

Abstract
Silkworm ovarian germ cells produce the Siwi‐piRNA‐induced silencing complex (piRISC) through two consecutive mechanisms, the primary pathway and the secondary ping‐pong cycle. Primary Siwi‐piRISC production occurs on the outer mitochondrial membrane in an Ago3‐independent manner, where Tudor domain‐containing Papi binds unloaded Siwi via its symmetrical dimethylarginines (sDMAs). Here, we now show that secondary Siwi‐piRISC production occurs at the Ago3‐positive nuage Ago3 bodies, in an Ago3‐dependent manner, where Vreteno (Vret), another Tudor protein, interconnects unloaded Siwi and Ago3‐piRISC through their sDMAs. Upon Siwi depletion, Ago3 is phosphorylated and insolubilized in its piRISC form with cleaved RNAs and Vret, suggesting that the complex is stalled in the intermediate state. The Ago3 bodies are also enlarged. The aberrant morphology is restored upon Siwi re‐expression without Ago3‐piRISC supply. Thus, Siwi depletion aggregates the Ago3 bodies to protect the piRNA intermediates from degradation until the normal cellular environment returns to re‐initiate the ping‐pong cycle. Overall, these findings reveal a unique regulatory mechanism controlling piRNA biogenesis.
Keywords: Ago3, nuage, phosphorylation, Siwi, Vreteno
Subject Categories: Development & Differentiation, RNA Biology
Temporary reduction of Siwi levels during silkworm piRNA biogenesis alters Ago3 phosphorylation and Ago3 body structure, which is thought to protect piRNA intermediates until normal cellular conditions are restored.

Introduction
PIWI‐interacting RNAs (piRNAs) repress transposons to maintain germline genome integrity (Czech et al, 2018; Yamashiro & Siomi, 2018; Ozata et al, 2019). piRNAs interact with PIWI proteins to assemble the piRNA‐induced silencing complex (piRISC), which is the core engine of the piRNA‐mediated gene silencing pathway. Failure to form the piRISC, loss of piRISC function, or both causes DNA damage in the germline genome because of unrestricted transposon expression and transposition, causing impairments in gametogenesis and infertility (Girard et al, 2006; Klenov et al, 2007; Khurana & Theurkauf, 2010).
piRNAs are produced from their own precursors transcribed from piRNA clusters, intergenic elements rich in transposon remnants (Vagin et al, 2006; Brennecke et al, 2007; Malone et al, 2009). piRNA processing from the precursors requires numerous factors (Czech et al, 2018; Yamashiro & Siomi, 2018; Ozata et al, 2019). The molecular functions of the piRNA factors and the mechanism underlying piRNA biogenesis have been studied extensively in Drosophila, mice and other organisms, including zebrafish, using gonadal tissues and cultured cell lines such as OSCs/OSSs (OSCs) and BmN4 cells (Houwing et al, 2007; Iwasaki et al, 2015; Sakakibara & Siomi, 2018). OSCs are composed of Drosophila ovarian somatic cells (Niki et al, 2006; Saito et al, 2009), whereas BmN4 cells are germ cells isolated from Bombyx ovaries (Kawaoka et al, 2009). These cells produce piRISC fully functional in transposon repression and are therefore suitable for analyzing the mechanism underlying the piRNA pathway.
The number of PIWI members differs among species. Drosophila expresses three members, Piwi, Aubergine (Aub), and Ago3, whereas Bombyx expresses two members, Siwi and Ago3 (Kawaoka et al, 2008). Siwi is the counterpart to Aub in Drosophila, and Bombyx lacks the homologue of Piwi. Piwi in Drosophila is unique because it localizes to the nucleus upon piRISC formation in the cytoplasm and represses transposons co‐transcriptionally through local heterochromatinization (Czech et al, 2018; Sato & Siomi, 2020). In contrast, Aub and Ago3 in Drosophila and Siwi and Ago3 in Bombyx remain in the cytosol even after piRISC assembly and repress transposons post‐transcriptionally by cleaving the RNA transcripts (Brennecke et al, 2007; Gunawardane et al, 2007; Kawaoka et al, 2009). Thus, Bombyx relies solely on post‐transcriptional silencing to repress transposons. This trend may be present in other organisms expressing two PIWI members, such as zebrafish (Houwing et al, 2008). As observed for Drosophila, mice express three PIWI members: cytoplasmic Mili and Miwi repress transposons post‐transcriptionally but Miwi2 represses transposons through DNA methylation (Carmell et al, 2007; Aravin et al, 2008; Kuramochi‐Miyagawa et al, 2008; De Fazio et al, 2011; Reuter et al, 2011).
PIWI‐dependent RNA cleavage represses transposons and produces piRNAs. This coupling system is termed the ping‐pong cycle and is highly conserved in many species, and the mechanism has been studied extensively in Drosophila (Brennecke et al, 2007; Gunawardane et al, 2007; Li et al, 2009; Malone et al, 2009). Briefly, piRNAs arising from piRNA clusters are predominantly loaded onto Aub, which shows a strong bias in direction to be antisense to transposon mRNAs. Thereby, Aub‐piRISC targets transposon mRNAs and cleaves them into two pieces. Of those, the 3′‐side fragment is then loaded onto Ago3 through the 5′‐end, giving rise to Ago3‐pre‐piRISC. The 3′‐end of the RNA fragment is further processed to produce mature Ago3‐piRISC. Because Ago3‐loaded piRNAs are the products of transposon mRNAs, Ago3 in turn targets and cleaves transposon transcripts in the antisense orientation. The 3′‐side fragment of the cleaved RNA is loaded onto Aub and upon 3′‐end processing Aub‐piRISC is produced. Aub and Ago3 continue these cleavage (slicer)‐dependent reactions reciprocally, producing a large number of Aub‐ and Ago3‐piRISCs. In parallel, the ping‐pong cycle activates a downstream piRNA biogenesis system to produce phasing piRNAs, which are nearly exclusively loaded onto Piwi and therefore Piwi‐bound piRNAs can be mapped on the genome in a tandem fashion (Han et al, 2015; Mohn et al, 2015).
In BmN4 cells, Siwi and Ago3 operate the ping‐pong cycle. In the absence of Siwi, Ago3‐piRISC is not produced but Siwi‐piRISC production still continues even in the absence of Ago3 because the primary pathway remains active irrespective of the presence or absence of Ago3 in cells (Nishida et al, 2015). Therefore, Siwi‐piRISC in BmN4 cells can be subgrouped into Ago3‐independent primary and Ago3‐dependent secondary piRISCs, whereas all Ago3‐piRISC is produced Siwi‐dependently through the ping‐pong pathway.
We recently proposed a model for piRNA biogenesis in silkworm ovarian germ cells (Nishida et al, 2018), where primary Siwi‐piRISC production is initiated when unloaded Siwi is anchored onto the Tudor domain‐containing protein Papi, which is located on the outer mitochondrial membrane. The primary piRNA precursor is loaded onto Papi‐bound Siwi. In turn, endonuclease Zucchini (Zuc), which is also located on the outer mitochondrial membrane, processes the 3′‐end of the piRNA precursor, liberating Siwi‐piRISC to the cytosol to initiate the ping‐pong cycle with the partner Ago3. The ping‐pong cycle also requires a DEAD‐box RNA helicase Vasa, which displaces cleaved RNAs from Siwi‐piRISC upon cleavage, facilitating the formation of Ago3‐piRISC. Without Vasa, cleaved RNAs remain on Siwi‐piRISC, which attenuates the ping‐pong cycle (Xiol et al, 2014; Nishida et al, 2015).
In this study, we show that Ago3‐piRISC in BmN4 cells is tightly associated with the silkworm homologue of Drosophila Vreteno (Vret), another Tudor protein required for piRNA biogenesis in ovaries (Handler et al, 2011; Zamparini et al, 2011). The closest homologue in mice and zebrafish is Tdrd1, which is necessary for piRNA production in the gonads (Reuter et al, 2009; Vagin et al, 2009; Wang et al, 2009; Huang et al, 2011). We also show that Ago3 and Vret collaboratively assemble Ago3‐positive nuage, which we term Ago3 bodies, and that Vret in the nuage interconnects Ago3‐piRISC with unloaded Siwi to facilitate secondary Siwi‐piRISC production. Nuage is a perinuclear non‐membranous organelle known as the center of piRNA biogenesis in germ cells (Mahowald, 1971, 2001). In the absence of Siwi, Ago3 was phosphorylated and insolubilized in the piRISC form with RNA fragments cleaved by Ago3‐piRISC, and Ago3 bodies were enlarged where Vret was encapsulated. These observations strongly suggest that the Ago3 complex is immediately stalled in the intermediate state upon sensing Siwi depletion, the ping‐pong partner of Ago3. This aberrant situation was restored by Siwi re‐expression without a new Ago3‐piRISC supply. These findings support the new concept that Ago3 bodies normally act as the center for secondary Siwi‐piRISC biogenesis, but upon Siwi loss, the nuage granulizes to store and protect the piRNA intermediates from degradation until the cellular circumstances return to re‐initiate the ping‐pong cycle.
Results
Vret interconnects Ago3‐piRISC and unloaded Siwi in secondary Siwi‐piRISC production
Immunoprecipitation and silver staining showed that Ago3 co‐immunoprecipitated with 150‐ and 130‐kDa proteins from BmN4 lysates (P150 and P130, respectively, in Fig 1A). Mass spectrometric analysis identified these proteins as the silkworm homologue of Drosophila Vret (Handler et al, 2011; Zamparini et al, 2011). Vret from silkworm has not been studied. Thus, we raised an anti‐Vret monoclonal antibody (Fig EV1A) and pursued our investigation. Western blotting using the antibody confirmed the presence of Vret in the Ago3 complex (Fig 1B). PIWI‐Vret association is conserved among species: Vret/Tdrd1 in flies, mice, and zebrafish also interacted with PIWI proteins (Reuter et al, 2009; Handler et al, 2011; Huang et al, 2011).
Figure 1. Vret interconnects Ago3‐piRISC and unloaded Siwi for secondary Siwi‐piRISC production.
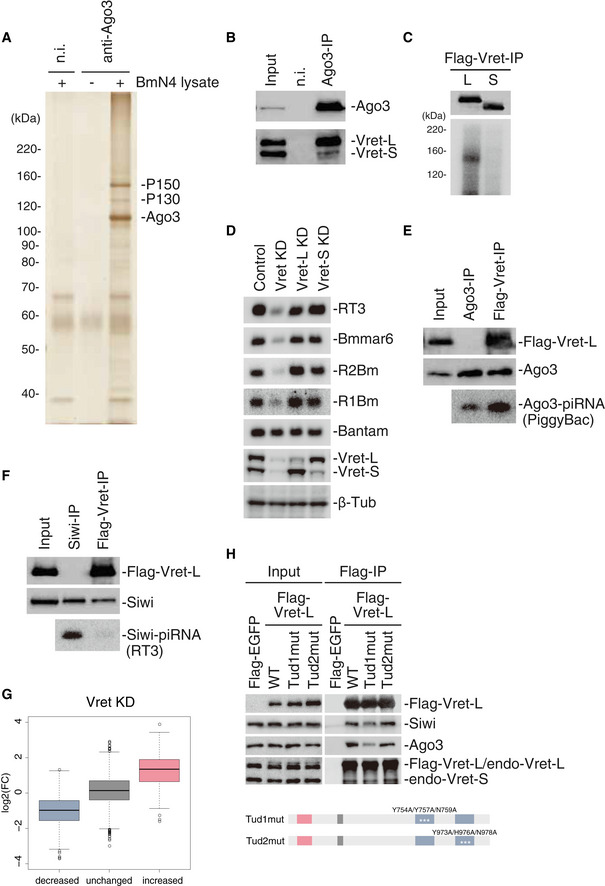
- Silver staining of protein components in the Ago3 complex immunoisolated from BmN4 cells. P150 and P130 co‐immunoprecipitated with Ago3. n.i.: non‐immune IgG.
- Western blots showing the presence of Vret‐L (P150) and Vret‐S (P130) in the Ago3 complex in A. n.i.: non‐immune IgG.
- Top: Western blot showing Vret‐L and Vret‐S immunoprecipitated from BmN4 cells upon UV irradiation. Bottom: CLIP signals showing that Vret‐L, but not Vret‐S, binds RNAs in vivo.
- Five upper panels: Northern blots showing the levels of RT3‐piRNA, Bmmar6‐piRNA, R2Bm‐piRNA, and R1Bm‐piRNA in normal (Control), Vret‐L/S-depleted (Vret KD), Vret‐L-depleted (Vret‐L KD), and Vret‐S-depleted (Vret‐S KD) cells. Bantam (miRNA) was detected as a loading control. Two lower panels: Western blots showing the efficiency of the Vret knockdown. β‐Tub was detected as a loading control.
- Two upper panels: Western blots showing the abundances of Flag‐Vret-L and Ago3 in both samples. Note that Flag‐Vret-L is present only in the Vret‐L complex but not with Ago3 isolated from total cell lysates (Fig EV1F). Bottom panel: Northern blot showing the level of PiggyBac‐piRNA associated with Ago3 in total lysate (Ago3‐IP) and in the Vret‐L complex (Flag‐Vret-IP). The ratio of the piRNA signal intensity in the two samples is indicated at the bottom.
- Two upper panels: Western blots showing the abundances of Flag‐Vret-L and Siwi in both samples. Bottom panel: Northern blot showing the level of RT3‐piRNA associated with Siwi in the total lysate (Siwi‐IP) and in the Vret‐L complex (Flag‐Vret-IP).
- Boxplot showing log2 fold‐change of Vret KD (RPM)/Control (RPM) values of piRNAs in increased, unchanged and decreased groups defined by Ago3‐KD (RPM)/Control (RPM) values shown in Fig EV1I. Boxplot represents median, first and third quartile, and maximum and minimum values of the log2 fold‐change of piRNAs (N = 2). Outliers are represented as open circles.
- Flag‐EGFP and ‐Vret-L WT and mutants were immunoprecipitated using the anti‐Flag antibody and then Western blotting was performed using anti‐Flag, anti‐Siwi, anti‐Ago3, and anti‐Vret antibodies. The domain structures of Tud1mut (Y753A/Y757A/N759A) and Tud2mut (Y973A/Y976A/N978A) are shown.
Source data are available online for this figure.
Figure EV1. Vret is a well‐conserved Tudor domain‐containing protein.
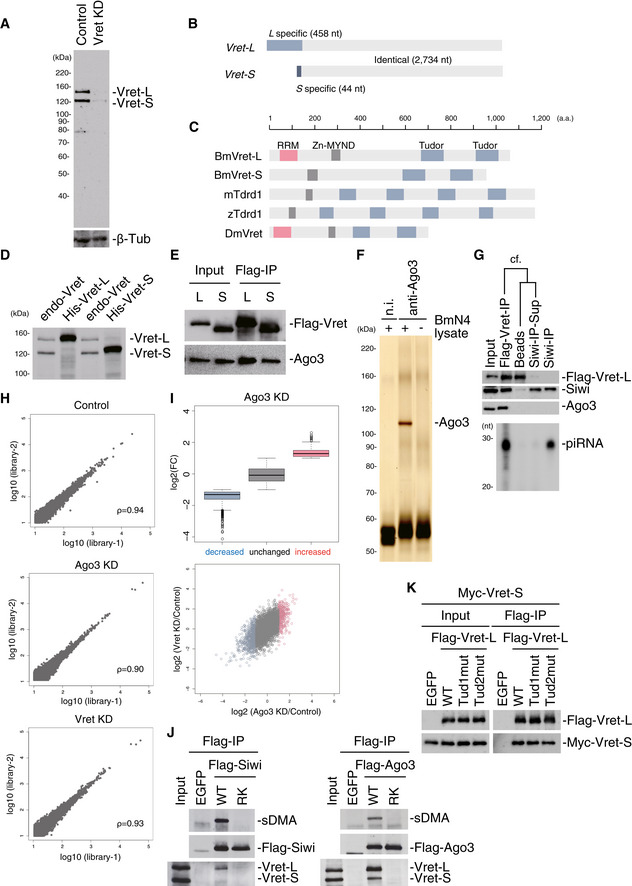
- The Western blot shows the specificity of the anti‐Vret monoclonal antibody raised in this study. β‐Tub was used as a loading control.
- Bombyx expresses two Vret isoforms, which have different 5′ terminal regions.
- Domain structures of BmVret‐L, BmVret‐S, mTdrd1, zTdrd1, and DmVret.
- Western blotting analysis showed that His‐tagged Vret‐L and Vret‐S co-migrate with endogenous Vret P150 and P130, respectively.
- Immunoprecipitation and Western blotting results show that Flag‐Vret-L and Flag‐Vret-S associate with Ago3 in BmN4 cells.
- Ago3 was immunoprecipitated from BmN4 cells using the anti‐Ago3 antibody. The cell lysate was prepared in Empigen‐containing cell lysis buffer. n.i.: non‐immune IgG.
- Tandem immunoprecipitation of Siwi from the Vret complex. The top three panels show Western blots performed using anti‐Flag, anti‐Siwi, and anti‐Ago3 antibodies. The bottom panel shows the abundance of piRNAs loaded onto Siwi.
- Scatter plots of normalized Flag‐Siwi-piRNA abundance in two replicates from Control, Ago3‐depleted, and Vret‐depleted BmN4 (Spearman's rho = 0.94, 0.90 and 0.93, respectively). Each dot represents a piRNA sequence.
- Boxplot showing log2 fold‐change of Ago3‐KD (RPM)/Control (RPM). piRNAs whose ratio was ≥ 2, 0.5 < ratio < 2 or ≤ 0.5 were categorized as increased, unchanged, and decreased, respectively. Boxplot represents median, first and third quartile, and maximum and minimum values of the log2 fold‐change of piRNAs (N = 2). Outliers are represented as open circles. Scatterplots for log2 fold‐change of piRNAs between Ago3‐KD (x axis) and Vret KD (y axis). Each dot represents a unique piRNA. Red dots indicate piRNAs in the increased group and blue dots indicate piRNAs from the decreased group.
- Immunoprecipitation and subsequent Western blotting using anti‐sDMA and anti‐Flag and anti‐Vret antibodies show that both Siwi and Ago3 RK mutants defective in sDMA modification failed to co‐purify with Vret.
- Flag‐EGFP and Flag‐Vret-L WT and mutants were immunoprecipitated using the anti‐Flag antibody and then Western blotting was performed using anti‐Flag and anti‐Myc antibodies. Myc‐Vret-S was co‐expressed in the cells prior to immunoprecipitation.
Source data are available online for this figure.
Vret appeared as two bands on Western blots (Figs 1B and EV1A), whereas Vret/Tdrd1 in other species was detected as a single band (Reuter et al, 2009; Huang et al, 2011; Zamparini et al, 2011). We assumed that Vret‐L in Fig 1B (P150 in Fig 1A) is full‐length Vret, whereas Vret‐S/P130 is a degraded form of Vret‐L. However, while cloning the cDNA, we realized that BmN4 cells express two Vret isoforms, which were distinct at the 5′ regions but identical for the remaining parts (Fig EV1B and Table EV1). The longer cDNA encoded a Vret containing one RNA recognition motif (RRM), one MYND family zinc‐finger (ZnF‐MYND), and two Tudor domains, whereas the shorter cDNA encoded a Vret containing one ZnF‐MYND and two Tudor domains but not the RRM (Fig EV1C). Exogenous Vret expressed via cDNA transfection co‐migrated with endogenous Vret on Western blots (Fig EV1D) and co‐immunopurified with Ago3, as did endogenous Vret (Fig EV1E).
The RRM confers RNA‐binding activity to host proteins (Maris et al, 2005). We therefore hypothesized that Vret‐L, but not Vret‐S, accommodates RNA‐binding activity. Crosslink immunoprecipitation (CLIP) experiments confirmed that Vret‐L, but not Vret‐S, binds RNAs in vivo (Fig 1C). Knockdown (KD) experiments showed that simultaneous depletion of Vret‐L and Vret‐S reduced the levels of Siwi‐bound piRNAs (Nishida et al, 2015), confirming the requirement of Vret in piRNA biogenesis in BmN4 cells (Fig 1D). In contrast, single depletion of Vret‐L or Vret‐S barely changed the piRNA levels, indicating that the RNA‐binding activity of Vret‐L through the RRM is dispensable for piRNA biogenesis in BmN4 cells, although Ago3 preferred to interact with Vret‐L (Fig 1B).
Comparison of Vret/Tdrd1 domain structures among species showed that Drosophila Vret (DmVret) contains one RRM, one ZnF‐MYND, and two Tudor domains (Handler et al, 2011), whereas Tdrd1 in mice (mTdrd1) and zebrafish (zTdrd1) has one ZnF‐MYND and four Tudor domains but lacks the RRM (Huang et al, 2011; Fig EV1C). These structural differences suggest functional differences of Vret/Tdrd1 among species although their requirement in the piRNA pathway is conserved.
We then examined the piRNA‐loading status of Vret‐associating Ago3 by Northern blotting. Total RNAs isolated from the immunoprecipitated Vret complex and piRNAs loaded onto Ago3 immunoisolated from total lysates were probed for detecting PiggyBac‐piRNA, one of the Ago3‐loaded piRNAs (Nishida et al, 2015). The Ago3 immunopurification was conducted under harsh conditions and so no other proteins co‐purified with Ago3 (Fig EV1F). Western blotting showed that the abundance of Ago3 in the two samples was nearly equal (Fig 1E). However, the abundance of PiggyBac‐piRNA in the Vret complex was 3.7‐fold higher when compared with that of Ago3 (Fig 1E). We repeated the experiment three times and obtained very similar data in each experiment. These results suggest that Ago3 in the Vret complex is predominantly in a piRISC form.
Siwi was also present in the Vret complex (Fig 1F). However, RT3‐piRNA, one of the most abundant Siwi‐bound piRNAs (Nishida et al, 2015), was hardly detected in the complex (Fig 1F). Siwi isolated from the Vret complex by tandem immunoprecipitation was devoid of piRNAs (Fig EV1G). These results support an intriguing concept that Vret interconnects Ago3‐piRISC and unloaded Siwi, and this connection likely facilitates Siwi‐piRISC production in the ping‐pong cycle. The latter inference is based on previous observations that Vasa interconnects Siwi‐piRISC and unloaded Ago3 to facilitate Ago3‐piRISC production through the ping‐pong cycle (Xiol et al, 2014; Nishida et al, 2015).
To gain further supportive evidence, we deep‐sequenced piRNAs loaded onto Flag‐Siwi that was expressed in Ago3‐ and Vret‐depleted cells. Two replicate libraries were generated individually in both KD and control cells. Strong correlations were observed in each replicate for normalized numbers of reads as reads per million (RPM) (Fig EV1H). Further analysis demonstrated that Siwi‐bound piRNAs reduced by the loss of Ago3 were downregulated by the loss of Vret, while those increased by the loss of Ago3 were upregulated by the loss of Vret (Figs 1G and EV1I). Siwi is higher in the piRNA biogenesis hierarchy than Ago3 (Nishida et al, 2015). Thus, loss of Ago3 barely disturbs primary Siwi‐piRNA production but attenuates secondary Siwi‐piRISC production. Based on these results, we propose that Vret functions in Ago3‐dependent, secondary Siwi‐piRISC production but is unnecessary for Ago3‐independent, primary Siwi‐piRISC production.
Vret in silkworm contains two Tudor domains (BmVret in Fig EV1C). Tudor domains often use symmetrical dimethylarginines (sDMAs) as an index to recognize and bind their partners (Siomi et al, 2010). Siwi and Ago3 were sDMA‐modified in BmN4 cells (Honda et al, 2013; Nishida et al, 2018). We then examined whether Vret association with Siwi and Ago3 requires sDMAs. We used Siwi and Ago3 RK mutants that were defective in sDMA modification (Nishida et al, 2018). Both mutants failed to co‐purify with Vret when compared with that of the wild‐type (WT) controls (Fig EV1J), indicating that the Vret interaction with Siwi and Ago3 is sDMA‐dependent.
To determine which of the two Tudor domains in Vret is responsible for the sDMA‐dependent PIWI association, we produced Vret‐L Tud1mut and Tud2mut mutants, where three conserved residues in Tudor1 (Tyr754, Tyr757, and Asn759) and Tudor2 (Tyr973, His976, and Asn978) were mutated to alanine (Fig 1H). Immunoprecipitation showed that Tud1mut weakly associated with Siwi and Ago3 while Tud2mut bound both to a similar extent to WT (Fig 1H). These results suggest that Vret interacts with Siwi and Ago3 through Tudor1. Endogenous Vret‐S was also detected in the Flag‐Vret‐L complex (Fig 1H). Additional experiments showed that Vret‐L co‐immunoprecipitated with Vret‐S (Fig EV1K). Thus, Vret can self‐associate. The interaction intensity did not change significantly by amino acid alterations (Fig 1H), indicating that Vret self‐association occurs through domains excluding Tudor1. Based on these findings, we propose that Vret assembles a high‐ordered structure with Ago3‐piRISC and unloaded Siwi, where each PIWI binds different Vret molecules via Tudor1‐sDMA association.
Ago3 bodies serve as the center of secondary Siwi‐piRISC production
Immunofluorescence showed that Vret is localized mostly to Ago3‐positive nuage (Figs 2A and EV2A). The percentage of Vret dotty signals merged with Ago3 signals (n = 52) was 80.8%. The Vret signals also overlapped with Siwi signals but to a lesser extent (Figs 2B and EV2B). The percentage of Vret signals that merged with Siwi signals (n = 66) was 19.7%. We also examined the overlapping status of Siwi and Ago3 (Fig EV2C), which was reasonably comparable with the data shown in Fig 2A and B. These findings indicate that Siwi‐positive nuage is not uniform but subgrouped by specific residences and possibly by their functions. We hereinafter term the Ago3/Vret‐positive nuage as Ago3 bodies.
Figure 2. Ago3 bodies serve as the center of secondary Siwi‐piRISC production.
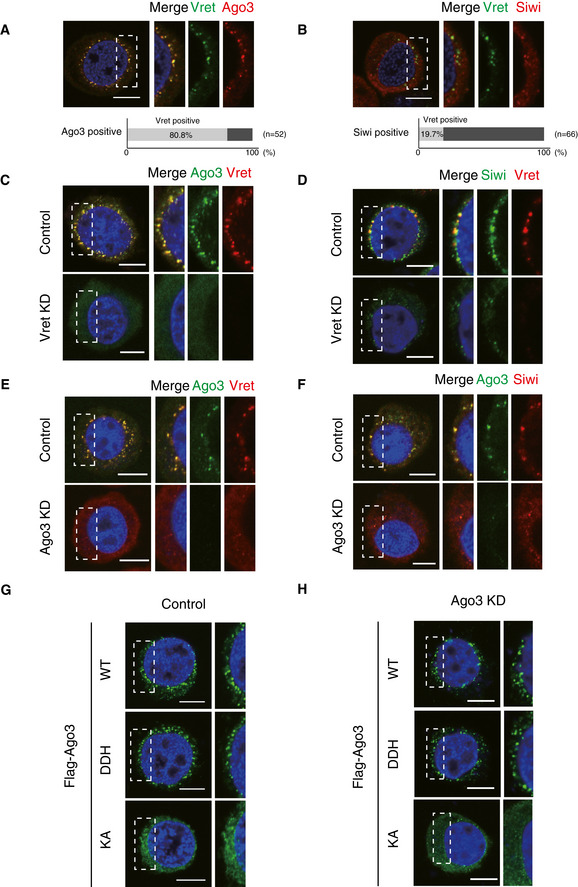
- Immunofluorescence shows that the Vret signals (green) co‐localize with the Ago3 signals (red). DAPI shows the nuclei (blue). Scale bar: 10 μm. The three panels on the right‐hand side show high magnification images of the part indicated by the white dashed line box in the image on the left‐hand side. This panel arrangement applies also for panels B–H. The bar chart shows the percentage of Vret‐positive nuage in Ago3‐positive nuage.
- Immunofluorescence shows that the Vret signals (green) partially co‐localize with the Siwi signals (red). DAPI shows the nuclei (blue). Scale bar: 10 μm. The bar chart shows the percentage of Vret‐positive nuage in Siwi‐positive nuage.
- Immunofluorescence showing the Ago3 signals (green) in control and Vret‐depleted BmN4 cells (Vret KD). Vret is shown in red. DAPI shows the nuclei (blue). Scale bar: 10 μm.
- Immunofluorescence showing the Siwi signals (green) in control and Vret‐depleted BmN4 cells (Vret KD). Vret is shown in red. DAPI shows the nuclei (blue). Scale bar: 10 μm.
- Immunofluorescence showing the Vret signals (red) in control and Ago3‐depleted BmN4 cells (Ago3 KD). Ago3 is shown in green. DAPI shows the nuclei (blue). Scale bar: 10 μm.
- Immunofluorescence showing the Siwi signals (red) in control and Ago3‐depleted BmN4 cells (Ago3 KD). Ago3 is shown in green. DAPI shows the nuclei (blue). Scale bar: 10 μm.
- Immunofluorescence showing Flag‐Ago3 WT, KA mutant, and DDH mutant signals (green) expressed in naïve BmN4 cells (Control). DAPI shows the nuclei (blue). Scale bar: 10 μm.
- Immunofluorescence showing Flag‐Ago3 WT, KA mutant, and DDH mutant signals (green) in Ago3‐depleted cells (Ago3 KD). DAPI shows the nuclei (blue). Scale bar: 10 μm.
Figure EV2. Ago3 bodies are required for the concentration of the Ago3‐Vret‐Siwi complex.
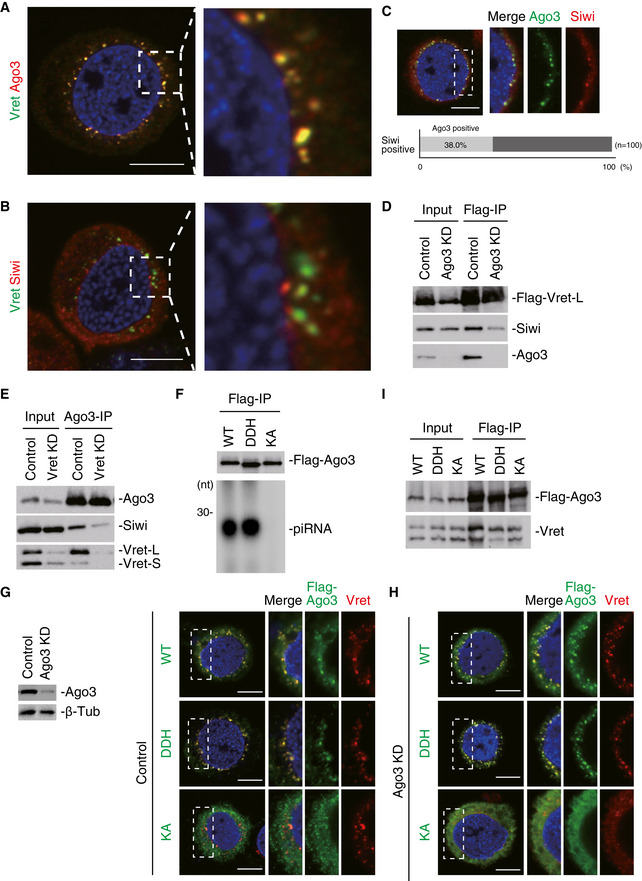
- Left: An enlarged view of the full cell image in Fig 2A. Right: A magnification image of the part indicated by the white dashed line box in the image of the left‐hand side.
- Left: An enlarged view of the full cell image in Fig 2B. Right: A magnification image of the part indicated by the white dashed line box in the image of the left‐hand side.
- Immunofluorescence shows that the Ago3 signals (green) co‐localize with the Siwi signals (red). DAPI shows the nuclei (blue). Scale bar: 10 μm. The three panels on the right‐hand side show high magnification images of the part indicated by the white dashed line box in the image on the left‐hand side. The bar chart shows the percentage of Ago3‐positive nuage in Siwi‐positive nuage.
- Immunoprecipitation and subsequent Western blotting show that Flag‐Vret-L only weakly associates with Siwi upon Ago3 depletion.
- Immunoprecipitation and subsequent Western blotting show that Ago3 associates with Siwi much less upon Vret depletion.
- 32P‐labeling showing the levels of piRNAs loaded onto Flag‐Ago3 WT and DDH and KA mutants. Lys637 was mutated to alanine in the KA mutant. Asp697, Asp767, and His901 were mutated to alanine in the DDH mutant.
- Immunofluorescence showing Flag‐Ago3 WT, KA mutant, and DDH mutant signals (green) expressed in naïve BmN4 cells (Control). Vret is shown in red. DAPI shows the nuclei (blue). Scale bar: 10 μm.
- Immunofluorescence showing Flag‐Ago3 WT, KA mutant, and DDH mutant signals (green) expressed in Ago3‐depleted BmN4 cells (Ago3 KD). Vret is shown in red. DAPI shows the nuclei (blue). Scale bar: 10 μm.
- Immunoprecipitation and subsequent Western blotting show that Flag‐Ago3 WT and DDH and KA mutants associate with Vret.
Source data are available online for this figure.
We depleted Vret and performed immunofluorescence to understand the molecular hierarchy in Ago3‐body formation. Ago3 was dispersed in the cytosol and failed to localize to any specific structures (Fig 2C). The Siwi signals were also broadly dispersed throughout the cytosol but some Siwi‐positive nuage remained (Fig 2D). The latter nuage might serve as the place for Siwi‐dependent Ago3‐piRISC production that occurs independent of Vret.
Similar outcomes were obtained upon Ago3 loss with Vret becoming essentially only cytosolic while some Siwi‐positive nuage remained in the cytoplasm (Fig 2E and F). The lack of Ago3 and Vret not only caused the disappearance of Ago3 bodies (Fig 2C and E) but also attenuated the Vret–Siwi and Ago3–Siwi interactions, respectively (Fig EV2D and E). These results suggest that Ago3‐body assembly requires both Vret and Ago3 and that Siwi is localized to the nuage, upon formation, through protein–protein interactions with Ago3 and Vret.
We then assessed whether the Ago3‐body assembly requires Ago3‐piRNA loading. To this end, an Ago3 mutant defective in piRNA loading was produced by mutating Lys637 to alanine based on our previous finding that the equivalent Siwi K611A mutant failed to bind piRNAs (Matsumoto et al, 2016). As expected, the K637A (KA) mutant failed to bind piRNAs (Fig EV2F). We also produced a slicer‐defective DDH mutant as a control, in which the active center residues (i.e., DDH motif composed of Asp697, Asp767, and His901) were substituted with alanine. This mutant was found to load with piRNAs as efficiently as WT (Fig EV2F).
The KA mutant was mostly cytosolic but weakly localized to Ago3 bodies in normal BmN4 cells (Figs 2G and EV2G). In contrast, the DDH mutant localized to the Ago3 bodies in a similar manner to WT (Figs 2G and EV2G). When we used BmN4 cells where endogenous Ago3 was depleted by RNAi, the KA mutant was nearly totally cytosolic while the DDH mutant assembled Ago3 bodies (Figs 2H and EV2H). Thus, piRNA loading onto Ago3 is required for Ago3‐body assembly. Both the KA and DDH mutants interacted with Vret as well as Ago3 WT (Fig EV2I). This indicates that Vret has an intrinsic capacity to interact with unloaded Ago3; although in normal cells, Vret preferentially bound Ago3‐piRISC over unloaded Ago3 (Fig 1E). However, interestingly, the Vret–unloaded Ago3 interaction was insufficient in Ago3‐body assembly (Figs 2G and H). The RNA targeting of Ago3‐piRISC may also be critical in Ago3‐body formation; however, we cannot provide direct evidence supporting this because we do not have an Ago3 mutant that binds piRNAs but does not target RNAs.
Lack of Siwi causes changes to Ago3‐body morphology and Ago3 characteristics
We finally conducted immunofluorescence using Siwi‐lacking cells, which was observed to cause Ago3‐body enlargement (Figs 3A and EV3A). The Vret dotty signals also increased in size as did the Ago3 signals (Figs 3B and EV3A). As expected, Vret was encapsulated in enlarged Ago3 bodies in the absence of Siwi (Fig EV3B).
Figure 3. Lack of Siwi causes Ago3 phosphorylation and insolubilization, causing Ago3‐body enlargement.
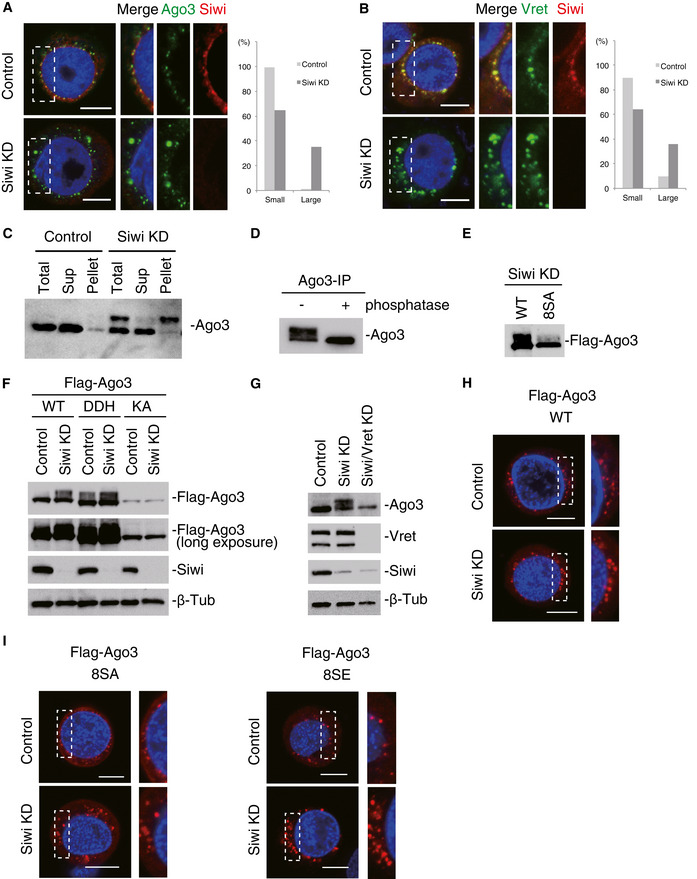
- Immunofluorescence showing the Ago3 signals (green) in control and Siwi‐depleted BmN4 cells (Siwi KD). Siwi is shown in red. DAPI shows the nuclei (blue). Scale bar: 10 μm. The three panels on the right‐hand side show high magnification images of the part indicated by the white dashed line box in the images on the left‐hand side. This panel arrangement applies also for panels B, H, and I. Bar chart indicates dot sizes of > 100 Ago3 particles in each case (Large: IntDen value > 106; Small: IntDen value < 106).
- Immunofluorescence showing the Vret signals (green) in control and Siwi‐depleted BmN4 cells (Siwi KD). Siwi is shown in red. DAPI shows the nuclei (blue). Scale bar: 10 μm. Bar chart indicates dot sizes of > 100 Ago3 particles in each case (Large: IntDen value > 105; Small: IntDen value < 105).
- Western blotting was performed before and after Siwi KD using the anti‐Ago3 antibody. Total BmN4 lysate (Total), supernatant (Sup), and pellet (Pellet) fractions upon centrifugation were used.
- Western blotting was performed using the anti‐Ago3 antibody on Ago3 immunoisolated from Siwi‐depleted cells. Ago3 immunoprecipitated was untreated (−) or treated with phosphatase (+) prior to Western blotting.
- Western blot showing the degree of phosphorylation of Flag‐Ago3 WT and the 8SA mutant in Siwi‐depleted cells (Siwi KD).
- Western blots showing the degree of phosphorylation of Flag‐Ago3 WT and its DDH and KA mutants in Siwi‐depleted cells (Siwi KD). The second panel from the top corresponds to the top panel with a longer exposure time. Siwi and β‐Tub as a loading control were also detected also by Western blotting.
- Western blotting was performed using anti‐Ago3, anti‐Vret, and anti‐Siwi antibodies on lysates of BmN4 cells before (Control) and after Siwi (Siwi KD) and Siwi/Vret (Siwi/Vret KD) depletion. β‐Tub was detected as a loading control.
- Immunofluorescence showing the Flag‐Ago3 signals (red) in normal (Control) and Siwi‐depleted BmN4 cells (Siwi KD). DAPI shows the nuclei (blue). Scale bar: 10 μm.
- Immunofluorescence showing the Flag‐Ago3 8SA and 8SE mutant signals (red) in normal (Control) and Siwi‐depleted BmN4 cells (Siwi KD). DAPI shows the nuclei (blue). Scale bar: 10 μm.
Source data are available online for this figure.
Figure EV3. Ago3 phosphorylation in Siwi‐depleted cells.
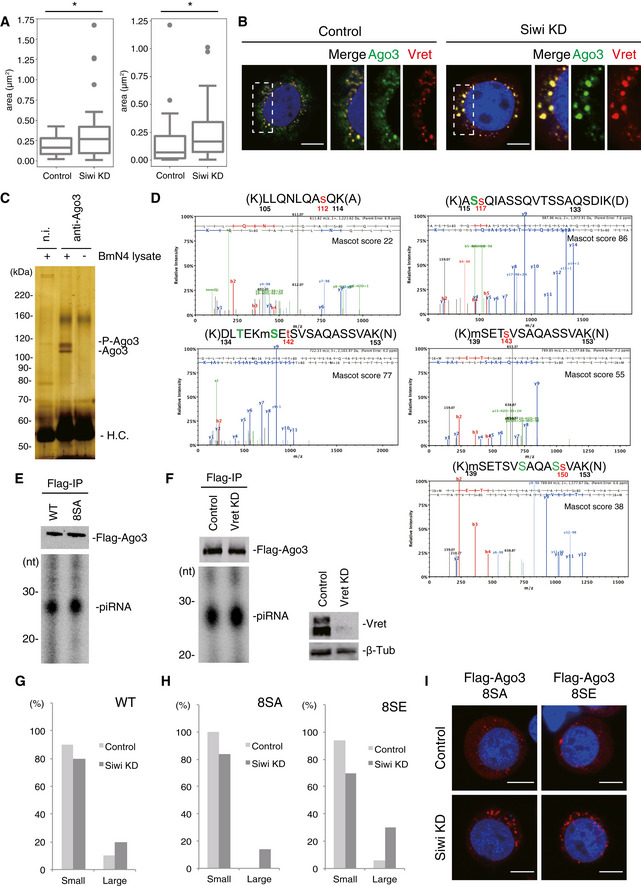
- Boxplot represents median, first and third quartile, and maximum and minimum values of the size of Ago3 particle (left panel; Control; n = 32, Siwi KD; n = 37) and Vret particle (right panel; Control; n = 54, Siwi KD; n = 26). Outliers are represented as filled circles. To calculate the size of nuage, we subtracted the background from the original images and generated binary images. After separating the adjacent object by the watershed algorithm and reducing noise, the area of nuage was obtained by Analyze Particles in ImageJ. P‐values were calculated by Brunner‐Munzel test using Scipy. *P < 0.05.
- Immunofluorescence showing the Ago3 signals (green) and the Vret signals (red) in control and Siwi‐depleted BmN4 cells (Siwi KD). DAPI shows the nuclei (blue). Scale bar: 10 μm. The three panels on the right‐hand side show high magnification images of the part indicated by the white dashed line box in the left‐hand side images.
- Silver staining showing unphosphorylated and phosphorylated Ago3 immunoisolated from Siwi‐depleted BmN4 cells.
- Representative ETD tandem mass spectra for Ago3 peptides, which include serine modifications. Mascot scores are shown on the top right of each spectrum.
- 32P‐labeling shows the levels of piRNAs loaded onto Flag‐Ago3 WT and 8SA.
- 32P‐labeling shows the level of piRNAs loaded onto Flag‐Ago3 is not affected by Vret depletion. Vret and β‐Tub as a loading control were also detected by Western blotting.
- Bar chart indicates dot sizes of > 100 Flag‐Ago3 WT particles under control and Siwi‐depleted conditions (Large: IntDen value > 105; Small: IntDen value < 105).
- Bar chart indicates dot sizes of > 100 Flag‐Ago3 8SA/8SE particles under control and Siwi‐depleted conditions (Large: IntDen value > 105; Small: IntDen value < 105).
- Immunofluorescence showing the Flag‐Ago3 8SA and 8SE mutant signals (red) in normal (Control) and Siwi‐depleted BmN4 cells (Siwi KD). DAPI shows the nuclei (blue). Scale bar: 10 μm.
Source data are available online for this figure.
In the absence of Siwi, Ago3 was detected as a doublet on Western blots (Fig 3C). After subfractionation of total lysates by centrifugation, the lower band in both Siwi KD and control samples was detected in the supernatant fraction, whereas the upper band in the Siwi KD sample appeared mostly in the pellet fraction (Fig 3C). Siwi depletion changed the molecular mass and solubility of Ago3 and the morphology of Ago3 bodies.
Upon phosphatase treatment, Ago3 was detected as a single band on Western blots (Fig 3D), suggesting that the difference in the molecular masses was caused by phosphorylation. We then determined the phosphorylation sites in Ago3 immunopurified from Siwi‐lacking lysates (Fig EV3C). Solubilization of Ago3 required the use of a buffer containing the zwitterionic detergent Empigen. Mass spectrometric analysis suggested that eight residues, Ser112, Ser116, Ser117, Ser140, Ser143, Ser145, Ser149, and Ser150, were the phosphorylation sites in Ago3 (Fig EV3D). The 8SA mutant, where all serines were mutated to alanine, was detected essentially as a single band on Western blots in Siwi‐lacking cells (Fig 3E). We consider the 8SA mutant as a phosphorylation‐defective Ago3 mutant.
Both the 8SA mutant and WT were loaded with piRNAs (Fig EV3E), suggesting that phosphorylation is not required for Ago3‐piRISC formation. However, piRNA loading was mandatory for Ago3 phosphorylation because the piRNA loading‐defective KA mutant was detected as a single band in the absence of Siwi (Fig 3F). This also indicates that Ago3 phosphorylation occurs after Ago3‐piRISC formation. Ago3 phosphorylation was barely detected when Vret was depleted along with Siwi (Fig 3G). Loss of Vret did not affect formation of the Ago3‐piRISC (Fig EV3F) but impaired Ago3‐body formation (Fig 2C). Thus, Ago3 phosphorylation in the absence of Siwi takes place exclusively on Ago3‐piRISC within Ago3 bodies.
We have so far obtained no data showing endogenous Ago3 being phosphorylated in naïve BmN4 cells. In contrast, Flag‐Ago3 WT was barely phosphorylated in control BmN4 cells (Fig 3F). Theoretically, Ago3 overexpression reduces the relative amount of Siwi to that of Ago3 albeit to a lesser extent when compared with cells lacking Siwi. Indeed, Flag‐Ago3 was more phosphorylated in the absence of Siwi (Fig 3F). We then examined the size of Ago3 bodies upon Ago3 overexpression in normal cells (Figs 3H and EV3G). The Ago3‐body morphology change was not obvious. However, depletion of Siwi caused the bodies to become clearly larger (Figs 3H and EV3G). These observations indicate that the Siwi vs. Ago3 ratio is crucial. We postulate that once the Siwi level relative to the Ago3 level becomes lower by any means, the germ cells sense the situation and induce Ago3 phosphorylation and insolubilization, resulting in Ago3‐body enlargement. Similar outcomes were obtained when the 8SA mutant and another mutant 8SE, which most likely serves as the phosphorylation‐mimicking mutant based on widely accepted knowledge in the post‐translational protein modification field (Pagès et al, 1994; Pearlman et al, 2011), were expressed (Figs 3I, and EV3H and I). This indicates that Ago3 phosphorylation and Ago3‐body enlargement are separable from each other.
Lack of Siwi rapidly stalls Ago3‐piRISC in a form bound with cleaved RNAs in the ping‐pong cycle
We then set out to examine RNAs co‐purifying with Ago3 from Siwi‐depleted BmN4 cells (Fig EV3C). 32P‐labeling confirmed the presence of piRNAs loaded onto Ago3 (Fig 4A). Additionally, a discrete 16‐nt band was observed (Fig 4A). We isolated 15–20‐nt RNAs, termed Target‐S, from the gel and sequenced them. Sequence reads matching to transposons were isolated from the primary dataset and forwarded for further analysis. Strand bias analysis showed that 80.4% of the sequences were opposite in direction to transposon mRNAs (Fig 4B). The most abundant size was 16 nt (Fig EV4A). We then measured the distance between the 3′‐end of the Target‐S and the 5′‐end of Ago3‐bound piRNAs and found that 48.67% were 10 nt (Fig 4C and Table EV2). These characteristics are reminiscent of the 16‐nt footprint RNAs found with Vasa in BmN4 cells. The footprint RNAs appeared during geldanamycin treatment to attenuate HSP90 function in Ago3‐piRNA loading, reducing the piRISC level (Xiol et al, 2014). However, neither Ago3‐body enlargement nor Ago3 phosphorylation was observed in that previous study. Thus, the effects of geldanamycin treatment (Xiol et al, 2014) and loss of Siwi (this study) are not identical.
Figure 4. The absence of Siwi rapidly stalls Ago3‐piRISC in a form bound with cleaved RNA fragments.
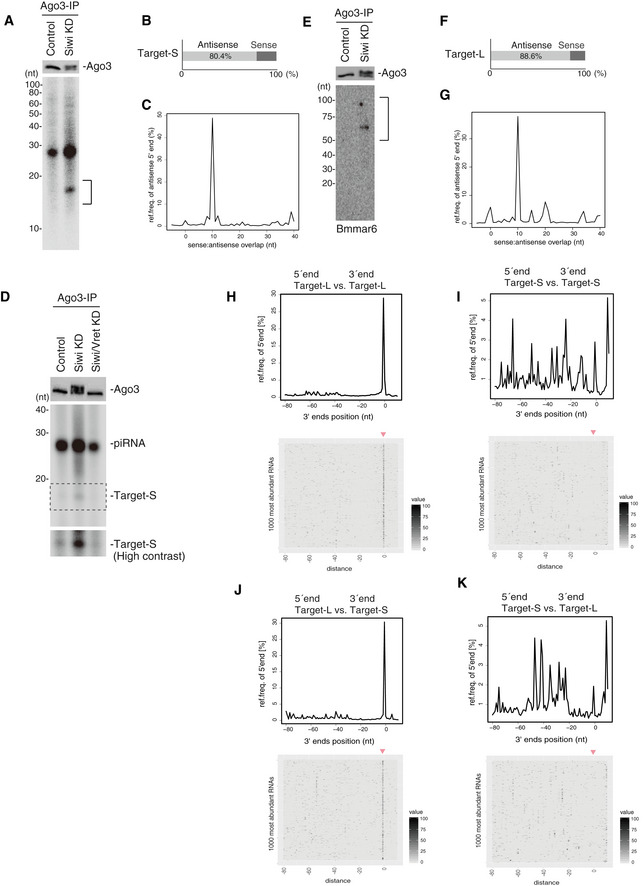
- Western blot showing Ago3 immunopurified from BmN4 cells before (Control) and after Siwi knockdown (Siwi KD). 32P‐labeling of RNAs isolated from Ago3 (a mixture of unphosphorylated and phosphorylated) immunopurified from Siwi‐depleted cells. 15–20‐nt RNAs (indicated by a black line) were isolated and sequenced.
- 80.4% of the 15–20‐nt RNAs corresponding to transposon sequences were antisense to transposon mRNAs.
- The measurement of distances between the 3′‐ends of Target‐S (15–20‐nt RNAs in (A)) and the 5′‐ends of Ago3‐bound piRNAs.
- 32P‐labeling of RNAs isolated from Ago3. Target‐Ss are isolated from Siwi‐depleted BmN4 cells but are not isolated from Siwi/Vret double knockdown cells.
- Western blot showing Ago3 immunopurified from BmN4 cells before (Control) and after Siwi knockdown (Siwi KD). Northern blot showing Bmmar6‐piRNA precursors in the Ago3 complex. 50–100‐nt RNAs (indicated by a black line) were isolated and sequenced.
- 88.6% of the transposon‐matching piRNA precursors were antisense to transposon mRNAs.
- The measurement of distances between the 5′‐ends of Target‐L and the 5′‐ends of Ago3‐bound piRNAs.
- The measurement of distances between the 5′‐ and 3′‐ends of Target‐L fragments on the same genomic strand. Heat map displays 5′‐ends of Target‐L in a window around the most abundant 1,000 3′‐ends of Target‐L.
- The measurement of distances between the 5′‐ends and 3′‐ends of Target‐S fragments on the same genomic strand. The heat map displays 5′‐ends of Target‐S in a window around the most abundant 1,000 3′‐ends of Target‐S.
- The measurement of distances between the 5′‐ends of Target‐L and the 3′‐ends of Target‐S on the same genomic strand. The heat map displays 5′‐ends of Target‐L in a window around the most abundant 1,000 3′‐ends of Target‐S.
- The measurement of distances between the 5′‐ends of Target‐S and the 3′‐ends of Target‐L on the same genomic strand. The heat map displays 5′‐ends of Target‐S in a window around the most abundant 1,000 3′‐ends of Target‐L.
Source data are available online for this figure.
Figure EV4. Bioinformatic analysis of RNAs bound with Ago3‐piRISCs in Siwi‐depleted cells.
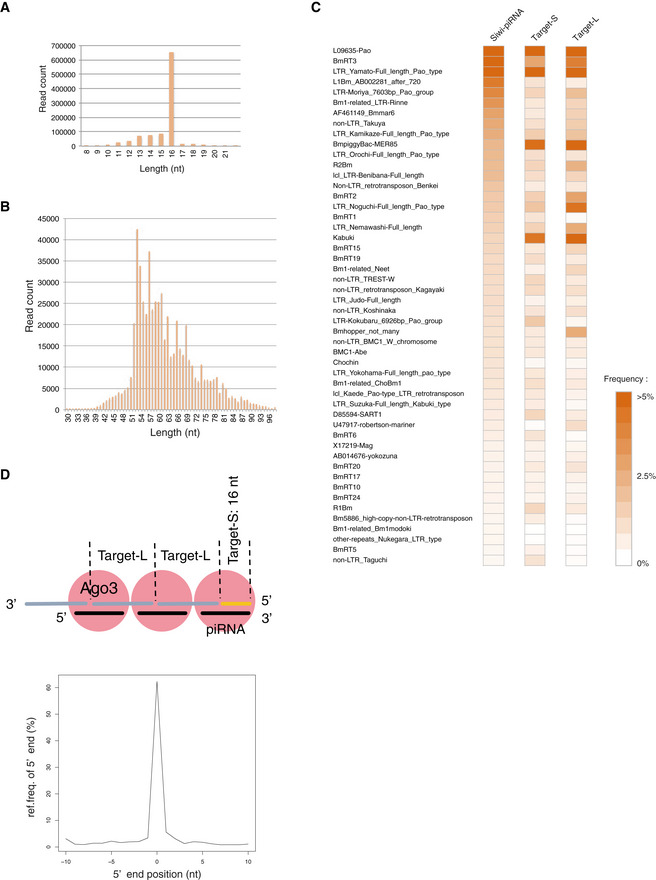
- The size distribution of Target‐S fragments.
- The size distribution of Target‐L fragments.
- Heat maps show the read frequency of transposon‐derived Ago3‐piRNA, Target‐S, and Target‐L. Color intensities indicate the degree of read frequency: (orange) higher and (white) lower.
- Characteristics of Target‐L and Target‐S. The 5′‐ends of Siwi‐associated piRNAs displayed a strong overlap with the 5′‐ends of Target‐L fragments.
We consider the presence of Target‐S as an index of RNA targeting of Ago3 and Ago3‐dependent RNA cleavage. The level of Target‐S was negligible upon Vret depletion in Siwi‐lacking cells (Fig 4D), suggesting that Ago3‐piRISC in the cytoplasm, without Vret, may target RNAs very inefficiently.
Northern blotting for detecting Bmmar6‐piRNA, one of the most abundant Siwi‐bound antisense piRNAs (Nishida et al, 2015), revealed the presence of piRNA precursors with Ago3‐piRISC (Fig 4E). We sequenced and analyzed 50–100‐nt RNAs termed Target‐L, and RNAs of 52 nt in length were the most abundant (Fig EV4B). The sequence reads matching transposons were biased to be antisense to the mRNAs (88.6%) (Fig 4F). The distances between the 5′‐end of Ago3‐bound piRNAs and the 5′‐end of antisense Target‐L were mostly 10 nt (37.86% in total) (Fig 4G and Table EV2). Heat mapping showed that Target‐L and Target‐S correspond to a wide range of transposons (Fig EV4C), indicating that the characteristics depicted here are not biased to particular types of transposons.
We also measured the distances between the 5′‐ends and the 3′‐ends of Target‐L as we did for Papi‐bound piRNA precursors (Nishida et al, 2018). This revealed that a large number of Target‐L, but not Target‐S, could be mapped tandemly to the Bombyx genome (downloaded from Silkbase; silkbase.ab.a.u‐tokyo.ac.jp) without any gaps between them, similarly to Papi‐bound piRNA precursors (Figs 4H and I, and Table EV3). The 3′‐ends of Target‐S matched frequently with 1‐nt upstream of the 5′‐ends of Target‐L (Fig 4J and Table EV3) but not vice versa (Fig 4K and Table EV3). These results support the notion that Ago3‐piRISC‐bound Target‐L, but not Target‐S, fragments are phasing (Fig EV4D). Indeed, 5′‐ends of Siwi‐associated piRNAs displayed a strong overlap with the 5′‐ends of Target‐L fragments (Fig EV4D). Target‐S may correspond to the very 5′‐ends of primary piRNA precursors targeted by Ago3‐piRISC, but the original 5′‐ends were likely cleaved by unknown enzymes during isolation, and so Target‐S becomes mostly 16‐nt artificially (Fig EV4D).
Siwi restoration reverses the aberrancy caused by Siwi loss to normal
The findings so far indicate that Ago3‐piRISC in the enlarged Ago3 bodies remains bound rigidly with target RNA fragments cleaved by Ago3. This suggests that Ago3‐piRISC was immediately stalled at the intermediate state in the ping‐pong cycle when unloaded Siwi, the partner of Ago3‐piRISC in the pathway, was not supplied. We next asked if this stalled state is reactivated by Siwi ectopic expression. Interestingly, the size of Ago3 bodies returned to a normal size upon supplying Siwi (Figs 5A and EV5A). We also observed in parallel that overexpression of the Siwi KA mutant attenuated Ago3 phosphorylation (Fig 5B) and normalized the body size (Fig 5C). The Siwi mutant was unloaded with piRNAs (Fig EV5B) as was the Ago3 KA mutant (Fig EV2F) and had presumably no activity to restart Ago3‐piRISC production (Fig EV5C), excluding the possibility that enlarged nuage decayed while new Ago3 bodies were supplied. These findings suggest that cells lacking Siwi cause Ago3 bodies to granulize to store and protect the piRNA intermediates from RNA degradation until cellular circumstances return to normal to re‐initiate the ping‐pong cycle.
Figure 5. The aberrant Ago3‐body morphology is rescued by Siwi restoration.
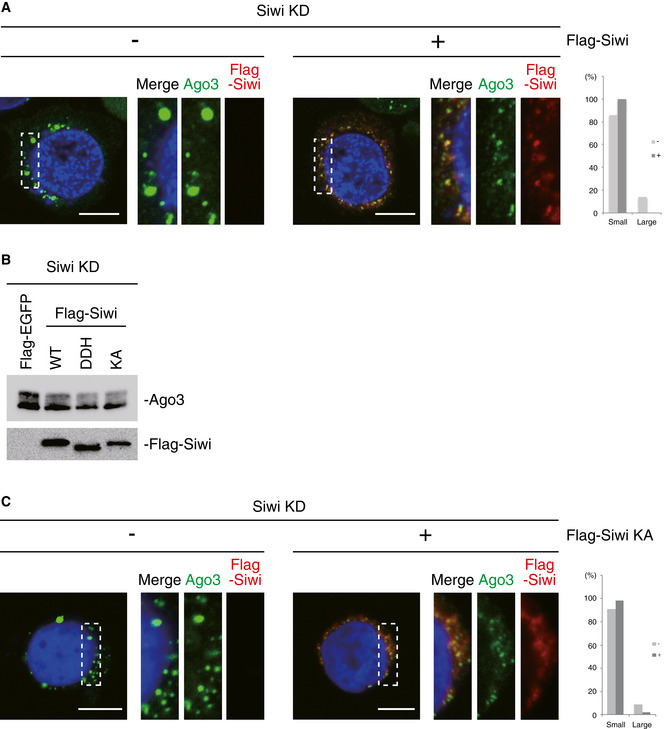
- Immunofluorescence showing the Ago3 signals (green) in Siwi‐depleted BmN4 cells (Siwi KD) and Siwi KD cells where Flag‐Siwi (red) was expressed. DAPI shows the nuclei (blue). Scale bar: 10 μm. The panels on the right‐hand side show high magnification images of the part indicated by the white dashed line box in the images on the left‐hand side. This arrangement applies also for panel C. Bar chart indicates dot sizes of > 100 Ago3 particles in each case (Large: IntDen value > 105; Small: IntDen value < 105). A significant difference was found plus/minus Flag‐Siwi expression by the Fisher's exact test (P < 0.05).
- Western blots show that the degree of Ago3 phosphorylation was reduced upon Siwi KA mutant as well as Siwi WT and DDH mutant expression.
- Immunofluorescence showing the Ago3 signals (green) in Siwi‐depleted BmN4 cells (Siwi KD) and Siwi KD cells where Flag‐Siwi KA mutant (red) was expressed. DAPI shows the nuclei (blue). Scale bar: 10 μm. Bar chart indicates dot sizes of > 100 Ago3 particles in each case (Large: IntDen value > 105; Small: IntDen value < 105). A significant difference was found plus/minus Flag‐Siwi expression by the Fisher's exact test (P < 0.05).
Source data are available online for this figure.
Figure EV5. A new model for the ping‐pong cycle in silkworm germ cells.
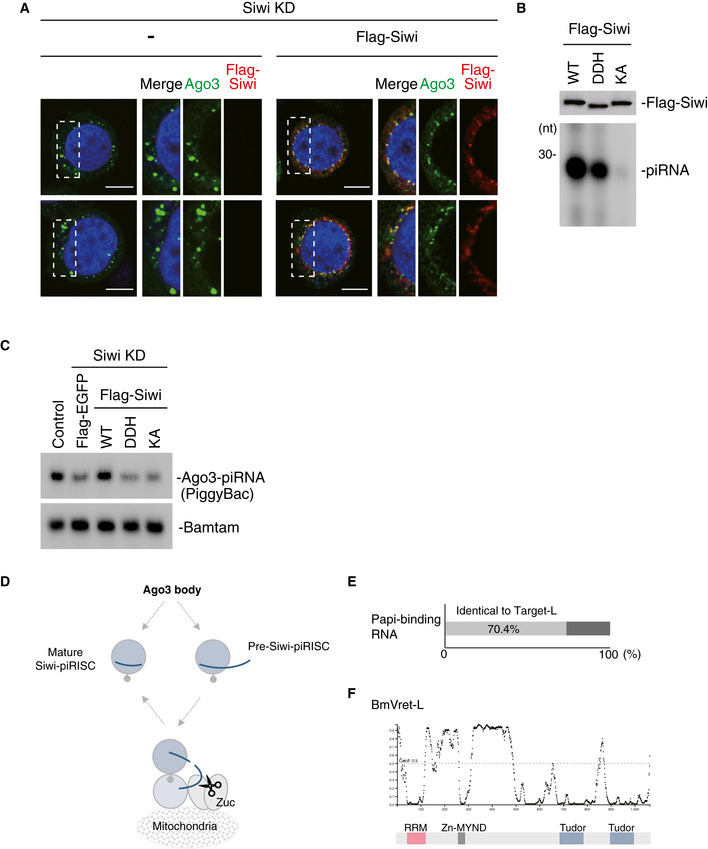
- Immunofluorescence showing the Ago3 signals (green) in Siwi‐depleted BmN4 cells (Siwi KD) and Siwi KD cells where Flag‐Siwi (red) was expressed. DAPI shows the nuclei (blue). Scale bar: 10 μm. The panels on the right‐hand side show high magnification images of the part indicated by the white dashed line box in the images shown on the left‐hand side.
- 32P‐labeling showing the levels of piRNAs loaded onto the Flag‐Siwi WT, DDH mutant, and KA mutant. Lys611 is mutated to alanine in the KA mutant, and Asp670, Asp740, and His874 are mutated to alanine in the DDH mutant.
- Northern blotting showing the levels of PiggyBac‐piRNA and Bantam miRNA in BmN4 cells. Flag‐Siwi WT and the DDH and KA mutants were individually expressed in Siwi‐depleted cells. Flag‐EGFP was expressed as a control.
- When RNA within secondary Siwi‐piRISC has extra bases at the 3′‐end, the complex relocates to mitochondria, where the piRISC precursor is docked onto Papi. Zuc processes the RNA to produce more secondary Siwi‐piRISC (see * in Fig 6).
- Bar chart indicates that 70.4% of Papi‐binding RNAs were identical to Target‐L.
- The DISOPRED plot analyzed by PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) predicted IDR regions in BmVret‐L.
Source data are available online for this figure.
Discussion
In this study, we revealed the regulatory mechanisms underlying secondary Siwi‐piRISC production in Bombyx ovarian germ cells and, in accordance with these results, updated the model of the piRNA biogenesis pathway that we proposed previously (Nishida et al, 2018; Fig 6).
Figure 6. Model of Siwi‐piRISC biogenesis in Bombyx germ cells.
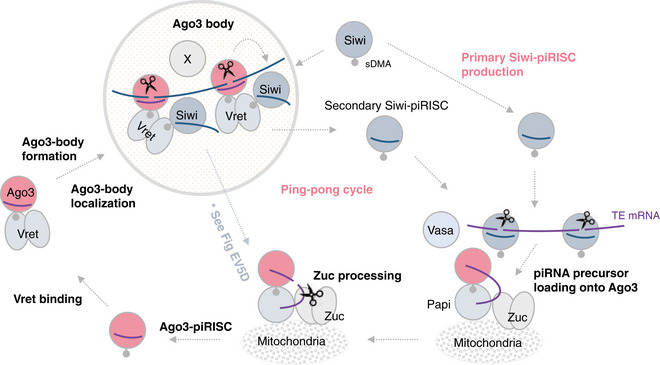
Primary Siwi‐piRISC is produced through the primary pathway (Nishida et al, 2018). The piRISC binds transposon mRNAs and cleaves them into pieces. Of those, one fragment is loaded onto unloaded Ago3, which is anchored to mitochondrial Papi with help from Vasa (Nishida et al, 2015). Zuc processes the RNA to give rise to mature Ago3‐piRISC. Vret associates with Ago3‐piRISC and assembles Ago3 bodies. During this process, the Vret–Ago3‐piRISC complex binds transposon transcripts in the antisense orientation. Unloaded Siwi enters into the bodies and joins the Vret–Ago3 complex. In this context, Ago3‐piRISC and unloaded Siwi are held in close proximity with assistance from two (or more) Vret molecules, facilitating secondary Siwi‐piRISC production in the ping‐pong cycle.
The new model shows that the Tudor protein Vret associates with Ago3‐piRISC in the cytoplasm and assembles a subset of nuage, termed Ago3 bodies, as the center of secondary Siwi‐piRISC generation. During the process, the Vret–Ago3‐piRISC complex binds target RNAs, i.e., transposon transcripts in antisense orientation that are complementary to Ago3‐bound piRNAs. In turn, unloaded Siwi is localized to the bodies to join the Vret–Ago3 complex. In this context, Ago3‐piRISC and unloaded Siwi are held in close proximity by the assistance from two (or more) Vret molecules to facilitate secondary Siwi‐piRISC production in the ping‐pong cycle.
The original model suggested that both primary and secondary Siwi‐piRISC production occurs on Papi located on mitochondria. However, we realized in this study that different organelles serve as the place for the reactions: primary Siwi‐piRISC production occurs on the outer mitochondrial membrane, whereas secondary Siwi‐piRISC production occurs within Ago3 bodies. The Siwi‐piRISC precursor might relocate to mitochondria for maturation when piRNA within the piRISC is still in its precursor form with extra bases at the 3′‐end (Fig EV5D). In fact, ~ 70% of Target‐L was found in the pool of RNAs bound with Siwi anchoring onto Papi (Nishida et al, 2018; Fig EV5E). The unique organization that employs two different organelles, mitochondria and Ago3 bodies, as primary and secondary Siwi‐piRISC production centers help to avoid collisions or interference between the two machineries. Currently, the control process that funnels unloaded Siwi to different organelle remains unknown. The expression levels of Papi and Vret likely affect the balance between primary and secondary (ping‐pong) reactions. Ago3‐piRISC biogenesis also takes place on Papi (Nishida et al, 2018), and thus, the relative amounts of Siwi and Ago3 that occupy Papi should also affect the outcome.
Vret/Tdrd1 plays an important role in piRNA biogenesis among species, although their domain arrangement varies (Fig EV1C). The steps where Vret/Tdrd1 functions also differ among species. For example, mTdrd1 is required for generating Miwi2‐piRISC, which corresponds to Ago3‐piRISC in flies and silkworm (Reuter et al, 2009; Vagin et al, 2009). DmVret was shown to play an important role in primary piRISC biogenesis in both germ and somatic cells (Handler et al, 2011; Zamparini et al, 2011). Furthermore, DmVret interacts with PIWI proteins through C‐terminal Tudor domains (Handler et al, 2011), whereas BmVret interacts through the N‐terminal Tudor domain. The function of zTdrd1 may be equivalent to that of BmVret because zTdrd1 is required for producing Ziwi‐piRISC, which corresponds to Siwi‐piRISC (Huang et al, 2011). Noteworthy, all Vret/Tdrd1 complexes act in generating piRISC possessing antisense piRNAs, which are capable of targeting transposon mRNAs directly. The commonality of Vret/Tdrd1 function among species is clear.
Earlier studies provided evidence that piRNA biogenesis takes place mostly at non‐membranous organelles formed through liquid–liquid phase separation (LLPS) (Hirakata & Siomi, 2019). The representatives are DDX4/MVH (MVH)‐positive nuage (Nott et al, 2015), Yb bodies (Hirakata et al, 2019), and Ago3 bodies (this study). The core components in MVH‐positive nuage and Yb‐body formation are MVH (a.k.a. Vasa in flies and silkworm) and Yb, respectively, and both proteins contain intrinsically disordered regions (IDRs) that trigger LLPS in vivo and in vitro (Banani et al, 2017). Ago3‐body assembly is dependent on Vret and Ago3‐piRISC. We examined both proteins for IDRs, revealing that Ago3 has an N‐terminal IDR that is also found in other PIWI proteins (Matsumoto et al, 2016; Yamaguchi et al, 2020) and Vret has two IDRs. One IDR in Vret is located between the RRM and ZnF‐MYND and the other between the ZnF‐MYND and Tudor domains (Fig EV5F). We are currently analyzing the dependency of Ago3‐body formation on the IDRs. Interestingly, Yb‐body formation is independent of the Yb IDR, whereas MVH‐body formation requires the MVH IDR (Nott et al, 2015; Hirakata et al, 2019).
Yb bodies are at the center of Piwi‐piRISC/pre‐Piwi‐piRISC formation in Drosophila ovarian somatic cells (Qi et al, 2011; Murota et al, 2014; Yamashiro et al, 2020). Our previous study showed that Yb‐body assembly requires not only the self‐association activity of Yb through the N‐terminal helicase‐C domain but also the RNA‐binding activity exhibited collaboratively by the central RNA helicase and C‐terminal extended Tudor (eTud) domains (Hirakata et al, 2019). Yb sequesters piRNA precursors to Yb bodies by binding to these precursors specifically through cis‐elements residing in RNA (Ishizu et al, 2015). In contrast, the RNA‐binding activity of Vret was dispensable for Ago3‐body formation but piRNA loading, i.e., RNA‐targeting of Ago3, was required. Thus, it appears that Ago3‐piRISC is the equivalent to the RNA helicase and eTud domains of Yb in Yb‐body assembly. As noted above, Yb selectively binds piRNA precursors through the cis‐trans action (Ishizu et al, 2015). By this way, only the piRNA precursors are consumed to produce piRNAs and the quality of piRNAs is maintained. Likewise, Ago3‐piRISC selectively binds Siwi‐bound piRNA precursors through RNA–RNA base‐pairings, thereby avoiding the situation where other RNAs are accidentally consumed in the piRNA processing event.
Upon Siwi depletion, the Ago3‐based machinery to generate secondary Siwi‐piRISC was found to stall in the intermediate state while Ago3 remains bound with RNA fragments. This stalled state was, however, reversed by Siwi restoration without new Ago3‐piRISC formation, revealing the plasticity of Ago3 bodies in response to the Siwi level. The germ cells could have alternatively destroyed all components of Ago3 bodies under Siwi‐depleted conditions and re‐initiated production of Ago3 bodies upon Siwi recovery. However, apparently, the germ cells prefer to store piRNA precursors in Ago3 bodies as stress granules store mRNAs till the cellular environment returns to normal conditions. Such a mechanism may save energy or facilitate a rapid response; however, providing supporting evidence to decipher this mechanistic feature is difficult experimentally.
Both Aub and Ago3 in Drosophila can perform the homotypic ping‐pong cycle; however, the efficiency is low and compromises the machinery (Li et al, 2009). To avoid such a deleterious situation, Drosophila employs two Tudor proteins Krimper (Krimp) and Qin/Kumo, which both play roles to ensure the heterotypic ping‐pong cycle by Aub and Ago3 (Zhang et al, 2011; Sato et al, 2015; Webster et al, 2015). Vret in BmN4 cells bound to unloaded Ago3 particularly when Ago3 was overexpressed (Fig EV2I), indicating that Ago3 may play a homotypic ping‐pong role in the absence of Siwi, as observed in Drosophila ovaries. The Krimp and Qin genes are found in the silkworm genome, but our preliminary data suggested that both genes barely contribute to piRNA biogenesis in BmN4 cells (Sato, Nishida and Siomi, unpublished). The silkworm germ cells may have acquired an alternative system to stall and insolubilize Ago3‐piRISC when Siwi levels are low to circumvent loss of functions to Krimp and Qin in the piRNA pathway.
Currently, the potential effect of Ago3 phosphorylation on piRNA biogenesis in BmN4 cells remains unresolved. We also have no understanding if at some point through gonadal development Siwi is naturally depleted. Future analysis is required to address these questions.
Materials and Methods
Cell culture
BmN4 cells were cultured in EX‐CELL(TM) 420 Serum‐Free Medium for Insect cells (Sigma) containing 10% FBS (Equitech‐Bio) and penicillin–streptomycin‐glutamine (Gibco).
RNAi and transgene expression
BmN4 cells (1 × 106 cells) were transfected with 500 pmol of the siRNA duplex in 100 μl of EP buffer [137 mM NaCl, 5 mM KCl, 0.5 mM Na2HPO4, and 2.1 mM HEPES‐KOH (pH 7.1)]. Transfection was conducted in electroporation cuvettes using a Nucleofector 2b device (Lonza). Upon transfection, cells were incubated at 26°C for 72 h. siRNA transfection was performed again to raise the RNAi effect. The sequences of siRNAs are summarized in Table EV4. Luc siRNA was used as a control. To exogenously express proteins in BmN4 cells, the cells (6 × 105 cells) were transfected with 2 μg of plasmid in 5 μl of FuGENE HD transfection reagent. After transfection, cells were incubated at 26°C for 48 h.
Rescue assay
BmN4 cells (5 × 105 cells) were transfected with 250 pmol of the siRNA duplex in 100 μl of EP buffer using a Nucleofector 2b device. Upon transfection, cells were incubated at 26°C for 48 h. To exogenously express proteins in cells, the cells (6 × 105 cells) were transfected with 2 μg of plasmid in 5 μl of FuGENE HD transfection reagent and then incubated at 26°C for 48 h. siRNA transfection was performed again to ensure the RNAi effect. Upon transfection, the cells were incubated at 26°C for 72 h. After 72 h, the cell medium was changed. The cells were further incubated at 26°C for 48 h.
Plasmid construction
Vectors for expressing Flag‐Vret‐L and ‐S were generated by Gateway Technology using the pIB‐3xFLAG vector (Thermo Fisher Scientific). A vector for expressing His‐Vret was generated by inserting Vret cDNA into the pET‐28a vector. Vectors for expressing Flag‐Ago3, Flag‐Siwi, Flag‐Vret mutants, and Myc‐Vret‐S were generated by inverse PCR using vectors for expressing Flag‐Ago3 WT, Flag‐Siwi WT, and Flag‐Vret WT, respectively [A vector for expressing the Flag‐Ago3 8SE mutant was generated by ligation with the NEBuilder HiFi DNA Assembly Master Mix (NEB)]. DNA fragments and PCR primers used are summarized in Table EV4. Exogenous Ago3 proteins used in this study were all RNAi‐resistant. For this, the cDNAs were mutated to be insensitive to Ago3 siRNA. The sequences of the oligos used for the mutagenesis are presented in Table EV4.
Cloning of Vret cDNA
Vret cDNAs were obtained by RT–PCR using total RNAs from BmN4 cells. The 5′‐ends of Vret‐L and Vret‐S mRNAs were determined by 5′ RACE with the SMARTer RACE 5′/3′ Kit (Clontech). PCR primers used are summarized in the Table EV4.
Production of monoclonal antibody
A mouse was immunized with bacterially produced glutathione S‐transferase (GST)‐tagged Vret‐L (amino acids 466–669). To produce GST‐Vret 466–669, Vret cDNA corresponding to the region was cloned into the pGEX5X vector. GST‐Vret 466–669 was expressed in Escherichia coli BL21 (DE3) cells and purified using glutathione sepharose (GE Healthcare) and the purification buffer [20 mM Tris–HCl (pH 8.0), 150 mM NaCl, and 1 mM DTT]. Fusing myeloma generated hybridomas as described previously (Nishida et al, 2015).
Northern blotting
Total RNAs were isolated from BmN4 cells using ISOGEN II (NIPPON GENE). 5 μg of total RNAs was resolved by electrophoresis. RNAs were transferred to Hybond‐N (GE Healthcare) and fixed with an EDC cross‐linking solution, as described previously (Pall & Hamilton, 2008). After pre‐hybridization with hybridization buffer [200 mM sodium phosphate (pH 7.2), 7% SDS, and 1 mM EDTA], hybridization was performed with 100 pM DNA probe in hybridization buffer at 42°C for 15 h. Membranes were washed three times with 2 × SSC buffer (30 mM sodium citrate, 300 mM NaCl, and 0.1% SDS). Upon exposure to an imaging plate, images were detected with Typhoon FLA 9500 (GE Healthcare). Sequences of DNA probes are shown in Table EV4.
Western blotting
Upon electrophoresis, proteins were transferred to a nitrocellulose or PVDF membrane (Wako). After blocking with 5% skim milk for 30 min, 1 ng/μl primary antibodies [anti‐Ago3 (Nishida et al, 2015), anti‐Siwi (Nishida et al, 2015), anti‐Vret (this study), anti‐DDDDK‐tag (Flag) (MBL), anti‐Myc (DSHB), and Y12 (Nishida et al, 2018) antibodies] in 0.1% Tween 20‐containing PBS (T‐PBS) were mounted on membranes for 1 h at room temperature. Membranes were washed three times with T‐PBS and 1 ng/μl anti‐mouse IgG (H + L) HRP (MP Biomedicals) in T‐PBS was mounted on membranes for 1 h at room temperature. Membranes were washed five times with T‐PBS, and images were obtained with ChemiDoc (Bio‐Rad) with the Clarity ECL substrate (Bio‐Rad).
Cell fractionation
6 × 105 BmN4 cells were lysed in immunoprecipitation (IP) buffer [30 mM HEPES‐KOH (pH 7.3), 150 mM KOAc, 2 mM Mg(OAc)2, 5 mM DTT, 0.1% NP‐40, 2 μg/ml pepstatin, 2 μg/ml leupeptin, and 0.5% aprotinin]. The total cell lysate was centrifuged at 600 g for 10 min at 4°C to obtain the supernatant and pellet fractions.
Immunoprecipitation
15 μl of dynabeads (Thermo Fisher Scientific) and 1.5 μg of antibodies were incubated in 500 μl PBS containing 0.1% Tween 20 for one reaction. 6 × 105 cells were lysed in IP buffer. The lysate was mixed with antibodies‐conjugated dynabeads at 4°C for 1 h. Phosphorylated Ago3 was immunoprecipitated from the pellet fraction dissolved in IP buffer containing 1% Empigen instead of 0.1% NP‐40. The beads were washed extensively with IP buffer. In Fig EV1G, Vret‐interacting proteins were eluted from the beads, from which Siwi was further immunopurified using the anti‐Siwi antibody in IP buffer containing 500 mM NaCl. The beads were washed extensively with IP buffer containing 500 mM NaCl.
32P‐labeling of RNAs
RNAs were purified from the beads using phenol/CHCl3. RNAs were then treated with Antarctic phosphatese (NEB) for dephosphorylation and radiolabeled using 32P‐gamma‐ATP by T4 polynucleotide kinase (NEB).
Immunofluorescence
BmN4 cells were placed on 0.075% poly‐l‐lysine‐coated cover glasses. Cells were fixed with 2% formaldehyde in PBS for 15 min and permeabilized with 0.1% Triton X‐100 in PBS for 15 min. After blocking with 3% BSA in PBS for 15 min, cells were incubated with 1 ng/μl primary antibodies [anti‐Ago3 (Nishida et al, 2015), anti‐Siwi (Nishida et al, 2015), anti‐Vret (this study), and anti‐Flag (Sigma) antibodies] for 1 h. The antibodies were diluted with 3% BSA in PBS. After washing with PBS, cells were incubated with secondary antibodies [Alexa 488/Alexa Fluor 555 goat anti‐mouse immunoglobulin G1 (IgG1)/IgG2a antibodies (Thermo Fisher Scientific)] in 3% BSA containing PBS for 1 h in a dark room. Cover glasses were mounted with VECTASHIELD Antifade Mounting Medium with DAPI (VECTOR LABORATORIES). For Flag‐tagged proteins and Siwi immunostaining, cells were treated with PBS containing 35 μg/ml digitonin for 5 min before fixation. Images were collected using a Zeiss LSM710 laser‐scanning microscope and an LSM980 laser‐scanning microscope with C‐Apochromat 40×/1.20 W Korr objective lens. The pixel dwell time was set to be 6.3 μs and eight sequentially scanned images were averaged.
CLIP
CLIP was performed essentially as described previously (Jaskiewicz et al, 2012). Dephosphorylation and 32P‐labeling of RNAs were performed using T4 polynucleotide kinase.
Dephosphorylation treatment of Ago3
Immunopurified Ago3 was incubated with Lambda Protein Phosphatase (NEB) at 30°C for 30 min.
Analysis of RNA sequences
Libraries were sequenced using Illumina MiSeq (single‐end, 51 cycles). After adaptor sequences were removed from obtained reads, for Ago3‐piRNAs, a total of 1,331,305 reads were obtained from the control sample, and 1,613,608 reads were obtained from the Siwi‐knockdown sample. For Siwi‐piRNAs, a total of 5,307,490 and 3,757,164 reads were obtained from the control sample (library‐1 and library‐2, respectively), 4,563,199 and 1,356,087 reads were obtained from the Ago3‐knockdown sample (library‐1 and library‐2, respectively) and 2,503,458 and 3,412,329 reads were obtained from the Vret‐knockdown sample (library‐1 and library‐2, respectively). For Ago3‐associated target RNAs, a total of 2,155,565 reads were obtained from the Target‐S sample, and 8,267,028 reads were obtained from the Target‐L sample. The reads were mapped to 121 Bombyx mori (B. mori) transposon consensus sequences (a gift from S. Kawaoka) using Bowtie2, allowing up to one mismatch for Target‐L. Target‐S, and Target‐L fragments were mapped 1,020,879 (47.36%) and 599,968 (7.26%) times to B. mori transposons, respectively. For others, reads were mapped to the transposons using bowtie and no mismatch was allowed. Using transposon‐mapped reads, length distribution, strand bias, and ping‐pong signatures were calculated. Calculations of the ping‐pong signature were performed as described previously (Brennecke et al, 2007). The reads were also mapped to the B. mori reference genome (downloaded from Silkbase; silkbase.ab.a.u‐tokyo.ac.jp) using Bowtie2 and bowtie as above. Using sequences mapped to the genome, phasing analyses were conducted. Calculations of the distance between intermediates were performed as described previously (Han et al, 2015). The statistical analyses were performed in the R software environment (version 3.1.3).
For Fig EV1H, the reads of library‐1 and library‐2 of Siwi‐piRNA from the Control, Ago3‐knockdown and Vret‐knockdown samples, as written above, were normalized to RPM against the total number of reads in that dataset. Extracting the reads more than 10 RPM, Spearman's correlation was calculated and a scatter plot was prepared for each sample. For Fig 1G, RPM scores of library‐1 and library‐2 were initially combined for each sample. Comparing Control and Ago3‐knockdown samples, the reads were classified into three groups based on the Ago3‐KD (RPM)/Control (RPM) value; namely, piRNAs whose Ago3‐KD (RPM)/Control (RPM) value was ≥ 2, 0.5 < ratio < 2 or ≤ 0.5 were categorized as increased, unchanged, and decreased, respectively. The statistical analyses were performed in the R software environment (version 3.1.3).
Image analysis of nuage
Images of immunostaining were processed with ImageJ. Images were split into each channel. Nuage was defined using threshold. Using Analyze Particles, particles of nuage were counted as masks, and the centroid of each particle was calculated. After particle definition and centroid calculation against one cell of channel A and B, for example, Ago3 and Vret, the squares of the distance between centroids in channel A and B were calculated comprehensively. When the squares of the distance were less than 0.01, we defined that two particles were co‐localized. The numbers of Ago3/Vret‐positive and Siwi/Vret‐positive nuage were counted and the ratios were calculated. To calculate the size of nuage, the scores of integrated density (IntDen), which is the results of Analyze Particles, were used. For Fig 3A, particles whose IntDen scores were more than 106 were defined as Large and less than 106 were defined as Small. For others (Figs 3B and 5A, C, and EV3G and H), particles whose IntDen scores were more than 105 were defined as “Large” and less than 105 were defined as “Small”. One hundred particles were gathered per one sample and the small‐to‐large ratios were calculated.
Sample preparation for mass spectrometry
P130 and P150 were excised from the silver‐stained gel in Fig 1A. Trypsin digestion was performed using a robot (ProGest) (DigiLab) with the following protocol: washed with 25 mM ammonium bicarbonate followed by acetonitrile and reduced with 10 mM DTT at 60°C followed by alkylation with 50 mM iodoacetamide at room temperature. Digested with trypsin (Promega) at 37°C for 4 h. Quenched with formic acid and the supernatant was analyzed directly without further processing.
Mass spectrometry
Mass spectrometric analyses were conducted twice with different method. Both methods are described below.
Method 1: The gel digest was analyzed by nano LC/MS/MS with a Waters NanoAcquity HPLC system interfaced to a Thermo Fisher Q Exactive. Peptides were loaded onto a trapping column and eluted over a 75 μm analytical column at 350 nl/min. Both columns were packed with Luna C18 resin (Phenomenex). The mass spectrum was operated in the data‐dependent mode, with MS and MS/MS performed in the Orbitrap at 70,000 FWHM resolution and 17,500 FWHM resolution, respectively. The fifteen most abundant ions were selected for MS/MS.
Method 2: The gel digest was analyzed by nano LC/MS/MS with an AMR Zaplous nanoHPLC system interfaced to a Thermo Fisher Orbitrap ELITE. Peptides were loaded onto a trapping column and eluted over a 200 μm analytical column at 1 μl/min. Both columns were packed with L‐column2 C18 resin (CERI). The mass spectrum was operated in the data‐dependent mode, with MS performed in the Orbitrap at 240,000 FWHM resolution and MS/MS used the linear ion trap mode. The seven most abundant ions were selected for MS/MS.
Data processing of mass spectrometry
Data were searched using a local copy of Mascot with the following parameters:
Enzyme: Trypsin. Database: Uniprot B. mori. Fixed modification: Carbamidomethyl (C). Variable modifications: Oxidation (M), Acetyl (Protein N‐term, K), Deamidation (NQ), Phospho (STY), GG (K) and Methyl (K). Mass values: Monoisotopic. Peptide Mass Tolerance: 10 ppm. Fragment Mass Tolerance: 0.8‐Da. Max Missed Cleavage: 2.
Mascot DAT files were parsed into the Scaffold software for validation, filtering and to create a nonredundant list per sample. Data were filtered using a minimum protein value of 90%, a minimum peptide value of 50% (Prophet scores) and requiring at least two unique peptides per protein.
Author contributions
KMN and KS performed biochemical analyses of RNAs and piRNA factors and immunofluorescence analyses. KMN and TM generated monoclonal Vret antibody. TS and HY performed bioinformatics analyses. TKa and TKo performed LC‐MS/MS analysis. MCS designed the experiments with other authors, supervised, and discussed the work, and wrote the manuscript. All authors commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank T. Watanabe, M. Komai, T. Miyoshi, A. Shibuya, Y. Shirasaki, and Y. Namba for technical support. We also thank H. Siomi, S. Yamanaka, and S. Hirakata for reading the manuscript and useful comments. We also thank all other members of the Siomi laboratory at the University of Tokyo. This study was supported by research grants from Ministry of Education, Culture, Sports, Science and Technology (MEXT) to M.C.S. (19H05466) and K.M.N. (20K06483). K.S. and T.S. are supported by the Japan Society for the Promotion of Science.
The EMBO Journal (2020) 39: e105130
Data availability
The datasets produced in this study are available in the following databases: RNA‐seq data: Gene Expression Omnibus GSE150444 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE150444).
References
- Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ (2008) A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31: 785–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18: 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ (2007) Discrete small RNA‐generating loci as master regulators of transposon activity in Drosophila . Cell 128: 1089–1103 [DOI] [PubMed] [Google Scholar]
- Carmell MA, Girard A, van de Kant HJG, Bourc'his D, Bestor TH, de Rooij DG, Hannon GJ (2007) MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12: 503–514 [DOI] [PubMed] [Google Scholar]
- Czech B, Munafò M, Ciabrelli F, Eastwood EL, Fabry MH, Kneuss E, Hannon GJ (2018) piRNA‐guided genome defense: from biogenesis to silencing. Annu Rev Genet 52: 131–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Fazio S, Bartonicek N, Di Giacomo M, Abreu‐Goodger C, Sankar A, Funaya C, Antony C, Moreira PN, Enright AJ, O'Carroll D (2011) The endonuclease activity of Mili fuels piRNA amplification that silences LINE1 elements. Nature 480: 259–263 [DOI] [PubMed] [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA (2006) A germline‐specific class of small RNAs binds mammalian Piwi proteins. Nature 442: 199–202 [DOI] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC (2007) A slicer‐mediated mechanism for repeat‐associated siRNA 5′ end formation in Drosophila . Science 315: 1587–1590 [DOI] [PubMed] [Google Scholar]
- Han BW, Wang W, Li C, Weng Z, Zamore PD (2015) Noncoding RNA. piRNA‐guided transposon cleavage initiates Zucchini‐dependent, phased piRNA production. Science 348: 817–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler D, Olivieri D, Novatchkova M, Gruber FS, Meixner K, Mechtler K, Stark A, Sachidanandam R, Brennecke J (2011) A systematic analysis of Drosophila TUDOR domain‐containing proteins identifies Vreteno and the Tdrd12 family as essential primary piRNA pathway factors. EMBO J 30: 3977–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakata S, Ishizu H, Fujita A, Tomoe Y, Siomi MC (2019) Requirements for multivalent Yb body assembly in transposon silencing in Drosophila . EMBO Rep 20: e47708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakata S, Siomi MC (2019) Assembly and function of gonad‐specific non‐membranous organelles in Drosophila piRNA biogenesis. Noncoding RNA 5: E52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda S, Kirino Y, Maragkakis M, Alexiou P, Ohtaki A, Murali R, Mourelatos Z, Kirino Y (2013) Mitochondrial protein BmPAPI modulates the length of mature piRNAs. RNA 19: 1405–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houwing S, Kamminga LM, Berezikov E, Cronembold D, Girard A, van den Elst H, Filippov DV, Blaser H, Raz E, Moens CB et al (2007) A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in zebrafish. Cell 129: 69–82 [DOI] [PubMed] [Google Scholar]
- Houwing S, Berezikov E, Ketting RF (2008) Zili is required for germ cell differentiation and meiosis in zebrafish. EMBO J 27: 2702–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HY, Houwing S, Kaaij LJT, Meppelink A, Redl S, Gauci S, Vos H, Draper BW, Moens CB, Burgering BM et al (2011) Tdrd1 acts as a molecular scaffold for Piwi proteins and piRNA targets in zebrafish. EMBO J 30: 3298–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizu H, Iwasaki YW, Hirakata S, Ozaki H, Iwasaki W, Siomi H, Siomi MC (2015) Somatic primary piRNA biogenesis driven by cis‐acting RNA elements and trans‐Acting Yb. Cell Rep 12: 429–440 [DOI] [PubMed] [Google Scholar]
- Iwasaki YW, Siomi MC, Siomi H (2015) PIWI‐interacting RNA: its biogenesis and functions. Annu Rev Biochem 84: 405–433 [DOI] [PubMed] [Google Scholar]
- Jaskiewicz L, Bilen B, Hausser J, Zavolan M (2012) Argonaute CLIP–a method to identify in vivo targets of miRNAs. Methods 58: 106–112 [DOI] [PubMed] [Google Scholar]
- Kawaoka S, Minami K, Katsuma S, Mita K, Shimada T (2008) Developmentally synchronized expression of two Bombyx mori Piwi subfamily genes, SIWI and BmAGO3 in germ‐line cells. Biochem Biophys Res Commun 367: 755–760 [DOI] [PubMed] [Google Scholar]
- Kawaoka S, Hayashi N, Suzuki Y, Abe H, Sugano S, Tomari Y, Shimada T, Katsuma S (2009) The Bombyx ovary‐derived cell line endogenously expresses PIWI/PIWI‐interacting RNA complexes. RNA 15: 1258–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana JS, Theurkauf W (2010) piRNAs, transposon silencing, and Drosophila germline development. J Cell Biol 191: 905–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenov MS, Lavrov SA, Stolyarenko AD, Ryazansky SS, Aravin AA, Tuschl T, Gvozdev VA (2007) Repeat‐associated siRNAs cause chromatin silencing of retrotransposons in the Drosophila melanogaster germline. Nucleic Acids Res 35: 5430–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi‐Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri TW et al (2008) DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 22: 908–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Vagin VV, Lee S, Xu J, Ma S, Xi H, Seitz H, Horwich MD, Syrzycka M, Honda BM et al (2009) Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell 137: 509–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahowald AP (1971) Polar granules of Drosophila. 3. The continuity of polar granules during the life cycle of Drosophila . J Exp Zool 176: 329–343 [DOI] [PubMed] [Google Scholar]
- Mahowald AP (2001) Assembly of the Drosophila germ plasm. Int Rev Cytol 203: 187–213 [DOI] [PubMed] [Google Scholar]
- Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ (2009) Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 137: 522–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris C, Dominguez C, Allain FHT (2005) The RNA recognition motif, a plastic RNA‐binding platform to regulate post‐transcriptional gene expression. FEBS J 272: 2118–2131 [DOI] [PubMed] [Google Scholar]
- Matsumoto N, Nishimasu H, Sakakibara K, Nishida KM, Hirano T, Ishitani R, Siomi H, Siomi MC, Nureki O (2016) Crystal structure of Silkworm PIWI‐Clade Argonaute Siwi bound to piRNA. Cell 167: 484–497 [DOI] [PubMed] [Google Scholar]
- Mohn F, Handler D, Brennecke J (2015) Noncoding RNA. piRNA‐guided slicing specifies transcripts for Zucchini‐dependent, phased piRNA biogenesis. Science 348: 812–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murota Y, Ishizu H, Nakagawa S, Iwasaki YW, Shibata S, Kamatani MK, Saito K, Okano H, Siomi H, Siomi MC (2014) Yb integrates piRNA intermediates and processing factors into perinuclear bodies to enhance piRISC assembly. Cell Rep 8: 103–113 [DOI] [PubMed] [Google Scholar]
- Niki Y, Yamaguchi T, Mahowald AP (2006) Establishment of stable cell lines of Drosophila germ‐line stem cells. Proc Natl Acad Sci USA 103: 16325–16330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida KM, Iwasaki YW, Murota Y, Nagao A, Mannen T, Kato Y, Siomi H, Siomi MC (2015) Respective functions of two distinct Siwi complexes assembled during PIWI‐interacting RNA biogenesis in Bombyx germ cells. Cell Rep 10: 193–203 [DOI] [PubMed] [Google Scholar]
- Nishida KM, Sakakibara K, Iwasaki YW, Yamada H, Murakami R, Murota Y, Kawamura T, Kodama T, Siomi H, Siomi MC (2018) Hierarchical roles of mitochondrial Papi and Zucchini in Bombyx germline piRNA biogenesis. Nature 555: 260–264 [DOI] [PubMed] [Google Scholar]
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett‐Jones DP, Pawson T, Forman‐Kay JD et al (2015) Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell 57: 936–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozata DM, Gainetdinov I, Zoch A, O'Carroll D, Zamore PD (2019) PIWI‐interacting RNAs: small RNAs with big functions. Nat Rev Genet 20: 89–108 [DOI] [PubMed] [Google Scholar]
- Pagès G, Brunet A, L'Allemain G, Pouysségur J (1994) Constitutive mutant and putative regulatory serine phosphorylation site of mammalian MAP kinase kinase (MEK1). EMBO J 13: 3003–3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pall GS, Hamilton AJ (2008) Improved northern blot method for enhanced detection of small RNA. Nat Protoc 3: 1077–1084 [DOI] [PubMed] [Google Scholar]
- Pearlman SM, Serber Z, Ferrell JE Jr (2011) A mechanism for the evolution of phosphorylation sites. Cell 147: 934–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Watanabe T, Ku HY, Liu N, Zhong M, Lin H (2011) The Yb body, a major site for Piwi‐associated RNA biogenesis and a gateway for Piwi expression and transport to the nucleus in somatic cells. J Biol Chem 286: 3789–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Chuma S, Tanaka T, Franz T, Stark A, Pillai RS (2009) Loss of the Mili‐interacting Tudor domain–containing protein‐1 activates transposons and alters the Mili‐associated small RNA profile. Nat Struct Mol Biol 16: 639–646 [DOI] [PubMed] [Google Scholar]
- Reuter M, Berninger P, Chuma S, Shah H, Hosokawa M, Funaya C, Antony C, Sachidanandam R, Pillai RS (2011) Miwi catalysis is required for piRNA amplification‐independent LINE1 transposon silencing. Nature 480: 264–267 [DOI] [PubMed] [Google Scholar]
- Saito K, Inagaki S, Mituyama T, Kawamura Y, Ono Y, Sakota E, Kotani H, Asai K, Siomi H, Siomi MC (2009) A regulatory circuit for piwi by the large Maf gene traffic jam in Drosophila . Nature 461: 1296–1299 [DOI] [PubMed] [Google Scholar]
- Sakakibara K, Siomi MC (2018) The PIWI‐interacting RNA molecular pathway: insights from cultured silkworm germline cells. BioEssays 40: 1700068 [DOI] [PubMed] [Google Scholar]
- Sato K, Iwasaki YW, Shibuya A, Carninci P, Tsuchizawa Y, Ishizu H, Siomi MC, Siomi H (2015) Krimper enforces an antisense bias on piRNA pools by binding AGO3 in the Drosophila germline. Mol Cell 59: 553–563 [DOI] [PubMed] [Google Scholar]
- Sato K, Siomi MC (2020) The piRNA pathway in Drosophila ovarian germ and somatic cells. Proc Japan Acad Ser B 96: 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Mannen T, Siomi H (2010) How does the royal family of Tudor rule the PIWI‐interacting RNA pathway? Genes Dev 24: 636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD (2006) A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313: 320–324 [DOI] [PubMed] [Google Scholar]
- Vagin VV, Wohlschlegel J, Qu J, Jonsson Z, Huang X, Chuma S, Girard A, Sachidanandam R, Hannon GJ, Aravin AA (2009) Proteomic analysis of murine Piwi proteins reveals a role for arginine methylation in specifying interaction with Tudor family members. Genes Dev 23: 1749–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Saxe JP, Tanaka T, Chuma S, Lin H (2009) Mili interacts with tudor domain‐containing protein 1 in regulating spermatogenesis. Curr Biol 19: 640–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster A, Li S, Hur JK, Wachsmuth M, Bois JS, Perkins EM, Patel DJ, Aravin AA (2015) Aub and Ago3 are recruited to nuage through two mechanisms to form a ping‐pong complex assembled by krimper. Mol Cell 59: 564–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiol J, Spinelli P, Laussmann MA, Homolka D, Yang Z, Cora E, Couté Y, Conn S, Kadlec J, Sachidanandam R et al (2014) RNA clamping by Vasa assembles a piRNA amplifier complex on transposon transcripts. Cell 157: 1698–1711 [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Oe A, Nishida KM, Yamashita K, Kajiya A, Hirano S, Matsumoto N, Dohmae N, Ishitani R, Saito K et al (2020) Crystal structure of Drosophila Piwi. Nat Commun 11: 858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro H, Siomi MC (2018) PIWI‐interacting RNA in Drosophila : biogenesis, transposon regulation, and beyond. Chem Rev 118: 4404–4421 [DOI] [PubMed] [Google Scholar]
- Yamashiro H, Negishi M, Kinoshita T, Ishizu H, Ohtani H, Siomi MC (2020) Armitage determines Piwi−piRISC processing from precursor formation and quality control to inter‐organelle translocation. EMBO Rep 21: e48796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamparini AL, Davis MY, Malone CD, Vieira E, Zavadil J, Sachidanandam R, Hannon GJ, Lehmann R (2011) Vreteno, a gonad‐specific protein, is essential for germline development and primary piRNA biogenesis in Drosophila . Development 138: 4039–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Xu J, Koppetsch BS, Wang J, Tipping C, Ma S, Weng Z, Theurkauf WE, Zamore PD (2011) Heterotypic piRNA ping‐pong requires qin, a protein with both E3 ligase and tudor domains. Mol Cell 44: 572–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
The datasets produced in this study are available in the following databases: RNA‐seq data: Gene Expression Omnibus GSE150444 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE150444).