

Abstract
While the microtubule end‐binding protein, EB1 facilitates early stages of HIV‐1 infection, how it does so remains unclear. Here, we show that beyond its effects on microtubule acetylation, EB1 also indirectly contributes to infection by delivering the plus‐end tracking protein (+TIP), cytoplasmic linker protein 170 (CLIP170) to the cell periphery. CLIP170 bound to intact HIV‐1 cores or in vitro assembled capsid–nucleocapsid complexes, while EB1 did not. Moreover, unlike EB1 and several other +TIPs, CLIP170 enhanced infection independently of effects on microtubule acetylation. Capsid mutants and imaging revealed that CLIP170 bound HIV‐1 cores in a manner distinct from currently known capsid cofactors, influenced by pentamer composition or curvature. Structural analyses revealed an EB‐like +TIP‐binding motif within the capsid major homology region (MHR) that binds SxIP motifs found in several +TIPs, and variability across this MHR sequence correlated with the extent to which different retroviruses engage CLIP170 to facilitate infection. Our findings provide mechanistic insights into the complex roles of +TIPs in mediating early stages of retroviral infection, and reveal divergent capsid‐based EB1 mimicry across retroviral species.
Keywords: +TIP, CLIP170, HIV‐1, trafficking, uncoating
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Membrane & Intracellular Transport; Microbiology, Virology & Host Pathogen Interaction
Microtubule binding protein CLIP170 facilitates HIV‐1 infection via interaction with the EB1‐mimicking motif in the HIV‐1 capsid.
Introduction
Intracellular cargo transport involves directed movement on microtubules (MTs) by motor proteins. Generally, dynein mediates minus‐end directed (retrograde) movement toward the nucleus, while plus‐end directed (anterograde) transport is mediated by kinesin family members (Reck‐Peterson et al, 2018). MTs themselves are composed of α/β tubulin heteropolymers arranged into polarized filaments. While their minus‐ends are anchored at a MT organizing center (MTOC), their plus‐ends extend toward the cell periphery and continuously grow and shrink to explore the intracellular environment (Akhmanova & Steinmetz, 2015). However, MT subsets can become stabilized and acquire post‐translational modifications (PTMs) that include detyrosination and acetylation (Janke & Bulinski, 2011). Acetylation confers mechanical strength, while detyrosination enables preferential binding of motors, allowing stable MTs to act as specialized transport networks (Gundersen, 2002; Portran et al, 2017; Xu et al, 2017). While MTs are regulated by a range of MT‐associated proteins (MAPs) that bind along the filament lattice, a specialized subset of MAPs recognize the plus‐end and are termed plus‐end tracking proteins (+TIPs) (Akhmanova & Steinmetz, 2008; Jiang & Akhmanova, 2011). Of these, the end‐binding protein (EB1) acts as a master regulator by recognizing the plus‐end to both directly regulate MT dynamics and recruit other +TIPs (Honnappa et al, 2006; Galjart, 2010; Jiang & Akhmanova, 2011). The N‐terminal calponin homology (CH) domain of EB1 interacts with the growing MT end, while the C‐terminus of EB1 harbors an EB homology (EBH) domain and an EEY motif that mediate binding to two major families of +TIPs, namely those with a SxIP motif (where x is any amino acid) or a cytoskeleton‐associated protein glycine‐rich (CAP‐Gly) motif, respectively (Honnappa et al, 2006; Bjelic et al, 2012; Jiang et al, 2012; Nehlig et al, 2017; Fig 1A). While over 50 +TIPs with SxIP motifs are known, one of the most widely studied and prototypical CAP‐Gly proteins is cytoplasmic linker protein 170 (CLIP170) that controls both cargo loading and MT behavior (Bjelic et al, 2012; Duellberg et al, 2014; Reck‐Peterson et al, 2018). CLIP170 can interact with EB1 in two ways. While its CAP‐Gly domain dominates interactions with EB1's EEY motif, CLIP170 also harbors an SxIP motif that interacts with EB1's EBH domain (Chen et al, 2019) (Fig 1A).
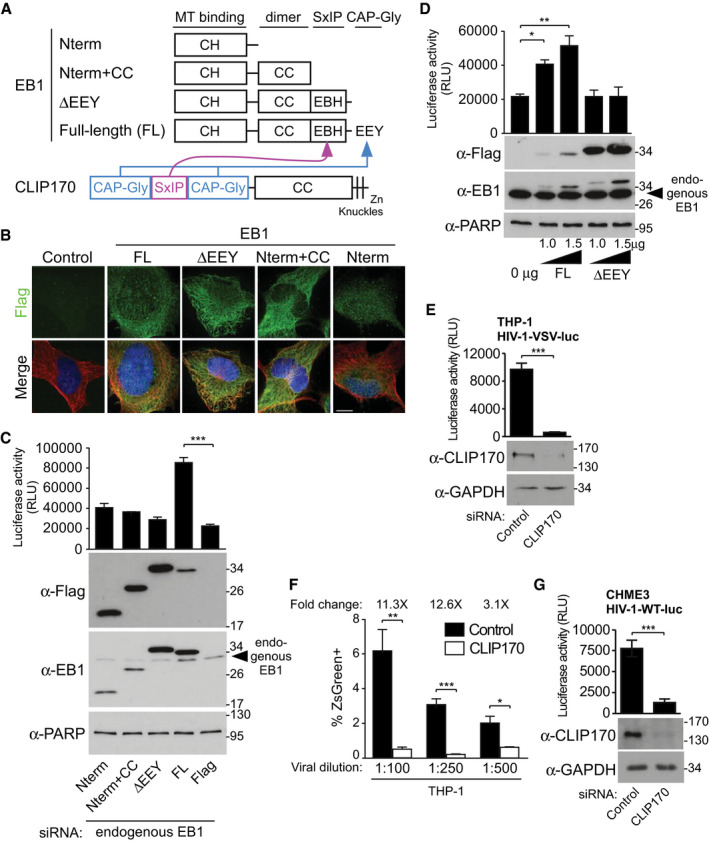
Figure 1. EB1‐associated +TIP, CLIP170 promotes early HIV‐1 infection.
-
ASchematic representation of EB1 constructs and domain functions. CLIP170 (lower) uses CAP‐Gly domains to interact with EB1's EEY motif, and secondary SxIP‐mediated interactions with EB1's EBH domain.
-
BRepresentative IF images in CHME3 stably expressing Flag alone control or the Flag‐tagged EB1 constructs depicted in (A) co‐stained for Tyrosinated‐MTs (Tyr‐MTs), and the nucleus (Hoechst). Scale bar, 10 μm.
-
CHIV‐1-VSV‐luc infection is enhanced in CHME3 depleted of endogenous EB1 and stably expressing full‐length (FL) EB1, but not in control Flag or any of the other EB1 constructs depicted in (A).
-
DIncreasing amount of FL‐EB1, but not ΔEEY results in a dose‐dependent enhancement of HIV-1‐VSV-luc infection in transiently transfected CHME3 cells.
-
E, FHIV-1‐VSV-luc (E) or HIV‐1-VSV‐ZsGreen at various dilutions (corresponding to multiplicities of infection (m.o.i) 0.06, 0.03, and 0.02), (F) infection is reduced in differentiated THP‐1 depleted of CLIP170 determined by luciferase assays or FACS analysis, respectively.
-
GHIV‐1-WT-luc infection is decreased in CLIP170‐depleted CHME3 cells.
Viruses such as human immunodeficiency virus type 1 (HIV‐1) hijack MT‐based transport systems to reach the nucleus (Dodding & Way, 2011; Naghavi & Walsh, 2017). The HIV‐1 core is composed of a complex of the nucleocapsid (NC) protein and genomic RNA, surrounded by the viral capsid (CA) protein (Pornillos et al, 2009). Upon release into the cytosol, the HIV‐1 core is not only transported to the nucleus, but it also undergoes a poorly understood process of disassembly, known as uncoating. However, the precise timing and location of uncoating is still hotly debated. While some propose that uncoating occurs entirely in the nucleus, there is no direct evidence that the cores observed inside the nucleus are fully intact (Burdick et al, 2020). Indeed, intact cores are physically too large to pass through nuclear pores and many studies have reported loss of CA protein and structural changes in the core at various stages prior to entry into the nucleus. This gradual loss of CA, known as partial uncoating, has been observed during cytoplasmic transport, at the nuclear pore complex and inside the nucleus (Miller et al, 1997; Fassati & Goff, 2001; McDonald et al, 2002; Arhel et al, 2007; Schaller et al, 2011; Zhou et al, 2011; Koh et al, 2013; Rasaiyaah et al, 2013; Ambrose & Aiken, 2014; Peng et al, 2014; Campbell & Hope, 2015; Hulme et al, 2015; Chen et al, 2016; Mamede et al, 2017; Francis & Melikyan, 2018; Marquez et al, 2018; Bejarano et al, 2019; Fernandez et al, 2019; Summers et al, 2019; Zila et al, 2019; Blanco‐Rodriguez et al, 2020; Dharan et al, 2020; Ingram et al, 2020; Zurnic Bonisch et al, 2020). As such, it seems that uncoating is a complex process that begins very early after entry into the cytosol, and completes inside the nucleus. Indeed, uncoating and trafficking are functionally interconnected. Interfering with the MT network itself, chemically or by targeting MAPs, or perturbing the function of MT motors or adaptors inhibits viral trafficking and delays uncoating (McDonald et al, 2002; Arhel et al, 2006; Sabo et al, 2013; Lukic et al, 2014; Pawlica & Berthoux, 2014; Jayappa et al, 2015; Malikov et al, 2015; Delaney et al, 2017; Carnes et al, 2018). However, how HIV‐1 actively controls MT‐based processes is poorly understood (Naghavi & Walsh, 2017).
We previously showed that HIV‐1 exploits a number of EB1‐associated +TIPs to stabilize MTs and control both the trafficking and uncoating of incoming viral cores (Sabo et al, 2013; Delaney et al, 2017; Mitra et al, 2020). Yet precisely how EB1 functions in early HIV‐1 infection remains unclear. Here, we show that beyond its role in regulating MT stability, EB1 also functions to deliver CLIP170 to the cell periphery. Unlike EB1, CLIP170 binds to incoming HIV‐1 cores and does so in a manner distinct from that of the well‐known capsid‐binding cofactor, CypA. We further reveal a functional +TIP‐binding motif within the major homology region (MHR) in HIV‐1 capsid and show that the extent to which MHRs of different retroviruses functionally mimic EB1 varies, and correlates with their induction of Ac‐MTs and dependence on CLIP170. Cumulatively, our findings provide a mechanistic explanation for how HIV‐1 engages several +TIP family members and highlights their distinct roles in early infection.
Results
The CAP‐Gly domain is required for EB1 to promote early HIV‐1 infection
Given that distinct EB1 domains mediate its interactions with MTs and +TIPs, we tested their contributions to early HIV‐1 infection. To do this, we generated human microglia CHME3 cells, a natural target cell type for HIV‐1 infection, stably expressing Flag‐tagged forms of EB1 containing various functional domains (Fig 1A). Importantly, such approaches are widely used as each domain retains functionality independently of the others. For example, the N‐terminal CH domain (Nterm) is sufficient to bind MTs, while the addition of CC domains (Nterm+CC) enables dimerization on MTs despite not binding +TIPs (Akhmanova & Steinmetz, 2015). Immunofluorescence (IF) staining using anti‐flag antibodies confirmed these flag‐tagged forms of EB1 retained their expected cellular localization, with the Nterm form exhibiting more limited localization on MTs due to the lack of a CC dimerization domain that enhances MT recognition, while all other forms localized to MTs (Fig 1B). To initially avoid potential confounding effects from endogenous EB1 when assessing the effects of these EB1 mutants on infection, CHME3 lines were depleted of endogenous EB1 using siRNAs and then infected with HIV‐1 carrying a luciferase reporter and pseudotyped with a vesicular stomatitis virus G (VSV‐G) envelope (HIV‐1‐VSV‐luc). Unlike full‐length (FL) EB1, neither the Nterm nor Nterm+CC domains could enhance infection over Flag‐expressing controls (Fig 1C). Moreover, removal of the EEY motif (ΔEEY) from EB1, leaving the SxIP motif in place, also failed to promote HIV‐1 infectivity (Fig 1C). To independently confirm the importance of the EEY motif identified in these stable lines, CHME3 cells were transiently transfected with increasing amounts of EB1 FL or ΔEEY constructs followed by infection with HIV‐1‐VSV‐luc. EB1 FL transfection resulted in a dose‐dependent enhancement of HIV‐1 infection while the ΔEEY mutant had no effect (Fig 1D). This demonstrated that EB1's ability to dimerize and bind MTs was not sufficient while its CAP‐Gly binding activity was critical to promote early HIV‐1 infection.
CLIP170 promotes early HIV‐1 infection
Given these findings, we next tested the dependence of HIV‐1 on one of the most prominent CAP‐Gly containing +TIP, CLIP170. To do this, we treated THP1 cells differentiated to macrophages with non‐targeting control or CLIP170 siRNAs followed by infection with HIV‐1‐VSV‐luc. Luciferase assays revealed that CLIP170 depletion potently suppressed HIV‐1 infection (Fig 1E). To independently validate luciferase‐based infection assays, THP1 cells depleted of CLIP170 were infected with VSV‐pseudotyped HIV‐1 carrying a ZsGreen fluorescent marker followed by FACS analysis. Similar to luciferase assays, FACS analysis of ZsGreen infections performed at various dilutions showed that infection was significantly reduced in CLIP170‐depleted THP1 cells compared with control siRNA‐treated cultures (Fig 1F). Similar results were observed in CHME3 microglia infected with HIV‐1‐luc pseudotyped with WT envelope (HIV‐1‐WT‐luc) (Fig 1G) or in primary normal human dermal fibroblasts (NHDFs) infected with either HIV‐1‐VSV‐luc or HIV‐1‐luc pseudotyped with a murine leukemia virus (MuLV) amphotropic envelope (HIV‐1‐Ampho‐luc) (Fig EV1A and B). NHDFs offer a convenient primary cell system to complement findings in natural target CHME3 and THP1 cell lines. Moreover, pseudotyping with different envelope proteins that mediate entry by endocytosis or surface fusion, respectively (Miyauchi et al, 2009; Herold et al, 2014), demonstrated that CLIP170 was required for early HIV‐1 infection in a range of human cell types independently of the route of viral entry, and these RNAi‐based findings are independently confirmed using overexpression approaches later.
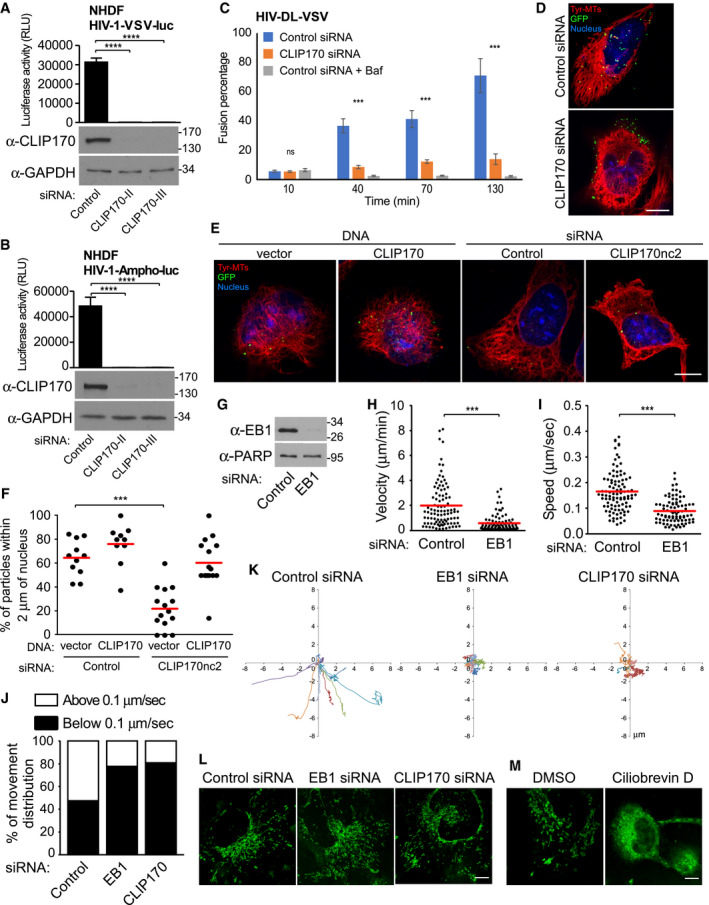
Figure EV1. EB1‐associated +TIP, CLIP170, promotes post‐entry stages of HIV‐1 infection.
-
A, BNHDF cells were treated with control siRNAs or siRNAs specific to CLIP170 (CLIP170‐II or CLIP170‐III). Cells were either lysed and analyzed by WB analysis using antibodies against CLIP170 and GAPDH (loading control) or infected with HIV-1‐VSV-luc (A) or HIV‐1-Ampho‐luc (B) as indicated, followed by measurements of luciferase activity. Statistical significance was determined by one‐way ANOVA. ****P < 0.0001. Data are mean values from three independent experiments ± SD.
-
CControl or CLIP170 siRNA‐treated CHME3 cells were left untreated or treated with bafilomycin A1 for 2 h during spinoculation followed by infection with HIV‐1-VSV containing Vpr‐GFP and S15 Tomato (HIV‐1-DL-VSV). Percentage fused viral particles (GFP‐Vpr+/S15-tomato−) is shown at the indicated times post‐infection. Data represent mean of ≥ 815 virus particles analyzed in ≥ 10 fields of view (≥ 14 cells) for each condition. ***P < 0.001. One‐way ANOVA was used to calculate statistical significance (mean ± SEM).
-
DCHME3 treated with control or CLIP170 siRNAs were infected with HIV‐1-WT-GFP‐Vpr. Representative images of Tyr‐MTs, viral particles (GFP), and the nucleus (Hoescht) from Fig 2F are shown. Scale bar, 10 μm.
-
E, FExogenous expression of CLIP170 enhances the translocation of HIV‐1 cores and rescues the effects of siRNA‐mediated CLIP170 depletion. 293A cells treated with control siRNA or a siRNA targeting the 3′UTR of CLIP170 (CLIP170nc2) were transfected with either empty vector (pQCXIN) or Flag‐tagged CLIP170 constructs after 24 h, followed by infection with Mock (not shown) or HIV‐1-VSV‐GFP-Vpr. (E) Representative staining images for tyrosinated tubulin (Tyr‐MTs), viral particles (GFP), and the nucleus (Hoescht). Scale bar, 10 μm. (F) Percentage viral particles within 2 μm of the nucleus at 4 h.p.i, scatterplot with mean of ≥ 62 virus particles quantified in ≥ 10 cells per sample. Statistical significance was determined by one‐way ANOVA. ***P < 0.001.
-
G–KEB1 and CLIP170 facilitate trafficking of HIV‐1 particles toward the nucleus. (G‐I) CHME3 cells treated with control or EB1‐specific siRNAs were infected with HIV‐1-WT-GFP‐Vpr then imaged live at 1 fps for 5–10 min using a Zeiss motorized spinning‐disk confocal microscope with MetaMorph imaging software. Viral particles were tracked to quantify virus movements in trajectories using Fiji software with Mosaic plug‐in. (G) Depletion of EB1 was confirmed by WB analysis. PARP was used as loading control. (H, I) Scatterplots representing displacement over time tracked (velocity) (H) and cumulative distance travelled over time (speed) (I) in cultures from (G). Each dot represents one viral particle tracked, and the red line indicates the median. A total of 183 trajectories were analyzed from 22 movies. Statistical significance was determined by t‐test. ***P < 0.001. (J) The percent of viral particles in (H, I) or from Fig 2G and H with movement > 0.1 μm per second (indicative of MT‐based movement) or < 0.1 μm per second (indicative of actin‐based movement or free diffusion). (K) Representative viral particle trajectories in (H, I) or from Fig 2G and H are depicted on XY axis.
-
L, MIntegrity of cellular cargo trafficking was confirmed by mitochondria staining. (L) CHME3 cells treated with control, EB1, or CLIP170 siRNAs were incubated with MitoTracker® Green, and images were captured at 3 fps for 3 min. (M) Dynein‐dependent trafficking was confirmed using ciliobrevin D (dynein inhibitor) and mitochondria staining. Representative still images are shown. Similar results were obtained in three independent experiments. Scale bar, 10 μm (L, M).
To understand how CLIP170 regulated infection, we determined whether virus fusion into the cytosol was affected. Compared to control siRNA‐treated CHME3 cells, depletion of CLIP170 decreased the fusion of HIV‐1 β‐lactamase‐Vpr reporter virus pseudotyped with VSV envelope (HIV‐1‐VSV‐BlaM‐Vpr) (Fig 2A). To confirm this independently, siRNA‐treated CHME3 cells were infected with double‐labeled HIV‐1 (S15‐Tomato/GFP‐Vpr) carrying a VSV envelope. Analysis of fixed samples showed that depletion of CLIP170 decreased the number of fused (S15‐Tomato−/GFP‐Vpr+) particles compared with controls over time, but not to levels seen in bafilomycin A‐treated cells (Fig EV1C) (Malikov et al, 2015). This suggests that loss of CLIP170 reduces the efficiency of, but does not completely block, endocytic entry of VSV‐pseudotyped virus. As such, of the ~12‐fold reduction in infectivity that is observed for VSV‐G‐pseudotyped viruses in CLIP170‐depleted cells, ~3‐fold of this may be attributable to effects on fusion alone. Reciprocally, overexpression of Flag‐tagged CLIP170 promoted infection by HIV‐1‐VSV‐luc in CHME3 cells compared with Flag control lines (Fig 2B and C). However, this enhancement occurred in the absence of any effect of Flag‐CLIP170 on fusion of HIV‐1‐VSV into CHME3 cells compared with Flag controls (Fig 2D). It should be noted that to avoid saturation of any given assay, different viral doses were used in luciferase versus GFP‐based FACS assays but the overall effects on infection were consistent throughout. This suggested that although very low CLIP170 levels affect fusion, CLIP170 also functions at a post‐entry level. Such a post‐entry function would explain why CLIP170 depletion affects early infection by HIV‐1 pseudotyped with a range of envelopes, as shown above. Indeed, while WT HIV‐1 can enter into some cell types by endocytosis, its primary mode of entry is membrane fusion. In line with this, CLIP170 depletion did not significantly reduce the fusion kinetics of HIV‐1 pseudotyped with WT envelope into CHME3 cells compared with control siRNAs (Fig 2E), yet suppressed early infection by HIV‐1‐WT‐luc (Fig 1F). Moreover, we have shown previously that fusion of HSV‐1 into cells is unaffected (Jovasevic et al, 2015). Cumulatively, these findings demonstrated that CLIP170 plays an important role in post‐entry stage(s) of infection.
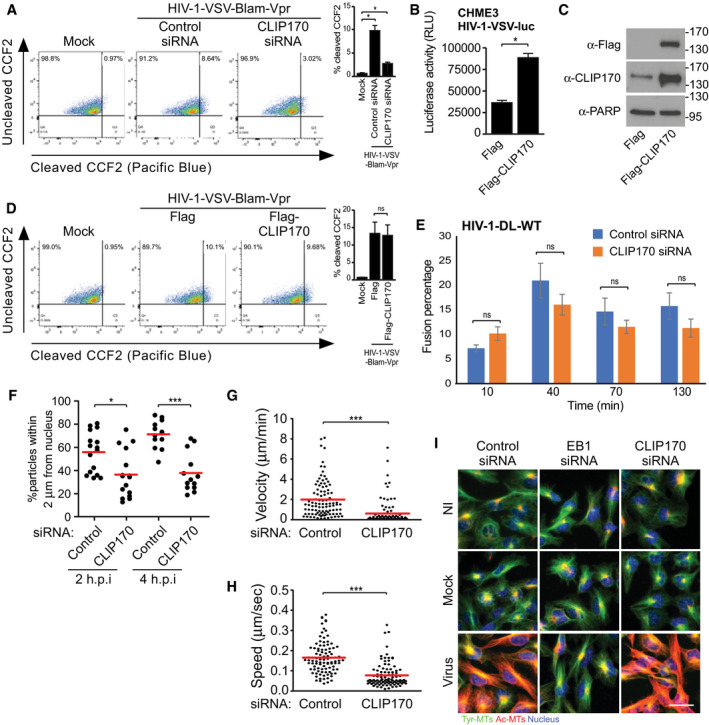
Figure 2. CLIP170 promotes multiple stages of early HIV‐1 infection.
-
A–DEffects of CLIP170 on fusion of viral particles by endocytosis. CHME3 cells either treated with control or CLIP170 siRNAs (A) or expressing Flag control or Flag‐CLIP170 (D) were infected with Mock or HIV‐1-VSV‐BlaM-Vpr. Representative FACS analysis of cells (n = 3) showing ˜9% (control siRNA) and 3% (CLIP170 siRNA) shift (A) or no difference (D) from green (uncleaved CCF2) to blue (cleaved CCF2). % infected cells is indicated at the top left corner of each plot, and quantification from independent repeats is presented as bar graphs at the right side of each FACS plot. (B, C) HIV-1‐VSV-luc infection is enhanced in Flag‐CLIP170, but not in Flag control, expressing CHME3 cells. Statistical significance was determined by t‐test (mean ± SEM). *P < 0.05. Molecular weight markers (in kDa) are shown to the right of WBs in (C).
-
EThe percentage fused double‐labeled (GFP‐Vpr+/S15‐tomato−) HIV‐1-DL-WT are not affected in CHME3 cells depleted of CLIP170. Data represent mean of ≥ 895 virus particles analyzed in ≥ 11 fields of view (≥ 17 cells) for each condition. One‐way ANOVA was used to calculate statistical significance (mean ± SEM).
-
FPercentage HIV-1‐WT-GFP‐Vpr particles within 2 μm of the nucleus is reduced in CHME3 depleted of CLIP170 at 2 and 4 h.p.i, scatterplot with mean of ≥ 354 virus particles quantified in ≥ 13 cells per sample. Statistical significance was determined by one‐way ANOVA *P < 0.05, ***P < 0.001.
-
G, HInfected cells in F analyzed by live‐cell imaging. Scatterplots representing displacement over time (velocity) (G) and cumulative distance travelled over time (speed) (H). Each dot represents one viral particle tracked, and red line indicates the mean. 188 trajectories were analyzed from 25 movies. Statistical significance was determined by t‐test. ***P < 0.001.
-
IThe induction of Ac‐MTs is blocked in HIV-1‐VSV-luc (Virus) infected, but not in uninfected (NI) or Mock infected CHME3 depleted of EB1, but not CLIP170 at 6 h.p.i. Tyrosinated‐MTs (Tyr‐MTs) are not affected. Representative fields are shown. Similar results were obtained in three independent experiments. Scale bar, 50 μm.
Source data are available online for this figure.
To better understand how CLIP170 affected post‐fusion steps of viral infection, we tested whether CLIP170 affected HIV‐1 transport to the nucleus. siRNA‐treated CHME3 cells were infected with HIV‐1 carrying wild‐type (WT) envelope and whose core was labeled using GFP‐tagged Vpr (HIV‐1‐WT‐GFP‐Vpr). Fixed samples were then stained for tyrosinated‐MTs, GFP (viral particles) and the nucleus, and the number of viral particles within 2 μm of the nucleus was measured (Malikov et al, 2015). While the number of viral particles within 2 μm of the nucleus increased over time in controls, the majority of particles remained distant from the nucleus in CLIP170‐depleted cells (Figs 2F and EV1D). Defects in viral particle translocation to the nucleus in CLIP170 siRNA‐treated cultures were rescued by the expression of siRNA‐resistant CLIP170 (Fig EV1E and F). Moreover, CLIP170 expression alone was sufficient to enhance virus accumulation around the nucleus in control siRNA‐treated cells (Fig EV1F). To verify that virus motility was affected, CHME3 cells treated with control or CLIP170 siRNAs were infected with HIV‐1‐WT‐GFP‐Vpr followed by live‐cell microscopy (Malikov et al, 2015). Compared to the rapid, long‐range movements in control siRNA‐treated cultures, depletion of CLIP170 reduced HIV‐1 motility (Movie EV1, panels 1 and 2, respectively). Analysis of individual viral trajectories further revealed a significant reduction in average velocity and average speed of viral particles in CLIP170‐depleted CHME3 cells relative to controls (Fig 2G and H, respectively, and Movie EV1, panels 1 and 2). Particle tracking further confirmed that, while 52.5% of viral particles exhibited MT‐based movement (> 0.1 μm/s) in control siRNA‐treated cultures, only 19.1% of particles exhibited movement > 0.1 μm/s in CLIP170‐depleted cells. Most particles exhibited slower, short‐range movement characteristic of either free diffusion or actin‐based motility in the absence of CLIP170 (Fig EV1J). While we previously used fixed imaging to show that EB1 affects HIV‐1 accumulation at the nucleus (Sabo et al, 2013), live‐cell imaging confirmed that EB1 exerts similar effects as CLIP170 on the trafficking of incoming HIV‐1 particles (Movie EV1, panels 1 and 3 and Fig EV1G–K). Importantly, depletion of CLIP170 or EB1 did not affect the motility of mitochondria, unlike the dynein inhibitor ciliobrevin D (Movies EV2 and EV3, Fig EV1L and M). This suggested that CLIP170 specifically affected transport mechanisms utilized by HIV‐1 for infection without broadly affecting cargo transport.
Given that EB1 is required for HIV‐1‐induced MT stabilization and virus transport (Sabo et al, 2013), we next tested whether CLIP170 functioned similarly. IF imaging revealed that EB1 depletion blocked Ac‐MT induction upon infection with HIV‐1‐VSV‐luc in CHME3 cells, as shown previously (Sabo et al, 2013), while depletion of CLIP170 had no detectable effect (Fig 2I). It is important to note that although CLIP170 depletion suppresses, it does not completely block fusion, thereby allow incoming virus particles to continue to induce the formation of Ac‐MTs. Imaging also showed that Ac‐MT networks and overall MT architecture were not grossly altered in CLIP170‐depleted cells. These findings identify CLIP170 as the first +TIP to promote HIV‐1 infection independently of effects on Ac‐MTs, suggesting that it has roles in infection that are distinct from those of previously studied +TIPs (Sabo et al, 2013; Delaney et al, 2017), yet is likely contribute to infection on multiple levels as we discuss later.
EB1 and CLIP170 facilitate HIV‐1 uncoating
To extend our comparative analysis of EB1 and CLIP170, we next determined whether they influenced HIV‐1 uncoating. Two independent uncoating assays, the fluorescence microscopy‐based in situ uncoating assay and the infectivity‐based fate‐of‐capsid (FoCA) assay, were used to assess loss of CA. In the in situ uncoating assay, control, EB1, or CLIP170 siRNA‐treated CHME3 cells were infected with double‐labeled HIV‐1 (S15‐Tomato/GFP‐Vpr) and fixed at various times before staining for p24, as described previously (Delaney et al, 2017). To determine effects on uncoating, we quantified p24 staining intensities associated only with fused (GFP‐Vpr+/S15‐Tomato−) viral particles at 30, 60, and 120 min post‐infection. Depletion of either +TIP resulted in a significant increase in the amount of p24 associated with fused virions compared with control cells for all three time points (Figs 3A and B, and EV2A–C), suggesting CA loss (uncoating) was delayed in EB1 or CLIP170‐depleted CHME3 cells.
Figure 3. EB1 and CLIP170 facilitate uncoating of incoming HIV‐1 cores.
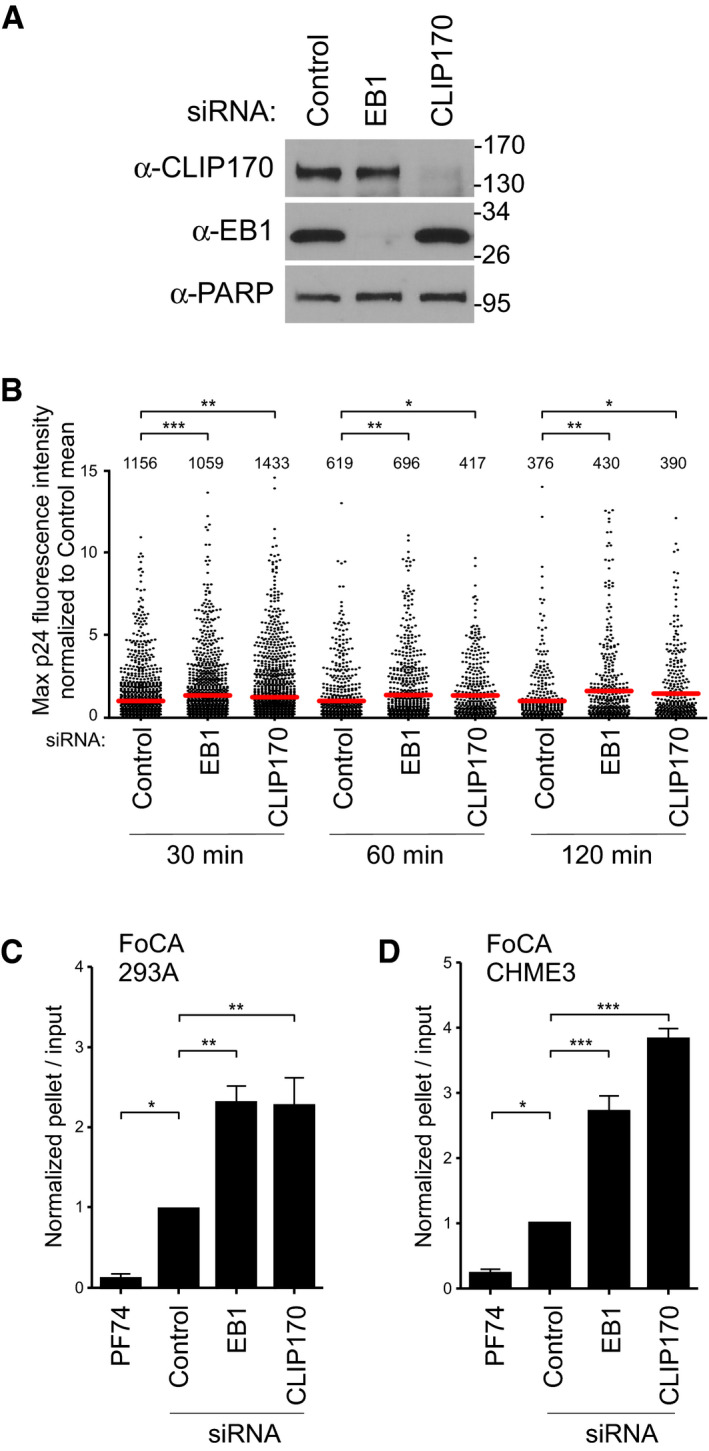
-
A, BCHME3 treated with negative control, EB1, or CLIP170 siRNAs was either lysed and analyzed by WB (A) or infected with GFP‐Vpr/S15‐tomato‐labeled VSV‐G pseudotyped HIV‐1 (B). Molecular weight markers (in kDa) are shown to the right of WBs in (A). Scatterplot of maximum p24 intensity of fused viral particles normalized to the control mean. Each dot represents one virion, and red lines indicate means. The number of viral particles analyzed per condition from three independent experiments is shown. Statistical significance was determined by one‐way ANOVA (*P < 0.05, **P < 0.01, ***P < 0.001).
-
C, DAmounts of pelleted p24 CA normalized to the input CA at 3 h.p.i. with HIV-1‐VSV-luc in 293A (C) or CHME3 (D) treated with control, EB1, or CLIP170 siRNAs. Mean ± SEM from three independent experiments is shown. Statistical significance was determined by one‐way ANOVA. Error bars = standard error of the mean (*P‐value ≤ 0.05, **P‐value ≤ 0.01, ***P‐value ≤ 0.001).
Source data are available online for this figure.
Figure EV2. EB1 and CLIP170 facilitate uncoating of incoming HIV‐1 cores.
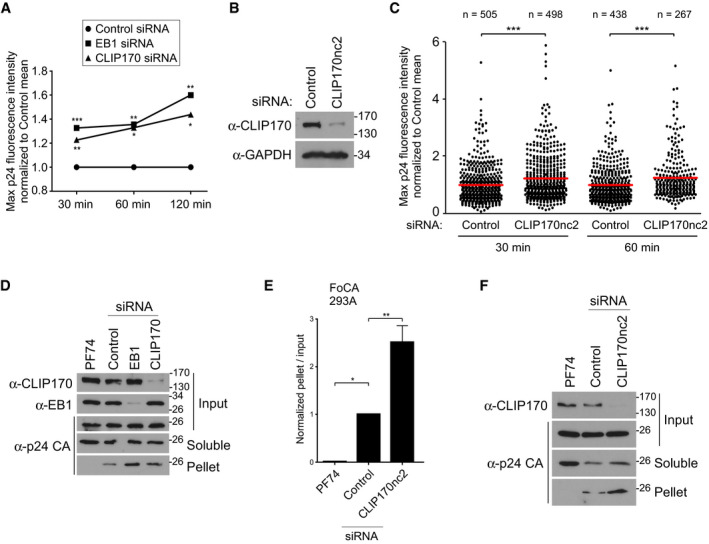
-
AIn situ uncoating assay in siRNA‐treated cells infected with GFP‐Vpr/S15‐tomato‐labeled VSV‐G pseudotyped HIV‐1 from Fig 3C. The means of the maximum fluorescence intensities for each time point from three independent experiments were plotted on a line graph. Statistical significance was determined by one‐way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
-
B, CConfirmation of the effect of CLIP170 on HIV‐1 uncoating using an independent siRNA. CHME3 cells treated with control siRNA or a siRNA targeting 3′UTR of CLIP170 (CLIP170nc2) were either lysed and analyzed by WB using antibodies against CLIP170 and GAPDH (loading control) (B) or infected and subjected to in situ uncoating assay as in (A). (C) Scatterplot of maximum p24 intensity of fused viral particles normalized to the control mean is shown. Each dot represents one virion, and red lines indicate means. The number of viral particles analyzed per condition from three independent experiments is indicated. Statistical significance was determined by t‐test. ***P < 0.001.
-
DRepresentative WB (n = 3) showing the amounts of soluble and particulate p24 CA in Fate‐of-capsid assay (FoCA) in siRNA‐treated 293A cells infected with HIV-1‐VSV-luc from Fig 3D.
-
E, F293A cells transfected with control or CLIP170nc2 siRNAs were infected and subjected to FoCA assay as in (D). (E) Graph shows amounts of pelleted p24 CA normalized to the input CA at 3 h.p.i. Statistical significance was determined by one‐way ANOVA. *P‐value ≤ 0.05, **P‐value ≤ 0.01. Mean ± SEM from three independent experiments is shown. (F) Representative WB is shown.
To independently verify these findings, a FoCA uncoating assay was used wherein soluble HIV‐1 CA proteins are separated from particulate (pelletable) CA by sedimentation through a sucrose cushion. To do this, 293A or CHME3 cells depleted for EB1 or CLIP170 were infected for 3 hours (h). High concentrations of the small molecule inhibitor, PF74, were used as a control for HIV‐1 core destabilization. In line with the in situ uncoating assay, depletion of either +TIP increased the amount of intact CA in the pellet compared with control siRNA‐treated cultures in both cell types, while PF74 destabilized HIV‐1 cores (Figs 3C and D, and EV2D–F). This demonstrated that both EB1 and CLIP170 were required for uncoating of incoming HIV‐1 cores, but that CLIP170 does so in the absence of effects on Ac‐MT induction.
CLIP170, but not EB1, binds to intact HIV‐1 cores
We next tested whether EB1 or CLIP170 interacts with HIV‐1 particles by using in vitro assembled capsid–nucleocapsid (CA‐NC) complexes. These are a mixture of cylinders and cones composed of hexamers and are closed by incorporation of pentamers at both ends similar to authentic viral cores (Ganser et al, 1999). Extracts of cells expressing Flag‐tagged EB1 or CLIP170 were incubated with or without in vitro assembled CA‐NC complexes that were then sedimented over a 70% sucrose cushion. Neither +TIP pelleted in the absence of CA‐NC complexes. Binding specificity to CA‐NC complexes was additionally confirmed using extracts from cells expressing the housekeeping protein GAPDH or a known CA interactor FEZ1 as negative and positive controls, respectively (Fig 4A) (Malikov et al, 2015; Delaney et al, 2017). Notably, while EB1 failed to associate with HIV‐1 CA‐NC complexes, CLIP170 pelleted with CA‐NC complexes (Fig 4B–D, respectively). To validate the relevance of CA‐NC assemblies, lysates from Flag and Flag‐CLIP170 expressing cells were incubated with fractions harboring intact HIV‐1 cores (fractions #8 and #9, Fig 4E) and subjected to co‐sedimentation (Shah & Aiken, 2011). Similar to CA‐NC assemblies, Flag‐CLIP170 specifically associated with fractions containing intact viral cores but not with mock fractions (Fig 4F). IF staining of Flag‐CLIP170‐expressing CHME3 cells infected with HIV‐1‐VSV‐luc further confirmed that CLIP170 was present in the cytosol and therefore likely to be accessible to incoming viral cores (Fig EV3A). Significant colocalization was not observed, likely because data below suggest that individual cores bind to only a small number of CLIP170 molecules. Cumulatively, these data validated the use of CA‐NC assemblies to study host protein interactions with viral cores and suggested that CLIP170 bound HIV‐1 cores independently of EB1, establishing a second divergence in how these two proteins functioned during early infection.
Figure 4. CLIP170, but not EB1, binds to HIV‐1 cores.
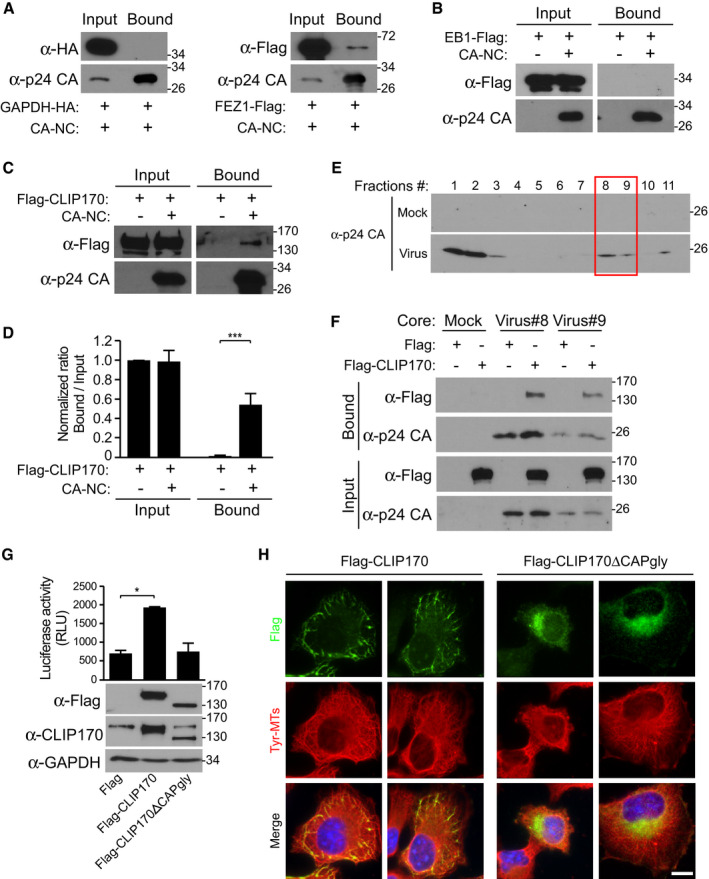
-
A–CRepresentative (n = 3) WB analysis demonstrating that while GAPDH‐HA control and EB1‐Flag do not bind ((A), upper panels and (B), respectively) FEZ1‐Flag control and Flag‐CLIP170 ((A), lower panels and (C), respectively) pelleted with in vitro assembled HIV‐1 CA-NC complexes.
-
DQuantification of the levels of bound CLIP170 as a ratio of total protein across experimental replicates (n = 3) from (C). Statistical significance was determined by t‐test (mean ± SEM) ***P < 0.001.
-
ERepresentative WB analysis (n = 3) showing the levels of HIV‐1 p24 CA in mock and virus fractions separated by sucrose gradient centrifugation. Fractions 1‐3 contain free p24 not associated with the intact cores, while fractions #8 and # 9 harboring intact HIV‐1 cores.
-
FFlag‐CLIP170, but not Flag control, also binds to fractions containing intact HIV‐1 cores (viruses #8 and #9) but not with Mock fractions.
-
GHIV-1‐WT-luc infection is increased in CHME3 cells stably expressing Flag‐CLIP170, but not control Flag or Flag‐CLIP170ΔCAP‐gly. Statistical significance was determined by one‐way ANOVA. *P < 0.05. Mean ± SEM from three independent experiments is shown.
-
HRepresentative images (n = 3) of CHME3 expressing Flag‐CLIP170 or Flag‐CLIP170ΔCAP‐gly stained for Flag and Tyr‐MTs. Scale bar, 10 μm.
Figure EV3. CLIP170 localizes proximal to incoming HIV‐1 particles.
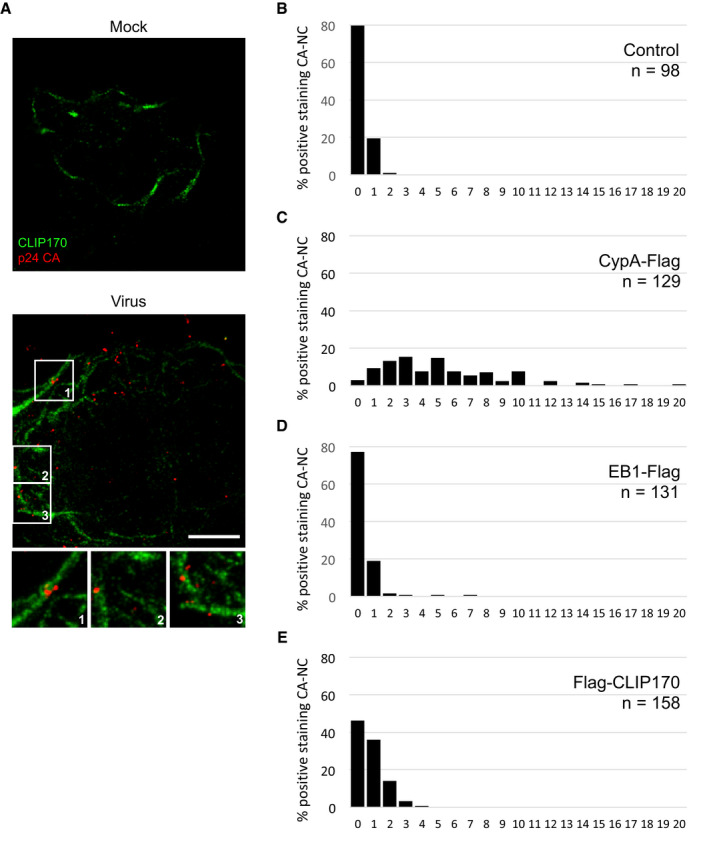
-
ACHME3 cells stably expressing CLIP170‐Flag were infected with Mock or HIV-1‐VSV-luc for 20 min then stained with anti‐Flag and anti‐p24 CA (AG3.0) antibodies. Maximum projection of a confocal stack of representative images (n = 3) is shown. Scale bar, 10 μm.
-
B–ECLIP170 binds to HIV‐1 CA-NC. Quantification of amounts of staining associated with each CA‐NC complex in samples from Fig 5A–C. Number of CA‐NC complexes (n) is indicated per sample.
Yet, earlier results showed that its CAP‐Gly‐binding EEY motif was important for EB1 to promote infection, raising the possibility that EB1 delivers +TIPs to the cell periphery that promote infection. Indeed, the CAP‐Gly domain has been shown to be required for CLIP170 transport to MT plus‐ends through its interaction with EB1 or α‐tubulin (Honnappa et al, 2006). To test this, CHME3 cells stably expressing Flag‐tagged WT CLIP170 or a CLIP170 mutant lacking its two consecutive CAP‐Gly domains (CLIP170ΔCAP‐gly) were infected with HIV‐1‐WT‐luc. Overexpression of WT CLIP170 significantly increased early HIV‐1 infection (Fig 4G) in line with our earlier findings using depletion approaches. By contrast, CLIP170ΔCAP‐gly expressing cells exhibited similar levels of infectivity to Flag controls. It should be noted that the lower expression of the CLIP170 mutant compared to that of the WT CLIP170 is not likely to explain the inability of this mutant to promote early infection for a number of reasons. First, the mutant still expresses to a relatively high level. As such, if it were functional in any way one would expect to see a partial increase in infection with this mutant, but it had no effect (Fig 4G). Second, this finding is in agreement with earlier data showing that increasing expression of the EB1 ΔEEY mutant lacking the CAP‐Gly binding domains also failed to promote early infection (Fig 1D), independently supporting the notion that CAP‐Gly‐based interactions are functionally important to infection. IF staining of Flag‐tagged CLIP170 or CLIP170ΔCAP‐gly in CHME3 cells together with tyrosinated‐MTs further revealed that in contrast to CLIP170 that accumulated on peripheral ends of MTs, CLIP170ΔCAP‐gly exhibited diffuse staining at perinuclear regions (Fig 4H). While we cannot rule out a role for α‐tubulin association in the failure of the CLIP170ΔCAP‐gly mutant to localize to MT plus‐ends, our cumulative findings support the idea that EB1 functions, in part at least, by enhancing the accumulation of CLIP170 at the cell periphery where it then binds incoming virus particles. Moreover, the interaction of CA‐NC assemblies and/or intact HIV‐1 cores with multiple +TIPs, including CLIP170, but not with EB1 itself, provided the first hints that HIV‐1 particles may interact with +TIPs in a manner similar to EB1 itself.
CLIP170 binds to HIV‐1 cores in a manner distinct from currently known capsid binders
To better understand how CLIP170 interacts with HIV‐1, extracts from cells expressing Flag‐CLIP170 along with control Flag‐tagged CypA, a known HIV‐1 CA interactor (Luban et al, 1993), were incubated with CA‐NC assemblies. These were then pelleted and stained using anti‐Flag, anti‐CLIP170, or anti‐p24 CA antibodies followed by super‐resolution microscopy (SIM). These assemblies appear in images as both individual tubes and overlapping tubes and aggregates that stain for CA protein (Fig 5A). Whereas CypA bound to sites located along the length of tubular CA‐NC structures, CLIP170 bound to more discrete sites (Fig 5A). We further quantified the number of CA‐NC tubes that were positive for either protein, using untransfected cells and EB1‐Flag‐expressing cells as controls for background non‐specific staining in this assay (Fig 5B and C, EV3B–E). Quantification showed that most tubular CA‐NC structures bound CypA, while 50–60% bound CLIP170. By contrast, EB1 did not interact with these complexes above background levels (Figs 5B and C, and EV3B–E). While it is possible that these different staining patterns between CypA and CLIP170 reflect differences in CA‐NC binding affinity in these assays, the pattern of CLIP170 staining was consistent and usually amounted to one or two bound CLIP170 molecules per tube. This suggests that regardless of any affinity differences, the site to which CLIP170 binds is also likely to be different to that of CypA.
Figure 5. CLIP170 binds to HIV‐1 CA‐NC in a manner distinct from currently known capsid binders.
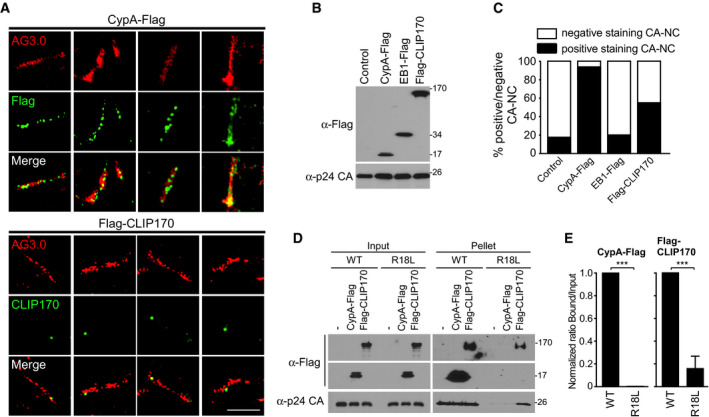
- Super‐resolution microscopy (SIM) imaging of Flag‐tagged proteins, cyclophilin A (CypA‐Flag), EB1‐Flag, or Flag‐CLIP170 bound to in vitro assembled CA‐NC complexes, stained with anti‐p24 AG3.0 (Red), anti‐Flag, or anti‐CLIP170 (Green). Scale bar, 2 μm. Representative images from different fields of view are shown.
- WB analysis confirming protein expression in samples from (A) and (C).
- Quantification (n = 3) of the association of each protein with 97 to 137 CA‐NC complexes, using untransfected lysates (Control) and EB1‐Flag as controls for non‐specific background staining.
- Lysates from untransfected, CypA‐Flag-, or Flag‐CLIP170‐transfected cells were subjected to CA‐NC binding assays and WB analysis.
- Levels of CypA or CLIP170 bound to WT or R18L CA‐NC assemblies in samples from (D) were quantified using Fiji (n = 4 experimental replicates; data are presented as a ratio of CypA or CLIP170 in bound versus input for each CA‐NC assembly, normalized to WT CA‐NC arbitrarily set to 1). Statistical significance was determined by t‐test (mean ± SEM). ***P < 0.001.
Notably, as CA‐NC tubes nucleate they occasionally incorporate pentamers rather than hexamers, which alters curvature in the assembled tube (Ganser‐Pornillos et al, 2004). Local curvature may influence how and where host factors bi nd to viral cores. To test this, we examined the ability of CypA or CLIP170 to bind to WT or Arg18‐Leu mutant (R18L) CA‐NC assemblies, as R18 mutant CA has been shown to have a higher propensity to incorporate pentamers into the assembling hexameric tubes (Ganser‐Pornillos et al, 2004). Binding assays showed that CypA bound to WT CA‐NC tubes more efficiently than CLIP170, which was in line with SIM imaging showing that fewer CLIP170 molecules decorate CA‐NC tubes (Fig 5D and E). However, CLIP170 both bound to and increased the amount of pelletable R18L CA‐NC assemblies. By contrast, CypA did not interact with R18L CA‐NC assemblies, as shown by others (Liu et al, 2016), and did not enhance the levels of pelletable R18L compared with untransfected controls (Fig 5D and E). This further supports the notion that CLIP170 regulates the stability of viral cores, and suggests that its interaction is based on curvature within the core. Moreover, these findings demonstrated that CLIP170 binds to HIV‐1 cores in a manner that is fundamentally different from currently known capsid‐binding cofactors, such as CypA.
The MHR of HIV‐1 capsid contains a functional EB1‐mimetic motif
Prompted by these observations, amino acid alignments and structure modeling revealed that the MHR of HIV‐1 CA assemblies had significant similarity to the N‐terminus of EB1's EBH domain, which contains binding sites for both CAP‐Gly and SxIP motifs (Figs 6A and B, and EV4A). While the MHR domain is oriented inside the capsid core, partial uncoating would explain its accessibility to CLIP170 in native cores observed earlier (Fig 4E). Moreover, CA‐NC tubular structures elongate from both ends as an open cylinder (Barklis et al, 2009), effectively mimicking partial uncoating and providing similar accessibility to host cofactors. Accessibility to the MHR is likely different within curved regions of the core, as discussed below, and may therefore explain why CLIP170's CA‐NC binding pattern is more specific and very distinct from that of CypA. To test the functionality of this motif, we fused the HIV‐1 MHR to HA‐tagged GAPDH (GAPDH‐MHRhiv) and tested if it conferred GAPDH with the ability to interact with CLIP170. To do this, we co‐transfected 293T cells with Flag‐CLIP170 along with either HA‐tagged GAPDH or GAPDH‐MHRhiv followed by anti‐HA co‐immunoprecipitation (co‐IP). In doing so, we found that GAPDH‐MHRhiv‐HA bound to CLIP170, while GAPDH‐HA did not (Fig 6C). Notably, CLIP170 was recently shown to use its SxIP motifs, rather than its CAP‐Gly domains, for its interaction with the EB1 EBH domain (Chen et al, 2019). In line with this, viral particles did not reach the nucleus as efficiently in cells expressing an SxIP mutant of CLIP170 (CLIP170ΔSxIP) compared with WT CLIP170 (Fig EV4B–D). Particle translocation was only partially impaired in cells expressing the CLIP170ΔSxIP mutant but this is not unexpected given the complex nature by which CLIP170 uses multiple domains to interact with EB1. However, these findings further validated the importance of the SxIP motif in early infection.
Figure 6. The MHR of HIV‐1 capsid encodes an EB1‐mimetic motif.
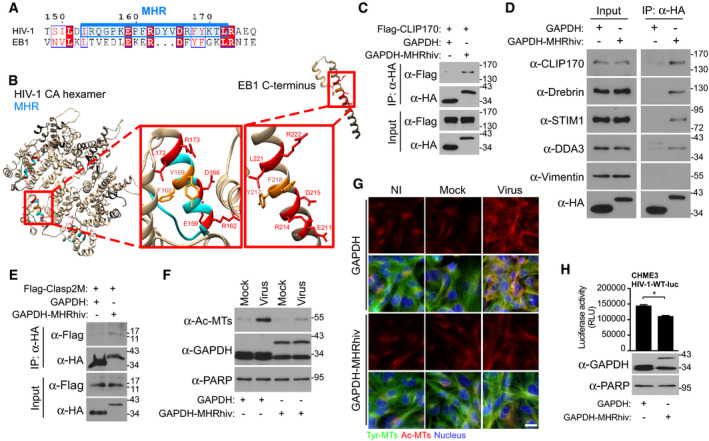
-
ASequence similarities of the EB1 C‐terminus with MHR (highlighted in blue) in HIV‐1 (NL4‐3). Identical residues are highlighted in red, and similar residues are printed in red.
-
BStructure modeling of HIV‐1 capsid hexamers with focus on MHR alongside C‐terminus of EB1 in red boxes. Identical residues are indicated in red, and similar residues are indicated in dark orange. UCSF Chimera 1.12 software was used for structure modeling of NL4‐3 hexamer (PDB 3J3Q) and EB1 (PDB 2HKQ).
-
C–EAnti‐HA co‐IP showing that GAPDH fused to the HIV‐1 MHR (GAPDH‐MHRhiv), but not HA‐tagged GAPDH (GAPDH) binds to Flag‐CLIP170 (C), endogenous forms of various human SxIP‐containing +TIPs, but not vimentin (D) or to Flag‐tagged SxIP fragment (Flag‐Clasp2M) (E) in 293T cells. Results are representative of three experimental replicates.
-
F, GHIV-1‐VSV-luc (Virus), but not uninfected (NI) or Mock, infection induces the formation of Ac‐MTs in CHME3 expressing GAPDH‐MHRhiv, but not GAPDH, as shown by WB analysis (F) or imaging at 6 h.p.i (G). Scale bar, 15 μm. Representative fields are shown. Results are representative of three experimental replicates.
-
HGAPDH‐MHRhiv expression in CHME3 reduces infection with HIV-1‐WT-luc. Statistical significance was determined by t‐test. *P < 0.05. Data are mean values from three independent experiments ± SEM.
Figure EV4. The MHR of HIV‐1 capsid encodes an EB1‐mimetic motif.
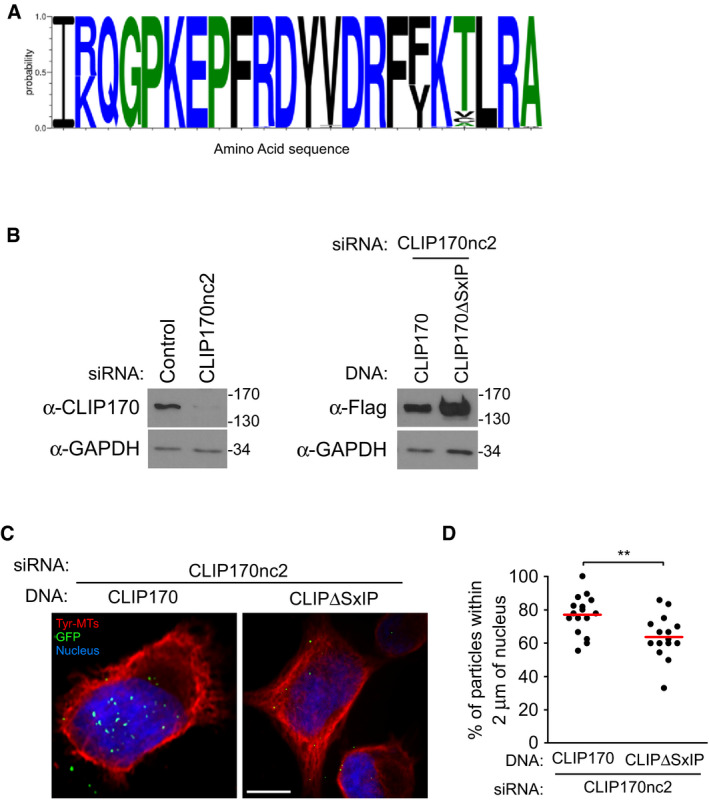
-
AEB1 mimetic residues within the MHR of HIV‐1 capsid display a significant degree of conservation across different HIV‐1 strains. Sequence similarities of 88 HIV‐1 MHR from different subtypes obtained from the Los Alamos database. Color scheme indicates hydrophobicity.
-
B–DAn SxIP mutant CLIP170 is impaired in its ability to promote viral translocation to the nucleus. 293A cells were treated with siRNA targeting the 3′UTR of CLIP170 (CLIP170nc2) to limit the influence of endogenous CLIP170 on assays, followed by transfection with either Flag‐tagged WT (Flag‐CLIP170) or mutant CLIP170 lacking SxIP motif (Flag‐CLIP170ΔSxIP) constructs after 24 h. Cells were then infected with HIV‐1-VSV‐GFP-Vpr. (B) WB analysis using anti‐CLIP170 or anti‐Flag antibodies confirms depletion of endogenous and expression of exogenous CLIP170, respectively. (C) Representative staining images for tyrosinated tubulin (Tyr‐MTs), viral particles (GFP), and the nucleus (Hoescht) are shown. Scale bar, 10 μm. (D) Percentage viral particles within 2 μm of the nucleus at 4 h.p.i, scatterplot with mean of ≥ 80 virus particles quantified in ≥ 16 cells per sample. Statistical significance was determined by t‐test. **P < 0.01.
Given these findings, we next tested whether the HIV‐1 MHR could interact with a broader range of +TIPs with SxIP motifs. Co‐IP assays revealed that GAPDH‐MHRhiv‐HA, but not GAPDH‐HA control, interacted with the endogenous forms of several SxIP‐containing +TIPs tested, CLIP170, drebrin, stromal interaction molecule 1 (STIM1), and DDA3 (Jiang et al, 2012; van de Willige et al, 2016), but not a non‐SxIP protein, vimentin (Fig 6D). To independently validate the specific HIV MHR interaction with the SxIP motif, we synthesized a Flag‐tagged peptide (Clasp2M) containing two SxIP motifs (Chen et al, 2019). In co‐transfected cells, GAPDH‐MHRhiv‐HA, but not GAPDH‐HA control, bound to the SxIP peptide (Fig 6E). As such, the presence of an EBH‐like motif would explain how HIV‐1 can engage several +TIPs during early infection, some of which regulate MT stabilization while others, like CLIP170 operate independently of Ac‐MTs. We therefore tested whether GAPDH‐MHRhiv‐HA expression might affect HIV‐1 infection, potentially by acting in a dominant negative manner over incoming viral particles. In line with this, while HIV‐1 infection induced the formation of Ac‐MTs in control GAPDH‐HA‐expressing CHME3 cells, expression of GAPDH‐MHRhiv‐HA caused a notable reduction in Ac‐MT formation during infection (Fig 6F and G). Moreover, when these cells were infected with HIV‐1‐WT‐luc reporter viruses, we found that GAPDH‐MHRhiv expression caused a significant reduction in infectivity (Fig 6H). While these effects were not as robust as depleting +TIPs such as EB1 or CLIP170, this would not be unexpected for a dominant negative. Collectively, these findings highlight that the HIV‐1 capsid MHR contains a +TIP‐binding motif that enables it to bind a variety of +TIPs and control early events in infection.
Divergent EB1 mimicry across retroviruses
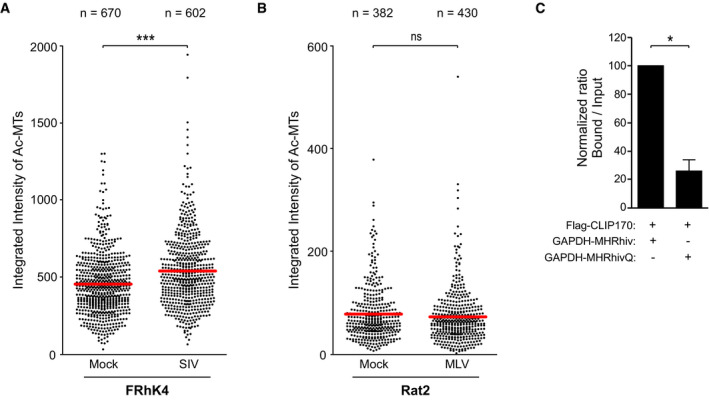
While certain residues in the MHR motif display significant conservation across retroviruses and function later in retroviral assembly, why other MHR residues vary and whether this is functionally significant remains unknown (Tanaka et al, 2016). We therefore examined similarities to EB1 in MHRs from HIV‐1, simian immunodeficiency virus (SIV) and MuLV. Sequence analysis revealed that the MHRs of SIV and MuLV CA exhibited differences in key amino acids from that of HIV‐1 MHR and the core SxIP‐binding motif within the EBH domain of EB1 from different species (Honnappa et al, 2009), with the greatest divergence being in the MuLV MHR (Fig 7A). To test the potential functional significance of this, CHME3 cells stably expressing HA‐tagged GAPDH fused with HIV‐1, SIV, or MuLV MHRs were tested for interaction with CLIP170 by co‐IP analysis (Fig 7B and C). Results showed that only GAPDH‐MHRhiv was able to bind endogenous human CLIP170 in CHME3 cells to any significant extent. CLIP170 is highly conserved both structurally and functionally across species. Indeed, rat CLIP170 was used in our earlier overexpression studies while the same functionality of its human counterpart was confirmed by the effects of depleting endogenous human CLIP170 in various human cell types, and the HIV‐1 MHR bound both rat CLIP170 and human CLIP170 (Fig 6C and D). However, it remained possible that SIV or MuLV MHRs might exhibit more restricted species‐specific functionality. To test this, we examined rhesus macaque FRhK4 cells stably expressing GAPDH‐HA or GAPDH‐MHRsiv‐HA for interaction with CLIP170 by co‐IP analysis. In doing so, we noted that the SIV MHR did indeed bind CLIP170 in monkey cells (Fig 7D). By contrast, we were unable to detect interactions between the GAPDH‐MHRmulv‐HA and CLIP170 in rat fibroblast Rat2 cell line, despite recovery of CLIP170 in anti‐EB1 co‐IPs (Fig 7E). While this does not rule out some degree of binding below our detection limits, it is in line with the greater degree of sequence divergence across the MuLV EBH‐like region. Moreover, we found that the level of CLIP170 binding activity that these assays detected also correlated with the extent to which each virus exploits +TIPs. We tested this first by measuring the levels of Ac‐MTs in FRhK4 or Rat2 cells infected with VSV‐G pseudotyped SIV or MuLV luciferase reporter viruses, respectively. Both IF and WB analyses showed that SIV induced formation of Ac‐MTs in macaque cells (Figs 7F and H, and EV5A respectively), albeit not as robustly as HIV‐1 in human cells (Fig 6F), while MuLV did not induce detectable levels of Ac‐MTs in rodent cells (Figs 7G and I, and EV5B respectively). To test whether these viruses depended on CLIP170 for infection, FRhK4 or Rat2 cells were depleted of CLIP170 and then infected with SIV‐VSV‐luc or MuLV‐VSV‐luc, respectively. Infectivity assays revealed that CLIP170 depletion in FRhK4 resulted in a significant decrease in SIV infection compared with controls, while it had no effect on MuLV infection in Rat2 cells (Fig 7J and K, respectively). Together, our findings show that the lentiviruses, HIV‐1 and to a lesser extent SIV, encode EBH‐like MHR domains that enable them to exploit +TIPs and control induction of Ac‐MTs, while the retrovirus MuLV does not.
Figure 7. Differential EB1‐mimicry in MHR domains across retroviruses.
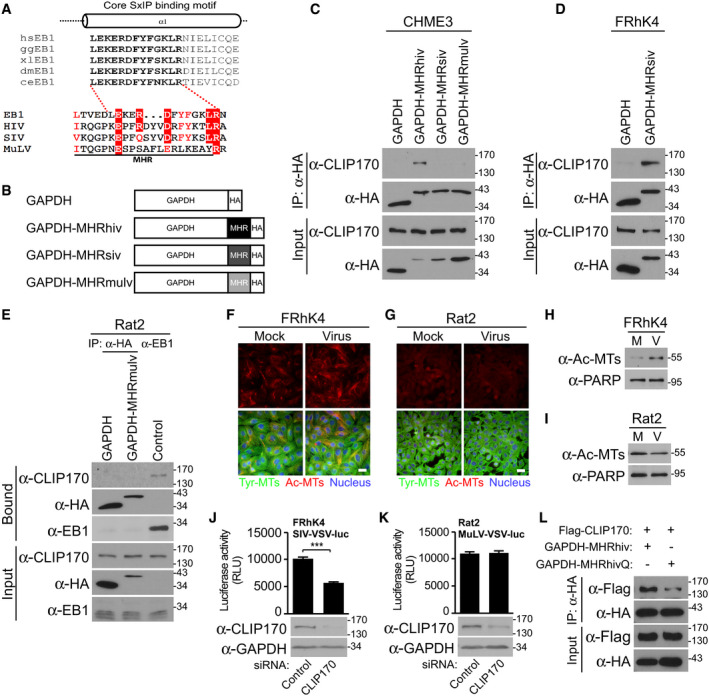
-
ASequence similarities of MHR in HIV‐1 (NL4‐3), SIVmac239, and MuLV with the core SxIP‐binding motif (bold text) within the EBH domain of EB1 from different species, adapted from Honnappa et al (2009). Residues identical to those in EB1 are highlighted in red, and similar residues are printed in red.
-
BSchematic representation of HA‐tagged GAPDH constructs, with MHR from HIV‐1 (GAPDH‐MHRhiv), SIVmac239 (GAPDH‐MHRsiv), or MuLV (GAPDH‐MHRmulv).
-
CAnti‐HA co‐IP in CHME3 expressing GAPDH‐MHRhiv, GAPDH‐MHRsiv, or GAPDH‐MHRmulv showing binding to endogenous CLIP170. Results are representative of three experimental replicates.
-
D, EAnti‐HA co‐IP in Rhesus macaque FRhK4 (D) or rat fibroblast Rat2 (E) cells expressing GAPDH alongside GAPDH‐MHRsiv or GAPDH‐MHRmulv, respectively. Anti‐EB1 co‐IP was included as positive control to demonstrate CLIP170 binding by EB1 in Rat2 cells. Results are representative of three experimental replicates.
-
F–IFRhK4 cells infected with SIV‐VSV-luc (F) or Rat2 cells infected with MuLV-VSV‐luc (G) stained for Ac‐MTs, Tyr‐MTs, and the nucleus (Hoechst) at 6 h.p.i, or analyzed by WB (H and I, respectively). Scale bar, 20 μm. Representative fields are shown. Results are representative of three experimental replicates.
-
J, KMeasurements of SIV‐VSV-luc or MuLV‐VSV-luc infectivity in FRhK4 (J) or Rat2 (K) cells depleted of CLIP170. Statistical significance was determined by t‐test. ***P < 0.001. Data are mean values from two independent experiments ± SEM.
-
L293T transfected with GAPDH‐MHRhiv or R162Q mutant (GAPDH‐MHRhivQ) was subjected to anti‐HA co‐IP. Representative WB analysis is shown (n = 2).
Figure EV5. Differential EB1 mimicry in MHR domains across retroviruses.
-
A, BQuantification of Ac‐MT fluorescence intensity in FRhK4 cells infected with SIV‐VSV-luc (A) or Rat2 cells infected with MuLV-VSV‐luc (B) and stained for Ac‐MTs and Tyr‐MTs in Fig 6F and G, respectively. Each dot represents one cell. The number of cells quantified (n) is indicated for each sample. The red line indicates the mean. Statistical significance was determined by t‐test. ***P < 0.001.
-
CQuantification of the levels of bound Flag‐CLIP170 as a ratio of total protein across experimental replicates (n = 3) in Fig 7L. Statistical significance was determined by t‐test, and data are shown as mean ± SEM. *P < 0.05.
The fact that the SIV MHR was more divergent from the EBH domain and did not bind CLIP170 in human cells suggested that it had less optimal EBH mimetic activity than its HIV counterpart (Fig 7A). Among the most conserved residues with homology to EB1, the HIV‐1 and SIV MHRs only differed by a positively charged arginine (R) substitution to an uncharged glutamine (Q) at position 162. To test whether this specific amino acid difference was functionally significant, we replaced R162 with Q in GAPDH‐MHRhiv‐HA (MHRhivQ) and tested its ability to bind Flag‐CLIP170 by co‐IP in 293T cells. Compared to MHRhiv, the R162 mutant exhibited reduced binding to CLIP170 (Figs 7L and EV5C). This identifies R162 as an important residue underlying the optimized EB1‐mimetic activity of the HIV‐1 MHR.
Discussion
While we recently showed that HIV‐1 exploits EB1 and associated +TIPs to stabilize MTs and promote early infection (Sabo et al, 2013; Delaney et al, 2017), here we reveal new insights into EB1's complex multifunctionality and the roles played by different +TIPs. Indeed, our data suggest that beyond its direct effects on MT dynamics, a critical function for EB1 is the delivery of +TIPs such as CLIP170 to the cell periphery where they are subsequently engaged by incoming viral cores to facilitate early HIV‐1 infection.
Our data suggest that CLIP170 plays a complex, multifunctional role in early HIV‐1 infection. Like EB1, CLIP170 influences MT dynamics, yet our data suggest that unlike EB1 and other +TIPs identified to date, CLIP170 regulates infection without significantly affecting the formation of acetylated MTs. Although our data do not rule out contributions from its effects on MT dynamics, CLIP170's established roles in actin‐MT linkage and cargo capture may make it particularly important for the loading of incoming viral cores onto MTs to initiate transport (Honnappa et al, 2006; Weisbrich et al, 2007; Jovasevic et al, 2015). Indeed, we observed that CLIP170 bound viral cores in vitro and colocalized with incoming viral particles, while mutants revealed that it needed to be peripherally localized in order to promote infection.
In terms of how CLIP170 concentrated at the periphery to engage incoming viral particles, we find a critical role for EB1's EEY motif that dominates CAP‐Gly interactions over its EBH domain, which contains a less well‐understood binding site for CAP‐Gly‐ and SxIP‐containing +TIPs (Chen et al, 2019). It is interesting that the HIV‐1 MHR mimics this latter EBH domain, and interacts with a range of SxIP‐containing +TIPs including CLIP170. This may avoid competition with EB1 itself so as to gain access to CLIP170 once delivered to the periphery, or distinct binding strategies may enable an efficient “hand‐over” from EB1 to HIV‐1 cores. Regardless of the functional reasons for its choice, the fact that HIV‐1 mimics the EBH domain reveals an unexpected biological context in which this domain is functionally important. Mimicking a +TIP‐binding motif in EB1 would enable virus particles to engage a range of peripherally localized +TIPs and control various aspects of cytoskeletal dynamics, precisely as EB1 does (Gouveia & Akhmanova, 2010; Jiang & Akhmanova, 2011).
Most strikingly perhaps, CLIP170 was found to bind cores in an unusual manner, distinct from that of CypA, and regulate their stability. Our cumulative data suggest that this involves the MHR and is influenced by curvature in the viral core that is determined by hexamer and pentamer ratios. Notably, areas of capsid with a higher concentration of pentamers have been suggested to be less stable than hexamer‐rich regions (Pornillos et al, 2011), and we find that CLIP170 specifically binds and stabilizes R18L assemblies. Recent reports also reveal novel interactions between neighboring CA monomers within pentamers that are critical for HIV‐1 core assembly, suggesting pentamers may form binding pockets that could serve as antiviral drug targets (Craveur et al, 2019). Such configurations or localized organization of pentamers and hexamers at sites of curvature may expose the MHR domain in a manner that enables its recognition by CLIP170, once partial uncoating has begun. Indeed, given the instability of cores with higher levels of pentamers (Pornillos et al, 2011), it is tempting to suggest that pentamer‐rich regions might initiate the first stages of uncoating and are recognized by factors such as CLIP170, to prevent more complete disassembly from occurring prematurely. While this is currently challenging to test directly, single‐particle uncoating kinetics of authentic HIV‐1 capsids show that the capsid‐binding inhibitor PF74 accelerates capsid opening but stabilizes the remaining lattice, with rapid uncoating in the absence of capsid binders (Marquez et al, 2018). As such, our findings lend further support to the idea that uncoating is a highly dynamic multi‐step process that begins in the cytoplasm, before completing in the nucleus, and is regulated by distinct host cofactors, such as CLIP170.
Finally, divergence in the extent to which the MHR from HIV‐1, SIV, and MuLV functionally mimic the EBH domain speaks to the broader functional importance of this region and differences in how retroviruses exploit +TIPs. This functional divergence was reflected in both the requirement of these viruses for CLIP170 and their ability to induce Ac‐MT formation, which is mediated by other +TIPs. MHR differences may have evolved for specific reasons. For example, MT stabilization prevents cell division, and unlike lentiviruses, MuLV requires cell division for infection (Yamashita & Emerman, 2006). As such, although stable MTs provide efficient tracks for virus transport, their effects on cell division may have driven divergence in this specific MHR function. However, despite its reduced level of EB1 mimicry it is possible that other, as yet unidentified +TIPs or a completely different class of host cofactors bind the MuLV MHR to fulfill this capsid‐binding function for other retroviruses. Future studies will undoubtedly extend our understanding of the diverse nature and functionality of capsid‐binding host factors in early retroviral infection.
Materials and Methods
Cells
Human embryonic kidney cells (293T and 293A), rat fibroblast cells (Rat2), and CHME3 and NHDF cells were previously described (Naghavi et al, 2007; Malikov et al, 2015). The monocytic line THP‐1 and rhesus macaque kidney cells (FRhK4) were kindly provided by Thomas Hope. THP‐1 cells were differentiated into macrophages by addition of PMA as described (Delaney et al, 2017). FRhK4 were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS and glutamine (4 mM).
Virus production and infections
HIV‐1 carrying a luciferase reporter or ZsGreen marker and pseudotyped with VSV‐G (HIV‐1‐VSV‐luc or HIV‐1‐VSV‐ZsGreen, respectively), WT (HIV‐1‐WT‐luc), or MuLV amphotropic envelope (HIV‐1‐Ampho‐luc) were generated by co‐transfection of 293T cells with pNL4‐3.luc.R‐.E‐ (AIDS Reagent Repository #3418) or pNL4‐3.ZsGreen.R‐.E‐ together with pVSV‐G, pC1‐env, or pHit456, respectively, as previously described (Delaney et al, 2017). To generate VSV‐G pseudotyped SIV or MuLV viruses carrying a luciferase reporter (SIV‐VSV‐luc and MuLV‐VSV‐luc, respectively), 293T cells were co‐transfected with pVSV‐G along with either SIVmac239.luc‐E‐ or MoMuLV Gag‐Pol expression vector (pCMV‐intron) and pFB‐luc (Sabo et al, 2013), respectively. All transfections were carried out using polyethylenimine (PEI from Polysciences) (Delaney et al, 2017). For live imaging, GFP‐Vpr‐labeled viral particles pseudotyped with WT envelope (HIV‐1‐WT‐GFP‐Vpr) were generated by co‐transfection of pNL4‐3.luc.R‐.E‐ with GFP‐Vpr and pC1‐env into 293T cells using PEI. Double‐labeled virus used in in situ uncoating assay were produced by co‐transfecting 293T cells with 6 μg pNL4‐3.luc.R‐.E‐, 3 μg pVSV‐G, 1 μg GFP‐Vpr, and 0.44 μg S15‐tomato plasmid as described (Malikov et al, 2015). All infections were carried out using 10 μg/ml polybrene (Santa Cruz). Cells were lysed 48 h.p.i. and assayed for luciferase activity or detached and fixed with 4% paraformaldehyde (PFA) for FACS analysis. HSV‐1 virus production and infection have been previously described (Jovasevic et al, 2015).
Generation of expression constructs and stable pools
Expression constructs containing the N‐terminal CH domain of EB1 (Nterm), CH‐CC domains (Nterm+CC), or CH‐CC‐EBH domains (Nterm+CC+EBH) were generated by Gibson Assembly (Gibson et al, 2009). Double‐stranded genomic DNA fragments (gBlocks) deleted of the domains of interest were synthesized by Integrated DNA Technologies (IDT). gBlocks and linearized pQCXIN‐EB1‐Flag (Sabo et al, 2013) were incubated with Gibson Assembly Master Mix (New England Biolabs) at 50°C for 1 h to ensure homologous recombination. The GFP‐tagged rat CLIP170 construct (Procter et al, 2018) was subcloned into pQCXIN (Clontech) using AgeI and EcoRI, while Flag was introduced by PCR in the N‐terminus (Flag‐CLIP170). Gibson assembly was also used to generate a Flag‐tagged CLIP170 construct depleted of the second C‐terminal zinc knuckle (Flag‐CLIP170ΔZn2). The IDT‐synthesized gBlock was incubated with the linearized pQCXIN‐Flag‐CLIP170 and the Gibson Assembly Master Mix as described above. In order to delete both CAP‐Gly domains from CLIP170, consecutive overlapping PCR was performed on the pQCXIN‐Flag‐CLIP170. Following primers were used as follows: Fw_Clip_NotI (5′‐ACGCTGCAGGAATTGATCCG‐3′) and Re_delCAP‐gly‐Clip (5′‐ACTTTGTGGACAAGAAACTGGATAAATCCAGGTTTATTCCCGTTCACCCAAACACG‐3′), as well as Fw_delCAP‐gly‐Clip (5′‐TCCAGTTTCTTGTCCACAAAGTGACAAAGATTGGCTTCCCTTCTACCACCCCA‐3′) and Re_clipEco6 (5′‐CTGCTGCCTGCTTTAGGGTTTCT‐3′). Both PCR amplicons were then amplified using Fw_Clip_NotI and Re_clipEco6 and double‐digested using NotI and BsaBI followed by introduction into a NotI‐BsaB1‐digested pQCXIN‐Flag‐CLIP170. A similar approach was used to delete the SxIP motif of CLIP170 (amino acids S‐N‐I‐P). Following primers were used as follows: Fw_Clip_NotI and Rev‐delSXIP‐A (5′‐TTTCTGGGGAGTGGCTGGAGAAGAG‐3′), as well as Fw‐delSXIP‐B (5′‐ACTCCCCAGAAACCGTCCCAGCCAGTG‐3′) and Rev‐delSXIP‐B (5′‐ CTCCAGGCTGTCCTCCTTCTCT‐3′). Both PCR amplicons were then amplified using Fw_Clip_NotI and Rev‐delSXIP‐B and double‐digested using NotI‐HF and BsaBI followed by introduction into a NotI‐BsaB1‐digested pQCXIN‐Flag‐CLIP170. Introduction of the HIV‐1, SIV, or MuLV Major Homology Region (MHR) into HA‐tagged GAPDH was achieved by Gibson assembly (same procedure as described above). IDT‐synthesized gBlocks were homologously recombined with pcDNA3.1(‐)‐GAPDH‐HA (Delaney et al, 2017). The MHRs were fused in the C‐terminus of GAPDH upstream of the HA tag. QuikChange Lightning Site‐Directed Mutagenesis kit (Agilent Technologies) was used to mutate MHRhiv using following primers: Fw_mhrhivQ (5′‐GAC CAA AGG AAC CCT TTC AAG ACT ATG TAG ACC GAT TC‐3′) and its reverse complement. Flag‐tagged CypA was introduced into pQCXIN by PCR amplification of dsRed‐CypA (Francis & Melikyan, 2018) using following primers: CypA‐AgeI‐Fw (5′‐ATGTAGCAACCGGTATGGTCAACCCCACCGTG‐3′) and CypA‐Bam‐Flag‐Re (5′‐TGTACACTGGATCCTTACTTGTCGTCATCGTCTTTGTAGTCTTCGAGTTGTCCACAGTCAGCAATGGTG‐3′). Flag‐tagged Clasp2M construct was generated by PCR amplification of pEGFP‐N1‐CLASP2γ (Akhmanova et al, 2001) construct using following primers: F_ClaspMflag (5′‐ATGgattacaaggatgacgacgataagGGAAGAAGTCGAACAAAAATGGTG‐3′) with Re_ClaspMeco (5′‐TGCTTCATGGGAATTCCTATCGACTTGATTGGCTGATC‐3′). The PCR product was then again amplified using F_ClaspMNot (5′‐acggaagctaGCGGCCGCATGgattacaaggatgacgac‐3′) with Re_Clasp2Meco primer. PCR product was digested by NotI and EcoRI and introduced into pQCXIN. Sequence integrity of each construct was verified by sequencing. pQCXIN‐based retroviruses encoding the gene of interest were generated and used to infect cells, followed by selection to generate stably expressing pools as previously described (Malikov et al, 2015). Protein expression was confirmed by WB analysis with the adequate antibody.
siRNA and DNA transfections
Following siRNA duplexes from Ambion were used to deplete cells of specific proteins: ID# 3891 targeting endogenous EB1, ID# s501556 targeting a non‐coding region of endogenous EB1, ID# 12224 and 142517 targeting CLIP170 (referred to as CLIP170‐II and CLIP170‐III, respectively), ID# s537340 targeting a non‐coding region of endogenous CLIP170 (referred to as CLIP170nc2), and a control non‐targeting siRNA (#AM46357). Knockdown experiments were carried out with CLIP170‐III, unless indicated otherwise. Cell were transfected with 100 pmol siRNA or 10 pmol of Select siRNA using Oligofectamine RNAiMAX transfection reagent (Invitrogen) as previously described (Delaney et al, 2017). Transient overexpression in CHME3 was performed in 12‐well plates with 0, 1, or 1.5 μg DNA along with 1.6 μl PEI. Protein overexpression for binding experiments was carried out in 293T plated in 10‐cm dishes transfected with 10 μg DNA and 22.5 μl PEI. 48 to 72 h post‐transfection, cells were used in different assays as described below. In binding experiments requiring knockdown, 293T cells were plated in 6‐well plates, treated with siRNA for 24 h, then transfected with 7.5 μl PEI and 3.3 μg DNA for another 48 h.
Western blotting
Western blots (WB) were carried out as previously described (Delaney et al, 2017). Following primary antibodies were used to detect specific proteins: rabbit anti‐CLIP170 (Santa Cruz, sc‐25613 or Cell Signaling, #8977), mouse anti‐EB1 (Santa Cruz, sc‐47704), mouse anti‐acetylated tubulin (Sigma, T7451), rat anti‐tyrosinated tubulin YL1/2 (Abcam, 6160), rabbit anti‐Flag (Sigma, F7425), mouse anti‐HIV‐1 p24 CA (Abcam, ab9071), rabbit anti‐HIV‐1 p24 CA (Abcam, ab63917), rabbit anti‐GAPDH (Santa Cruz, sc‐25778), mouse anti‐HA (Sigma, H6098), rabbit anti‐Drebrin (Abcam, ab11068), rabbit anti‐DDA3 (also known as PSRC1) (Abcam, ab187199), rabbit anti‐STIM1 (Cell Signaling, D88E10), rabbit anti‐Vimentin (Cell Signaling, 5741), rabbit anti‐PARP (Cell Signaling, 9542), anti‐HSV, ICP0 (Abcam, ab6513), and anti‐HSV, ICP4 (Abcam, ab6514). Primary antibodies were diluted 1:1,000, while secondary antibodies (HRP‐conjugated anti‐mouse and anti‐rabbit from GE Healthcare UK Limited, HRP‐conjugated anti‐goat from Thermo Scientific, or HRP‐conjugated anti‐rat from Cell Signaling) were diluted 1:10,000.
Live imaging and particle tracking
Live imaging and particle tracking were performed as previously described (Delaney et al, 2017). Briefly, CHME3 cells were grown on 35‐mm glass culture dishes (MatTek) coated with gelatin. Cells were incubated with HIV‐1‐WT‐GFP‐Vpr in CO2‐independent media containing 5% FBS and supplemented with sodium pyruvate and 10 μg/ml polybrene. After spinoculation at 1,200 g for 30 min at 16°C, cells were washed twice with fresh CO2‐independent media to remove non‐attached viral particles, and incubated with polybrene, sodium DL‐lactate (Sigma), and oxyrase (Oxyrase Company). Cells were then imaged in an environmental chamber (InVivo) at 37°C using a 100× oil objective with a Hamamatsu EM‐CCD camera and a Yokogawa CSU‐X1 A1 confocal head on a Leica DMI6000B motorized spinning‐disk confocal microscope run by MetaMorph imaging software (Molecular Devices). For live imaging of viral particles, images were captured every second for a period of 5–10 min. Particle tracking was analyzed with Fiji software using the Mosaic plug‐in. Velocity (displacement over time) was defined as the distance between the starting point and the end point divided by the time that the particle was tracked. Particles tracked for < 30 s were excluded from the calculations. At least three independent experiments were carried out for each condition, and each experiment consisted of at least 7 cells with 4–10 particles per cell. For mitochondria imaging, siRNA‐treated CHME3 cells were incubated with 100 nm MitoTracker® Green (Cell Signaling) for 10 min then imaged every 3 s for a period of 3 min. As a control, CHME3 cells were incubated with 200 μM ciliobrevin D (CbD) or DMSO in CO2‐independent media for 1 h at 37°C, before the addition of MitoTracker® Green for 10 min and imaging.
Immunofluorescence (IF) and Super‐resolution microscopy (SIM)
For IF imaging, cells were seeded on gelatin‐coated coverslips, then fixed with either ice‐cold methanol (when performing MT staining) or 4% PFA, blocked, permeabilized with saponin, and stained (Delaney et al, 2017; Mamede et al, 2017). The samples were incubated with primary antibodies over night at 4°C, then washed, and incubated with the appropriate Alexa Fluor‐conjugated secondary antibody for 1 h at room temperature. After another wash, samples were stained with Hoechst 33342 for 10 min. The coverslips were then mounted on slides using FluorSave (Calbiochem). Following antibodies were used in IF: rat anti‐tyrosinated tubulin (Tyr‐MTs) YL1/2 (Abcam, ab6160), chicken anti‐GFP (Abcam, ab13970), mouse anti‐acetylated tubulin (Sigma, T7451), rabbit anti‐Flag (Cell Signaling, D6W5B), anti‐EB1 (Santa Cruz, sc‐47704), and rabbit anti‐CLIP170 (Santa Cruz, sc‐25613 or Cell Signaling, #8977). All IF images were acquired using a Leica DMI6000 B microscope. Imaging of viral localization and in situ uncoating assay was carried out with a spinning‐disk confocal microscope with Yokogawa CSU‐X1 A1 confocal head and a Hamamatsu EM‐CCD camera run by MetaMorph imaging software. For imaging of MTs, widefield microscopy with a Photometrics Prime 95B camera run by MetaMorph software was used (Delaney et al, 2017). Ac‐MT quantification was assessed using CellProfiler version 3.1.5. For SIM, the bound fractions from CA‐NC binding assay were resuspended in PBS and then spun on poly‐L‐lysine‐coated coverslips (Sigma) for 1 h at 1,200 g at 4°C. The coverslips were extensively washed with PBS and fixed with 4% PFA for 5 min. After incubation with blocking buffer (10% normal donkey serum, 0.01% NaN3 and 0.1% Triton X‐100 in PBS) (Mamede et al, 2017) for 20 min, coverslips were washed with PBS and incubated overnight with primary antibodies diluted 1:1,000 in blocking buffer. Coverslips were stained with Alexa Fluor 488‐conjugated and Alexa Fluor 568‐conjugated secondary antibodies diluted 1:400 for 30 min and then mounted on slides using ProLong™ Gold antifade reagent (Invitrogen). SIM images were taken with a Nikon SIM microscope with a 100× oil objective lens (NA 1.49) at the Cell Imaging Facility & Nikon Imaging Center, Northwestern University in Chicago. Images were reconstructed using NIS‐Elements software (Nikon), and structures were analyzed and quantified using Fiji.
Uncoating assays
The in situ uncoating experiments were performed as previously described (Delaney et al, 2017). siRNA‐treated CHME3 cells were seeded on gelatin‐coated glass coverslips prior to incubation with S15‐tomato‐labeled HIV‐1‐VSV‐GFP‐Vpr (HIV‐1‐DL‐VSV) (> 90% of virus particles were double labeled) followed by spinoculation at 1,200 g for 1 h at 16°C. Infectious media was removed after spinoculation, and cells were incubated with warm media at 37°C for 30 min, 1, or 2 h before fixation with 4% PFA for 15 min. Cells were then washed with PBS, blocked, and permeabilized. Coverslips were incubated overnight at 4°C with monoclonal anti‐HIV‐1 p24 (AG3.0). The next day, cells were washed and incubated with AlexaFluor 647 donkey anti‐mouse for 1 h at room temperature. Images were acquired in an environmental chamber at 37°C using a 100× oil objective with a Hamamatsu EM‐CCD digital camera with a Yokogawa CSU‐X1 A1 confocal head on a Leica DMI6000 B confocal microscope with motorized spinning‐disk. For each time point, a minimum of 20 cells was analyzed. MetaMorph imaging software was used to quantify maximal fluorescence intensity of p24. Background was removed from each analyzed viral particle. Images were analyzed as previously described (Delaney et al, 2017). The fate‐of‐capsid assay was performed as previously described (Delaney et al, 2017). Briefly, 293A or CHME3 cells were seeded into 6‐well plates and treated with siRNA for 72 h. Cells were incubated with HIV‐1‐VSV‐luc in complete media complemented with 10 μg/ml polybrene and spinoculated for 1 h at 1,200 g and 16°C. Fresh virus‐free media containing polybrene and 10 μM MPF74 (Sigma) or DMSO was added to the cells. After a 3 h of incubation at 37°C, cells were incubated for 5 min on ice with 7 mg/ml pronase (Sigma) to remove any virus attached to the cell membrane. Cells were detached, washed 3 times with cold PBS, and lysed in 1 ml hypotonic lysis buffer (10 mM Tris–HCl [pH 8.0], 10 mM KCl, 1 mM EDTA completed with protease inhibitor cocktail [Roche]) on ice for 15 min. Cellular disruption was achieved using a 7 ml dounce‐homogenizer with the “LOOSE” pestle and performing 30 strokes for 293A cells or 80 strokes for CHME3 cells. Lysate was clarified for 5 min at 2,000 g. A 30 μl fraction was removed to analyze in WB as input, while the remaining was loaded on 3.5 ml of 50% sucrose cushion (w/v in PBS) and spun down in a Beckman Coulter SW T55i rotor at 4°C for 2 h at 100,000 g. After ultracentrifugation, 30 μl of the top layer was collected and mixed with 2× Laemmli (referred to as soluble) while the pellet was resuspended in 30 μl 1× Laemmli (referred to as pellet). Samples were subjected to WB analysis to detect HIV‐1 p24 using rabbit anti‐p24 CA antibody (Abcam, ab63917). Knockdown efficiency was assessed by WB analysis using anti‐EB1 or anti‐CLIP170 antibodies.
In vitro binding assay
Expression, purification, and assembly of HIV‐1 CA‐NC or CA‐NC R18L as well as binding experiments were performed as previously described (Ganser et al, 1999; Ganser‐Pornillos et al, 2007; Delaney et al, 2017). Briefly, HIV‐1 CA‐NC proteins were assembled in vitro by resuspending CA‐NC in 50 mM Tris–HCL [pH 8.0], 0.5 M NaCl, and 2 mg/ml DNA oligo‐(TG)50 overnight (Malikov et al, 2015). 293T transfected with expression constructs for 48 h were washed with PBS prior to lysis in 200 μl hypotonic buffer (10 mM Tris–HCl [pH 7.4], 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM DTT in PBS). Clarified lysates were incubated with the in vitro assembled CA‐NC (referred to as “Input”) for 1 h at room temperature. The mixture was spun down on a 70% sucrose cushion prepared in 1× PBS with 0.5 mM DTT for 1 h at 100,000 g at 4°C in a SW 55Ti Beckman Coulter high speed rotor. The pellet was then resuspended in Laemmli buffer (referred to as “bound”) for WB analysis, or in PBS for further SIM imaging. Input and bound mixtures were analyzed by WB using indicated antibodies.
HIV‐1 core binding assay
HIV‐1 cores were isolated from concentrated HIV‐VSV‐Luc or Mock separated into fractions as previously described (Shah & Aiken, 2011). Briefly, 293T were transfected with Mock or proviral DNA using PEI (as described above). The virus‐containing supernatant was collected 48 h post‐transfection, filtered, and concentrated by ultracentrifugation. It was spun through a solution of 20% (w/v) sucrose in PBS using a SW32Ti Beckman Coulter rotor for 3 h at 174,900 g and 4°C. Pelleted viral particles were resuspended in STE buffer (10 mM Tris–HCl [pH 7.4], 100 mM NaCl, 1 mM EDTA) and run through a layer of 1% Triton X‐100 into a linear sucrose gradient (30 to 70%) in STE buffer using a SW41Ti Beckman Coulter rotor for 20 h at 174,900 g and 4°C. Fractions of 1 ml were collected from the top of the gradient, aliquoted into 0.2 ml aliquots, and used for binding assays or flash‐frozen and store at −80°C. 30 μl of each fraction was collected and lysed in Laemmli for subsequent WB analysis. Protocol for binding to intact viral cores was adapted from CA‐NC assay (Ganser et al, 1999; Delaney et al, 2017). As mentioned above, transfected 293T cells were lysed in hypotonic buffer (10 mM Tris–HCl [pH 7.4], 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM DTT in PBS). Clarified lysates expressing Flag or Flag‐tagged CLIP170 were incubated with the Mock or core‐containing fractions diluted in STE buffer with BSA (10 μg/ml) for 2 h on ice (“Input”). The mixture was spun down on 30% sucrose in STE buffer for 1 h at 100,000 g at 4°C in a SW 55Ti Beckman Coulter rotor. The pellet was then resuspended in Laemmli buffer (referred to as “bound”) for WB analysis.
Immunoprecipitation (IP)
IPs were carried out as previously described (Malikov et al, 2015). Briefly, cells were rinsed with cold PBS and then scraped with 1 ml of ice‐cold NP‐40 lysis buffer (50 mM HEPES [pH 7.4], 150 mM NaCl, 0.5 mM MgCl2, 2 mM EDTA, 2 mM Na3VO4, 25 mM glycerophosphate, 1.5% NP‐40, and protease inhibitor). Samples were lysed by rocking at 4°C for 40 min, then clarified by centrifugation for 10 min at 10,000 g. Samples were pre‐cleared with Protein G Sepharose beads (GE Healthcare), and 30 μl aliquot was kept as input. The remaining pre‐cleared samples were incubated with the indicated mouse antibodies for 1–2 h followed by the incubation with Protein G beads for another hour at 4°C. The beads were extensively washed with NP‐40 lysis buffer and lysed in Laemmli buffer (referred to as bound).
Fusion assay (BlaM‐Vpr)
CHME3 stably expressing Flag or Flag‐tagged CLIP170 was incubated with HIV‐1‐VSV‐Luc containing the BlaM‐Vpr fusion protein at 37°C for 3 h. After washing with CO2‐independent media, cells were loaded with CCF2/AM (Life Technologies) for 1 h in accordance with manufacturer's recommendations. Samples were then washed and incubated with CO2‐independent media supplemented with 10% NU Serum for 16 h at room temperature before fixation with 1.2% paraformaldehyde. Cleavage of CCF2 was detected by FACS using the Pacific Blue filter set.
Statistical analysis
Experiments were analyzed for statistical significance using one‐way ANOVA or Student's t‐test. Statistical significance is denoted by asterisks, as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Statistical tests were performed using GraphPad Prism software. Data are shown as the mean of at least three independent experiments ± SEM, unless stated otherwise.
Author contributions
ESDS, SS, VM, FG, MKD, and MHN designed research; ESDS, SS, VM, FG, and MKD performed research; ESDS, SS, VM, FG, MKD, and MHN analyzed data; and ESDS, and MHN wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We are grateful to Derek Walsh, Barbie Ganser‐Pornillos, and Owen Pornillos for reagents and advice. We also thank Gregg Gundersen, Thomas Hope, Gregory Melikian, Vladimir Rodionov, and Vladimir Jovasevic for reagents. We thank Kayla Schipper and Amy Hulme, for technical assistance. The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: pNL4‐3.Luc.R. −.E− from Dr. Nathaniel Landau, and HIV‐1 p24 (AG3.0) antibody from Dr. Jonathan Allan. This work was supported by NIH grant P01GM105536, and R01AI150559 (to M.H.N.), and NIH Grant T32AI007476‐19 (to M.K.D.).
The EMBO Journal (2020) 39: e104870
Data availability
This study includes no data deposited in external repositories.
References
- Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, Vermeulen W, Burgering BM, De Zeeuw CI, Grosveld F et al (2001) Clasps are CLIP‐115 and ‐170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell 104: 923–935 [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz MO (2008) Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol 9: 309–322 [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz MO (2015) Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 16: 711–726 [DOI] [PubMed] [Google Scholar]
- Ambrose Z, Aiken C (2014) HIV‐1 uncoating: connection to nuclear entry and regulation by host proteins. Virology 454–455: 371–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhel N, Genovesio A, Kim KA, Miko S, Perret E, Olivo‐Marin JC, Shorte S, Charneau P (2006) Quantitative four‐dimensional tracking of cytoplasmic and nuclear HIV‐1 complexes. Nat Methods 3: 817–824 [DOI] [PubMed] [Google Scholar]
- Arhel NJ, Souquere‐Besse S, Munier S, Souque P, Guadagnini S, Rutherford S, Prevost MC, Allen TD, Charneau P (2007) HIV‐1 DNA Flap formation promotes uncoating of the pre‐integration complex at the nuclear pore. EMBO J 26: 3025–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barklis E, Alfadhli A, McQuaw C, Yalamuri S, Still A, Barklis RL, Kukull B, Lopez CS (2009) Characterization of the in vitro HIV‐1 capsid assembly pathway. J Mol Biol 387: 376–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejarano DA, Peng K, Laketa V, Borner K, Jost KL, Lucic B, Glass B, Lusic M, Muller B, Krausslich HG (2019) HIV‐1 nuclear import in macrophages is regulated by CPSF6‐capsid interactions at the nuclear pore complex. Elife 8: e41800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelic S, De Groot CO, Scharer MA, Jaussi R, Bargsten K, Salzmann M, Frey D, Capitani G, Kammerer RA, Steinmetz MO (2012) Interaction of mammalian end binding proteins with CAP‐Gly domains of CLIP‐170 and p150(glued). J Struct Biol 177: 160–167 [DOI] [PubMed] [Google Scholar]
- Blanco‐Rodriguez G, Gazi A, Monel B, Frabetti S, Scoca V, Mueller F, Schwartz O, Krijnse‐Locker J, Charneau P, Di Nunzio F (2020) Remodeling of the core leads HIV‐1 pre‐integration complex in the nucleus of human lymphocytes. J Virol 94: e00135‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick RC, Li C, Munshi M, Rawson JMO, Nagashima K, Hu WS, Pathak VK (2020) HIV‐1 uncoats in the nucleus near sites of integration. Proc Natl Acad Sci USA 117: 5486–5493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EM, Hope TJ (2015) HIV‐1 capsid: the multifaceted key player in HIV‐1 infection. Nat Rev Microbiol 13: 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes SK, Zhou J, Aiken C (2018) HIV‐1 engages a dynein‐dynactin‐bicd2 complex for infection and transport to the nucleus. J V irol 92: e00358‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen NY, Zhou L, Gane PJ, Opp S, Ball NJ, Nicastro G, Zufferey M, Buffone C, Luban J, Selwood D et al (2016) HIV‐1 capsid is involved in post‐nuclear entry steps. Retrovirology 13: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang P, Slep KC (2019) Mapping multivalency in the CLIP‐170‐EB1 microtubule plus‐end complex. J Biol Chem 294: 918–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craveur P, Gres AT, Kirby KA, Liu D, Hammond JA, Deng Y, Forli S, Goodsell DS, Williamson JR, Sarafianos SG et al (2019) Novel intersubunit interaction critical for HIV‐1 core assembly defines a potentially targetable inhibitor binding pocket. MBio 10: e02858‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney MK, Malikov V, Chai Q, Zhao G, Naghavi MH (2017) Distinct functions of diaphanous‐related formins regulate HIV‐1 uncoating and transport. Proc Natl Acad Sci USA 114: E6932–E6941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharan A, Bachmann N, Talley S, Zwikelmaier V, Campbell EM (2020) Nuclear pore blockade reveals that HIV‐1 completes reverse transcription and uncoating in the nucleus. Nat Microbiol 5: 1088–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodding MP, Way M (2011) Coupling viruses to dynein and kinesin‐1. EMBO J 30: 3527–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duellberg C, Trokter M, Jha R, Sen I, Steinmetz MO, Surrey T (2014) Reconstitution of a hierarchical +TIP interaction network controlling microtubule end tracking of dynein. Nat Cell Biol 16: 804–811 [DOI] [PubMed] [Google Scholar]
- Fassati A, Goff SP (2001) Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J Virol 75: 3626–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez J, Machado AK, Lyonnais S, Chamontin C, Gartner K, Leger T, Henriquet C, Garcia C, Portilho DM, Pugniere M et al (2019) Transportin‐1 binds to the HIV‐1 capsid via a nuclear localization signal and triggers uncoating. Nat Microbiol 4: 1840–1850 [DOI] [PubMed] [Google Scholar]
- Francis AC, Melikyan GB (2018) Single HIV‐1 imaging reveals progression of infection through CA‐dependent steps of docking at the nuclear pore, uncoating, and nuclear transport. Cell Host Microbe 23: 536–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galjart N (2010) Plus‐end‐tracking proteins and their interactions at microtubule ends. Curr Biol 20: R528–R537 [DOI] [PubMed] [Google Scholar]
- Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI (1999) Assembly and analysis of conical models for the HIV‐1 core. Science 283: 80–83 [DOI] [PubMed] [Google Scholar]
- Ganser‐Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI (2004) Assembly properties of the human immunodeficiency virus type 1 CA protein. J Virol 78: 2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser‐Pornillos BK, Cheng A, Yeager M (2007) Structure of full‐length HIV‐1 CA: a model for the mature capsid lattice. Cell 131: 70–79 [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345 [DOI] [PubMed] [Google Scholar]
- Gouveia SM, Akhmanova A (2010) Cell and molecular biology of microtubule plus end tracking proteins: end binding proteins and their partners. Int Rev Cell Mol Biol 285: 1–74 [DOI] [PubMed] [Google Scholar]
- Gundersen GG (2002) Evolutionary conservation of microtubule‐capture mechanisms. Nat Rev Mol Cell Biol 3: 296–304 [DOI] [PubMed] [Google Scholar]
- Herold N, Anders‐Osswein M, Glass B, Eckhardt M, Muller B, Krausslich HG (2014) HIV‐1 entry in SupT1‐R5, CEM‐ss, and primary CD4 + T cells occurs at the plasma membrane and does not require endocytosis. J Virol 88: 13956–13970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honnappa S, Okhrimenko O, Jaussi R, Jawhari H, Jelesarov I, Winkler FK, Steinmetz MO (2006) Key interaction modes of dynamic +TIP networks. Mol Cell 23: 663–671 [DOI] [PubMed] [Google Scholar]
- Honnappa S, Gouveia SM, Weisbrich A, Damberger FF, Bhavesh NS, Jawhari H, Grigoriev I, van Rijssel FJ, Buey RM, Lawera A et al (2009) An EB1‐binding motif acts as a microtubule tip localization signal. Cell 138: 366–376 [DOI] [PubMed] [Google Scholar]
- Hulme AE, Kelley Z, Okocha EA, Hope TJ (2015) Identification of capsid mutations that alter the rate of HIV‐1 uncoating in infected cells. J Virol 89: 643–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram Z, Taylor M, Okland G, Martin R, Hulme AE (2020) Characterization of HIV‐1 uncoating in human microglial cell lines. Virol J 17: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Bulinski JC (2011) Post‐translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 12: 773–786 [DOI] [PubMed] [Google Scholar]
- Jayappa KD, Ao Z, Wang X, Mouland AJ, Shekhar S, Yang X, Yao X (2015) Human immunodeficiency virus type 1 employs the cellular dynein light chain 1 protein for reverse transcription through interaction with its integrase protein. J Virol 89: 3497–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Akhmanova A (2011) Microtubule tip‐interacting proteins: a view from both ends. Curr Opin Cell Biol 23: 94–101 [DOI] [PubMed] [Google Scholar]
- Jiang K, Toedt G, Montenegro Gouveia S, Davey NE, Hua S, van der Vaart B, Grigoriev I, Larsen J, Pedersen LB, Bezstarosti K et al (2012) A Proteome‐wide screen for mammalian SxIP motif‐containing microtubule plus‐end tracking proteins. Curr Biol 22: 1800–1807 [DOI] [PubMed] [Google Scholar]
- Jovasevic V, Naghavi MH, Walsh D (2015) Microtubule plus end‐associated CLIP‐170 initiates HSV‐1 retrograde transport in primary human cells. J Cell Biol 211: 323–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh Y, Wu X, Ferris AL, Matreyek KA, Smith SJ, Lee K, KewalRamani VN, Hughes SH, Engelman A (2013) Differential effects of human immunodeficiency virus type 1 capsid and cellular factors nucleoporin 153 and LEDGF/p75 on the efficiency and specificity of viral DNA integration. J Virol 87: 648–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Perilla JR, Ning J, Lu M, Hou G, Ramalho R, Himes BA, Zhao G, Bedwell GJ, Byeon IJ et al (2016) Cyclophilin A stabilizes the HIV‐1 capsid through a novel non‐canonical binding site. Nat Commun 7: 10714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP (1993) Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73: 1067–1078 [DOI] [PubMed] [Google Scholar]
- Lukic Z, Dharan A, Fricke T, Diaz‐Griffero F, Campbell EM (2014) HIV‐1 uncoating is facilitated by dynein and kinesin 1. J Virol 88: 13613–13625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikov V, da Silva ES, Jovasevic V, Bennett G, de Souza Aranha Vieira DA, Schulte B, Diaz‐Griffero F, Walsh D, Naghavi MH (2015) HIV‐1 capsids bind and exploit the kinesin‐1 adaptor FEZ1 for inward movement to the nucleus. Nat Commun 6: 6660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamede JI, Cianci GC, Anderson MR, Hope TJ (2017) Early cytoplasmic uncoating is associated with infectivity of HIV‐1. Proc Natl Acad Sci USA 114: E7169–E7178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez CL, Lau D, Walsh J, Shah V, McGuinness C, Wong A, Aggarwal A, Parker MW, Jacques DA, Turville S et al (2018) Kinetics of HIV‐1 capsid uncoating revealed by single‐molecule analysis. Elife 7: e34772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, Hope TJ (2002) Visualization of the intracellular behavior of HIV in living cells. J Cell Biol 159: 441–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MD, Farnet CM, Bushman FD (1997) Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J Virol 71: 5382–5390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Shanmugapriya S, Santos da Silva E, Naghavi MH (2020) HIV‐1 exploits CLASP2 to induce microtubule stabilization and facilitate virus trafficking to the nucleus. J Virol 94: e00404‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB (2009) HIV enters cells via endocytosis and dynamin‐dependent fusion with endosomes. Cell 137: 433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naghavi MH, Valente S, Hatziioannou T, de Los Santos K, Wen Y, Mott C, Gundersen GG, Goff SP (2007) Moesin regulates stable microtubule formation and limits retroviral infection in cultured cells. EMBO J 26: 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naghavi MH, Walsh D (2017) Microtubule regulation and function during virus infection. J Virol 91: e00538‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehlig A, Molina A, Rodrigues‐Ferreira S, Honore S, Nahmias C (2017) Regulation of end‐binding protein EB1 in the control of microtubule dynamics. Cell Mol Life Sci 74: 2381–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlica P, Berthoux L (2014) Cytoplasmic dynein promotes HIV‐1 uncoating. Viruses 6: 4195–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K, Muranyi W, Glass B, Laketa V, Yant SR, Tsai L, Cihlar T, Muller B, Krausslich HG (2014) Quantitative microscopy of functional HIV post‐entry complexes reveals association of replication with the viral capsid. Elife 3: e04114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser‐Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M (2009) X‐ray structures of the hexameric building block of the HIV capsid. Cell 137: 1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser‐Pornillos BK, Yeager M (2011) Atomic‐level modelling of the HIV capsid. Nature 469: 424–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portran D, Schaedel L, Xu Z, Thery M, Nachury MV (2017) Tubulin acetylation protects long‐lived microtubules against mechanical ageing. Nat Cell Biol 19: 391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procter DJ, Banerjee A, Nukui M, Kruse K, Gaponenko V, Murphy EA, Komarova Y, Walsh D (2018) The HCMV assembly compartment is a dynamic golgi‐derived MTOC that controls nuclear rotation and virus spread. Dev Cell 45: 83–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M et al (2013) HIV‐1 evades innate immune recognition through specific cofactor recruitment. Nature 503: 402–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck‐Peterson SL, Redwine WB, Vale RD, Carter AP (2018) The cytoplasmic dynein transport machinery and its many cargoes. Nat Rev Mol Cell Biol 19: 382–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo Y, Walsh D, Barry DS, Tinaztepe S, de Los Santos K, Goff SP, Gundersen GG, Naghavi MH (2013) HIV‐1 induces the formation of stable microtubules to enhance early infection. Cell Host Microbe 14: 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN et al (2011) HIV‐1 capsid‐cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog 7: e1002439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah VB, Aiken C (2011) In vitro uncoating of HIV‐1 cores. J Vis Exp 8: 3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers BJ, Digianantonio KM, Smaga SS, Huang PT, Zhou K, Gerber EE, Wang W, Xiong Y (2019) Modular HIV‐1 capsid assemblies reveal diverse host‐capsid recognition mechanisms. Cell Host Microbe 26: 203–216 e206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Robinson BA, Chutiraka K, Geary CD, Reed JC, Lingappa JR (2016) Mutations of conserved residues in the major homology Region arrest assembling HIV‐1 Gag as a membrane‐targeted intermediate containing genomic RNA and cellular proteins. J Virol 90: 1944–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrich A, Honnappa S, Jaussi R, Okhrimenko O, Frey D, Jelesarov I, Akhmanova A, Steinmetz MO (2007) Structure‐function relationship of CAP‐Gly domains. Nat Struct Mol Biol 14: 959–967 [DOI] [PubMed] [Google Scholar]
- van de Willige D, Hoogenraad CC, Akhmanova A (2016) Microtubule plus‐end tracking proteins in neuronal development. Cell Mol Life Sci 73: 2053–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Schaedel L, Portran D, Aguilar A, Gaillard J, Marinkovich MP, Thery M, Nachury MV (2017) Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science 356: 328–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Emerman M (2006) Retroviral infection of non‐dividing cells: old and new perspectives. Virology 344: 88–93 [DOI] [PubMed] [Google Scholar]
- Zhou L, Sokolskaja E, Jolly C, James W, Cowley SA, Fassati A (2011) Transportin 3 promotes a nuclear maturation step required for efficient HIV‐1 integration. PLoS Pathog 7: e1002194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zila V, Muller TG, Laketa V, Muller B, Krausslich HG (2019) Analysis of CA content and CPSF6 dependence of early HIV‐1 replication complexes in SupT1‐R5 cells. mBio 10: e02501‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurnic Bonisch I, Dirix L, Lemmens V, Borrenberghs D, De Wit F, Vernaillen F, Rocha S, Christ F, Hendrix J, Hofkens J et al (2020) Capsid‐labelled HIV to investigate the role of capsid during nuclear import and integration. J Virol 94: e01024‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
This study includes no data deposited in external repositories.