of oxamflatin and salermide caused increased levels of histone H4 acetylation in zebrafish larvae. Overall, our study provides novel and effective methods for performing a pooled chemical-combination screen and gives further evidence that epigenetic small molecules may be good candidates for successfully treating DMD.
Methods
Zebrafish husbandry
All experiments involving live zebrafish (Danio rerio) were carried out in compliance with Seattle Children’s Research Institute’s and the University of Maine’s Institutional Animal Care and Use Committee guidelines. Zebrafish were raised and staged as previously described [53]. Time (hpf or dpf) refers to hours or days post-fertilization at 28.5 °C. Eggs were collected from 20-30-min spawning periods and raised in Petri dishes in ICS water (300 mg Instant Ocean/L, 0.56 mM CaCl2, 1.2 mM NaHCO3), in a dark 28.5 °C incubator, up to 5 dpf. After 5 dpf, fish were maintained on a recirculating water system (Aquaneering) under a 14-h on, 10-h off light cycle. From 6-30 dpf, fish were raised in 2.8 L tanks with a density of no more than 50 fish per tank and fed a standard diet of paramecia (Carolina) one time per day and Zeigler AP100 dry larval diet two times per day. From 30 dpf on, fish were raised in 6 L tanks with a density of no more than 50 fish per tank and fed a standard diet of Artemia nauplii (Brine Shrimp Direct) and Zeigler adult zebrafish feed, each two times per day. The wild-type stock and genetic background used was AB. The zebrafish dmdta222a mutant line (also known as sapje; hereafter, referred to as dmd) has been described and is a null allele [39, 44]. dmdta222a genotyping was performed as previously described [54].
Small molecules
Epigenetic small molecule library screening was performed using the Cayman Chemical Epigenetics Screening Library (Item No. 11076, Batch No. 0455098). The library was received as 10 mM stocks of each chemical dissolved in dimethyl sulfoxide (DMSO). The composition of the Cayman Chemical Epigenetics Screening Library can vary. The version we obtained contained 94 chemicals distributed over two 96-well plates. The identities of the chemicals and plate layouts are shown in Table 1 and Fig. 1a. Additional chemicals were purchased individually from Cayman Chemical and were dissolved in DMSO (Sigma).
Table 1.
Cayman chemical epigenetics screening library
| Plate 1 | Plate 2 | ||
|---|---|---|---|
| Well | Small molecule | Well | Small molecule |
| A2 | 3-amino benzamide | A2 | AGK2 |
| A3 | SB 939 | A3 | CAY10603 |
| A4 | PCI 34051 | A4 | Chaetocin |
| A5 | 4-iodo-SAHA | A5 | Splitomicin |
| A6 | Sirtinol | A6 | CBHA |
| A7 | C646 | A7 | M 344 |
| A8 | Ellagic acid | A8 | Oxamflatin |
| A9 | Scriptaid | A9 | Salermide |
| A10 | Suberohydroxamic acid | A10 | Mirin |
| A11 | Apicidin | A11 | Pimelic diphenylamide 106 |
| B2 | UNC0321 (trifluoroacetate salt) | B2 | (S)-HDAC-42 |
| B3 | (−)-Neplanocin A | B3 | MS-275 |
| B4 | Cl-Amidine | B4 | HNHA |
| B5 | F-Amidine (trifluoroacetate salt) | B5 | RG-108 |
| B6 | JGB1741 | B6 | 2′,3′,5′-triacetyl-5-azacytidine |
| B7 | UNC0638 | B7 | S-Adenosylhomocysteine |
| B8 | Phthalazinone pyrazole | B8 | UNC0224 |
| B9 | Isoliquiritigenin | B9 | Chidamide |
| B10 | CCG-100602 | B10 | 3-Deazaneplanocin A |
| B11 | CAY10669 | B11 | Sinefungin |
| C2 | Zebularine | C2 | Pyroxamide |
| C3 | Delphinidin chloride | C3 | WDR5-0103 |
| C4 | ITF 2357 | C4 | AMI-1 (sodium salt) |
| C5 | PFI-1 | C5 | UNC1215 |
| C6 | 5-Azacytidine | C6 | GSK 343 |
| C7 | Decitabine | C7 | SIRT1/2 inhibitor IV |
| C8 | (+)-JQ1 | C8 | I-CBP112 (hydrochloride) |
| C9 | (−)-JQ1 | C9 | UNC1999 |
| C10 | BSI-201 | C10 | PFI-3 |
| C11 | 1-Naphthoic acid | C11 | trans-Resveratrol |
| D2 | Sodium 4-phenylbutyrate | D2 | 2,4-DPD |
| D3 | IOX1 | D3 | DMOG |
| D4 | MI-2 (hydrochloride) | D4 | Trichostatin A |
| D5 | MI-nc (hydrochloride) | D5 | CAY10398 |
| D6 | Gemcitabine | D6 | RSC-133 |
| D7 | Lomeguatrib | D7 | KD 5170 |
| D8 | Octyl-α-ketoglutarate | D8 | CAY10433 |
| D9 | Daminozide | D9 | Piceatannol |
| D10 | GSK-J1 (sodium salt) | D10 | CAY10591 |
| D11 | GSK-J2 (sodium salt) | D11 | EX-527 |
| E2 | GSK-J4 (hydrochloride) | E2 | SAHA |
| E3 | GSK-J5 (hydrochloride) | E3 | 2-PCPA (hydrochloride) |
| E4 | CPTH2 | E4 | Nicotinamide |
| E5 | Valproic acid (sodium salt) | E5 | BIX01294 |
| E6 | Tenovin-1 | E6 | N-Oxalylglycine |
| E7 | Tenovin-6 | E7 | Suramin (sodium salt) |
| E8 | Sodium butyrate | ||
| E9 | Anacardic acid | ||
The identities of the chemicals and composition of the library, 94 chemicals distributed over two 96-well plates, are shown. The library (Cayman Chemical Item No. 11076, Batch No. 0455098) was received as 10 mM stocks of each chemical in DMSO
Fig. 1.

Cayman Chemical Epigenetic Screening Library chemical-combination pooling scheme. a The library consists of 2 96-well plates (P1, P2). See Table 1 for the list of chemicals. Gray X’s mark empty wells. Blue X’s mark wells with chemicals not used in our screen: CAY10669, (−)-JQ1, MI-nc, GSK-J1, GSK-J2, and GSK-J5. Pooling process for plate 1 well A2 chemical (3-amino benzamide; yellow well) is illustrated. b Grid representation of the library chemicals and pools, in which each chemical (88 out of 94 total library chemicals) is represented along both the x- and y-axes. CAY10669, (−)-JQ1, MI-nc, GSK-J1, GSK-J2, and GSK-J5 are removed from this grid representation. Shading (dark blue/light blue, dark gray/light gray) represents rows of chemicals in the library plates. Each horizontal, alternately shaded row represents a candidate chemical pool. Blue shading represents the 93 pools tested. Gray shading represents the remaining candidate pools (403 total pools on this grid)
Small molecule treatments
To test toxicity and assess working doses of the Epigenetics Screening Library chemicals, wild-type AB embryos were treated with individual chemicals beginning at 4 hpf. Embryos were not dechorionated prior to these dose tests. Three concentrations of each chemical were tested: 10 μM, 1 μM, and 100 nM. Embryos were treated with small molecules with 1% DMSO in embryo medium (EM; 14.97 mM NaCl, 0.50 mM KCl, 0.98 mM CaCl2.2H2O, 0.15 mM KH2PO4, 0.99 mM MgSO4.7H2O, 0.05 mM Na2HPO4, 0.83 mM NaHCO3), or with 1% DMSO in EM as a vehicle control. One chemical from the library, JGB1741, exhibited precipitation from EM at 10 μM but not at lower concentrations; all other chemicals appeared soluble with DMSO in EM. Twelve-well plates were used for treatments; each well contained 25 embryos and 3 mL of chemical treatment or vehicle control. The treatment media was changed every 24 h until 4 dpf. Over the 4-day treatments, developmental abnormalities and survival were noted. Most chemical treatments showed little or no effect on embryo health and survival at 1 μM doses, so we determined 1 μM to be the working dose for pooled screening for most of the library chemicals. C646 and apicidin were toxic and caused embryo lethality at 1 μM but not at 100 nM, so these compounds were used at 100 nM. CAY10669 exhibited strong toxicity and GSK-J1 is reported to be non-cell permeable, so these chemicals were removed from chemical pool screening. (−)-JQ1, MI-nc, GSK-J2, and GSK-J5 are designated as negative control compounds in the library and thus were also removed from chemical pool screening.
For chemical screening and dmd rescue tests, embryos from dmd+/− crosses were used. At about 24 hpf, the chorions were manually removed from the embryos, and embryos were sorted into wells of 12-well plates for treatments. Each well contained 25 embryos and 3 mL of chemical treatment or vehicle control. 1000X stocks of chemicals were made in DMSO just prior to each treatment experiment. Embryos were treated with chemicals with 1% DMSO in embryo medium (EM), or with 1% DMSO in EM as a vehicle control. Treatments were started at about 24 hpf and continued for 3 days, changing the treatment or vehicle control EM solution every 24 h. Larvae were fixed in 4% paraformaldehyde (PFA) in PBS at 4 dpf and stored at 4 °C. To obtain tissue for genotyping, larval heads were removed with a scalpel at the level of the pectoral fins. Larval tails with trunk skeletal muscle remained intact and were maintained in 4% PFA until imaging was performed.
Imaging and scoring muscle lesions
Larvae fixed in 4% PFA were rinsed in PBS with 0.01% Tween (PBSTw) prior to imaging. Animals were placed in PBSTw in a 60-mm glass Petri dish. An Olympus SZX16 stereomicroscope with an attached Olympus DP72 camera was set up with one sheet of polarizing film over the trans-illumination base and another sheet of the film placed over the objective lens such that the two films were crossed [55]. For chemical pool screening, larvae were sorted under birefringence into muscle lesion positive or negative groups. The number of larvae in each group was recorded.
For quantitative birefringence measurements, we adapted an approach based on previously described methods [47, 55]. In this approach, larvae were placed in a glass-bottom Petri dish in 2.5% methyl cellulose and oriented to maximize the brightness of the muscle tissue through the crossed polarizers. If necessary, pectoral fins and any remaining yolk tissue were removed so that the larvae could lie flat, to ensure consistent birefringence across the length of the trunk. Birefringence was imaged in individual animals as above. Images were acquired using the Olympus Cellsens Dimensions software. Exposure time was adjusted so that only a few saturated pixels were present. Adjusting exposure time in this way should control for any variation in brightness caused by larvae being at slightly different orientations. All other microscope and camera settings were kept constant. ImageJ was used to outline the trunk musculature, using the wand tool, and to calculate the average pixel intensity within the resulting selection. Average pixel intensity values for experimental conditions were normalized to wild-type control values. We see similar relative average pixel intensity values for control and mutant samples from experiment to experiment, indicating that our procedure is highly reproducible.
Wholemount immunocytochemistry and phalloidin staining
Whole-animal immunostaining was performed with the primary antibodies anti-dystrophin (1:100, Sigma D8043) and anti-beta dystroglycan (1:50, Novocastra NCL-b-DG). The secondary antibody was either goat anti-mouse AlexaFluor-488 or goat anti-mouse AlexaFluor-568 (1:200, Life Technologies/Molecular Probes). Four dpf larvae were fixed in 4% PFA in PBS for 4 h at room temperature and then washed into PBSTw. For anti-dystrophin staining, larvae were dehydrated through a methanol series and stored in 100% methanol at −20 °C overnight. They were then re-hydrated into PBSTw, washed 5× in PBSTw containing 1% DMSO, and blocked overnight at 4 °C in 2% BSA, 2% heat inactivated normal goat serum, and 1% DMSO in PBSTw. The samples were then incubated with primary antibody diluted in the above block for 24 h at 4 °C, washed with PBSTw +1% DMSO, re-blocked for 6 h at room temperature, incubated with secondary antibody for 16 h at 4 °C, washed with PBSTw +1% DMSO, and stored in 4% PFA in PBS. For anti-beta dystroglycan staining, the fixed larvae were permeabilized with 2% Triton X-100 in PBS for 2 h at room temperature, then washed into PBSTw, and then blocked in 5% BSA, 2% heat-inactivated normal goat serum, 1% DMSO, 1% Triton X-100, and 0.2% saponin in PBS overnight at 4 °C. The samples were then processed as for the anti-dystrophin stained samples, except the block including Triton X-100 and saponin was used. To visualize actin in the muscle fibers of the beta-dystroglycan-stained animals, CF488A-conjugated phalloidin (1:50, Biotium Cat. # 00042) was included with the secondary antibody. For confocal imaging, the heads and yolk were removed from representative larvae and the trunks/tails were mounted laterally in 80% glycerol containing 4% propyl gallate. Images were collected using a Leica TCS SP5 II confocal on a DMI6000 stand with a 20× NA 0.7 air objective.
Immunoblotting
Chemical treatments of embryos from dmd+/− crosses were performed as above. At 4 dpf, heads were cut from anesthetized animals and were arrayed in 96-well plates for genotyping. The remaining trunk and tail portions of the animals were arrayed in separate 96-well plates, to which LDS sample buffer (Invitrogen NP0007) was added and stored at −200C until genotyping was completed. Trunk lysates were then pooled by genotype. Approximately, 0.75 animal equivalent per lane was separated on 12% NuPAGE Bis-Tris gels run in NuPAGE MES buffer (Invitrogen) and blotted. Blots were probed separately with anti-pan acetylated histone H4 (1:2000, rabbit polyclonal, Millipore Cat. # 06-866) and anti-acetylated histone H4 K16 (1:2500, rabbit polyclonal, Millipore Cat. # 07-329). Anti-actin was included with both anti-acetylated histone antibodies as a loading control (1:2000; mouse monoclonal Clone C4, MP Biomedical Cat. # 69100). Infrared-dye labeled secondary antibodies (Rockland) were visualized using a Licor Odyssey infrared scanner.
Statistical analyses
Statistical tests used are provided in the figure legends. Statistical analyses were performed and graphs were constructed using GraphPad Prism 8.
Results
Epigenetic small-molecule library pooling approach
In a previous study, we showed that the pan-HDACi TSA could improve the zebrafish dmd muscle lesion phenotype [27]. We wanted to test whether we could identify new epigenetic small molecules, or epigenetic small molecule combinations, that improve the zebrafish dmd muscle lesion phenotype. We thus decided to test the effects of small molecules from the commercially available Cayman Chemical Epigenetics Screening Library. The version of the library that we obtained contained 94 chemicals distributed over two 96-well plates (Table 1 and Fig. 1a). Prior to screening these chemicals on dmd mutant embryos, we performed dose and toxicity testing of each individual library compound on wild-type embryos (see the “Methods” section). Most chemicals showed little or no detrimental effect on embryo health and survival at a 1 μM dose, so we selected 1 μM to be the working dose for chemical screening for most of the library compounds. C646 and apicidin were toxic and caused embryo lethality at 1 μM but not at 100 nM, so these compounds were used at 100 nM. CAY10669 exhibited strong toxicity and embryonic lethality by 3 dpf (days post-fertilization) and was removed from further screening. GSK-J1 is reported to be non-cell permeable and was removed from further screening (Fig. 1a). We also removed library chemicals that are designated as negative control compounds from further screening ((−)-JQ1, MI-nc, GSK-J2, and GSK-J5; Table 1; Fig. 1a).
Previous large-scale chemical screens in dmd mutant zebrafish either tested individual chemicals or tested chemical pools by subdividing libraries into unique chemical pools containing eight compounds each [45, 48, 49]. In contrast to these previous screens, we wanted a strategy that would test each compound in combination with every other library compound. In order to efficiently test combinations of epigenetic small molecules in the library, we designed a grid system consisting of a set of 403 different chemical pools (Fig. 1a, b). In this system, compound A2 of plate 1 is pooled with the remaining compounds of row A of plate 1 for combination pool 1 (Fig. 1a, b). Compound A2 is then pooled with compounds from plate 1 row B (pool 2), plate 1 row C (pool 3), and the remaining rows from the library for pools 4-10 (Fig. 1a, b). Compound A3 of plate 1 is then combined with compounds of plate 1 row B for pool 11, and so on (Fig. 1a, b). In this grid system, each compound would be tested in combination with each of the other library chemicals in at least 1 pool (Fig. 1a, b).
To determine if screening pools of epigenetic small molecules was sensitive enough to identify compounds that could improve the zebrafish dmd muscle phenotype, we tested the pool of 10 chemicals from plate 2 row D, which contains TSA (Table 1). Animals from dmd+/− crosses were treated from 1-4 days and then scored for muscle birefringence (as in [27] and illustrated in Fig. 2). Treatment with the pool of chemicals from plate 2 row D significantly decreased the number of affected animals exhibiting abnormal muscle birefringence (Fig. 3). These results indicate that a pool of compounds containing a known beneficial epigenetic small molecule, TSA, can improve the zebrafish dmd muscle phenotype. These results suggest that the epigenetic chemical-pooling design of our screen is sensitive enough to pick up positive hits of small molecules that improve the zebrafish dmd muscle phenotype.
Fig. 2.

Timeline of dmd mutant zebrafish small molecule treatments. Eggs are collected from crosses of dmd+/− fish. dmd is autosomal recessive in zebrafish, so about 25% of embryos will be dmd−/−. At 1 dpf (days post-fertilization), embryos are sorted into dishes for treatments, and DMSO or small molecule compounds are added to the embryo bath (as in [27]). Treatment media are replaced daily. At 4 dpf, animals are fixed and muscle birefringence is scored. dmd−/− animals exhibit dark lesions in the larval trunk muscle (arrow in image), as visualized using polarized light birefringence
Fig. 3.
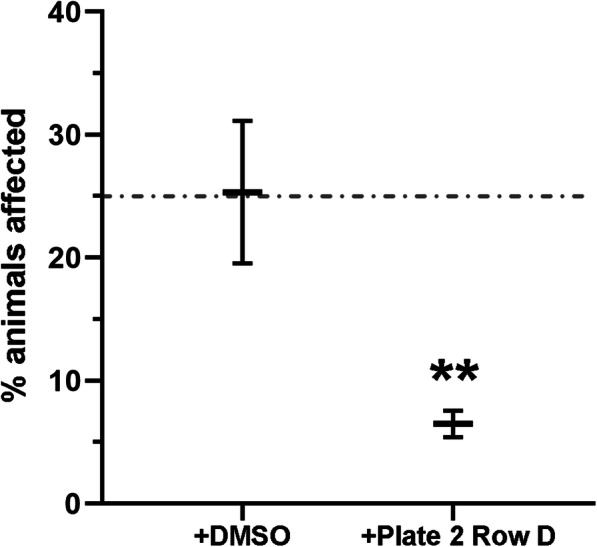
Plate 2 row D chemical pool, which includes TSA, significantly reduces the frequency of zebrafish dmd mutant animals exhibiting abnormal muscle birefringence. Control treatment is 1% DMSO. The dashed line represents the expected 25% affected animals. See Table 1 for the 10 chemicals in plate 2 row D. Each chemical was used at 1 μM. For each treatment condition, n = 3 replicates, with 23-27 embryos in each replicate. Error bars represent standard error. Significance was determined using a Mantel-Haenszel test. **p < 0.004 compared to DMSO control
Pilot screen of epigenetic small-molecule combination pools
We then proceeded to test epigenetic small molecule pools from our chemical-combination grid system (Fig. 1). We tested 93 of the 403 possible chemical pool combinations (Figs. 1b and 4a). Animals from dmd+/− crosses were treated from 1-4 days and then scored for muscle birefringence. Our approach for assessing the dystrophic phenotype using birefringence in our pilot library screen followed the approach used in previous zebrafish dmd mutant chemical screens [45, 48, 49], in which control and drug-treated animals are scored as “affected,” or showing a clear dmd mutant muscle lesion phenotype by birefringence, or “unaffected,” or showing largely normal birefringence. Each pool was tested in duplicate, with 25 animals per well for each replicate. We determined the average percentage of affected animals from each chemical pool, and from each DMSO control tested, and we plotted the results as a heat map on our combination pool grid (Fig. 4a). This heat map grid reveals that the majority of pools tested that included plate 2 row A reduce the percentage of animals affected (column 5 in Fig. 4a). To test whether the plate 2 row A pools are significantly improving the percentage of animals affected, we plotted the combined results from all pools containing each plate row compared with the combined results from DMSO control treatments (Fig. 4b). This analysis shows that treatment pools using plate 2 row A led to a significant reduction in the percentage of affected animals relative to the DMSO controls (Fig. 4b). On average, treatment with chemical pools that included plate 2 row A resulted in only about 10% affected animals, compared to the 25% affected average observed in the DMSO controls (Fig. 4b). Figure 4c and d shows an example of the improved birefringence from animals treated with a representative pool from the plate 2 row A pools (pool 135 in Fig. 1b; pilot screen pool #59), compared to sibling DMSO controls. While these results do not rule out beneficial effects of compounds or treatment pools from our screen that did not include plate 2 row A, these results do highlight that compounds within plate 2 row A are having significant beneficial effects on the zebrafish dmd muscle lesion phenotype.
Fig. 4.

Pilot screen identifies the ability of plate 2 row A chemical pools to lower the frequency of affected dmd animals. (a) Heat map representation of the average percentage of affected animals observed from each of the 93 pools tested. Individual chemical wells tested were from plate 1 (left column, plate number and well number shown). Library rows tested are rows A-D from plate 1 and row A from plate 2. See Table 1 for the chemicals in each well and row. Arrows point to pilot screen pool #59 (pool 135 in Fig. 1b), shown in (d), and pilot screen pool #72 (pool 173 in Fig. 1b), analyzed in Fig. 5 below. (b) Graph of combined average percentages of affected animals from all tested pools that included each plate row. Control treatments are 1% DMSO. The dashed line represents the average of the control DMSO treatments (25%). Each dot represents a tested drug pool’s average of affected animals. Error bars represent standard error. Significance was determined using a one-way ANOVA test comparing each chemical pool group to the DMSO control group with Dunnett’s correction for multiple comparisons. **p = 0.0021 compared to DMSO control. (c, d) Example of birefringence images of 4 dpf larvae from a replicate of (c) DMSO control and (d) pool #59 (plate 2 row A + UNC0638) treatments. DMSO control animals are siblings of pool #59 animals. Asterisks mark affected animals. Scale bar = 1 mm
Analysis of plate 2 row A small molecules identifies a beneficial combination of oxamflatin and salermide
While useful for screening purposes, the approach of scoring animals as “affected” versus “unaffected” may only provide an approximate measure of the effects of small molecules on the dmd mutant phenotype, as animals scored as “affected” could still be improved over typical dmd mutants, and “unaffected” larvae could still have dystrophic lesions but are substantially less severe than is typical for dmd mutants. To determine which compounds mediate the effect of plate 2 row A, we wanted an approach that would allow us to more quantitatively assess the effects of small molecules on zebrafish dmd mutant birefringence. Adapting previously described methods [47, 55], we used gray value measurements to quantitate the brightness level of the muscle birefringence of each animal (Fig. 5a). For each animal, we outlined a unilateral area of trunk muscle birefringence and measured the average pixel brightness within that area (Fig. 5a). Zebrafish dmd mutant animals show reduced birefringence, as measured by lower pixel intensity, compared to their control siblings (Fig. 5c, e). As a test of this approach, we confirmed that TSA treatment significantly improved the average pixel brightness of dmd mutant birefringence (Fig. 5d, e).
Fig. 5.

Individual compounds from plate 2 row A do not significantly improve zebrafish dmd mutant muscle birefringence. a-d Four dpf zebrafish trunk muscle, with trunk skeletal muscle birefringence outlined in red. Lateral views, anterior to the left. Average pixel brightness values are shown for (a) wild-type (WT) + DMSO, (b) WT + TSA, (c) dmd + DMSO, and (d) dmd + TSA. Scale bar = 500 μm. e Graph of normalized birefringence pixel intensities for 200 nM TSA treatments vs DMSO controls. n = 3 biological replicates for each treatment. Plot shows the average normalized pixel intensity for the 3 replicates for each treatment (4-9 genotyped animals per replicate). The dashed line represents the average normalized pixel intensity for all of the DMSO-treated dmd animals (n = 17). Error bars represent standard error. p value determined by Student’s t test. (f) Graph of normalized birefringence pixel intensities for treatments of zebrafish dmd mutants with the 10 individual chemicals from plate 2 row A. Control treatment is 1% DMSO. All chemicals were tested at 1 μM. For each treatment condition, n = 3 replicates, with 2-9 dmd embryos in each replicate. Plot shows the normalized birefringence pixel intensity for the 3 replicates for each treatment. The dashed line represents the average normalized pixel intensity for all of the DMSO-treated dmd animals (n = 15). Error bars represent standard error. Significance was determined using a one-way ANOVA test comparing each treatment group to the dmd DMSO control group with Dunnett’s correction for multiple comparisons. p > 0.2 for all individual chemicals compared to dmd DMSO control
We, next tested each compound from plate 2 row A to see if any of the individual chemicals could improve the dmd birefringence phenotype. Animals from dmd+/− crosses were treated from 1-4 days and then scored for muscle birefringence. When tested individually, none of the compounds showed a significant effect on the DMSO-treated dmd muscle birefringence brightness (Fig. 5f). These results indicate that a combination of chemicals from plate 2 row A is needed to improve the dmd mutant birefringence phenotype.
To identify the compounds required for the effects of plate 2 row A, we next decided to remove individual chemicals from the drug pool. We chose to test pilot screen pool #72 (pool 173 in Fig. 1b; plate 2 row A + plate 1 well C3; 11 total chemicals), because this pool represented an average effect from the plate 2 row A pools (Fig. 4a, b). As above, animals from dmd+/− crosses were treated from 1-4 days and then scored for muscle birefringence. Treatment with the full 11 chemicals from pool #72 showed significant improvement of the dmd mutant birefringence using our quantitative analysis (Fig. 6a). However, the removal of four different chemicals (chaetocin, oxamflatin, salermide, and delphinidin chloride) each inhibited the ability of pool #72 to improve dmd birefringence (Fig. 6a), indicating that these four compounds are involved in producing the dmd rescue effect of pool #72.
Fig. 6.

Oxamflatin and salermide mediate the effects of plate 2 row A. a Graph of average pixel intensities for treatments of zebrafish dmd mutants with pilot screen pool #72 and with pool #72 with each individual chemical removed. Control treatment is 1% DMSO. All chemicals included in the pools were tested at 1 μM. For each condition, 3 replicates of 25 embryos each were treated, with 2-9 dmd−/− embryos in each replicate. Plotted are the average normalized pixel intensities of the embryos from all 3 replicates for that treatment (n = 14-26 total dmd−/− embryos per treatment). Treatment with pool #72 without chaetocin, oxamflatin, salermide, or delphinidin chloride did not achieve the rescue effect seen with the full pool #72. The dashed line represents the average normalized pixel intensity for all of the DMSO-treated dmd animals (n = 26). Error bars represent standard error. Significance was determined using a one-way ANOVA test comparing each treatment group to the dmd DMSO control group with Dunnett’s correction for multiple comparisons. *p ≤ 0.04 compared to dmd DMSO control. b Graph of average normalized pixel intensities for treatments of dmd mutants with pool #72 and with combinations of chaetocin, oxamflatin, and salermide. Control treatment is 1% DMSO. All chemicals were used at 1 μM. For each treatment condition, n = 3 replicates, with 1-11 dmd−/− embryos in each replicate. Plot shows the average normalized pixel intensity for the 3 replicates for each treatment. The dashed line represents the average normalized pixel intensity for all of the DMSO-treated dmd animals (n = 14). Error bars represent standard error. Significance was determined using a one-way ANOVA test comparing each treatment group to the dmd DMSO control group with Dunnett’s correction for multiple comparisons. c-f 4 dpf zebrafish trunk muscle birefringence. Lateral views, anterior to the left. Representative animals are shown from treatments in (b). c WT + DMSO, d dmd + DMSO, e dmd + pool #72, and f dmd + oxamflatin and salermide. Scale bar = 500 μm. g Validation of oxamflatin and salermide treatment effects. Graph of average normalized pixel intensities for treatments of dmd fish with oxamflatin and salermide, performed in the Henry Lab. Treatments were performed from 1-4 dpf. Control treatment is 1% DMSO. Compounds were each used at 1 μM. Dashed line represents the average normalized pixel intensity for all of the DMSO-treated dmd−/− animals (n = 11). WT + DMSO, n = 10; WT + ox+sal, n = 9; dmd−/− + ox+sal, n = 14. Error bars represent standard error. p value determined by Student’s t test. h Validation of TSA treatment effects. Graph of average normalized pixel intensities for treatments of dmd fish with TSA, performed in the Henry Lab. Treatments were performed from 1-4 dpf. Control treatment is 1% DMSO. TSA was used at 200 nM. Dashed line represents the average normalized pixel intensity for all of the DMSO-treated dmd−/− animals (n = 31). WT + DMSO, n = 6; WT + TSA, n = 6; dmd−/− + TSA, n = 28. Error bars represent standard error. p value determined by Student’s t test
We next focused on the roles of chaetocin, oxamflatin, and salermide. We decided not to further test the role of delphinidin chloride primarily because it is not a component of the plate 2 row A compounds on which we decided to focus (delphinidin chloride is the plate 1 well C3 compound in pool #72) and also because it did not exhibit beneficial activity on its own (data not shown). To further test the roles of chaetocin, oxamflatin, and salermide in improving dmd muscle birefringence, we tested these three compounds together and in each pair-wise combination (Fig. 6b). The combination of oxamflatin and salermide significantly improved dmd mutant muscle birefringence, similarly to pool #72, whereas the other pair-wise combinations and the three-chemical combination did not (Fig. 6b). Figure 6c-f shows examples of the improved birefringence from animals treated with pool #72 (Fig. 6e) and the combination of oxamflatin+salermide (Fig. 6f). While these findings do not rule out possible contributions of delphinidin chloride and chaetocin, these results show that the combination of oxamflatin and salermide can mediate the beneficial effects of pool #72 in improving dmd mutant muscle birefringence.
An independent laboratory test validates the beneficial effects of oxamflatin+salermide
To validate these effects of oxamflatin and salermide, we performed treatments from 1 dpf-4 dpf at an independent site, in the laboratory of Dr. Henry. We observed similar improvement of zebrafish dmd mutant muscle birefringence with oxamflatin+salermide treatments performed in the Henry Lab (Fig. 6g) as we observed with treatments performed in the Maves Lab (Fig. 6b). Dr. Henry’s lab also validated that TSA-improved zebrafish dmd mutant birefringence (Fig. 6h). By demonstrating reproducibility, these independent tests are a strong validation of the benefits of these small molecules, and in particular the novel oxamflatin+salermide combination, for dmd mutant zebrafish.
Oxamflatin and salermide have dose-dependent effects, independent of dystrophin expression
To further address the effects of oxamflatin and salermide, we performed a dose-response analysis. We tested concentrations of oxamflatin and salermide, from 0.5 μM to 4 μM, both individually and in combination (Fig. 7a). As above, animals from dmd+/− crosses were treated from 1-4 days and then scored for muscle birefringence. We found that oxamflatin and salermide in combination at doses lower than 1 μM do not improve the dmd mutant birefringence phenotype, while a combination dose of 2 μM oxamflatin+salermide improves birefringence to a degree similar to that seen with the 1 μM combination dose (Fig. 7a). Four micromolar and 2 μM doses of oxamflatin alone improve the birefringence phenotype, but not as well as the 1 μM and 2 μM combination doses (Fig. 7a). Salermide alone did not improve muscle birefringence at any dose tested. We also found that a combination dose of 4 μM oxamflatin+salermide was toxic and caused lethality by 4 dpf (data not shown). These results reveal that, while oxamflatin alone can have beneficial effects, the combination of oxamflatin+salermide appears more effective at improving the dmd mutant birefringence phenotype.
Fig. 7.

Oxamflatin and salermide have dose-dependent effects, independent of dystrophin expression. a Graph of average normalized pixel intensities for treatments of dmd mutants with doses of oxamflatin and salermide. Control treatment is 1% DMSO. Chemicals were used between 0.5 μM and 4 μM, over two separate experiments. For each treatment condition, n = 4 replicates, with 2-11 dmd−/− embryos in each replicate. Plot shows the average normalized pixel intensity for each of the 4 replicate pools for each treatment. The vertical line separates the treatment conditions from the two experiments, each of which has its own WT + DMSO and dmd + DMSO controls. The dashed lines represent the average normalized pixel intensity for all of the DMSO-treated dmd animals (n = 26 and n = 30). Error bars represent standard error. Significance was determined using a one-way ANOVA test comparing each treatment group to the dmd DMSO control group with Dunnett’s correction for multiple comparisons. *p ≤ 0.029, **p = 0.0029, ***p = 0.0007 compared to dmd DMSO control. b-e Confocal images of anti-dystrophin staining in the trunk musculature of 4 dpf b WT + DMSO, c WT + oxamflatin and salermide, d dmd + DMSO, and e dmd + oxamflatin and salermide larvae. Lateral views, anterior to the left. Arrow points to dystrophin expression in the vertical myoseptum. All dmd+/+ animals showed normal dystrophin expression (WT + DMSO, n = 16; WT + ox+sal, n = 14) and all dmd−/− animals lacked detectable dystrophin expression (dmd−/− + DMSO, n = 23; dmd−/− + ox+sal, n = 18). Scale bar = 50 μm. f-i Confocal images of anti-β-dystroglycan (βDG) and phalloidin staining in the trunk musculature of 4 dpf f WT + DMSO, g WT + oxamflatin and salermide, h dmd + DMSO, i dmd + oxamflatin and salermide. Lateral views, anterior to the left. Arrow points to βDG expression (white) in the vertical myoseptum. Phalloidin staining of filamentous actin (magenta) shows the disrupted muscle structure in dmd mutants (* in h). All wild type animals (+/+ and +/−) showed normal β-dystroglycan expression (WT + DMSO, n = 27; WT + ox+sal, n = 26), and dmd−/− animals showed largely maintained β-dystroglycan expression (dmd−/− + DMSO, n = 9; dmd−/− + ox+sal, n = 14). Scale bar = 50 μm
We next asked whether oxamflatin and salermide are acting by upregulating dystrophin expression. Dystrophin expression is lost in dmdta222a mutants (Fig. 7d) [39]. One to four dpf treatments with 1 μM oxamflatin+salermide do not cause any noticeable upregulation of dystrophin expression (Fig. 7e). We also examined the expression of β-dystroglycan (βDG), a marker of the DAPC at the vertical myosepta in the zebrafish trunk [39, 56]. Previous studies have shown that βDG expression is largely still intact at the myosepta in dmd mutants (Fig. 7h) [39, 57], although see [58]. βDG-labeled myosepta, and overall muscle fiber structure, appear improved by 1 μM oxamflatin+salermide treatments in dmd mutants, and we do not observe any obvious upregulation of βDG or actin filaments (Fig. 7i). These results suggest that oxamflatin+salermide are acting independently of dystrophin upregulation and further demonstrate the improved muscle structure of dmd mutants caused by oxamflatin+salermide treatments.
Oxamflatin and salermide together increase histone acetylation
We next wanted to address the mechanisms by which oxamflatin and salermide might be working to improve the dmd mutant phenotype. Oxamflatin is an HDACi that inhibits class I and II HDACs [59, 60]. Salermide is a class III HDACi that inhibits the NAD + -dependent deacetylases SIRT1 and SIRT2 [61]. Oxamflatin and salermide have each been shown to cause increased histone acetylation in mammalian cell culture models [60, 61]. Salermide has also been shown to cause increased acetylation of lysine 16 of histone H4 (H4K16ac), a histone modification target of SIRT1 and SIRT2 [61–63]. We therefore asked whether oxamflatin and salermide treatments cause increased histone acetylation in dmd mutant zebrafish. We treated animals from dmd+/− crosses from 1-4 days and then collected animals at 4 dpf for protein lysates. Western analysis showed that treatments of 1 μM oxamflatin+salermide caused increased levels of pan-acetyl histone H4 and H4K16ac in both control and dmd mutant animals (Fig. 8a-d). However, 1 μM treatments of either oxamflatin or salermide did not increase levels of these histone marks (Fig. 8a-d). These results show that these small molecules are indeed acting as HDACi in zebrafish larvae and begin to reveal a possible mechanism for the activity of this chemical combination.
Fig. 8.

Oxamflatin and salermide together increase histone acetylation. a-b Representative Western blots of (a) histone H4 acetylated at lysines 5, 8, 12, and 16 (pan-H4ac) and (b) histone H4 acetylated specifically at lysine 16 (H4K16ac). Lysates are from genotyped 4 dpf larvae. c-d Quantification of Western blot analyses of levels of (c) pan-H4ac and (d) H4K16ac. Lysates were prepared from 4 replicate pools of animals for each treatment condition, with n = 4-10 animals per genotype per replicate. For each sample, pan-H4ac or H4K16ac signal was normalized to actin as a loading control. This value was then normalized to the WT + DMSO value within each set of samples. The plots show the average normalized signals of the 4 replicates. Error bars represent standard error. Significance was determined using a Student’s t test with Welch’s correction for unequal sample variances. In (c), *p < 0.05. In (d), *p = 0.015, **p = 0.003 compared to WT + DMSO control. Pan-H4ac levels are increased an average of 2.4× in oxamflatin+salermide-treated animals, and H4K16ac levels are increased an average of 1.6×
Discussion
In this study, we performed a screen of epigenetic-modifying small molecules to identify compounds that prevented muscle degeneration in the zebrafish model of DMD. We developed a novel chemical-pooling approach to screen combinations of epigenetic small molecules, and we performed a pilot screen of 93 novel epigenetic small molecule pools. While our pilot screen identified several new candidate small molecule pools that have beneficial effects on zebrafish dmd mutants, our chemical-pooling strategy and subsequent analysis highlighted the activity of compounds from plate 2 row A (Fig. 4). We were able to resolve a novel small molecule combination, oxamflatin and salermide that significantly reduced the number of dmd animals with muscle lesions when compared to DMSO-treated controls. Our results provide support for further investigations of oxamflatin and salermide as potential therapeutic compounds for DMD. Our results also support additional screening of epigenetic small molecules for their beneficial effects in dmd mutant zebrafish.
Several previous large-scale chemical screens have demonstrated the potential of dmd mutant zebrafish for identifying new therapeutic compounds and targets [43, 45, 48, 49]. These studies, and our work here, took advantage of two key approaches, the birefringence assay and drug pooling, to efficiently screen large numbers of compounds. Assessing the percentage of “affected” animals in large-scale dmd mutant zebrafish chemical screens is efficient because the numbers of animals involved preclude performing genotyping. While there is potential for false negatives from these chemical screens, validation studies that incorporate animal genotyping and quantitative birefringence analysis, such as we use here with oxamflatin and salermide treatments, can alleviate concerns of false positives from chemical screens.
In contrast to previous screens that subdivided chemical libraries into unique chemical pools containing eight compounds each [45, 48], we wanted a strategy that would test each compound in combination with every other library compound. We designed a grid system in which each compound would be tested in combination with each of the other library compounds in at least 1 chemical pool (Fig. 1). By taking this novel drug pooling approach, we wanted to identify epigenetic small molecule combinations that are beneficial for dmd mutant zebrafish, and we identified the new combination of oxamflatin and salermide. While our particular drug pooling approach may not be easily feasible for larger library screens, taking similar systematic approaches to testing combinations of chemical library compounds could be fruitful for DMD and other diseases.
Based on results from our pilot screen, we focused our studies here on oxamflatin and salermide. Oxamflatin is an HDACi that inhibits class I and II HDACs and is chemically similar to TSA [59, 60]. Salermide is a class III HDACi that inhibits the NAD + -dependent deacetylases SIRT1 and SIRT2 [61], and so represents a new class of HDACi for DMD. In mammalian cell culture models, oxamflatin and salermide have each been shown to cause increased histone acetylation [60, 61]. Oxamflatin has been shown to inhibit cell proliferation and induce cell shape changes in cancer cell lines [60, 64, 65]. Salermide has been shown to inhibit cell proliferation and induce apoptosis in cancer cell lines, acting through reactivation of pro-apoptotic genes that are repressed by SIRT1-mediated H4K16ac deacetylation [61, 66, 67]. The H4K16ac mark is highly conserved, inhibits chromatin compaction, and has been associated with cell cycle progression, dosage compensation, cancer, and lifespan [68]. Our finding that oxamflatin+salermide caused increased levels of pan-acetyl histone H4 and H4K16ac in both wild-type and dmd mutant zebrafish (Fig. 8) shows that these HDACi compounds are having the expected biochemical effects on zebrafish larvae and also reveals that the combination of oxamflatin+salermide may be synergizing to activate specific histone marks.
In previous studies, overexpression of SIRT1 has been shown to ameliorate DMD pathophysiology in mdx mice [69], while salermide inhibition of SIRT1 has been shown to protect muscle cells against oculopharyngeal muscular dystrophy in Caenorhabditis elegans [70]. SIRT1 and SIRT2 can have opposing effects in promoting angiogenesis and in providing neuroprotective effects in neurodegenerative disease models [71, 72]. Future studies are needed to determine whether oxamflatin and salermide are working additively or synergistically to provide skeletal muscle benefits in zebrafish dmd mutants and whether they are acting through additional mechanisms that are independent of histone acetylation.
While many DMD small molecule therapies are being developed and entering clinical trials, some have difficulties showing efficacy in patients, possibly due to inadequate preclinical evaluation or targeting mechanisms too late in disease progression [7, 16, 19]. Underscoring the significance of this issue, a recent study was unable to replicate the benefits of the serotonin pathway modulator fluoxetine for dmd mutant zebrafish [73]. Here, we take advantage of the zebrafish model for DMD drug discovery and initial mechanistic validation. We also validated the effects of the epigenetic small molecules TSA and oxamflatin and salermide on dmd mutant zebrafish in an independent laboratory (Fig. 6). A critical next step will be to validate the effects of oxamflatin and salermide in mammalian DMD models, such as mdx mice, the Dmd rat, or human-induced pluripotent stem cell models of DMD [74–76]. We predict that small molecule validation in independent laboratories and in multiple model systems will increase the potential for future translation of epigenetic small molecule therapies and other pharmacological approaches for DMD.
The development of significantly beneficial therapies for DMD will likely involve the simultaneous use of a combination of therapies that target dystrophin and/or downstream pathological mechanisms [19, 24]. Epigenetic small molecules may represent a promising component of a DMD combination therapy. Epigenetic small molecules are outstanding candidates for a DMD pharmacological therapy for many reasons. Certain epigenetic small molecules have been shown to benefit DMD and other neuromuscular disorders [25, 77–80]. Several HDACi, including SAHA, and other epigenetic small molecules are already FDA-approved for use in cancers [81–83]. While some epigenetic small molecules have off-target or solubility issues, there is potential for optimizing dosing or using these chemicals as lead compounds for further analyses [81, 83]. In addition, there are growing numbers of examples showing that epigenetic small molecules synergize with each other or with other compounds to target different diseases, including cancer and heart disease [59, 81–85]. In our work here, we show an example of a combination of two epigenetic small molecules that are beneficial for dmd mutant zebrafish. Because of the advantages of the zebrafish animal model, we expect that dmd mutant zebrafish will be an increasingly important model for investigating combination therapies for DMD.
Conclusions
We developed a new chemical-pooling approach for screening combinations of small molecules in dmd mutant zebrafish.
We identified a novel combination of epigenetic compounds, oxamflatin and salermide, that together improve skeletal muscle structural defects in dmd mutant zebrafish.
We have taken an important step in small molecule validation for DMD by demonstrating that oxamflatin and salermide show beneficial effects on zebrafish dmd mutants in two independent laboratories.
Acknowledgements
We would like to thank the SCRI Office of Animal Care for caring for the zebrafish. We thank Kimia Imani, Max Urbanek, and Ermias Yohannes for technical assistance.
Abbreviations
- βDG
β-dystroglycan
- DAPC
Dystrophin-associated protein complex
- DMD
Duchenne muscular dystrophy/dystrophin
- DMSO
Dimethyl sulfoxide
- dpf
Days post-fertilization
- EM
Embryo medium
- H4ac
Pan-acetylation of histone H4
- H4K16ac
Acetylation of lysine 16 of histone H4
- HDACi
Histone deacetylase inhibitors
- hpf
Hours post-fertilization
- PFA
Paraformaldehyde
- TSA
Trichostatin A
- WT
Wild-type
Authors’ contributions
GHFIII performed experiments, conducted data analyses, and prepared figures. MM, AG, TP, and EUP performed chemical screening and data analysis. EK and CH performed validation tests. GHFIII and SS developed the quantitative birefringence assay. SS provided technical assistance. LM conceived the project, obtained funding, and assisted and consulted on all experiments, data analysis, and figure preparation. LM wrote the manuscript with input from GHFIII and EUP. All authors read, edited, and approved the final manuscript.
Funding
This work was supported by grants 1R03AR065760 (NIH, NIAMS), 1R21AR068536 (NIH, NIAMS), and MDBR-15-107-DMD (Million Dollar Bike Ride Grant Program) to L.M. L.M. is a member of the Brotman Baty Institute for Precision Medicine (BBI) and this work was funded in part by a Catalytic Collaborations award from the BBI. We would like to thank the University of Washington School of Medicine Medical Student Research Training Program for financial support. Funding bodies played no role in the design of the study, in the collection, analysis, and interpretation of data, or in the writing of the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
All experiments involving live zebrafish (Danio rerio) were approved by and carried out in compliance with the Institutional Animal Care and Use Committee guidelines of Seattle Children’s Research Institute and the University of Maine.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Monaco AP, Neve RL, Colletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646–650. doi: 10.1038/323646a0. [DOI] [PubMed] [Google Scholar]
- 3.Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem. 1990;108:748–752. doi: 10.1093/oxfordjournals.jbchem.a123276. [DOI] [PubMed] [Google Scholar]
- 5.Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen DG, Whitehead NP, Froehner SC. Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol Rev. 2016;96:253–305. doi: 10.1152/physrev.00007.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spinazzola JM, Kunkel LM. Pharmacological therapeutics targeting the secondary defects and downstream pathology of Duchenne muscular dystrophy. Expert Opin Orphan Drugs. 2016;4:1179–1194. doi: 10.1080/21678707.2016.1240613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bodensteiner JB, Engel AG. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: a study of 567,000 muscle fibers in 114 biopsies. Neurology. 1978;28:439–446. doi: 10.1212/WNL.28.5.439. [DOI] [PubMed] [Google Scholar]
- 9.Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- 10.Guiraud S, Aartsma-Rus A, Vieira NM, Davies KE, van Ommen GJ, Kunkel LM. The pathogenesis and therapy of muscular dystrophies. Annu Rev Genomics Hum Genet. 2015;16:281–308. doi: 10.1146/annurev-genom-090314-025003. [DOI] [PubMed] [Google Scholar]
- 11.Dellorusso C, Crawford RW, Chamberlain JS, Brooks SV. Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J Muscle Res Cell Motil. 2001;22:467–475. doi: 10.1023/A:1014587918367. [DOI] [PubMed] [Google Scholar]
- 12.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reeve JL, McArdle A, Jackson MJ. Age-related changes in muscle calcium content in dystrophin-deficient mdx mice. Muscle Nerve. 1997;20:357–360. doi: 10.1002/(SICI)1097-4598(199703)20:3<357::AID-MUS14>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 14.Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-U. [DOI] [PubMed] [Google Scholar]
- 15.Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 2014;24:482–491. doi: 10.1016/j.nmd.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 16.Crone M, Mah JK. Current and emerging therapies for Duchenne muscular dystrophy. Curr Treat Options Neurol. 2018;20:31. doi: 10.1007/s11940-018-0513-6. [DOI] [PubMed] [Google Scholar]
- 17.Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 18.Kinnett K, Rodger S, Vroom E, Furlong P, Aartsma-Rus A, Bushby K. Imperatives for DUCHENNE MD: a simplified guide to comprehensive care for Duchenne muscular dystrophy. PLoS Curr. 2015;7. [DOI] [PMC free article] [PubMed]
- 19.Hoffman EP. Pharmacotherapy of Duchenne muscular dystrophy. Handb Exp Pharmacol. 2019 Aug 3. 10.1007/164_2019_256. [DOI] [PubMed]
- 20.Chamberlain JR, Chamberlain JS. Progress toward gene therapy for Duchenne muscular dystrophy. Mol Ther. 2017;25:1125–1131. doi: 10.1016/j.ymthe.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conboy I, Murthy N, Etienne J, Robinson Z. Making gene editing a therapeutic reality [version 1; peer review: 2 approved] F1000Res. 2018;7:1970. doi: 10.12688/f1000research.16106.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan D. Mol Ther. 2018;26:2337–2356. doi: 10.1016/j.ymthe.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Min YL, Bassel-Duby R, Olson EN. CRISPR correction of Duchenne muscular dystrophy. Annu Rev Med. 2019;70:239–255. doi: 10.1146/annurev-med-081117-010451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guiraud S, Davies KE. Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr Opin Pharmacol. 2017;34:36–48. doi: 10.1016/j.coph.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Consalvi S, Saccone V, Mozzetta C. Histone deacetylase inhibitors: a potential epigenetic treatment for Duchenne muscular dystrophy. Epigenomics. 2014;6:547–560. doi: 10.2217/epi.14.36. [DOI] [PubMed] [Google Scholar]
- 26.Bettica P, Petrini S, D’Oria V, D’Amico A, Catteruccia M, Pane M, et al. Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2016;26:643–649. doi: 10.1016/j.nmd.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Johnson NM, Farr GH 3rd, Maves L. The HDAC inhibitor TSA ameliorates a zebrafish model of Duchenne muscular dystrophy. PLoS Curr. 2013;5:ecurrents.md.8273cf41db10e2d15dd3ab827cb4b027. [DOI] [PMC free article] [PubMed]
- 28.Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12:1147–1150. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]
- 29.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 31.Chang NC, Sincennes MC, Chevalier FP, Brun CE, Lacaria M, Segalés J, et al. The dystrophin glycoprotein complex regulates the epigenetic activation of muscle stem cell commitment. Cell Stem Cell. 2018;22:755–768. doi: 10.1016/j.stem.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colussi C, Mozzetta C, Gurtner A, Illi B, Rosati J, Straino S, et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci U S A. 2008;105:19183–19187. doi: 10.1073/pnas.0805514105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colussi C, Gurtner A, Rosati J, Illi B, Ragone G, Piaggio G, et al. Nitric oxide deficiency determines global chromatin changes in Duchenne muscular dystrophy. FASEB J. 2009;23:2131–2141. doi: 10.1096/fj.08-115618. [DOI] [PubMed] [Google Scholar]
- 34.Saccone V, Consalvi S, Giordani L, Mozzetta C, Barozzi I, Sandoná M, et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. 2014;28:841–857. doi: 10.1101/gad.234468.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colussi C, Banfi C, Brioschi M, Tremoli E, Straino S, Spallotta F, et al. Proteomic profile of differentially expressed plasma proteins from dystrophic mice and following suberoylanilide hydroxamic acid treatment. Proteomics Clin Appl. 2010;4:71–83. doi: 10.1002/prca.200900116. [DOI] [PubMed] [Google Scholar]
- 36.Consalvi S, Saccone V, Giordani L, Minetti G, Mozzetta C, Puri PL. Histone deacetylase inhibitors in the treatment of muscular dystrophies: epigenetic drugs for genetic diseases. Mol Med. 2011;17:457–465. doi: 10.2119/molmed.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Consalvi S, Mozzetta C, Bettica P, Germani M, Fiorentini F, Del Bene F, et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol Med. 2013;19:79–87. doi: 10.2119/molmed.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bajanca F, Vandel L. Epigenetic regulators modulate muscle damage in Duchenne muscular dystrophy model. PLoS Curr. 2017;21:9. doi: 10.1371/currents.md.f1e2379fa632f8135577333dd92ca83b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bassett DI, Bryson-Richardson RJ, Daggett DF, Gautier P, Keenan DG, Currie PD. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development. 2003;130:5851–5860. doi: 10.1242/dev.00799. [DOI] [PubMed] [Google Scholar]
- 40.Bassett D, Currie PD. Identification of a zebrafish model of muscular dystrophy. Clin Exp Pharmacol Physiol. 2004;31:537–540. doi: 10.1111/j.1440-1681.2004.04030.x. [DOI] [PubMed] [Google Scholar]
- 41.Berger J, Berger S, Hall TE, Lieschke GJ, Currie PD. Dystrophin-deficient zebrafish feature aspects of the Duchenne muscular dystrophy pathology. Neuromuscul Disord. 2010;20:826–832. doi: 10.1016/j.nmd.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 42.Maves L. Recent advances using zebrafish animal models for muscle disease drug discovery. Expert Opin Drug Discov. 2014;14:1–13. doi: 10.1517/17460441.2014.927435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Widrick JJ, Kawahara G, Alexander MS, Beggs AH, Kunkel LM. Discovery of novel therapeutics for muscular dystrophies using zebrafish phenotypic screens. J Neuromuscul Dis. 2019;6:271–287. doi: 10.3233/JND-190389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Granato M, van Eeden FJ, Schach U, Trowe T, Brand M, Furutani-Seiki M, et al. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development. 1996;123:399–413. doi: 10.1242/dev.123.1.399. [DOI] [PubMed] [Google Scholar]
- 45.Kawahara G, Karpf JA, Myers JA, Alexander MS, Guyon JR, Kunkel LM. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2011;108:5331–5336. doi: 10.1073/pnas.1102116108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sztal TE, Ruparelia AA, Williams C, Bryson-Richardson RJ. Using touch-evoked response and locomotion assays to assess muscle performance and function in zebrafish. J Vis Exp. 2016;54431. [DOI] [PMC free article] [PubMed]
- 47.Berger J, Sztal T, Currie PD. Quantification of birefringence readily measures the level of muscle damage in zebrafish. Biochem Biophys Res Commun. 2012;423:785–788. doi: 10.1016/j.bbrc.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 48.Kawahara G, Gasperini MJ, Myers JA, Widrick JJ, Eran A, Serafini PR, et al. Dystrophic muscle improvement in zebrafish via increased heme oxygenase signaling. Hum Mol Genet. 2014;23:1869–1878. doi: 10.1093/hmg/ddt579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waugh TA, Horstick E, Hur J, Jackson SW, Davidson AE, Li X, et al. Fluoxetine prevents dystrophic changes in a zebrafish model of Duchenne muscular dystrophy. Hum Mol Genet. 2014;23:4651–4662. doi: 10.1093/hmg/ddu185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hightower RM, Reid AL, Gibbs DE, Wang Y, Widrick JJ, Kunkel LM, et al. The SINE compound KPT-350 blocks dystrophic pathologies in DMD zebrafish and mice. Mol Ther. 2020;28:189–201. doi: 10.1016/j.ymthe.2019.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li M, Andersson-Lendahl M, Sejersen T, Arner A. Muscle dysfunction and structural defects of dystrophin-null sapje mutant zebrafish larvae are rescued by ataluren treatment. FASEB J. 2014;28:1593–1599. doi: 10.1096/fj.13-240044. [DOI] [PubMed] [Google Scholar]
- 52.Winder SJ, Lipscomb L, Angela Parkin C, Juusola M. The proteasomal inhibitor MG132 prevents muscular dystrophy in zebrafish. PLoS Curr. 2011;3:RRN1286. doi: 10.1371/currents.RRN1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Westerfield M. The zebrafish book, 5th edition; a guide for the laboratory use of zebrafish (Danio rerio) 2007. [Google Scholar]
- 54.Berger J, Berger S, Jacoby AS, Wilton SD, Currie PD. Evaluation of exon-skipping strategies for Duchenne muscular dystrophy utilizing dystrophin-deficient zebrafish. J Cell Mol Med. 2011;15:2643–2651. doi: 10.1111/j.1582-4934.2011.01260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith LL, Beggs AH, Gupta VA. Analysis of skeletal muscle defects in larval zebrafish by birefringence and touch-evoke escape response assays. J Vis Exp. 2013;50925. [DOI] [PMC free article] [PubMed]
- 56.Goody MF, Kelly MW, Reynolds CJ, Khalil A, Crawford BD, Henry CA. NAD+ biosynthesis ameliorates a zebrafish model of muscular dystrophy. PLoS Biol. 2012;10:e1001409. doi: 10.1371/journal.pbio.1001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guyon JR, Goswami J, Jun SJ, Thorne M, Howell M, Pusack T, et al. Genetic isolation and characterization of a splicing mutant of zebrafish dystrophin. Hum Mol Genet. 2009;18:202–211. doi: 10.1093/hmg/ddn337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lipscomb L, Piggott RW, Emmerson T, Winder SJ. Dasatinib as a treatment for Duchenne muscular dystrophy. Hum Mol Genet. 2016;25:266–274. doi: 10.1093/hmg/ddv469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huber K, Doyon G, Plaks J, Fyne E, Mellors JW, Sluis-Cremer N. Inhibitors of histone deacetylases: correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J. Biol. Chem. 2011;286:22211–22218. doi: 10.1074/jbc.M110.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim YB, Lee KH, Sugita K, Yoshida M, Horinouchi S. Oxamflatin is a novel antitumor compound that inhibits mammalian histone deacetylase. Oncogene. 1999;18:2461–2470. doi: 10.1038/sj.onc.1202564. [DOI] [PubMed] [Google Scholar]
- 61.Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML, et al. Salermide, a sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene. 2009;28:781–791. doi: 10.1038/onc.2008.436. [DOI] [PubMed] [Google Scholar]
- 62.Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 63.Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006;20:1256–1261. doi: 10.1101/gad.1412706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sonoda H, Nishida K, Yoshioka T, Ohtani M, Sugita K. Oxamflatin: a novel compound which reverses malignant phenotype to normal one via induction of JunD. Oncogene. 1996;13:143–149. [PubMed] [Google Scholar]
- 65.Wang YL, Liui HL, Fu RG, Wang ZW, Ren HT, Dai ZJ, et al. HDAC inhibitor oxamflatin induces morphological changes and has strong cytostatic effects in ovarian cancer cell lines. Curr Mol Med. 2016;16:232–242. doi: 10.2174/1566524016666160225151408. [DOI] [PubMed] [Google Scholar]
- 66.Liu G, Su L, Hao X, Zhong N, Zhong D, Singhal S, et al. Salermide up-regulates death receptor 5 expression through the ATF4-ATF3-CHOP axis and leads to apoptosis in human cancer cells. J Cell Mol Med. 2012;16:1618–1628. doi: 10.1111/j.1582-4934.2011.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rotili D, Tarantino D, Nebbioso A, Paolini C, Huidobro C, Lara E, et al. Discovery of salermide-related sirtuin inhibitors: binding mode studies and antiproliferative effects in cancer cells including cancer stem cells. J Med Chem. 2012;55:10937–10947. doi: 10.1021/jm3011614. [DOI] [PubMed] [Google Scholar]
- 68.Bosch-Presegué L, Vaquero A. Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 2015;282:1745–1767. doi: 10.1111/febs.13053. [DOI] [PubMed] [Google Scholar]
- 69.Chalkiadaki A, Igarashi M, Nasamu AS, Knezevic J, Guarente L. Muscle-specific SIRT1 gain-of-function increases slow-twitch fibers and ameliorates pathophysiology in a mouse model of Duchenne muscular dystrophy. PLoS Genet. 2014;10:e1004490. doi: 10.1371/journal.pgen.1004490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pasco MY, Rotili D, Altucci L, Farina F, Rouleau GA, Mai A, et al. Characterization of sirtuin inhibitors in nematodes expressing a muscular dystrophy protein reveals muscle cell and behavioral protection by specific sirtinol analogues. J Med Chem. 2010;53:1407–1411. doi: 10.1021/jm9013345. [DOI] [PubMed] [Google Scholar]
- 71.Keskin-Aktan A, Akbulut KG, Yazici-Mutlu Ç, Sonugur G, Ocal M, Akbulut H. The effects of melatonin and curcumin on the expression of SIRT2, Bcl-2 and Bax in the hippocampus of adult rats. Brain Res Bull. 2018;137:306–310. doi: 10.1016/j.brainresbull.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 72.Shi Y, Xu X, Zhang Q, Fu G, Mo Z, Wang GS, et al. tRNA synthetase counteracts c-Myc to develop functional vasculature. Elife. 2014;3:e02349. doi: 10.7554/eLife.02349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spinazzola JM, Lambert MR, Gibbs DE, Conner JR, Krikorian GL, Pareek P, et al. Effect of serotonin modulation on dystrophin-deficient zebrafish. Biol Open. 2020;9:bio053363. doi: 10.1242/bio.053363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choi IY, Lim H, Estrellas K, Mula J, Cohen TV, Zhang Y, et al. Concordant but varied phenotypes among Duchenne muscular dystrophy patient-specific myoblasts derived using a human iPSC-based model. Cell Rep. 2016;15:2301–2312. doi: 10.1016/j.celrep.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 75.McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech. 2015;8:195–213. doi: 10.1242/dmm.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wells DJ. Tracking progress: an update on animal models for Duchenne muscular dystrophy. Dis Model Mech. 2018;11:dmm035774. [DOI] [PMC free article] [PubMed]
- 77.Consalvi S, Sandoná M, Saccone V. Epigenetic reprogramming of muscle progenitors: inspiration for clinical therapies. Stem Cells Int. 2016;2016:6093601. doi: 10.1155/2016/6093601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Campbell AE, Oliva J, Yates MP, Zhong JW, Shadle SC, Snider L, et al. BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet Muscle. 2017;7:16. doi: 10.1186/s13395-017-0134-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gordon BS, Delgado Díaz DC, Kostek MC. Resveratrol decreases inflammation and increases utrophin gene expression in the mdx mouse model of Duchenne muscular dystrophy. Clin Nutr. 2013;32:104–111. doi: 10.1016/j.clnu.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 80.Riessland M, Ackermann B, Förster A, Jakubik M, Hauke J, Garbes L, et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010;19:1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- 81.Abdelfatah E, Kerner Z, Nanda N, Ahuja N. Epigenetic therapy in gastrointestinal cancer: the right combination. Therap Adv Gastroenterol. 2016;9:560–579. doi: 10.1177/1756283X16644247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ahuja N, Sharma AR, Baylin SB. Epigenetic therapeutics: a new weapon in the war against cancer. Annu Rev Med. 2016;67:73–89. doi: 10.1146/annurev-med-111314-035900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bai Y, Li W, Wang T, Ahmad D, Cui G. Research advances in the use of histone deacetylase inhibitors for epigenetic targeting of cancer. Curr Top Med Chem. 2019. 10.2174/1568026619666190125145110. [DOI] [PubMed]
- 84.Alexanian M, Padmanabhan A, McKinsey TA, Haldar SM. Epigenetic therapies in heart failure. J Mol Cell Cardiol. 2019;130:197–204. doi: 10.1016/j.yjmcc.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sánchez-Rivera FJ, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1163–1171. doi: 10.1038/nm.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.