

Abstract
Introduction
Microglial cells play an important role in the development of Alzheimer's disease (AD). People with Down syndrome (DS) inevitably develop AD neuropathology (DSAD) by 40 years of age. We characterized the distribution of different microglial phenotypes in the brains of people with DS and DSAD.
Methods
Autopsy tissue from the posterior cingulate cortex (PCC) from people with DS, DSAD, and neurotypical controls was immunostained with the microglial marker Iba1 to assess five microglia morphological types.
Results
Individuals with DS have more hypertrophic microglial cells in their white matter. In the gray matter, individuals with DSAD had significantly fewer ramified microglia and more dystrophic microglia than controls and the younger individuals with DS. The DSAD group also exhibited more rod‐shaped and amoeboid cells than the AD group.
Discussion
Individuals with DS and DSAD show a microglial phenotype that distinguishes them from non‐DS controls.
Keywords: IBA‐1, microglial morphology, neuroinflammation, posterior cingulate cortex, trisomy 21
1. INTRODUCTION
Down syndrome (DS) or trisomy 21 is the most common genetic cause of intellectual disability. While improvements in medical care along with social enrichment in DS has led to a significant extension in lifespan and increased quality of life, it has also increased the prevalence of Alzheimer's disease (AD). Further, the overexpression of the amyloid beta (Aβ) precursor protein (APP) gene, located on chromosome 21, leads to an enhanced production of Aβ 1 and is likely to be one of the main causes for the increased risk of AD in this population. 2 , 3 , 4 As a result, by 40 years of age the majority of people with trisomy 21 have sufficient numbers of Aβ plaques and neurofibrillary tangles for a neuropathological diagnosis of AD. Notably, alterations in blood and cerebrospinal fluid (CSF) biomarkers consistent with changes in familial and sporadic AD are also found in people with DS. 5 , 6 , 7 , 8 , 9 It is estimated that 55% of individuals with DS between the ages of 40 to 49 show progressive cognitive impairment, while in individuals aged 60 to 69 this prevalence increases to 77%; 10 , 11 thus, there is a temporal disconnect between age when AD neuropathology is present and the presence of clinical symptoms of dementia. 11 , 12 A unique advantage to studying AD in people with DS is that there is a distinct age‐related pattern of neuropathology that may allow us to investigate and determine the first appearance of alterations in specific pathways or cells. 2
Neuroinflammation may be a key causative factor in the pathogenesis of AD in DS. 13 , 14 , 15 , 16 , 17 In the frontal cortex of people with DS and dementia, there is a dysregulation of numerous genes related to Aβ and tau biology, as well as other fundamental pathways related to intracellular signaling, cell homeostasis, and death, and the cholinergic and glutamatergic systems, among others, suggesting a specific genetic profile implicating cortical dysfunction in demented individuals with DS. 18 Interestingly, the dysregulation of immune‐related genes indicate an inflammatory state in the adult brain, as these genes are not altered in fetal brains, and could be directly linked to the development of AD pathology in DS. 19 There are at least six genes on chromosome 21 that may contribute to increased inflammation in DS. 16 Both human DS and DS mouse models present with marked neuroinflammation, with the activation of both microglia and astrocytes. 14 , 20 , 21 In adults with DS there is a correlation between inflammatory markers and other AD biomarkers as well as an increase in the circulating levels of inflammatory cytokines in blood. 22 , 23 In previous autopsy studies, we noted that the inflammatory phenotype of people with DS is different from age‐matched neurotypical controls reflecting an increase in pro‐inflammatory gene expression like interleukin (IL)‐6 and IL‐12. In the presence of AD neuropathology in the DS (DSAD) brain, there is additional inflammation present that overlaps with that seen in sporadic AD. 15 However, elevated CD86 and FcgR1 is observed in DSAD, which is different from sporadic AD, and suggests extravasation of serum proteins into the brain that may promote the formation of immune complexes. Consistent with increased inflammation in DS, our initial studies of microglial activation, using HLA‐DR immunostaining, showed higher microglial loads in DSAD relative to DS and controls, 15 consistent with reports in sporadic AD. 24
HIGHLIGHTS
Hypertrophic microglial cell numbers increase with age in the white matter.
Numbers of ramified microglia are lower with age in the gray matter in people with DS and controls.
People with DS and AD neuropathology have more dystrophic cells in the gray matter.
People with DSAD have more rod‐shaped and amoeboid cells than controls.
RESEARCH IN CONTEXT
Systematic Review: We reviewed the literature using PubMed. We searched for research studies describing microglial changes and characterization in autopsy tissue from brains of people with Down syndrome (DS) and/or Alzheimer's disease (AD), and these have been appropriately cited.
Interpretation: Our findings demonstrate that people with DS, and particularly those with Alzheimer's disease pathology (DSAD), present a microglial phenotype pattern that distinguishes them from non‐DS controls. In DSAD there are more dystrophic and fewer ramified microglial cells, suggesting more damage to microglia. Further, it reinforces the differences seen in individuals with DS with regard to their neuroinflammation response.
Future Directions: More studies are necessary to fully evaluate the role of each specific microglial phenotype in DS, and to understand the microglial pathways that contribute to the development and progression of AD in individuals with DS.
Microglial cells play an important role in AD development, with evidence evolving from studies of their close interaction with plaques and tangles, an increased risk of AD due to polymorphisms in microglial‐expressed genes such as TREM2 and CD33, as well as changes in their morphology in the presence of AD pathology. 25 , 26 , 27 Microglia morphology in humans can range from ramified to amoeboid cells. While a ramified shape is characterized by a small, round cell body with extended long branching processes, amoeboid microglia are at the extreme end of the spectrum with a large round cell body, rich in cytoplasm and without processes. Changes in microglial morphology and motility can be exacerbated in AD and other pathologies, which reflect the functional state of the cell, and potentially contribute to the development and progression of the disease. 28 , 29
Microglial morphology in DS, particularly in people with DSAD, remains largely unexplored. The first description of differences in microglia in DS reported significantly more IL‐1 positive microglia in brain tissue from young people with DS relative to age‐matched controls (postnatal to 41 years of age). 30 Moreover, a reduced number of resting microglia is accompanied by increased dystrophic microglial cells in a small study of autopsy cases with DS over 40 years of age. 31 Thus, the current study was intended to expand upon these studies and explore microglial morphology responses in the brains of people with DS with or without AD across the lifespan, with a focus on the posterior cingulate cortex (PCC). The rationale for selecting the PCC is three‐fold. First, older individuals with DS show lower glucose metabolic rates in the PCC relative to their control groups. 32 Second, the PCC, along with the precuneus and posterior parietotemporal lobes, is also one of the earliest sites of reduced glucose metabolism (fluorodeoxyglucose positron emission tomography) before the appearance of symptoms in sporadic AD, which can lead to neuronal and synaptic loss. 33 Third, neuronal health biomarkers in this region are lower as measured by magnetic resonance spectroscopy, while glial biomarkers are significantly higher, in demented adults with DS. 34 To further characterize microglia phenotypic changes in DS, we used Iba1 (ionized calcium binding adaptor molecule 1) immunostaining that labels all microglia cells and their processes, allowing for a better characterization of their morphology. Thus, the aim of this study was to characterize the distribution of different microglial phenotypes in the brains of people with DS and DSAD relative to age‐matched controls, and late onset AD.
2. METHODS
2.1. Human tissue samples
Fixed tissue samples were acquired from the PCC (Brodmann area 23 and 31) from the tissue repositories located at the Alzheimer's Disease Research Center at the University of California Irvine, the NIH NeuroBioBank, and the University of Kentucky Alzheimer's Disease Center. Human tissue collection and handling adhered to the University of Kentucky and University of California, Irvine, Institutional Review Board guidelines. Neuropathologic diagnosis of AD was in accordance with current National Institute on Aging‐Alzheimer's Association (NIA‐AA) guidelines. 35 , 36 Cases from the University of California Irvine were fixed in 4% paraformaldehyde, while cases from the NIH NeuroBioBank and the University of Kentucky were fixed in 10% formalin. A solution of phosphate buffered saline (PBS) with 0.02% sodium azide was used for long‐term storage. No information regarding the length of fixation is available.
Six autopsy groups were included in this study (Table 1): young controls (YC; age‐matched to young DS group; n = 10), middle‐aged controls (MC; age‐matched to Down syndrome‐Alzheimer's disease group; n = 10), aged controls (AC; age‐matched to AD group; n = 6), Down syndrome (DS, n = 10), Down syndrome with neuropathological AD (DSAD, n = 17), and sporadic AD (AD, n = 6). DSAD are DS cases that had sufficient neuropathology for a post mortem diagnosis of AD, according to Hyman et al. 35 Because people with DSAD come to autopsy at younger ages than those with sporadic AD, we were not able to match for age between these two groups. All control cases were selected to match for post mortem interval (PMI) to the DS, DSAD, and AD cases. Our groups contained both males and females, but due to the limited availability of cases, we were not able to match for sex across groups. Clinical data were not incorporated into the present study.
TABLE 1.
Case characteristics for posterior cingulate cortex tissue obtained from brain banks
| YC | DS | MC | DSAD | AC | AD | |
|---|---|---|---|---|---|---|
| N | 11 | 10 | 10 | 17 | 6 | 6 |
| Age at death, years |
17.89 (11.83) |
19.5 (15.54) |
53.5 (6.65) |
57.00 (8.28) |
78.67 (2.16) |
78.83 (1.83) |
| Male/female, n/n | 9/2 | 7/3 | 6/4 | 8/9 | 3/3 | 4/2 |
| Post mortem interval (PMI), hours |
20.45 (5.05) |
19.3 (6.15) |
16.2 (6.78) |
5.98 (6.41) |
9.00 (6.54) |
12.86 (10.13) |
Abbreviations: AC, aged control; AD, sporadic Alzheimer's disease; DS, Down syndrome; DSAD, Down syndrome with neuropathological Alzheimer's disease; MC, middle‐aged control; YC, young control.
Note: Mean + standard deviation.
2.2. Immunohistochemistry
Tissue was sectioned on a vibratome (Leica Biosystems, Buffalo Grove, Illinois, USA) at 50 μm. Sequential sections were collected and stored in PBS with 0.02% sodium azide until used. Following standard immunohistochemistry protocols and using Iba‐1 antibody (Abcam, Cambridge, Massachusetts, USA; 1:800) the sections were incubated in the primary antibody overnight and followed by incubation in an anti‐rabbit secondary antibody (Vector Laboratories, Burlingame, California, USA). This was followed by amplification of the signal using an avidin‐biotin complex peroxidase kit, and 3,3′ diaminobenzidine substrate kit. After immunohistochemistry, each tissue section was mounted on a glass slide and coverslipped with Depex mounting media. All tissue sections were immunostained within a single experiment to reduce variability.
2.3. Image analysis
The Aperio ScanScope XT digital slidescanner was used to scan the entire slide at 20x magnification. The Aperio ImageScope (v11.1.2.752) software was used to draw five random 250 × 250 micron boxes in the white matter (WM) and five random 250 × 250 micron boxes in the gray matter (GM) for each tissue section (Figure 1A). Five microglia morphological types were assessed, as defined in previous work and illustrated in Figure 1B–F, 37 , 38 , 39 , 40 : (B) ramified microglia, which have thin, branched processes that are thought to actively “survey” changes in their environments; (C) hypertrophic microglia (also known as activated microglia), which may have enlarged, short processes and thicker bodies; (D) dystrophic microglia that have fragmented or “beaded” processes possibly due to microglial dysfunction due to aging 41 , 42 ; (E) rod‐shaped microglia that have elongated nuclei, few processes, and are most notable in chronic disorders; and (F) amoeboid microglia that have a round body, may lack processes, and appear in response to acute destruction of central nervous system tissue. The number of ramified, hypertrophic, dystrophic, rod‐shaped, and amoeboid microglia in each annotation box was counted by hand and averaged for each case, separating WM and GM. The categorical analysis used to quantify the microglia expanded upon previously established guidelines and figures. 38 , 43 , 44 Counts were made by two investigators blinded to all samples groups and case histories. Cohen's κ was run to determine whether there was agreement between investigators.
FIGURE 1.
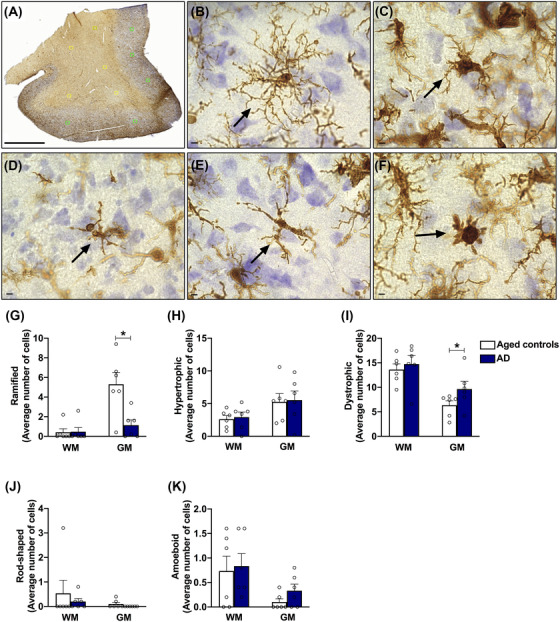
Microglia morphologies evaluated in the posterior cingulate cortex, and microglia cell counts in the Alzheimer's disease (AD) brain. A, Characterization of microglia was performed in five random 250 × 250 micron boxes in the white matter and five random 250 × 250 micron boxes in the gray matter for each tissue section Scale: 3 μm. Five microglia morphologies were analyzed: (B) ramified microglia, which have thin, branched processes to actively “survey” changes in their environment; (C) hypertrophic microglia (also known as activated microglia), which may have enlarged, short processes and thicker bodies; (D) dystrophic microglia, that present fragmented or “beaded” processes possibly due to microglial dysfunction as a result of aging; (E) rod‐shaped microglia, that have elongated nuclei, few processes, and are most notable in chronic disorders; (F) amoeboid microglia, which have a round body, lack processes, and appear in response to acute destruction of central nervous system tissue. Scale: 5 μm. G–K, In the white matter, no significant changes were observed in any of the microglial phenotypes analyzed. In the gray matter, a significant reduction was observed in the number of ramified cells in the AD group, while the number of dystrophic cells was increased, compared to aged controls. Data are presented as mean ± standard error of the mean, * P ≤ 0.05 (Student's t test)
2.4. Statistics
IBM SPSS Statistics Software (Version 24) was used for statistical analysis. Overall group differences were assessed for the GM and WM separately using a two‐way analysis of variance to examine interactions between age groups and genotype (DS vs non‐DS). Comparisons between two groups were made using Student's t test. Differences between means were considered significant at P ≤ .05. Graphs were generated using GraphPad Prism software version 8.0, with values expressed as mean ± standard error of the mean.
3. RESULTS
3.1. Microglial phenotypes in AD versus age‐matched controls
Quantification of Iba1‐positive cells in the GM of patients with AD demonstrate a significant decrease of ramified cells, while dystrophic cells are increased, compared to age‐matched controls (Figure 1G, I, respectively), consistent with previous reports. 38 , 40 , 42 , 45 Also, the numbers of hypertrophic microglia did not vary significantly across groups (Figure 1H) nor did the numbers of rod‐shaped or amoeboid cells (Figure 1J, K), highlighting the robustness and reliability of the methodology used in this study.
3.2. Microglial morphology differences with age and genotypes
Age and the presence of AD pathology are completely intertwined in DS. By 40 years of age, virtually all adults with DS have enough neuritic plaques and neurofibrillary tangles for a diagnosis of AD pathology. 2 , 46 Deposition of Aβ happens decades earlier in DS, compared to aged‐matched controls and AD brains. 1 Thus, we believe that our rationale of comparing DSAD with middle‐aged controls without controlling for age is the most appropriate way to conduct these analyses. Comparing DS to DSAD while controlling for age would eliminate all possible outcomes as the DSAD are always older than the DS group.
Therefore, in PCC WM, the number of hypertrophic microglia is consistently higher in DS groups (main effect of genotype, independent of age [F(1,46) = 6.239, P = .016]), but none of the other microglial phenotypes are significantly different (Figure 2A).
FIGURE 2.
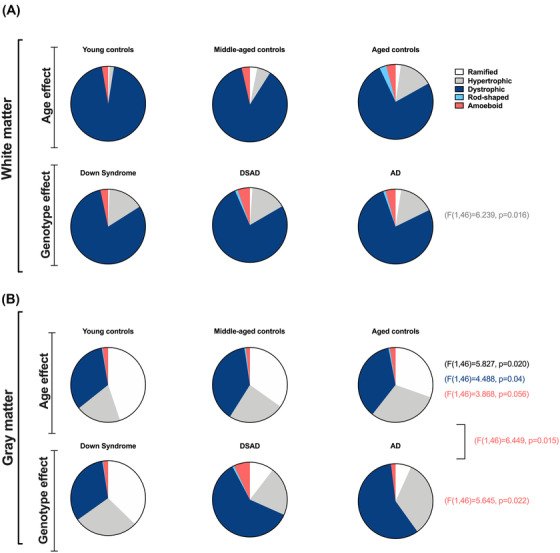
Distribution of microglia phenotypes across different groups in white and gray matter. The average number of microglia in each of the five classifications according to each group in the white (A) and gray (B) matter, demonstrating the age and genotype effects related to the morphology of cells. Data are presented as means. A, In the white matter, hypertrophic microglia increase as a main effect of genotype. B, In the gray matter, the age effect is significant for ramified and dystrophic cells. Amoeboid cells have an effect of age, genotype, and a significant interaction between these factors
In the GM, both age group and genotype have different effects for individual microglial phenotypes. Comparing ramified microglial counts, we find a main effect of age group independent of genotype (F[1,46] = 5.827, P = 0.020), reflecting an overall decrease in the numbers of ramified microglial cells independent of the presence of DS (Figure 2B). While the number of ramified cells is lower in cases over 40 years of age, dystrophic microglia counts are higher, demonstrating also a significant effect of age independent of genotype in the gray matter of PCC (F[1,46] = 4.488, P = 0.04). The amoeboid cell quantification demonstrate a main effect of genotype overall (F[1,46] = 5.645, P = 0.022) as well as age (F[1,46] = 3.868, P = 0.056), and a significant interaction between age and genotype (F[1,46] = 6.449, P = 0.015). The number of amoeboid cells was higher in DS overall and primarily driven by high numbers in DSAD PCC.
3.3. Microglial morphology in DS
Given that we cannot separate age from the presence of AD neuropathology in DS, we compared four groups divided between genotype (DS vs control) and age group (<40 vs >40 years), reflecting absence and presence of AD neuropathology, respectively (Figure 3).
FIGURE 3.
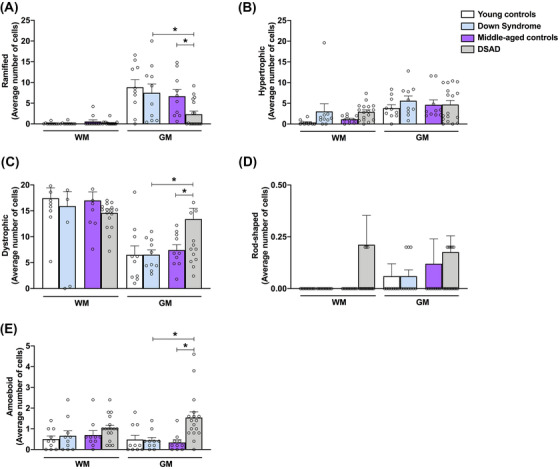
Characterization of microglia phenotypes in the white and gray matter in Down syndrome (DS) groups. Microglial counts are provided for the white (WM) and gray matter (GM) for (A) ramified, (B) hypertrophic, (C) dystrophic, (D) rod‐shaped, and (E) amoeboid morphologies. No significant changes were observed in any of the microglial phenotypes analyzed in the white matter. In the gray matter, the DS with neuropathological Alzheimer's disease group presented a significant reduction in the number of ramified cells and an increase in the counts of dystrophic and amoeboid cells, when compared to DS and middle‐aged control groups. Data are presented as mean ± standard error of the mean, * P ≤ .05 (two‐way analysis of variance)
No significant differences were found in the WM between the groups (Figure 3A ‐ E). In the GM, DSAD was associated with the fewest numbers of ramified microglial cells, both in comparison to MC (P = 0.023) or DS (P = 0.009) (Figure 3A). While the number of ramified cells is lower in cases over 40 years of age, dystrophic microglia counts are higher in the DSAD group when compared to the DS group (P = 0.007) and middle‐aged controls (P = 0.019; Figure 3C). The average number of rod‐shaped cells is consistently low, so the apparent increase in DSAD groups is not sufficient to achieve statistical significance (Figure 3D). Counts for amoeboid microglia are also consistently low across all groups; however, higher numbers of amoeboid microglia are present in a subset of our DSAD cases . While we found a significant increase in the number of amoeboid cells overall in the DSAD group in comparison to the DS group (P = 0.008) and middle‐aged controls (P = 0.003), the numbers of these cells are low and this result needs to be cautiously interpreted (Figure 3E).
3.4. Microglial phenotype comparing DSAD and sporadic AD
No significant differences are observed for any of the microglial phenotypes in the WM comparing DSAD to AD (Figure 4). Further, the numbers of ramified, hypertrophic, and dystrophic microglia were similar across groups (Figure 4A–C). There are, however, individual DSAD cases that have higher numbers of ramified and dystrophic microglia. Consistent with the literature, 38 , 47 few rod‐shaped microglia were found, thus the average counts are relatively low. Interestingly, in the GM of DSAD cases there are more rod‐shaped cells (P = 0.0387) compared to AD cases (Figure 4D). Moreover, DSAD also presents with more amoeboid cells than the AD group (P = 0.0009; Figure 4E).
FIGURE 4.
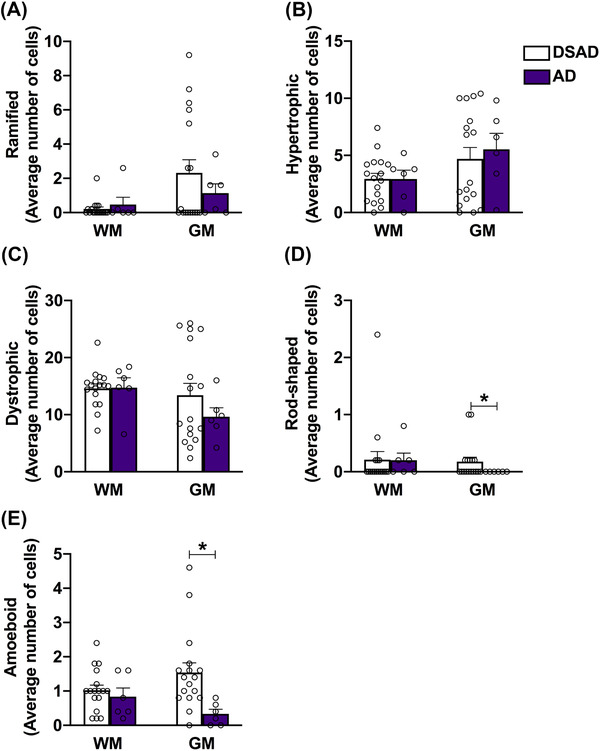
Down syndrome with neuropathological Alzheimer's disease (DSAD) presents more rod‐shaped and amoeboid cells than AD. Differences in microglial counts for the white matter (WM) and gray matter (GM) between DSAD and AD groups are shown for (A) ramified, (B) hypertrophic, (C) dystrophic, (D) rod‐shaped, and (E) amoeboid phenotypes. While no significant changes were seen in the WM, the DSAD group presented significant increase in the number of rod‐shaped and amoeboid microglia cells in the GM, compared to AD alone. Data are presented as mean ± standard error of the mean, * P ≤ 0.05 (Student's t test)
FIGURE 5.
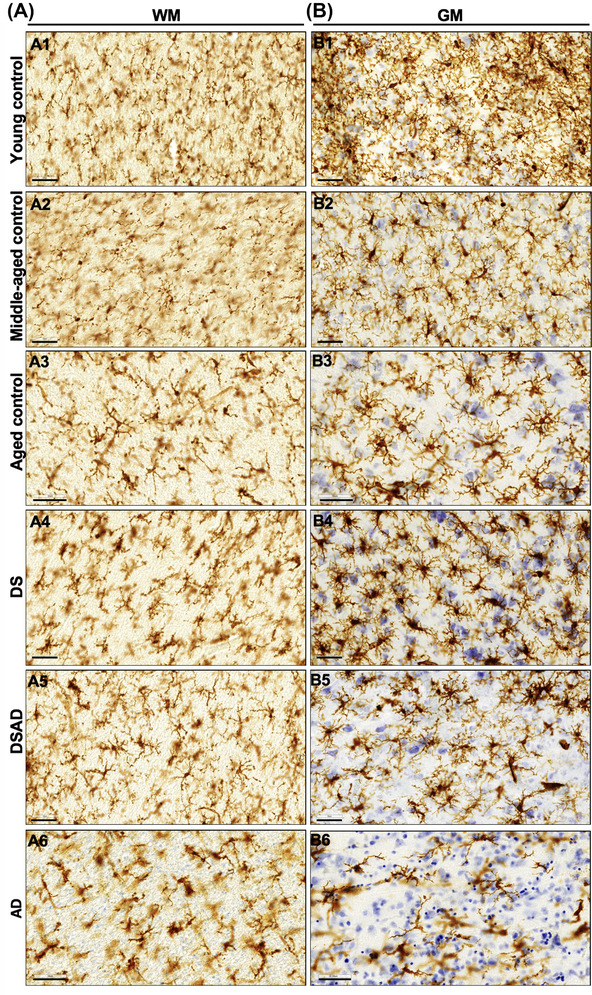
Group specific patterns in Iba1 microglia staining. Representative images of microglia morphologies observed in the white (A) and gray (B) matter of young controls (A1, B1), middle‐aged controls (A2, B2), aged controls (A3, B3), Down syndrome (A4, B4), Down syndrome with neuropathological Alzheimer's disease (A5, B5), and AD (A6, B6), respectively. Scale: 50 μm
4. DISCUSSION
The aim of this study was to characterize the distribution of different microglial phenotypes in the brains of people with DS and DSAD. We used autopsy tissue from the PCC to characterize microglia in the white and gray matter by manually assessing morphology based upon immunostaining with Iba1, a marker of all microglia cells and their processes.
In our study, we observe a significant genotype effect (DS vs non‐DS) in the number of hypertrophic microglia in the WM. Hypertrophic microglia have enlarged, short processes, and thicker bodies, and are thought to be actively responding to injury. 38 , 40 This increase in hypertrophic microglia contrasts with other published findings on microglia in DS, which show individuals with DS having fewer hypertrophic microglia than controls. 31 However, these studies were focused on the GM in the temporal cortex, highlighting the importance of examining both the white and gray matter. In addition to a unique inflammatory response, people with DS also show a significant reduction in WM integrity compared to controls. 48 Moreover, significant vascular pathology is also observed with increasing age in DS, which could also potentially elicit hypertrophied microglial cells observed in the WM in our study. 49 , 50 Our results do not demonstrate significant changes in any of the other microglial phenotypes in the WM, which could be due to lower numbers overall, leading to a floor effect. Interestingly, we observed an increase, although not significant, in the number of dystrophic cells in the WM of all groups, including young controls. Microglia is a dynamic cell that can adapt and change its morphology according to different pathological situations. 51 In our study, different causes of death could potentially explain the pattern changes observed in young groups; unfortunately, this information is not available for further investigation. Observations in our cases with DS are consistent with studies in other autopsy series of AD. Recent studies shed some light into aspects of WM changes during aging, where activated microglia respond to age‐related pathologic changes, 52 and microglia proteins increase in an age‐dependent manner already in middle‐aged people. 53 Even more interesting is the detection of increased microglia activity in the WM of human post mortem early‐onset AD (EOAD) brain tissue, detected through non‐invasive positron emission tomography imaging using [11C]‐(R)‐PK11195, a ligand that binds to activated microglia. 53 Findings from other studies demonstrate a relationship between the increase of activated microglia in the WM and GM atrophy in other dementias, 54 suggesting abnormalities in WM as an early event preceding GM atrophy, especially in EOAD. 55
We found a significant reduction of the number of ramified microglial cells in the GM with increasing age, as well as a significantly lower number of ramified microglial phenotype in the AD group. Ramified microglia, which have thin, branched processes are thought to be actively “surveying” changes in their environment. 37 , 39 Previous studies also found reduced ramified microglia in individuals with DS relative to age‐matched controls, 31 and fewer ramified microglia are also observed in individuals with non‐DS dementia (including AD, hippocampal sclerosis of aging, hippocampal sclerosis of aging with AD, and dementia with Lewy bodies) than age‐matched controls without dementia. 38
Descriptions of rod‐shaped microglia, which present elongated nuclei, few processes, and are most notable in chronic disorders, are sparse in the literature. Based on previous studies showing an increase in rod‐shaped microglia with age 38 , 47 and the presence of rod cells in the hippocampus of people with DS 56 we hypothesized that we would see an increase in rod‐shaped microglia with age in our controls and DS autopsy cases. However, we only found a significant increase in rod‐shaped microglia in our DSAD cohort in comparison to AD. The function of these cells remains under investigation, but it is believed that they could provide neuroprotection to neurons and trophic support. 57 , 58 The role of rod‐shaped microglia in aging and the development of AD in DS is still unclear and encourages more studies; however, our findings suggest that DSAD is associated with a microglial phenotype that distinguishes them from non‐DS cases.
Amoeboid microglia are most commonly found in the developing brain, before transforming into ramified microglia. 39 , 59 They have an increased level of motility, facilitated by their reduced and retracted processes, and are phenotypically very similar to macrophages found in the rest of the body. 60 It is still unknown whether these microglia are senescing microglia found in the brain, or if they originate from monocytes entering the brain in response to injury. 37 We found that our DSAD cases had significantly more amoeboid microglia than age matched controls or young individuals with DS. It is possible that monocytes from the blood are entering the brains of people with DS as vascular neuropathology may lead to small microhemorrhages in the brain 49 , 50 and the presence of more amoeboid microglial cells; however, further work needs to be done to explore the clinical significance of amoeboid microglia in DSAD.
Finally, and most importantly, dystrophic microglia counts are significantly increased in older individuals with DS (age effect), and in AD. The concept of a hypofunctional microglia, as opposed to activated, came from morphological analysis of Iba1‐positive microglia in autopsy samples from aged humans, where dystrophic microglia preceded neurofibrillary pathology in AD. 61 This suggested that the loss of microglial function might be as important as the activation of a toxic, reactive cell. Other studies that aimed to quantify and characterize microglial morphology in AD also found increased dystrophic cells. 38 , 40 , 42 , 45 Individuals with DSAD showed a higher number of dystrophic microglia than the AD group, albeit their numbers did not reach statistical difference. Additionally, young controls with DS have high numbers of dystrophic microglia in the WM, suggesting that this region in the DS brain is particularly vulnerable to degeneration, and that some aspects of aging might be accelerated in this population. 62 , 63 Dystrophic microglia have fragmented or “beaded” processes that may be the result of microglial dysfunction or senescence due to aging or AD neuropathology, and are thought of as important markers for AD pathology. It is well accepted that human microglial processes shorten with age, leading to a reduced arborization area and reduced capacity for GM surveillance. 27 , 51 Importantly, tau‐positive neurons are associated with dystrophic microglia, supporting a fundamental role for lack of microglial support in the disease progression. 61
Overall, individuals with DS, particularly those with DSAD, have a distinct microglial phenotype pattern. In DSAD, there is a shift toward the presence of higher numbers of dystrophic microglial cells and fewer ramified microglial cells suggesting fewer resting state microglia and more damage to microglia, as well as increased amoeboid cells. Further, individuals with DS appear to have important microglial changes that could be related to the development and progression of AD. Our data suggest that microglial changes progress gradually over time as DS individuals are aging and, more importantly, provide specific information related to the presence of AD in DS. These data reinforce the differences in the neuroinflammation phenotype seen in individuals with DS and, given the large variety of inflammatory genes located on chromosome 21, suggest that these different gene expression profiles could also be impacting the morphology of microglia cells, and consequently, their response to the presence of AD pathology. Neuropathological observations from autopsy cases with DS allows us to directly assess early events in AD, addressing a critical gap in our knowledge regarding early events, which are targetable, in sporadic AD pathogenesis.
AUTHOR CONTRIBUTIONS
Conception and design of the study: Alessandra C. Martini, Alex M. Helman, Frederick A. Schmitt, Elizabeth Head. Data acquisition and analysis: Alessandra C. Martini, Alex M. Helman, Katie L. McCarty. Manuscript preparation: Alessandra C. Martini, Alex M. Helman, Ira T. Lott, Eric Doran, Frederick A. Schmitt, Elizabeth Head. Study supervision: Elizabeth Head.
CONFLICTS OF INTEREST
The authors declare no competing interests.
ACKNOWLEDGMENTS
This study was supported by grants from the National Institutes of Health (NIH; NIH/NICHD HDR01064993 [E.H., E.A., F.A.S.]). Autopsy cases from UC Irvine were supported by the NIH (NIH/NIA P50AG16573 [I.T.L., E.D.]), R01AG21912 (E.D., I.T.L.), U01AG051412 (I.T.L, E.D, EH), and Brightfocus Grant #CA2018010 (E.H.). We also would like to thank the NIH NeuroBioBank for a subset of the autopsy cases used in this study (https://neurobiobank.nih.gov/). The authors would also like to thank Morgan Siever for her collaboration throughout the study.
Martini AC, Helman AM, McCarty KL, et al. Distribution of microglial phenotypes as a function of age and Alzheimer's disease neuropathology in the brains of people with Down syndrome. Alzheimer's Dement. 2020;12:e12113 10.1002/dad2.12113
Alessandra C Martini and Alex M Helman contributed equally to this study.
REFERENCES
- 1. Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, Martins R, et al. Amyloid A4 protein and its precursor in Down's syndrome and Alzheimer's disease. N Engl J Med. 1989;320:1446‐1452. [DOI] [PubMed] [Google Scholar]
- 2. Lott IT, Head E Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat Rev Neurol. 2019;15:135‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doran E, Keator D, Head E, Phelan MJ, et al. Down Syndrome, partial trisomy 21, and absence of Alzheimer's disease: the role of APP. J Alzheimers Dis. 2017;56:459‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prasher VP, Farrer MJ, Kessling AM, et al. Molecular mapping of Alzheimer‐type dementia in Down's syndrome. Ann Neurol. 1998;43:380‐383. [DOI] [PubMed] [Google Scholar]
- 5. Schupf N, Zigman WB, Tang MX, et al. Change in plasma Ass peptides and onset of dementia in adults with Down syndrome. Neurology. 2010;75:1639‐1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schupf N, Lee A, Park N, et al. Candidate genes for Alzheimer's disease are associated with individual differences in plasma levels of beta amyloid peptides in adults with Down syndrome. Neurobiol Aging. 2015;36:2907 e1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dekker AD, Fortea J, Blesa R, De Deyn PP Cerebrospinal fluid biomarkers for Alzheimer's disease in Down syndrome. Alzheimers Dement (Amst). 2017;8:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Startin CM, Ashton NJ, Hamburg S, et al. Plasma biomarkers for amyloid, tau, and cytokines in Down syndrome and sporadic Alzheimer's disease. Alzheimers Res Ther. 2019;11:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fortea J, Carmona‐Iragui M, Benejam B, et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer's disease in adults with Down syndrome: a cross‐sectional study. Lancet Neurol. 2018;17:860‐869. [DOI] [PubMed] [Google Scholar]
- 10. Ballard C, Mobley W, Hardy J, Williams G, Corbett A Dementia in Down's syndrome. Lancet Neurol. 2016;15:622‐636. [DOI] [PubMed] [Google Scholar]
- 11. Sinai A, Mokrysz C, Bernal J, et al. Predictors of age of diagnosis and survival of Alzheimer's disease in Down syndrome. J Alzheimers Dis. 2018;61:717‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schupf N, Sergievsky GH Genetic and host factors for dementia in Down's syndrome. Br J Psychiatry. 2002;180:405‐410. [DOI] [PubMed] [Google Scholar]
- 13. Royston MC, McKenzie JE, Gentleman SM, et al. Overexpression of s100beta in Down's syndrome: correlation with patient age and with beta‐amyloid deposition. Neuropathol Appl Neurobiol. 1999;25:387‐393. [DOI] [PubMed] [Google Scholar]
- 14. Griffin WS, Sheng JG, McKenzie JE, et al. Life‐long overexpression of S100beta in Down's syndrome: implications for Alzheimer pathogenesis. Neurobiol Aging. 1998;19:401‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilcock DM, Hurban J, Helman AM, et al. Down syndrome individuals with Alzheimer's disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer's disease. Neurobiol Aging. 2015;36:2468‐2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilcock DM, Griffin WS Down's syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflammation. 2013;10:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilcock DM Neuroinflammation in the aging down syndrome brain; lessons from Alzheimer's disease. Curr Gerontol Geriatr Res. 2012; 2012:170276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perez SE, Miguel JC, He B, et al. Frontal cortex and striatal cellular and molecular pathobiology in individuals with Down syndrome with and without dementia. Acta Neuropathol. 2019;137:413‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90:647‐660. [DOI] [PubMed] [Google Scholar]
- 20. Hartley D, Blumenthal T, Carrillo M, et al. Down syndrome and Alzheimer's disease: common pathways, common goals. Alzheimers Dement. 2015;11:700‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stoltzner SE, Grenfell TJ, Mori C, et al. Temporal accrual of complement proteins in amyloid plaques in Down's syndrome with Alzheimer's disease. Am J Pathol. 2000;156:489‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Naude PJ, Dekker AD, Coppus AM, et al. Serum NGAL is Associated with distinct plasma amyloid‐beta peptides according to the clinical diagnosis of dementia in Down syndrome. J Alzheimers Dis. 2015;45:733‐743. [DOI] [PubMed] [Google Scholar]
- 23. Rodrigues R, Debom G, Soares F, et al. Alterations of ectonucleotidases and acetylcholinesterase activities in lymphocytes of Down syndrome subjects: relation with inflammatory parameters. Clin Chim Acta. 2014;433:105‐110. [DOI] [PubMed] [Google Scholar]
- 24. Hopperton KE, Mohammad D, Trepanier MO, Giuliano V, Bazinet RP Markers of microglia in post‐mortem brain samples from patients with Alzheimer's disease: a systematic review. Mol Psychiatry. 2018;23:177‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Serrano‐Pozo A, Gomez‐Isla T, Growdon JH, Frosch MP, Hyman BT A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am J Pathol. 2013;182:2332‐2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Calderon D, Bhaskar A, Knowles DA, et al. Inferring Relevant cell types for complex traits by using single‐cell gene expression. Am J Hum Genet. 2017;101:686‐699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davies DS, Ma J, Jegathees T, Goldsbury C Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer's disease. Brain Pathol. 2017;27:795‐808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Torres‐Platas SG, Comeau S, Rachalski A, et al. Morphometric characterization of microglial phenotypes in human cerebral cortex. J Neuroinflammation. 2014;11:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Franco‐Bocanegra DK, McAuley C, Nicoll JAR, Boche D Molecular Mechanisms of microglial motility: changes in ageing and Alzheimer's Disease. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Griffin WS, Stanley LC, Ling C, et al. Brain interleukin 1 and S‐100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:7611‐7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xue QS, Streit WJ Microglial pathology in Down syndrome. Acta Neuropathol. 2011;122:455‐466. [DOI] [PubMed] [Google Scholar]
- 32. Azari NP, Pettigrew KD, Pietrini P, Horwitz B, Schapiro MB Detection of an Alzheimer disease pattern of cerebral metabolism in Down syndrome. Dementia. 1994;5:69‐78. [DOI] [PubMed] [Google Scholar]
- 33. Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol. 1997;42:85‐94. [DOI] [PubMed] [Google Scholar]
- 34. Lin AL, Powell D, Caban‐Holt A, et al. (1)H‐MRS metabolites in adults with Down syndrome: effects of dementia. Neuroimage Clin. 2016;11:728‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boche D, Perry VH, Nicoll JA Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013;39:3‐18. [DOI] [PubMed] [Google Scholar]
- 38. Bachstetter AD, Van Eldik LJ, Schmitt FA, et al. Disease‐related microglia heterogeneity in the hippocampus of Alzheimer's disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun. 2015;3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A Physiology of microglia. Physiol Rev. 2011;91:461‐553. [DOI] [PubMed] [Google Scholar]
- 40. Streit WJ, Xue QS, Tischer J, Bechmann I Microglial pathology. Acta Neuropathol Commun. 2014;2:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Streit WJ Microglial senescence: does the brain's immune system have an expiration date? Trends Neurosci. 2006;29:506‐510. [DOI] [PubMed] [Google Scholar]
- 42. Streit WJ, Sammons NW, Kuhns AJ, Sparks DL Dystrophic microglia in the aging human brain. Glia. 2004;45:208‐212. [DOI] [PubMed] [Google Scholar]
- 43. Bachstetter AD, Norris CM, Sompol P, et al. Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age‐dependent progression of Alzheimer's disease‐related pathology. J Neurosci. 2012;32:10201‐10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bachstetter AD, Rowe RK, Kaneko M, Goulding D, Lifshitz J, Van Eldik LJ. The p38alpha MAPK regulates microglial responsiveness to diffuse traumatic brain injury. J Neurosci. 2013;33:6143‐6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paasila PJ, Davies DS, Kril JJ, Goldsbury C, Sutherland GT The relationship between the morphological subtypes of microglia and Alzheimer's disease neuropathology. Brain Pathol. 2019;29:726‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Head E, Lott IT, Wilcock DM, Lemere CA Aging in Down Syndrome and the development of Alzheimer's disease neuropathology. Curr Alzheimer Res. 2016;13:18‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bachstetter AD, Ighodaro ET, Hassoun Y, et al. Rod‐shaped microglia morphology is associated with aging in 2 human autopsy series. Neurobiol Aging. 2017;52:98‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Powell D, Caban‐Holt A, Jicha G, et al. Frontal white matter integrity in adults with Down syndrome with and without dementia. Neurobiol Aging. 2014;35:1562‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Head E, Phelan MJ, Doran E, et al. Cerebrovascular pathology in Down syndrome and Alzheimer disease. Acta Neuropathol Commun. 2017;5:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Helman AM, Siever M, McCarty KL, et al. Microbleeds and cerebral amyloid angiopathy in the brains of people with Down syndrome with Alzheimer's disease. J Alzheimers Dis. 2019;67:103‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wolf SA, Boddeke HW, Kettenmann H Microglia in Physiology and Disease. Annu Rev Physiol. 2017;79:619‐643. [DOI] [PubMed] [Google Scholar]
- 52. Gefen T, Kim G, Bolbolan K, et al. Activated microglia in cortical white matter across cognitive aging trajectories. Front Aging Neurosci. 2019;11:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Raj D, Yin Z, Breur M, Doorduin J, et al. Increased white matter inflammation in aging‐ and Alzheimer's disease brain. Front Mol Neurosci. 2017;10:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ohm DT, Kim G, Gefen T, et al. Prominent microglial activation in cortical white matter is selectively associated with cortical atrophy in primary progressive aphasia. Neuropathol Appl Neurobiol. 2019;45:216‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Caso F, Agosta F, Mattavelli D, et al. White matter degeneration in atypical Alzheimer disease. Radiology. 2015;277:162‐172. [DOI] [PubMed] [Google Scholar]
- 56. Head E, Azizeh BY, Lott IT, Tenner AJ, Cotman CW, Cribbs DH Complement association with neurons and beta‐amyloid deposition in the brains of aged individuals with Down syndrome. Neurobiol Dis. 2001;8:252‐265. [DOI] [PubMed] [Google Scholar]
- 57. Ziebell JM, Taylor SE, Cao T, Harrison JL, Lifshitz J Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J Neuroinflammation. 2012;9:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Streit WJ, Walter SA, Pennell NA Reactive microgliosis. Prog Neurobiol. 1999;57:563‐581. [DOI] [PubMed] [Google Scholar]
- 59. Harry GJ, Kraft AD Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology. 2012;33:191‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Karperien A, Ahammer H, Jelinek HF Quantitating the subtleties of microglial morphology with fractal analysis. Front Cell Neurosci. 2013;7:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Streit WJ, Braak H, Xue QS, Bechmann I Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Auge E, Bechmann I, Llor N, Vilaplana J, Krueger M, Pelegri C Corpora amylacea in human hippocampal brain tissue are intracellular bodies that exhibit a homogeneous distribution of neo‐epitopes. Sci Rep. 2019;9:2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nishimura A, Sawada S, Ushiyama I, et al. Lectin‐histochemical detection of degenerative glycoconjugate deposits in human brain. Forensic Sci Int. 2000;113:265‐269. [DOI] [PubMed] [Google Scholar]