

Abstract
The metabolism of the non-essential amino acid L-proline is emerging as a key pathway in the metabolic rewiring that sustains cancer cells proliferation, survival and metastatic spread. Pyrroline-5-carboxylate reductase (PYCR) and proline dehydrogenase (PRODH) enzymes, which catalyze the last step in proline biosynthesis and the first step of its catabolism, respectively, have been extensively associated with the progression of several malignancies, and have been exposed as potential targets for anticancer drug development. As investigations into the links between proline metabolism and cancer accumulate, the complexity, and sometimes contradictory nature of this interaction emerge. It is clear that the role of proline metabolism enzymes in cancer depends on tumor type, with different cancers and cancer-related phenotypes displaying different dependencies on these enzymes. Unexpectedly, the outcome of rewiring proline metabolism also differs between conditions of nutrient and oxygen limitation. Here, we provide a comprehensive review of proline metabolism in cancer; we collate the experimental evidence that links proline metabolism with the different aspects of cancer progression and critically discuss the potential mechanisms involved.
Subject terms: Cancer metabolism, Cancer
Facts
Proline metabolism is widely rewired during cancer development.
The rewiring of proline metabolism influences numerous physiological pathways, including mitochondrial metabolism, apoptosis, protein synthesis.
PRODH acts as a tumor suppressor or an oncogene depending on the tumor type and the environmental, metabolic context.
PYCR1 acts as an oncogene and is overexpressed in a wide variety of malignancies.
Open questions
What is the impact of the interplay between proline biosynthesis and degradation in cancer?
How is the rewiring of proline metabolism regulated depending on cancer type and cancer subtype?
Is it possible to develop successful pharmacological inhibitor of proline metabolism enzymes for anticancer therapy? And to what extent would these inhibitors be useful in conjunction with standard of care chemotherapies?
Does the rewiring of proline metabolism affect the anti-tumor immune response and cancer immune evasion?
To what extent and with what consequences are proline metabolism enzymes post-translational modified?
Introduction
In their revised version of the hallmarks of cancer, Hanahan and Weinberg introduced the new hallmark ‘deregulating cellular energetics’1 in acknowledgement of the growing amount of literature that flourished around the rediscovery of Otto Warburg’s observation of deregulated glucose metabolism in cancer cells. During the 1920s, Warburg demonstrated that cultured tumor tissues display high rates of glucose uptake and lactate secretion, even in the presence of oxygen, a phenomenon known as aerobic glycolysis or the Warburg effect2,3. In the wake of the revival of Warburg’s work, numerous discoveries led to the appreciation that cancer cells undergo extensive metabolic reprogramming to preserve redox and energetic balance while meeting the metabolic demand imposed by rapid proliferation4,5. Depending on the cancer type and its tissue of origin, alterations in diverse metabolic pathways have been identified that are involved in the metabolism of the main biological macromolecules: lipids, nucleic acids, carbohydrates, and amino acids, including non-essential amino acids (NEAAs)6.
The rewiring of NEAA metabolism bears clinically relevant consequences7. L-asparaginase has been used to treat acute lymphoblastic leukemia and non-Hodgkin lymphomas since the 1970s. Asparaginase enzymes deaminate L-asparagine to aspartic acid and ammonia. Since leukemia cells require external supplementation of the NEAA L-asparagine, its depletion by asparaginase treatment is lethal to tumor cells. The efficacy and selectivity of this treatment result from cancer cells downregulating the expression of the asparagine synthetase ASNS gene8. Similarly, some cancers silence the expression of the argininosuccinate synthase-1 ASS1 gene, which encodes the enzyme that catalyzes the condensation of citrulline and aspartate to form argininosuccinate in the urea cycle9. ASS1 silencing sustains proliferation by diverting aspartate away from the urea cycle toward nucleotide biosynthesis10, but, concomitantly, results in cancer cells becoming auxotrophic (i.e., depending on external supplementation) for arginine. Preclinical studies and clinical trials have confirmed that ASS1-negative cancers are susceptible to arginine deprivation therapies using mycoplasma-derived arginine deiminase or recombinant human arginase11–26.
In recent years, adaptations of other NEAA metabolic pathways have been associated with cancer progression7,27. Enthusiasm and research efforts have grown with regard to the possibility of targeting those pathways for developing new cancer therapeutics7. This review focuses on the NEAA proline and the experimental evidence that associates its unique metabolism to cancer.
The essential NEAA L-proline
L-proline has a distinctive structure compared to other proteinogenic amino acids, as it lacks the primary amine group and instead has a secondary amine due to the nitrogen group covalently binding the alpha carbon to form a five-membered imino ring (Fig. 1). This unique conformation grants L-proline essential properties in influencing the 3D structure of proteins28. L-proline is known as a ‘helix breaker’ due to its ability to disrupt the α-helix conformation by introducing a kink. Proline kinks play important roles in influencing the 3D structure of proteins, including transmembrane helices29,30. In addition, proline-rich motifs within proteins mediate critical protein–protein interactions31.
Fig. 1. The proline metabolic pathway.
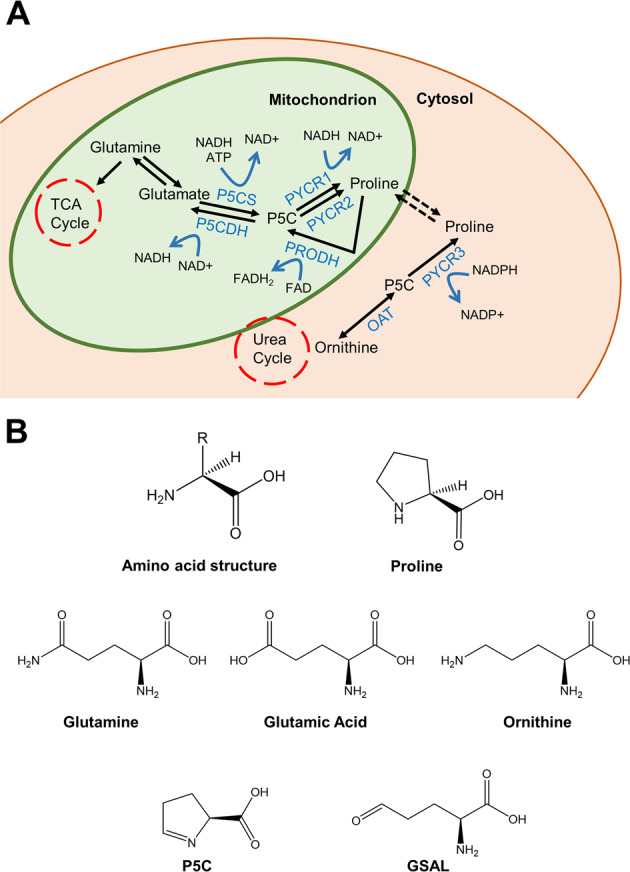
A The proline metabolic pathway. The NEAA L-proline is formed through reduction of precursor P5C that is obtained via two pathways, from glutamine in the mitochondria and from ornithine in the cytosol. B Chemical structures of the proline metabolic pathway intermediates. Note the secondary amine group in proline is different from that of other amino acids. Produced in Chemdraw.
L-proline can also be modified for regulatory purposes. For example, cis–trans-isomerization of L-proline residues by peptidyl-prolyl isomerases controls protein folding and function, with important implications for tumorigenesis32,33. L-proline hydroxylation is a well know post-translational modification of proteins. Hydroxylation of L-proline can have critical regulatory functions, as in the case of the transcription factor hypoxia-inducible factor (HIF) 1, which mediates cellular response to oxygen deprivation. HIF1 is a dimer of a constitutively expressed HIF1-β subunit and an oxygen-sensitive HIF1-α subunit34 that accumulates in hypoxic cells. HIF1-α protein contains two proline residues (P402/P564) that are hydroxylated in the presence of oxygen by prolyl hydroxylase domain-containing proteins (PHDs). Hydroxylation of the proline residues is a pre-requisite for von Hippel–Lindau-mediated ubiquitination and degradation of HIF1-α, preventing its accumulation in oxygenated tissues34.
L-proline is also the most abundant amino acid contributing to the composition of the extracellular matrix (ECM). Collagen, the main structural protein of the ECM, contains ~25% of L-proline residues. When L-proline is incorporated into collagen, its hydroxylation by prolyl hydroxylases generates 4-hydroxyproline. This high L-proline content stabilizes collagen molecules with 4-hydroxyproline having the biggest stabilizing effect. The positioning of these residues is also important for recognition and subsequent degradation of collagen by matrix metalloproteinases35–37. Moreover, intracellular hydroxyproline can originate from collagen digestion by prolidase, a cytosolic dipeptidase that breaks down dipeptides to produce free L-proline and L-hydroxyproline36.
Proline metabolism
The metabolic pathways that control proline biosynthesis and catabolism are distinct from that of other NEAAs, as the common pyridoxal-phosphate-coupled transaminases cannot metabolize the L-proline secondary amine.
The biosynthesis of L-proline occurs from glutamine in the mitochondria and ornithine in the cytosol with both precursors converging to the intermediate glutamate-γ-semialdehyde (GSAL), which exists in spontaneous equilibrium with the ring-structured pyrroline-5-carboxylate (P5C) (Fig. 1). In the cytosol, ornithine is converted to GSAL/P5C by the action of the pyridoxal-phosphate dependent, mitochondrial enzyme ornithine aminotransferase (OAT), which is also responsible for catalyzing the reverse reaction. In the mitochondria, glutamate is converted to GSAL/P5C by the mitochondrial enzyme 1-pyrroline-5-carboxylate synthase (P5CS, encoded by the ALDH18A1 gene, chromosome 10q24.1). In eukaryotes, P5CS is a bifunctional enzyme, which contains an N-terminal glutamate kinase domain and a C-terminal γ-glutamyl phosphate reductase (GPR) domain. P5CS phosphorylates and then reduces glutamate to P5C, and requires both ATP and NADH as cofactors38. The P5CS protein has two alternative spliced forms. The shorter isoform is highly expressed in the gut, whereas the longer isoform, which differs from the shorter by the addition of two amino acids in the GPR domain, is ubiquitous39.
In the final reaction, P5C is converted to L-proline by the activity of NAD(P)H-dependent PYCR enzymes. There are three homologous PYCR isoforms: PYCR1, PYCR2, PYCR3 (aka PYCRL), which are encoded for by separate genes, namely PYCR1 (chromosome 17q25.3), PYCR2 (chromosome 1q42.12), and PYCR3 (chromosome 8q24.3). In addition, each isoform is encoded by several poorly characterized splice variants (Table 1). PYCR3 is the only proline metabolism enzyme to be localized in the cytoplasm, and its similarity to the other PYCR isoforms is around 45%. PYCR1 and 2 share 85% sequence similarity and have been reported to heterodimerize and to be localized to the mitochondria40–42. However, the exact localization of PYCR1 and 2 within the mitochondrion is not known, and PYCR1 has been suggested to localize to the outer mitochondrial membrane and, partially, to the cytoplasm42,43. PYCR isoforms differ in their cofactor and substrate affinities. Current data indicate that PYCR1 and 2 have a higher affinity for the cofactor NADH, whereas the rate of conversion of P5C by PYCR3 is higher in the presence of NADPH41,44. Moreover, isotope enrichment experiments in melanoma cell lines showed that PYCR1 and PYCR2 primarily catalyze the synthesis of L-proline from glutamate, although, in the presence of high levels of ornithine, PYCR1 can also produce L-proline through ornithine. The same experiments also indicated that PYCR3 works exclusively along the ornithine route41.
Table 1.
Splice variants of the three PYCR isoforms as identified on NCBI.
| Isoform | Gene location | NCBI reference | Transcript variant | mRNA length | Amino acid length |
|---|---|---|---|---|---|
| PYCR1 | 17q25.3 | NP_008838.2 | 1 | 2269 | 319 |
| NP_722546.1 | 2 | 1926 | 316 | ||
| NP_001269208.1 | 3 | 1776 | 288 | ||
| NP_001269209.1 | 4 | 1869 | 319 | ||
| NP_001269210.1 | 5 | 1951 | 346 | ||
| NP_001317452.1 | 6 | 1705 | 217 | ||
| PYCR2 | 1q42.12 | NP_037460.2 | 1 | 1680 | 320 |
| NP_001258610.1 | 2 | 1458 | 246 | ||
| PYCR3 | 8q24.3 | NP_075566.3 | 1 | 3342 | 274 |
| NP_001316795.2 | 2 | 3282 | 254 |
PYCR1 transcript variants 1 and 4 code for the same protein.
A detailed 3D structure of the human PYCR1 protein has been solved by Christensen and colleagues (Fig. 2)44,45. This work confirmed previous observations that in vivo monomeric PYCR1 proteins coalesce into a decameric structure46 or, more accurately, a pentamer of dimers of over 350 kDa. Each monomeric enzyme consists of an N-terminal Rossmann-fold domain for binding of NAD(P)H and a six α-helix C-terminal dimerization domain. The substrate P5C binds at the interface between two dimers with its ring positioned in parallel to nicotinamide, such that the hydride acceptor atom of P5C lies in proximity to the C4 carbon donor of nicotinamide, suggesting a direct hydride-transfer mechanism for the reduction of P5C44.
Fig. 2. Detailed 3D structure of the human PYCR1 enzyme.
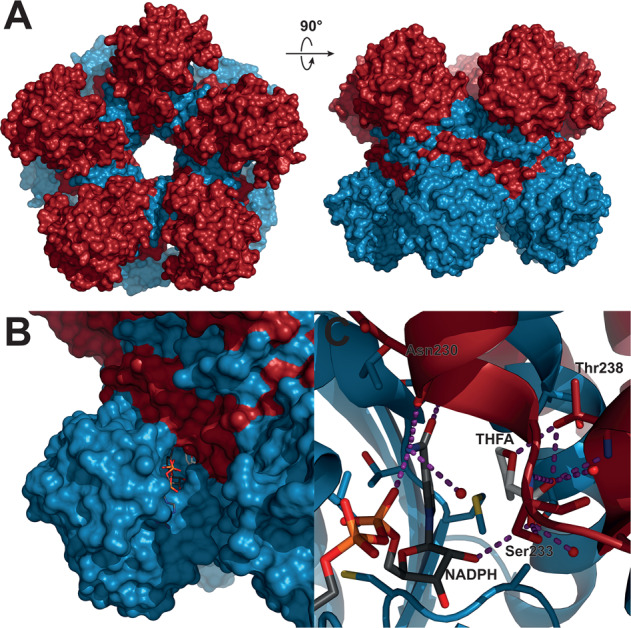
A Overview of space-filling model of PYCR1 (PDB ID 5UAV, ref. 44). PYCR1 forms a pentamer of dimers, with individual dimer subunits colored blue and red, respectively. B Zoomed view of the NADPH binding site and the active site. NADPH is bound to the N-terminal Rossmann-fold dinucleotide-binding domain, which forms the outer part of the protomer, whereas the P5C/proline analog L-tetrahydrofuroic acid (THFA) interacts with the C-terminal dimerization domains of both protomers. NADPH and THFA are shown as sticks with dark gray and light gray carbons, respectively. C Close-up cartoon of the active site of PYCR1. Residues within 4 Å of THFA or the nicotinamide moiety of NADPH are shown as sticks, water oxygens are shown as red spheres, and hydrogen bonds are highlighted with purple dashes. The substrate analog is mainly stabilized by a hydrogen bond network with the backbone and side-chain atoms of Ser233 and Thr238 and 2 water molecules. The images were prepared using PyMOL 2.3.5 (ref. 45).
The catabolism of L-proline follows an opposite pathway to its biosynthesis catalyzed by different enzymes. The first step is mediated by the PRODH (aka proline oxidase, POX) enzymes, which catalyze the FADH-dependent oxidation of proline to P5C. There are two PRODH enzymes; PRODH1, the primary enzyme in the conversion of L-proline back to P5C, coded by the PRODH1 gene (chromosome 22q.11.21), and PRODH2, coded by the PRODH2 gene (chromosome 19q13.12), which catalyzes hydroxyproline conversion to pyrroline-3-hydroxy-5-carboxylate47. PRODH1 is conserved throughout evolution although, in some prokaryotes, it is fused with pyrroline-5-carboxylate dehydrogenase (P5CDH) to form the Proline Utilization A (PutA) flavoprotein. A study of the Escherichia coli homolog of the PRODH portion of PutA showed a dimeric structure with each subunit containing a dimerization domain, a three-helix bundle, and a β/α-barrel catalytic PRODH domain48,49. The residues that are involved in proline binding are conserved across different organisms, suggesting that structural properties of the prokaryotic enzymes likely apply to their eukaryotic counterpart50. L-proline catabolism is coupled to mitochondrial respiration through the FADH-mediated transfer of electrons to the electron transport chain (ETC) or direct interaction between PRODH and coenzyme Q51–53.
PRODH activity generates GSAL/P5C. GSAL/P5C can be metabolized to ornithine by OAT, linking proline metabolism to the urea cycle54. Alternatively, GSAL is oxidized by NAD-dependent mitochondrial enzyme P5CDH (ALDH4A1, chromosome 1p36.13) to yield glutamate55–57. P5CDH displays a high degree of conservation across prokaryotes and eukaryotes and has a typical ALDH structure, consisting of a NAD+-binding non-canonical Rossmann fold, a catalytic β/α-barrel domain, and a β structured oligomerization domain58. Two subunits interact via the β sheets of the catalytic domain to form a dimeric structure50. In Thermus thermophiles, bacterial P5CDH was shown to form a hexameric complex with two other dimers59.
From this summary, it emerges that the metabolism of proline is wired with other key metabolic pathways, including the urea cycle through ornithine and the tricarboxylic acid (TCA) cycle through glutamate, which can be promptly metabolized to the TCA cycle intermediate α-ketoglutarate by glutamate dehydrogenase or through transamination. In addition, through redox regulation of cofactors NADH and FADH, proline metabolism influences the activity of mitochondrial TCA and ETC51,52. Although it remains to be fully elucidated, the ability of PYCR3 and, possibly, PYCR1 to metabolize NADPH within the cytoplasm might stimulate the pentose phosphate pathway (PPP) with important implication for ribose biosynthesis and nucleotide metabolism60. It is noteworthy that, other than de novo biosynthesis or nutrient uptake, breakdown of collagen is an important reservoir of L-proline, which, as outlined below, can be exploited by cancer cells43,61,62.
Regulation of the proline metabolic pathway
When L-proline levels are high, negative feedback loops are activated in order to decrease biosynthesis. Both P5CS and PYCR1 are inhibited by the final product of the pathway, L-proline, though the mechanism of this inhibition is still elusive (Fig. 3). In vitro data indicate that, of the three PYCR isoforms, PYCR2 is the most sensitive to proline inhibition, showing a robust decline in enzymatic activity at physiological concentrations of L-proline (0.1–0.3 mM)41, whereas PYCR1 and PYCR3 showed limited product feedback inhibition41. It will be important to ascertain the implications of these dissimilar behaviors for proline metabolism rate in vivo. Moreover, PYCR enzymes have been reported to be sensitive to inhibition by ATP63. On the other hand, studies in renal cell carcinoma cell lines showed that reducing the level of glutamine in cell culture causes increased transcription of the PYCR1 gene, but not its paralogues64. Glutamine can be deaminated to glutamate by glutaminase enzymes and, therefore, it is a major precursor of proline biosynthesis. Hence, increased expression of biosynthesis enzymes compensates for reduced availability of L-proline precursors.
Fig. 3. Proline metabolism is regulated by many cellular pathways and influence key cellular signaling nodes.

A Regulation of the proline metabolic pathway is supported by the activity of numerous signaling pathways (→). Feedback inhibition loops (--|) also contribute to regulate the rate of the proline metabolism pathway. B The proline metabolic pathway enzymes regulate signaling activity of numerous pathways both positively (→) and negatively (--|).
Ornithine can inhibit P5CS activity in an isoform-dependent fashion38,39. Whereas the short P5CS isoform is inhibited non-competitively by ornithine, the two amino acid insertion that mark the long isoform abolishes the feedback inhibition by ornithine. It is thought that ornithine inhibition rewires proline metabolism to favor arginine biosynthesis38,39. Indeed, high ornithine levels have been shown to inhibit the biosynthesis of L-proline from glutamate through reduced activity of P5CS38,39. Since the long isoform of P5CS is ubiquitous, whereas the short one is preferentially expressed in the gut, a channeling of proline metabolism toward arginine biosynthesis likely occurs in the gut, whereas other tissues primarily synthesize L-proline38,65.
PRODH has been shown to be inhibited by lactate66. Lactate is the product of anaerobic respiration and is produced by cancer cells that display enhanced aerobic glycolysis. Therefore, the inhibition of PRODH by lactate indicates PRODH activity might be negatively affected in cancer cells.
The ambivalent role of PRODH in cancer
Currently, no mutations have been described in the proline metabolism genes that have been functionally linked to tumor development. However, changes in gene expression and metabolic flux through the pathway have been extensively reported. The majority of the experimental evidence that associates the metabolism of L-proline to cancer development revolves around the two genes that catalyze the last step in L-proline biosynthesis and the first step of its catabolism, namely PYCR1 and PRODH1. A simplified view of those genes’ modus operandi in cancer would implicate proline biosynthesis by the PYCR enzymes in cancer progression and its catabolism by PRODH in the suppression of tumorigenesis. However, the regulation and outcome of these enzymes’ activities in cancer progression are mostly context-dependent and exceedingly more complex (Fig. 3).
PRODH1 was identified in 1997 as PIG6, a pro-oxidative gene upregulated in response to the ectopic expression of the tumor suppressor p53 in colorectal cancer cell lines67. PRODH was thus implicated in the ROS-dependent apoptotic response to p53 induction68. Notably, a follow-up work indicated that overexpression of PRODH1 was sufficient to induce apoptosis in a p53-resistant bladder cancer cell line68. Subsequent reports confirmed the ability of PRODH1 and, to some extent PRODH2, to induce ROS, in particular anion superoxide, trigger apoptosis in a ROS-dependent fashion and act as a tumor suppressor69–72. Intriguingly, ectopic expression of PRODH1 in DLD-1 colorectal cancer cell lines triggers apoptosis through multiple mechanisms (Fig. 4). In addition to ROS-dependent intrinsic apoptosis73, PRODH induces expression of the death receptor TRAIL, and inhibition of caspase-8 activity is effective in restricting apoptosis in PRODH1-overexpressing cells, implicating activation of the extrinsic apoptotic pathway74,75. Finally, these apoptotic responses are enabled by a robust ROS-dependent downregulation of the ERK1/2 branch of the mitogen-activated protein kinase (MAPK) pathway, which has been widely implicated in malignant progression74. Indeed, expression of the ROS scavenging enzyme manganese superoxide dismutase (MnSOD) inhibits PRODH1-induced apoptosis74. However, the contribution of PRODH1-induced apoptosis to tumor suppression in vivo is unclear. Indeed, xenograft experiments performed using DLD-1 colorectal cancer cells engineered to overexpress PRODH1 showed that the reduction in tumor growth triggered by PRODH1 expression correlated only with marginal induction of cell death, but with markedly reduced proliferation71. A follow-up in vitro experiment led to the appreciation that the phenotypic outcome of PRODH1 expression depends on quantity: high levels of PRODH1 cause apoptosis, whereas lower levels of expression prompt a cell cycle arrest in the G2 phase71. Another study implicated PRODH1 in the regulation of the enzyme cyclooxygenase 2 (COX-2), whereby increased expression of PRODH1 inhibited expression of COX-2 (Fig. 3)76. COX-2 is an enzyme involved in prostaglandin biosynthesis and its expression correlates with worse prognosis in several malignancies77. Indeed, COX-2 inhibitors, such as celecoxib, have widespread anticancer activities and can induce apoptosis in cancer cells78. Of note, preliminary indications in oral squamous cell carcinoma suggest that celecoxib treatment increases levels of PRODH1, although it has yet to be established whether such an increase contributes to celecoxib-mediated suppression of cell growth and viability79. In agreement with its role as tumor suppressor gene, reduced expression of PRODH1 has been reported in several cancers, with more consistent downregulation observed in renal cancers and cancers arising from the digestive tract64,71,76,79–81.
Fig. 4. Proline metabolism in the regulation of cell survial and redox balance.
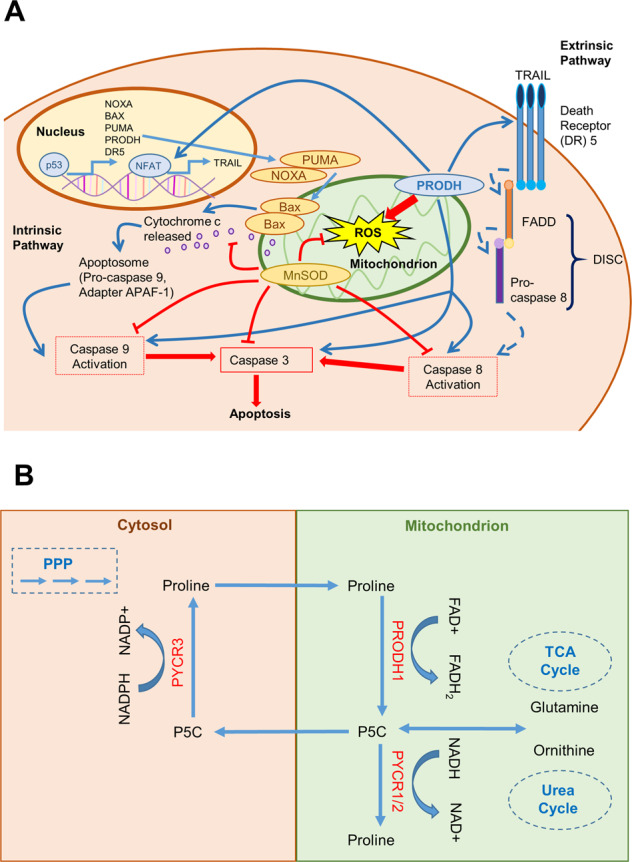
A PRODH has been implicated in the activation of apoptosis via both the intrinsic and extrinsic pathways. Activation of the intrinsic pathway by reactive oxygen species (ROS) is mediated by the p53-induced transcriptional activation of genes, including PRODH1. In the intrinsic pathway, the BH3-only proteins, such as NOXA and PUMA, inactivate the pro-survival proteins such as BCL-2 and lead to the release of pro-apoptotic proteins such as BAX. This allows the permeabilization of the mitochondrial outer membrane. Cytochrome c is released from the mitochondria and stimulates assembly of the apoptosome containing APAF-1 and pro-caspase 9. The apoptosome leads to the activation of caspase 9, which, in turn, activates the effector caspase 3 resulting in apoptosis. The extrinsic pathway of apoptosis is initiated by the activation of the death receptor of the TNF superfamily, such FAS and TRAIL. Receptor engagement and trimerization lead to the recruitment and activation of caspase-8 in the death inducing signaling complex (DISC), which in turn activates the effector caspase 3, resulting in apoptosis. PRODH1 has been shown to induce expression of the death receptor 5 (DR5) and its ligand TRAIL through stimulation of the nuclear factor of activated T cells (NFAT) transcription factor. B Schematic representation of the proposed proline cycle. L-proline is oxidized to P5C by PRODH1 in the mitochondria. P5C is then reduced back to L-proline through the action of the PYCR3 enzyme in the cytosol, a reaction coupled with the oxidation of NADPH. The cycle shuttles electrons from the cytosol to the mitochondria and stimulates flux through the PPP via oxidation of NADPH. In this way, the proline cycle can potentially contribute to nucleotide biosynthesis.
As aforementioned, the PRODH1 enzyme localizes in the mitochondrial inner membrane and transfers electrons directly to Coenzyme Q1 (CoQ1)51,82. In this context, the role of PRODH1 in cancer is double-faced. In conditions of nutrient limitation, PRODH1 reduces CoQ1 directly and contributes to ETC function and energy production51,82. However, as the levels and activity of PRODH1 increases, the consequent generation of ROS causes downregulation of several components of the ETC and compromises mitochondrial respiration51. Interesting as they are, these data also outline the limitations of interpreting experiments performed using non-physiological overexpression. Notwithstanding, in cancer cell lines from different tumors, PRODH1 is upregulated in response to energetic stress and nutrient deprivation triggered by hypoxia or glucose restriction82,83. In these challenging circumstances, cells engage the AMP-protein kinase (AMPK). AMPK is a sensor of cellular energy status, which is activated under conditions of high AMP/ATP ratio caused by stressors, including hypoxia or nutrient deprivation. Once activated, AMPK rewires cellular metabolism by stimulating catabolism to revert the energetic deficit and by triggering autophagy to counteract nutrient limitation84. AMPK also stimulates transcription from the PRODH1 gene and the accumulation of PRODH1 enzyme contributes to cell survival82,85. Under glucose restriction, PRODH1 degrades L-proline to sustain mitochondrial activity and ATP production, whereas, in hypoxic cells, PRODH1-mediated increase in ROS levels contributes to activation of autophagy (Fig. 3)82,85. Therefore, the outcome of ROS production by PRODH is context-dependent: it is pro-apoptotic in normoxia but stimulates protective autophagy in hypoxia83. The link between hypoxia and proline metabolism goes deeper; the main outcome of the hypoxia response protein HIF1-α is to promote expression of matrix metalloproteinase and degradation of the ECM. This causes an increase in hydroxyproline availability, which has been reported to contribute to HIF1-α stabilization and cancer cell survival in hepatocellular carcinoma cells exposed to hypoxia86.
The ability of PRODH1 to support mitochondrial function during nutrient stress proved essential in pancreatic cancer61. Pancreatic tumor cells organize into glandular structures surrounded by a dense, collagen-rich fibrotic tissue that hinders vascularization and reduces oxygen and nutrient availability87. In this instance, tumor cells maintain a functional TCA cycle through PRODH1 activity and collagen breakdown, as carbon obtained from the catabolism of collagen-derived L-proline feeds into the TCA cycle61. Chemical inhibition or genetic depletion of PRODH1 in pancreatic cancer cells compromise mitochondrial oxygen consumption, which can be recovered by external L-proline supplementation, and dramatically reduces cell growth in vitro or in vivo61.
PRODH1 has been shown to contribute to non-small cell lung cancer (NSCLC) progression through both cell-autonomous mechanisms and the promotion of a pro-tumorigenic inflammatory milieu88. Increased expression of PRODH1 occurs in a subset of NSCLC specimens. Consistent with previous findings67,69, this increased expression is p53-dependent, which begs the questions whether NSCLC reliance on PRODH1 persists in cancers in which p53 is mutated88. Functionally, PRODH activity drives cell proliferation and tumor growth in vivo and promotes cell migration and invasion88. Moreover, expression of PRODH1 triggers production of intracellular ROS, which, in turn, drives the expression of pro-inflammatory cytokines88. Whether those cytokines play a direct role in promoting carcinogenesis necessitates further investigation. However, their increased expression in lung adenocarcinoma specimens correlates with poor prognosis88.
A role for PRODH in promoting breast cancer has also been reported89. When grown in 3D culture, breast cancer cells increase the expression of PRODH1 and augment proline catabolism. Genetic depletion of PRODH1 or pharmacological inhibition of PRODH1 using the reversible inhibitor L-tetrahydro-2-furoic acid (L-THFA) impair growth of breast cancer cells89 in vitro. However, L-THFA treatment failed to inhibit growth of primary tumors in vivo when breast cancer cells were orthotopically injected in recipient mice89.
Overall, these reports unambiguously demonstrate that, in some relevant circumstances, PRODH1 supports tumor progression and is a potential anticancer drug target89,90. The attribution of PRODH1 to the category of tumor suppressor is, therefore, context-dependent, emphasizing the notion that metabolic adaptations are cancer-specific.
The unequivocal role of PYCR1 in cancer
PYCR1 was initially identified as a pro-tumorigenic gene in an RNA-interference screening in breast cancer91. More recently, a meta-analysis covering 1981 tumors from 19 different types of cancers highlighted that PYCR1 is one of the most commonly overexpressed metabolic genes in human cancer92. A wider rewiring of proline metabolism in favor of proline biosynthesis was suggested by Phang’s lab. The oncogenic transcription factor c-Myc is often upregulated in cancer, and it is known to rewire tumor cell metabolism, especially stimulating glutamine catabolism to fuel the TCA cycle93,94. Working with prostate cancer and Burkitt lymphoma cells, Phang’s team demonstrated that c-Myc triggers downregulation of the PRODH1 enzyme through increased expression of Mir-23b*, while enhancing transcription from the PYCR1 and ALDH18A1 (encoding P5CS) genes95. Therefore, c-Myc appears to promote a switch from proline catabolism to proline anabolism in cancer, reshuffling part of the cellular glutamine toward proline biosynthesis.
Increased expression of PYCR1 has since been confirmed in different cancers, including melanoma, NSCLC, renal cell carcinoma, breast cancer, colorectal cancer, prostate cancer, hepatocellular carcinoma, and isocitrate dehydrogenase 1 (IDH1)-mutant gliomas41,42,64,96–102. In some instances, PYCR1 expression is a reliable diagnostic biomarker which predicts poor prognosis in patients with melanoma96, breast cancer64,101, renal cell cancers100,103, and NSCLC98. Studies that employed genetic depletion of PYCR1 have confirmed a functional role for PYCR1 in promoting tumor progression and cancer cell survival in colorectal cancer cells104, NSCLC cells42,97,98, renal cancer cells64,103, prostate cancer cells105, and IDH1-mutant gliomas106.
Different mechanisms have been proposed to explain the ability of PYCR1 to support tumor progression, which could suggest a remarkable degree of pleiotropy and tissue specificity.
Proline biosynthesis and protein translation
Intuitively, PYCR1 activity might satisfy cancer cells’ increased demand for the NEAA L-proline. Indeed, in some experimental conditions, external supplementation of L-proline partially rescues the reduced cellular proliferation and survival caused by perturbations of the proline biosynthesis pathway42,64,107. A subset of clear cell renal cell carcinomas and the majority of invasive ductal breast carcinomas show increased expression of PYCR1 mRNA64,101. In these cancers, PYCR1 activity provides L-proline necessary for protein biosynthesis. Cancer cells depleted of PYCR1 display impaired progression in growth-limiting conditions, such as in vivo in recipient mice or in the presence of low glutamine in vitro64. This growth defect is associated with ribosome stalling at proline codons because of reduced aminoacylation of the corresponding tRNAs. Consistent with a direct role for L-proline production, external supplementation of L-proline is sufficient to rescue the growth defect64. That L-proline availability might be limiting to cancer cells is in agreement with the reduced fasting plasma levels of L-proline in patients with lymphoma and some sarcomas, a reduction that is not correlated to weight loss or circulating levels of other amino acids108.
Modulation of protein translation by PYCR1 activity has been reported, where disruption of proline metabolism affects the two main pathways that regulate protein synthesis: the PI3K/Akt/mTOR pathway and the amino acid response (AAR) pathway (Fig. 3). The mammalian (or mechanistic) target of rapamycin (mTOR) kinase is a major regulator of organismal response to nutrient availability. When nutrients are abundant and in response to receptor tyrosine kinase activation of the PI3K/Akt pathway, the mTOR complex 1 (mTORC1), which, together with the mTOR kinase and other components, includes the essential scaffolding protein regulatory-associated protein of mTOR (RAPTOR), stimulates protein translation through direct phosphorylation of downstream targets EF4B and S6K kinase109,110. In melanoma cells, knockdown of PYCR1 causes decreased activity of the Akt pathway and reduces the expression of RAPTOR, suggesting a downstream inhibition of protein synthesis96. Similarly, RNA-interference suppression of PYCR1 decreases levels of activated phospho-Akt and activated phospho-mTOR in renal cancer cells103.
The AAR pathway is engaged in response to amino acid starvation. The general control nonderepressible 2 (GCN2) kinase is an amino acid sensor activated by uncharged tRNAs that accumulate in response to low amino acid availability. Once activated, GCN2 inhibits translation through phosphorylation of the eukaryotic initiation factor eIF2 and, concomitantly, stimulates the Cap-independent translation of the transcription factor activating transcription factor 4 (ATF4)111,112. ATF4 drives a transcriptional program that enhances amino acid metabolism, including increasing the transcription of the proline metabolism genes ALDH18A1 and PYCR1113,114. Compared to normal tissue controls and non-transformed melanocytes, melanoma cells express higher levels of proline biosynthesis enzymes and display higher intracellular levels of L-proline41,107. Knockdown of P5CS reverts the increase in intracellular L-proline and, concomitantly, compromises cellular proliferation and viability, and severely reduces protein synthesis107. Notably, those effects depend on activation of the AAR pathway and are rescued by supplementation of the growth media with L-proline, which partially restores protein translation and cellular viability, preventing the activation of the AAR pathway107. Whether proline synthesis is associated with regulation of the mTOR pathway in melanoma cells is controversial107,115.
A remarkable example of direct reliance on L-proline production has been recently reported in lung adenocarcinoma. Guo and colleagues identified a cross talk between the ECM and proline metabolism mediated by the protein kindlin 242. Kindlin 2 is a ubiquitously expressed protein that localizes to focal adhesions to regulate integrin-mediated cell–ECM adhesion116,117. The authors show that a subset of kindlin 2 localizes to the mitochondria where it binds to PYCR1 (and PYCR2). Remarkably, this interaction stabilizes PYCR1 protein and increases proline biosynthesis. In addition, kindlin 2 localization to the mitochondria is stimulated by ECM stiffness: when cells are plated on a stiff ECM substrate more kindlin 2 is found in the mitochondria, bound to PYCR1. Consequently, the rate of proline biosynthesis responds to changes in ECM stiffness with functional implication for tumorigenesis. Lung adenocarcinoma cells where kindlin 2 is knocked-out using CRISPR/Cas9, together with impaired proline metabolism, display reduced proliferation and survival. Overexpression of PYCR1 or external supplementation of L-proline is sufficient to restore proliferative competence in kindlin 2-null cells42. Unfortunately, the authors did not investigate whether the primary role of L-proline was to sustain protein translation, and the function of L-proline biosynthesis in this context remains to be fully elucidated.
Proline biosynthesis and the TCA cycle
Similar to what has been observed for PRODH, the link between proline biosynthesis and the metabolism of glutamine and NAD(P)H necessarily affects mitochondrial function. This is the case for a subset of human gliomas characterized by mutations in the IDH1 gene, which codes for the NADP-dependent cytosolic IDH1 enzyme. Cancer-related mutations in IDH1 are heterozygous missense alterations most commonly generating an arginine to histidine substitution in residue 132 (R132H)118. This mutation has been shown to change the enzymatic activity of the IDH1 enzyme; IDH1 oxidation of isocitrate to α-ketoglutarate is replaced by NADPH-coupled reduction of α-ketoglutarate to (R)-2-hydroxyglutarate119. 2-Hydroxyglutarate is known as an oncometabolite due to its direct role in promoting tumor progression through inhibition of α-ketoglutarate-dependent dioxygenases, such as the TET-family of DNA demethylases and the HIF1-α prolyl hydroxylases, thereby altering the epigenetic landscape of tumor cells and the response to hypoxia120,121. IDH1-mutant gliomas express higher levels of PYCR1 than their wild-type counterparts and engage proline biosynthesis from glutamate106. In this case, the ability of PYCR1 to drive cancer progression does not rely on the end product L-proline, as IDH1-mutant glioma cells release most of the newly synthesized L-proline extracellularly106. Rather, it is the parametabolic regulation of NADH metabolism that confers on PYCR1 the ability to sustain TCA cycle activity. PYCR1 oxidation of NADH to NAD+ allows the TCA cycle to progress independently of oxygen consumption106. This activity might be essential in tumors, such as gliomas, that experience hypoxia and therefore limited ETC activity. Conditions that compromise mitochondrial ETC force cells to increase flux through the glutamine-proline pathway, in order to dissipate electron buildup through NADH oxidation by P5CS and PYCR enzymes122,123. This property of PYCR1 to act as a “mitochondrial vent” has wider implications. Recent findings demonstrate that, in fibroblasts, TGF-β signaling enhances L-proline production from glutamate through SMAD4-dependent transcription of L-proline metabolism genes, including ADH18A1, PYCR1, and PYCR2124. TGF-β signaling increases the synthesis of collagen and ECM deposition. It also enhances mitochondrial redox potential by channeling glucose and glutamine for oxidation via the TCA cycle, in order to increase energetic output. This leads to TCA cycle activity exceeding the capacity of the ETC to convert mitochondrial redox potential into ATP. In this circumstance, proline biosynthesis supports collagen production and alleviates mitochondrial hyperpolarization through oxidation of NADH and diversion of glutamine-derived carbon away from the TCA cycle124.
Additional pathways modulated by PYCR1
In the context of different cancers and experimental models, researchers have identified different signaling pathways that are influenced by proline biosynthesis (Fig. 3). In colorectal cancer cells, PYCR1 knockdown is associated with reduced activity of the MAPK pathway and NF-kB signaling, phenotypes that seem to depend on signal transducer and activator of transcription (STAT) 3-mediated signaling104. Indeed, Yan and colleagues found that ectopic expression of the transcription factor STAT3 reverses the proliferative reduction observed following PYCR1 knockdown104. According to their data, PYCR1 binds directly to STAT3 to promote its transcriptional activity. A role for PYCR1 in regulating the MAPK p38 was confirmed in NSCLC, where p38 MAPK activation depends on PYCR1 expression and contributes to PYCR1-mediated increase in cell survival and proliferation98,125. In hepatocellular cancer cells, shRNA-mediated knockdown of PYCR1 in vitro resulted in significantly decreased activation of the stress-activated protein kinase/c-Jun NH(2)-terminal kinase (SAPK/JNK) signaling pathway and the insulin receptor substrate 1102. However, the implications of these changes for tumorigenesis are unclear.
Other proline enzymes
Whilst it is generally acknowledged that PYCR1 plays a major role in cancer progression, some preliminary data suggest a pro-tumorigenic role for the PYCR2 gene as well. Confirming the high dependence of melanoma on proline metabolism, siRNA-mediated knockdown of PYCR2 was found to reduce proliferation and provoke a mild increase in apoptosis in melanoma cells115. Depletion of PYCR2 also caused activation of AMPK kinase. In line with AMPK ability to inhibit the mTOR pathway84, mTOR activation was impaired in PYCR2-knockdown cells with a concomitant increase in autophagy markers115. These data are notable as they unveil the regulation of the mTOR pathway by another PYCR isoform. However, the regulation of mTOR by proline biosynthesis enzymes in melanoma has not been consistently observed107.
Melanoma cells have increased PC5CS protein levels compared to melanocyte controls and, as previously reported, P5CS is essential to sustain protein translation and proliferation41,107. In Burkitt lymphoma and prostate cancer cell lines the oncogene c-Myc causes a significant increase in P5CS expression95. Other findings have reported that the expression of the P5CS gene ALDH18A1 is increased and contributes toward enhanced L-proline biosynthesis in fibroblasts stimulated with TGF-β and in hepatocellular carcinoma cells subject to hypoxic environment86,124. In the DLD-1 colorectal cell line, induction of p53 resulted in increased expression of the long isoform of P5CS38. However, whether this is functionally meaningful is doubtful, as ectopic expression of both the short and long P5CS isoforms in different cell lines had no overt effect on their proliferation or survival38. The role of P5CS is likely to be context-specific, as, in breast cancer, CRISPR/Cas9-mediated knockout of P5CS had no impact on tumor growth, but it sensitized cancer cells to pharmacological inhibition of lipogenesis, indicating that inhibition of proline biosynthesis could synergize with therapies targeting lipid metabolism122. Recently, increased expression of OAT has been shown to correlate with NSCLC progression, and OAT activity supports proliferation and metastatic spread of NSCLC cells126.
In addition to PRODH1, p53 also induces the expression of ALDH4, which codes for P5CDH127. Therefore, the p53 transcriptional program stimulates proline catabolism to glutamate. However, whereas PRODH expression has been linked to p53-induced apoptosis, induction of P5CDH has a protective role from oxidative stress. RNA-interference-mediated knockdown of P5CDH sensitizes HCT-116 colorectal cancer cell lines to p53-induced cell death, whereas, H1299 lung cancer cells that overexpress P5CDH showed significantly lower intracellular ROS levels than parental cells when challenged with hydrogen peroxide or UV127. In addition, analysis of RNA-seq data unmasked an exon skip (a common pattern of alternative splicing) in the P5CDH gene that predicts poorer survival in rectal cancer patients128, although the physiological implications of this alternative splicing event are unknown. Overall, the contribution of P5CDH to tumor progression is likely to be limited, as P5CDH has not been associated with progression of breast cancer and NSCLC89,128–131.
PYCR3 has been so far poorly investigated and any link to tumorigenesis remains unexplored. However, a recent association between PYCR3 and cells’ adaptation to L-proline starvation has been reported. Sahu and colleagues identified subsets of cancer cells that rely on external supplementation of L-proline for proliferation and survival and undergo robust ER stress if starved of L-proline. These cells express lower levels of both ALDH18A1 and PYCR3 gene and RNA-interference-mediated knockdown of PYCR3 reduced colony formation in L-proline starved cells. The authors conclude that increased L-proline biosynthesis through the P5CS-PYCR3 axis protects cells from ER stress and contributes to proliferation and survival in L-proline limiting conditions132. Increased expression of PYCR3 has also been identified in a pan-cancer systematic analysis of metabolic adaptations in response to hypoxic environment, again confirming the critical role of proline metabolism in response to oxygen limitation123. These findings indicate a potential role for PYCR3 in tumorigenesis that might be dependent on tumor cell adaptation to L-proline availability and the hypoxic tumor microenvironment.
Proline and ROS
The increase in intracellular L-proline in response to stress has been known for many years in plants, where it provides protection against oxidative stress133. Indeed, carbon atoms within the L-proline ring can efficiently react with and quench some species of radicals134. Cancer cells face the challenge of coping with oxidative stress and have developed several adaptive mechanisms to manage oxidative damage5. Notably, L-proline has been shown to mitigate photo-oxidative damage caused by exposure to UV light135, and supplementation of L-proline in the culture medium protects human cells from hydrogen-peroxide-induced cell death136. Importantly, this protection is substantial at a nearly physiological concentration of the amino acid (0.5 mM in culture media vs 0.2–0.3 mM normally found in the human serum)136. The role of L-proline in balancing cellular oxidative burden in cancer needs to be confirmed in physiologically relevant models. Alternatively, proline metabolism regulates ROS production directly in the mitochondria. As previously mentioned, PRODH is known to increase intracellular ROS69. In contrast, active proline biosynthesis activity has been shown to diminish ROS production, probably by easing the hyperpolarization of mitochondrial triggered by accumulation of reduced NADH or intense TCA cycle activity106,124. Emphasizing the anti-oxidative function of proline biosynthesis enzymes, Kuo and colleagues showed that PYCR1 and PYCR2 bind to the ribonucleotide reductase small subunit RRM2B and support its anti-oxidant activity40. RRM2B, also known as the p53 inducible p53R2, has been shown to protect cells from oxidative damage137,138. Kuo and colleagues found that PYCR1 and PYCR2 directly interact with RRMB2 in the mitochondria and confer resistance to hydrogen-peroxide-mediated cell death stimulating the anti-oxidant activity of RRM2B40. Intriguingly, together with the data on melanoma115, this work provides an additional indication that PYCR2 might play a significant role in carcinogenesis. PYCR1 has also been linked to protection from ROS in neuronal cells via direct binding to the protein DJ-1139. Beyond its known role in neuronal physiology and Parkinson’s disease, DJ-1 is overexpressed in several cancers and has been linked to worse prognosis140–142. Although any interaction between PYCR1 and DJ-1 in cancer is yet to be established, these data warrant further investigation into their possible cooperation in promoting tumorigenesis by conferring protection to oxidative stress.
However, exceptions to the anti-oxidant function of PYCR1 have been reported. Kuo and colleagues unveiled an interaction between PYCR1 and the mitochondrial chaperone protein Lon125. Lon expression is induced in response to stress, such as the unfolded protein response, hypoxia, and oxidative stress, to safeguard cells’ ability to proliferate and survive143–146. In cancer cells, Lon expression triggers mitochondrial ROS production and, surprisingly, this depends on Lon interaction with PYCR1. The authors showed that Lon expression induces accumulation of PYCR1 and that the two proteins bind to each other and cooperate in enhancing ROS generation125. Of note, overexpression of PYCR1 itself in cancer cells from different tumors is associated with increased intracellular ROS levels. The Lon/PYCR1-mediated induction of ROS has notable downstream effects, as it triggers ROS-dependent activation of the p38 MAPK and NF-κB pathways to promote an inflammatory milieu, which can contribute to tumorigenesis125. Similarly, in a subset of lung cancers that overexpress PRODH, tumor progression is driven by PRODH-mediated increase in ROS, which acts as signaling molecules stimulating the expression of pro-inflammatory cytokines88. In this instance, PRODH-mediated generation of ROS is blunted upon knockdown of proline biosynthesis enzymes PYCR1, PYCR2, and PYCR388. This suggests that by recycling P5C back to L-proline, PYCR enzymes fuel PRODH activity and, indirectly, act as pro-oxidants88. Indirect evidence in support of this possibility is the reported anti-oxidant effect of P5CDH expression triggered in response to p53 activation127. Indeed, by metabolizing P5C to glutamate, P5CDH could compete with PYCRs for substrate utilization, reducing L-proline availability for oxidation and ROS generation by PRODH.
These findings are puzzling to the extent that they contradict previous robust indications that PYCR1 activity would rather dampen cellular ROS. It will be necessary to establish whether the net effect of PYCR1 on mitochondrial ROS production is context-dependent and whether it depends on the concomitant expression of PRODH. Of course, it cannot be ruled out that yet-to-be identified mechanisms regulating PYCR1 interaction with mitochondrial metabolism are responsible for these conflicting outcomes.
The proline cycle in cancer
Hagedorn and Phang first suggested the existence of a proline cycle over 30 years ago147,148. In the cycle, PRODH oxidizes L-proline to P5C in the mitochondria, a reaction coupled with the transfer of electrons to the mitochondrial ETC. P5C is then reduced back to L-proline through the action of the PYCR enzymes in the cytosol, a reaction coupled with the oxidation of NADPH. The outcome of this cycle is to transfer electrons from the cytosol to the mitochondria, similarly to what observed with traditional mitochondrial shuttles, such as the malate–aspartate shuttle or the glycerol–phosphate shuttle (Fig. 4). However, the proline cycle entails another important metabolic consequence. Since proline biosynthesis in the cytosol is mostly dependent on NADPH oxidation, a sustained activity of the cycle will result in a higher NADP+/NADPH ratio60. As initially indicated by Krebs, the NADP+/NADPH ratio is the rate-limiting variable that controls flux through the PPP149, and an increase in NADP+ levels will stimulate the oxidative arm of the PPP leading to increase ribose and, consequently, nucleotide biosynthesis. Evidence exists that proline metabolism might accelerate the rate of PPP and the metabolic flux of nucleotide biosynthesis, fostering support to the proline cycle model81,82,150. Moreover, the aforementioned pro-tumorigenic role of PRODH in breast and lung cancers necessitates the concomitant activity of the PYCR enzymes, indicating that P5C recycling is important for sustaining PRODH activity88,89. These findings can be partially reconciled with the existence of a proline cycle, but an issue with the pathway compartmentalization emerges. The initial proline cycle model suggested a series of compartmentalized reactions of oxidation of L-proline in the mitochondria and reduction of P5C in the cytosol. This would suggest that the main PYCR enzyme involved should be the elusive cytosolic PYCR3, which also has the highest affinity for NADPH41. However, there is no sound evidence implicating PYCR3 in cancer or the PPP. Moreover, this would not explain why PRODH pro-tumorigenic properties rely on the expression of mitochondrial PYCR1 and PYCR2. The presence of PYCR1 in the cytosol, at least in minimal quantity, has been reported, as well as its potential localization on the outer mitochondrial membrane42,43. If confirmed, these possibilities could help reconcile the proline cycle model with our understanding of the compartmentalization of proline metabolism. Otherwise, at least in some circumstances, cancer cell proliferation might be sustained not by the proline cycle, but by recycling of P5C from L-proline, an alternative option that would weaken links to the PPP.
Emerging pathways linked to proline metabolism
Invasion and metastasis
PRODH expression has been reported to drive metastatic spread from breast cancer89. In clinical specimens, PRODH1 expression is significantly higher in metastatic versus non-metastatic breast cancers. This is reflected in the reduced content of proline in lung metastasis isolated from mice orthotopically implanted with the mouse breast cancer cells 4T189. Pharmacological inhibition of PRODH in vivo using L-THFA results in reduced metastatic spread to the lung89.
Tumor spread is thought to rely on the epithelial–mesenchymal transition (EMT). During EMT, epithelial tumor cells undergo a molecular reprogramming that causes the loss of epithelia adhesion molecules and the acquisition of a fibroblast-like morphology with increased ability to migrate and invade151. Together with its role in metastatic spread in breast cancer, PRODH has been shown to instigate EMT reprogramming and to increase migration and invasion of NSCLC cells88. Although not formally proved, the cross talk between proline catabolism and biosynthesis is likely to play a role also in regulation of invasiveness. Indeed, PYCR1 expression is increased in metastatic breast cancer. Its genetic depletion in breast, lung cancer, renal carcinoma cells, and melanoma reduces cells’ ability to migrate and invade in in vitro essays96,99,101,103. In addition, supplementing L-proline in the culture medium stimulates proliferation and induces a mesenchymal invasive phenotype in mouse embryonic stem cells (mESCs)152,153. mESCs experience an intrinsic L-proline starvation in traditional culture medium and activate the AAR pathway. Notably, L-proline is the only NEAA able to alleviate the AAR, a step necessary to induce EMT-like features153. Additional data indicate that L-proline supports protein synthesis and induces reprogramming of the cellular histone methylation profiles in mESCs, therefore reshaping the transcriptomic output152,153. It will be interesting to investigate whether these mechanisms persist in cancer cells, especially in the context of IDH-mutant tumors, where tumor-related epigenetic changes are induced by the oncometabolite 2-hydroxyglutarate120,121.
Cancer stem cells (CSCs)
Tumors are thought to arise from or to be driven by a subset of cells that exhibit properties of stem cells. CSCs possess the ability to self-renew, are highly tumorigenic and, being resistant to chemotherapy, are believed to be responsible for the persistence and relapse of cancer154. Recently, Sharif and colleagues reported an unexpected role for the p53 homolog TAp73 in safeguarding CSCs155. The transcription factor TAp73 was initially shown to possess tumor suppressor functions156,157. However, other findings implicated TAp73 in the regulation of cellular metabolism, showing that it promotes pro-proliferative anabolism, mitochondrial respiration, and metabolic adaptation in response to oxidative stress158–163. Sharif and colleagues demonstrated that depletion of TAp73 in CSCs reduces their survival and ability to form tumors in vivo155. This surprising phenotype is associated with a rewiring of amino acid metabolism: knockdown of TAp73 decreases the expression of PYCR1 and GLS (coding the glutamine metabolizing enzyme glutaminase) while increasing OAT expression155. The final outcome is a reshuffling of proline metabolism toward the urea cycle. The effective relevance of proline metabolism in maintaining CSCs survival remains to be elucidated, since external supplementation of L-proline is ineffective in restoring CSCs fitness following TAp73 depletion155. However, TAP73 is necessary to maintain redox balance and to regulate autophagy through the AMPK-mTOR axis in CSCs155, all pathways linked to proline metabolism. These data on CSCs could also be reconciled with the role of proline metabolism in EMT and invasion, since during the process of EMT cancer cells acquire features of stem cells164.
The tumor microenvironment (TME)
Rich in ECM and collagen the TME is an important reservoir of L-proline for cancer cells82,89. However, proline metabolism can also influence non-tumor cells within the stroma. Indeed, the transcription factor c-Myc triggers expression of PYCR1 and ALDH18A1 in activated T lymphocytes, and there is evidence that proline metabolism contributes to the reprogramming of tumor-infiltrating macrophages125,165,166. Two major molecular subtypes of macrophages populate the TME; M1 macrophages are very effective antigen-presenting cells and support anti-tumor immune response, whereas the alternative M2 phenotype exhibits anti-inflammatory and pro-tumorigenic activity. M2 cells influence multiple features of tumor development, including cell survival, proliferation, stemness, and invasiveness along with angiogenesis and immune evasion167. M2 polarized macrophages display an intense rewiring of NEAA metabolism, including arginine–glutamine–proline metabolism165. As previously discussed, the mitochondrial chaperon Lon interacts with PYCR1 in cancer cells to increase intracellular ROS production and activate a p38-NF-kB signaling axis125. The latter, in turn, stimulates cancer cells to secrete cytokines, including TGF-β, IL-13, IL-6, and VEGF-A, which trigger M2 macrophage polarization125. A role for PRODH in supporting a pro-inflammatory milieu has been reported also in NSCLC88. Notably, the metabolite P5C could also act as a signaling molecule between tumor cells and cells of the immune system. Prostate tumors release P5C to inhibit the proliferation of T lymphocytes and cytokine production, hence suppressing the immune response168. These data suggest that proline metabolism can have a major impact on shaping the tumor inflammatory milieu and promoting immune evasion.
Concluding remarks and future perspectives
The evidence provided here outlines a major, and perhaps so far under-appreciated, role for proline metabolism in influencing the process of carcinogenesis. Proline metabolic enzymes work downstream of p53 to promote cell death and tumor suppression, but, in other contexts, they behave as powerful oncogenic proteins in support of tumor growth and metastatic spread. Notwithstanding its relevance to tumorigenesis, on this day there are no clinically relevant pharmacological compounds to target the proline pathway.
The amount of literature on the subject has accumulated rapidly in the past few years; this has amplified the amount of information available, but not necessarily its clarity. Moving forward, it will be pivotal to develop appropriate tools to investigate the role of proline metabolism in cancer. The use of RNA-interference-based approaches to gene depletion has been informative, but mostly limited to cell culture work and potentially affected by the lack of selectivity when targeting conserved isoforms. The engineering of more physiologically relevant models, such as inducible knockout mouse models or CRISPR/Cas9 modified organoids, will be key to clarify the function of the different enzymes and their isoforms in vivo, as elegantly demonstrated by the generation by Liu and colleagues of a CRE-inducible P5CS knockout mouse model for the analysis of breast cancer tumorigenesis122. These genetically neat models could unravel the different functions of PYCR isoforms in cancer and finally shed light on the role of the neglected PYCR3 gene in physiology and disease, not to mention its potential contribution to the proline cycle. More generally, it will be key to address in which tumors and in which context the metabolism of proline is preferentially unidirectional, and which malignancies rely on the coexistence of PYCR and PRODH activities. In this regard, large cancer datasets should be mined to address expression of proline metabolism genes comprehensibly, rather than limiting the analysis to the expression of single isoforms. This will enable a stratification of tumors based on gene expression profiles of the whole pathway. Flux metabolomics should be extensively employed to aid discovery and inform about the cross talk between proline metabolism and the wider cellular metabolic network. These investigations will aid efforts to target proline metabolism pharmacologically and could inform on potential toxicities. Indeed, although preliminary evidence indicates that in vivo inhibition of PRODH is achievable89, mutations in proline metabolism genes have been implicated in metabolic syndromes known as prolinemia and impaired development of the nervous system, the epidermis and other organs169–176.
Efforts should also be dedicated to unmasking the interaction between proline metabolism and chemotherapy. Preliminary data suggest that inhibiting proline biosynthesis enhances sensitivity of colorectal cancer cells and hepatocellular carcinoma cells to standard of care chemotherapy agents 5-FU and sorafenib, respectively86,104. On the other hand, PRODH has been implicated in cell death induced by p53-inducing DNA-damaging drugs69 and to contribute to anti-tumor activity of celecoxib79 and troglitazone, a peroxisome proliferator-activated receptor γ (PPARγ) agonist that induces apoptosis in several cancers177. This information will help prioritize cancers for mechanistic studies and preclinical assessments of new compounds targeting proline enzymes.
Post-translational modifications of proline metabolic enzymes deserve a final mention as a research area likely to receive growing attention in the future. In their recent study, Chen and colleagues reported that PYCR1 activity is regulated through acetylation of lysine K228, located in the C-terminal dimerization domain. The CREB-binding protein (CBP) acetylation of K228 is counteracted by the activity of the NAD+-dependent, mitochondrial SIRT3 deacetylase. Importantly, acetylation impairs the ability of PYCR1 to polymerize and partially reduces its enzymatic activity, and impacts cellular proliferation178. Of note, the regulation of PYCR1 acetylation is dictated by a NAD+-dependent enzyme, suggesting that PYCR1 enzymatic output is wired to the NAD+/NADH balance. More generally, here we proffer the contention that, whilst the post-translation modifications of proline metabolizing enzymes are largely unknown, they very likely play major roles in the regulation of this metabolic pathway in cancer. How they influence proline metabolism during tumorigenesis warrants further investigation.
Acknowledgements
L.B. is supported by a PhD fellowship from the Cancer Prevention Research Trust (TM60004-CPRT). I.G. is supported by a grant from the Cancer Prevention Research Trust (RM60G0665-CPRT). C.T. is supported by a Wellcome Trust Research Career Re-entry Fellowship. R.P.G. is supported through a Development grant to A.R. and C.T. from the charity Hope Against Cancer (https://www.hopeagainstcancer.org.uk/, grant code RM60G0751). D.B. was funded by Cancer Research UK Accelerator Award C1362/A20263. We are thankful to Dr Sara Galavotti and Professor Andreas Gescher for their critical reading of the manuscript and insightful suggestions.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by Ivano Amelio
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J. Gen. Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 4.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 5.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Disco. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 6.Vander Heiden MG, et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. 2011;76:325–334. doi: 10.1101/sqb.2012.76.010900. [DOI] [PubMed] [Google Scholar]
- 7.Vettore L, Westbrook RL, Tennant DA. New aspects of amino acid metabolism in cancer. Br. J. Cancer. 2020;122:150–156. doi: 10.1038/s41416-019-0620-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards NG, Kilberg MS. Asparagine synthetase chemotherapy. Annu Rev. Biochem. 2006;75:629–654. doi: 10.1146/annurev.biochem.75.103004.142520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phillips MM, Sheaff MT, Szlosarek PW. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res. Treat. 2013;45:251–262. doi: 10.4143/crt.2013.45.4.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabinovich S, et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature. 2015;527:379–383. doi: 10.1038/nature15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexandrou C, et al. Sensitivity of colorectal cancer to arginine deprivation therapy is shaped by differential expression of urea cycle enzymes. Sci. Rep. 2018;8:12096. doi: 10.1038/s41598-018-30591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beddowes E, et al. Phase 1 dose-escalation study of pegylated arginine deiminase, cisplatin, and pemetrexed in patients with argininosuccinate synthetase 1-deficient thoracic cancers. J. Clin. Oncol. 2017;35:1778–1785. doi: 10.1200/JCO.2016.71.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szlosarek PW, et al. Metabolic response to pegylated arginine deiminase in mesothelioma with promoter methylation of argininosuccinate synthetase. J. Clin. Oncol. 2013;31:e111–e113. doi: 10.1200/JCO.2012.42.1784. [DOI] [PubMed] [Google Scholar]
- 14.Allen MD, et al. Prognostic and therapeutic impact of argininosuccinate synthetase 1 control in bladder cancer as monitored longitudinally by PET imaging. Cancer Res. 2014;74:896–907. doi: 10.1158/0008-5472.CAN-13-1702. [DOI] [PubMed] [Google Scholar]
- 15.Delage B, et al. Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death Dis. 2012;3:e342. doi: 10.1038/cddis.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feun L, Savaraj N. Pegylated arginine deiminase: a novel anticancer enzyme agent. Expert Opin. Investig. Drugs. 2006;15:815–822. doi: 10.1517/13543784.15.7.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang HY, et al. ASS1 as a novel tumor suppressor gene in myxofibrosarcomas: aberrant loss via epigenetic DNA methylation confers aggressive phenotypes, negative prognostic impact, and therapeutic relevance. Clin. Cancer Res. 2013;19:2861–2872. doi: 10.1158/1078-0432.CCR-12-2641. [DOI] [PubMed] [Google Scholar]
- 18.Long Y, et al. Cisplatin-induced synthetic lethality to arginine-starvation therapy by transcriptional suppression of ASS1 is regulated by DEC1, HIF-1alpha, and c-Myc transcription network and is independent of ASS1 promoter DNA methylation. Oncotarget. 2016;7:82658–82670. doi: 10.18632/oncotarget.12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savaraj N, et al. Targeting argininosuccinate synthetase negative melanomas using combination of arginine degrading enzyme and cisplatin. Oncotarget. 2015;6:6295–6309. doi: 10.18632/oncotarget.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Syed N, et al. Epigenetic status of argininosuccinate synthetase and argininosuccinate lyase modulates autophagy and cell death in glioblastoma. Cell Death Dis. 2013;4:e458. doi: 10.1038/cddis.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yau T, et al. Preliminary efficacy, safety, pharmacokinetics, pharmacodynamics and quality of life study of pegylated recombinant human arginase 1 in patients with advanced hepatocellular carcinoma. Investigational N. drugs. 2015;33:496–504. doi: 10.1007/s10637-014-0200-8. [DOI] [PubMed] [Google Scholar]
- 22.Tsai WB, et al. Resistance to arginine deiminase treatment in melanoma cells is associated with induced argininosuccinate synthetase expression involving c-Myc/HIF-1alpha/Sp4. Mol. Cancer Therapeut. 2009;8:3223–3233. doi: 10.1158/1535-7163.MCT-09-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McAlpine JA, Lu HT, Wu KC, Knowles SK, Thomson JA. Down-regulation of argininosuccinate synthetase is associated with cisplatin resistance in hepatocellular carcinoma cell lines: implications for PEGylated arginine deiminase combination therapy. BMC Cancer. 2014;14:621. doi: 10.1186/1471-2407-14-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miraki-Moud F, et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood. 2015;125:4060–4068. doi: 10.1182/blood-2014-10-608133. [DOI] [PubMed] [Google Scholar]
- 25.Mussai F, et al. Arginine dependence of acute myeloid leukemia blast proliferation: a novel therapeutic target. Blood. 2015;125:2386–2396. doi: 10.1182/blood-2014-09-600643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szlosarek PW, et al. In vivo loss of expression of argininosuccinate synthetase in malignant pleural mesothelioma is a biomarker for susceptibility to arginine depletion. Clin. Cancer Res. 2006;12:7126–7131. doi: 10.1158/1078-0432.CCR-06-1101. [DOI] [PubMed] [Google Scholar]
- 27.Choi, B. H. & Coloff, J. L. The diverse functions of non-essential amino acids in cancer. Cancers11. 10.3390/cancers11050675 (2019). [DOI] [PMC free article] [PubMed]
- 28.Woolfson DN, Williams DH. The influence of proline residues on alpha-helical structure. FEBS Lett. 1990;277:185–188. doi: 10.1016/0014-5793(90)80839-B. [DOI] [PubMed] [Google Scholar]
- 29.Cordes FS, Bright JN, Sansom MS. Proline-induced distortions of transmembrane helices. J. Mol. Biol. 2002;323:951–960. doi: 10.1016/S0022-2836(02)01006-9. [DOI] [PubMed] [Google Scholar]
- 30.Williams KA, Deber CM. Proline residues in transmembrane helices: structural or dynamic role? Biochemistry. 1991;30:8919–8923. doi: 10.1021/bi00101a001. [DOI] [PubMed] [Google Scholar]
- 31.Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. doi: 10.1096/fasebj.14.2.231. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, et al. Prolyl isomerase Pin1: a promoter of cancer and a target for therapy. Cell Death Dis. 2018;9:883. doi: 10.1038/s41419-018-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmidpeter PA, Schmid FX. Prolyl isomerization and its catalysis in protein folding and protein function. J. Mol. Biol. 2015;427:1609–1631. doi: 10.1016/j.jmb.2015.01.023. [DOI] [PubMed] [Google Scholar]
- 34.Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis. 2018;7:10. doi: 10.1038/s41389-017-0011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoulders MD, Raines RT. Collagen structure and stability. Annu Rev. Biochem. 2009;78:929–958. doi: 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu G, et al. Proline and hydroxyproline metabolism: implications for animal and human nutrition. Amino Acids. 2011;40:1053–1063. doi: 10.1007/s00726-010-0715-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krane SM. The importance of proline residues in the structure, stability and susceptibility to proteolytic degradation of collagens. Amino Acids. 2008;35:703–710. doi: 10.1007/s00726-008-0073-2. [DOI] [PubMed] [Google Scholar]
- 38.Hu CA, et al. Human Delta1-pyrroline-5-carboxylate synthase: function and regulation. Amino Acids. 2008;35:665–672. doi: 10.1007/s00726-008-0075-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu CA, Lin WW, Obie C, Valle D. Molecular enzymology of mammalian Delta1-pyrroline-5-carboxylate synthase. Alternative splice donor utilization generates isoforms with different sensitivity to ornithine inhibition. J. Biol. Chem. 1999;274:6754–6762. doi: 10.1074/jbc.274.10.6754. [DOI] [PubMed] [Google Scholar]
- 40.Kuo ML, et al. PYCR1 and PYCR2 interact and collaborate with RRM2B to protect cells from overt oxidative stress. Sci. Rep. 2016;6:18846. doi: 10.1038/srep18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Ingeniis J, et al. Functional specialization in proline biosynthesis of melanoma. PLoS ONE. 2012;7:e45190. doi: 10.1371/journal.pone.0045190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo L, et al. Kindlin-2 links mechano-environment to proline synthesis and tumor growth. Nat. Commun. 2019;10:845. doi: 10.1038/s41467-019-08772-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phang JM, Liu W, Zabirnyk O. Proline metabolism and microenvironmental stress. Annu Rev. Nutr. 2010;30:441–463. doi: 10.1146/annurev.nutr.012809.104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christensen EM, et al. Resolving the cofactor-binding site in the proline biosynthetic enzyme human pyrroline-5-carboxylate reductase 1. J. Biol. Chem. 2017;292:7233–7243. doi: 10.1074/jbc.M117.780288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delano WL. The Pymol Molecular Graphics System. New York, NY: Schrödinger, LLC; 2017. [Google Scholar]
- 46.Meng Z, et al. Crystal structure of human pyrroline-5-carboxylate reductase. J. Mol. Biol. 2006;359:1364–1377. doi: 10.1016/j.jmb.2006.04.053. [DOI] [PubMed] [Google Scholar]
- 47.Summitt CB, et al. Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. Biochem. J. 2015;466:273–281. doi: 10.1042/BJ20141159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee YH, Nadaraia S, Gu D, Becker DF, Tanner JJ. Structure of the proline dehydrogenase domain of the multifunctional PutA flavoprotein. Nat. Struct. Biol. 2003;10:109–114. doi: 10.1038/nsb885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu LK, Becker DF, Tanner JJ. Structure, function, and mechanism of proline utilization A (PutA) Arch. Biochem. Biophys. 2017;632:142–157. doi: 10.1016/j.abb.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanner JJ. Structural biology of proline catabolism. Amino Acids. 2008;35:719–730. doi: 10.1007/s00726-008-0062-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hancock CN, Liu W, Alvord WG, Phang JM. Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino Acids. 2016;48:859–872. doi: 10.1007/s00726-015-2134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boggess SF, Koeppe DE. Oxidation of proline by plant mitochondria. Plant Physiol. 1978;62:22–25. doi: 10.1104/pp.62.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson AB, Strecker HJ. The interconversion of glutamic acid and proline. IV. The oxidation of proline by rat liver mitochondria. J. Biol. Chem. 1962;237:1876–1882. [PubMed] [Google Scholar]
- 54.Ginguay, A., Cynober, L., Curis, E. & Nicolis, I. Ornithine aminotransferase, an important glutamate-metabolizing enzyme at the crossroads of multiple metabolic pathways. Biology6. 10.3390/biology6010018 (2017). [DOI] [PMC free article] [PubMed]
- 55.Forlani G, Scainelli D, Nielsen E. [delta]1-Pyrroline-5-carboxylate dehydrogenase from cultured cells of potato (purification and properties) Plant Physiol. 1997;113:1413–1418. doi: 10.1104/pp.113.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deuschle K, et al. The role of [Delta]1-pyrroline-5-carboxylate dehydrogenase in proline degradation. Plant Cell. 2004;16:3413–3425. doi: 10.1105/tpc.104.023622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu CA, Lin WW, Valle D. Cloning, characterization, and expression of cDNAs encoding human delta 1-pyrroline-5-carboxylate dehydrogenase. J. Biol. Chem. 1996;271:9795–9800. doi: 10.1074/jbc.271.16.9795. [DOI] [PubMed] [Google Scholar]
- 58.Liu ZJ, et al. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat. Struct. Biol. 1997;4:317–326. doi: 10.1038/nsb0497-317. [DOI] [PubMed] [Google Scholar]
- 59.Inagaki E, et al. Crystal structure of Thermus thermophilus Δ1-pyrroline-5-carboxylate dehydrogenase. J. Mol. Biol. 2006;362:490–501. doi: 10.1016/j.jmb.2006.07.048. [DOI] [PubMed] [Google Scholar]
- 60.Tanner JJ, Fendt SM, Becker DF. The proline cycle as a potential cancer therapy target. Biochemistry. 2018;57:3433–3444. doi: 10.1021/acs.biochem.8b00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olivares O, et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017;8:16031. doi: 10.1038/ncomms16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phang JM, Liu W, Hancock CN, Fischer JW. Proline metabolism and cancer: emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care. 2015;18:71–77. doi: 10.1097/MCO.0000000000000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith ME, Greenberg DM. Preparation and properties of partially purified glutamic semialdehyde reductase. J. Biol. Chem. 1957;226:317–327. [PubMed] [Google Scholar]
- 64.Loayza-Puch F, et al. Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature. 2016;530:490–494. doi: 10.1038/nature16982. [DOI] [PubMed] [Google Scholar]
- 65.Baumgartner MR, et al. Hyperammonemia with reduced ornithine, citrulline, arginine and proline: a new inborn error caused by a mutation in the gene encoding delta(1)-pyrroline-5-carboxylate synthase. Hum. Mol. Genet. 2000;9:2853–2858. doi: 10.1093/hmg/9.19.2853. [DOI] [PubMed] [Google Scholar]
- 66.Kowaloff EM, Phang JM, Granger AS, Downing SJ. Regulation of proline oxidase activity by lactate. Proc. Natl Acad. Sci. USA. 1977;74:5368–5371. doi: 10.1073/pnas.74.12.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 68.Maxwell SA, Davis GE. Differential gene expression in p53-mediated apoptosis-resistant vs. apoptosis-sensitive tumor cell lines. Proc. Natl Acad. Sci. USA. 2000;97:13009–13014. doi: 10.1073/pnas.230445997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Donald SP, et al. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001;61:1810–1815. [PubMed] [Google Scholar]
- 70.Liu Y, et al. MnSOD inhibits proline oxidase-induced apoptosis in colorectal cancer cells. Carcinogenesis. 2005;26:1335–1342. doi: 10.1093/carcin/bgi083. [DOI] [PubMed] [Google Scholar]
- 71.Liu Y, et al. Proline oxidase functions as a mitochondrial tumor suppressor in human cancers. Cancer Res. 2009;69:6414–6422. doi: 10.1158/0008-5472.CAN-09-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cooper SK, Pandhare J, Donald SP, Phang JM. A novel function for hydroxyproline oxidase in apoptosis through generation of reactive oxygen species. J. Biol. Chem. 2008;283:10485–10492. doi: 10.1074/jbc.M702181200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65–80. doi: 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu Y, Borchert GL, Surazynski A, Hu CA, Phang JM. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene. 2006;25:5640–5647. doi: 10.1038/sj.onc.1209564. [DOI] [PubMed] [Google Scholar]
- 75.Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14:32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- 76.Liu Y, Borchert GL, Surazynski A, Phang JM. Proline oxidase, a p53-induced gene, targets COX-2/PGE2 signaling to induce apoptosis and inhibit tumor growth in colorectal cancers. Oncogene. 2008;27:6729–6737. doi: 10.1038/onc.2008.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hashemi Goradel N, Najafi M, Salehi E, Farhood B, Mortezaee K. Cyclooxygenase-2 in cancer: a review. J. Cell Physiol. 2019;234:5683–5699. doi: 10.1002/jcp.27411. [DOI] [PubMed] [Google Scholar]
- 78.Harris RE, Beebe J, Alshafie GA. Reduction in cancer risk by selective and nonselective cyclooxygenase-2 (COX-2) inhibitors. J. Exp. Pharm. 2012;4:91–96. doi: 10.2147/JEP.S23826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Toloczko-Iwaniuk, N. et al. Proline-dependent induction of apoptosis in oral squamous cell carcinoma (OSCC)—the effect of celecoxib. Cancers12. 10.3390/cancers12010136 (2020). [DOI] [PMC free article] [PubMed]
- 80.Maxwell SA, Rivera A. Proline oxidase induces apoptosis in tumor cells, and its expression is frequently absent or reduced in renal carcinomas. J. Biol. Chem. 2003;278:9784–9789. doi: 10.1074/jbc.M210012200. [DOI] [PubMed] [Google Scholar]
- 81.Liu W, Hancock CN, Fischer JW, Harman M, Phang JM. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: involvement of pyridine nucleotides. Sci. Rep. 2015;5:17206. doi: 10.1038/srep17206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pandhare J, Donald SP, Cooper SK, Phang JM. Regulation and function of proline oxidase under nutrient stress. J. Cell Biochem. 2009;107:759–768. doi: 10.1002/jcb.22174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu W, et al. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012;72:3677–3686. doi: 10.1158/0008-5472.CAN-12-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu W, Phang JM. Proline dehydrogenase (oxidase), a mitochondrial tumor suppressor, and autophagy under the hypoxia microenvironment. Autophagy. 2012;8:1407–1409. doi: 10.4161/auto.21152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tang L, et al. Global metabolic profiling identifies a pivotal role of proline and hydroxyproline metabolism in supporting hypoxic response in hepatocellular carcinoma. Clin. Cancer Res. 2018;24:474–485. doi: 10.1158/1078-0432.CCR-17-1707. [DOI] [PubMed] [Google Scholar]
- 87.Feig C, et al. The pancreas cancer microenvironment. Clin. Cancer Res. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu Y, et al. Cancer progression is mediated by proline catabolism in non-small cell lung cancer. Oncogene. 2020;39:2358–2376. doi: 10.1038/s41388-019-1151-5. [DOI] [PubMed] [Google Scholar]
- 89.Elia I, et al. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017;8:15267. doi: 10.1038/ncomms15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scott GK, et al. Targeting mitochondrial proline dehydrogenase with a suicide inhibitor to exploit synthetic lethal interactions with p53 upregulation and glutaminase inhibition. Mol. Cancer Ther. 2019;18:1374–1385. doi: 10.1158/1535-7163.MCT-18-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Possemato R, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nilsson R, et al. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014;5:3128. doi: 10.1038/ncomms4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gao P, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wise DR, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl Acad. Sci. USA. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu W, et al. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl Acad. Sci. USA. 2012;109:8983–8988. doi: 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ye Y, Wu Y, Wang J. Pyrroline-5-carboxylate reductase 1 promotes cell proliferation via inhibiting apoptosis in human malignant melanoma. Cancer Manag Res. 2018;10:6399–6407. doi: 10.2147/CMAR.S166711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cai F, et al. Pyrroline-5-carboxylate reductase 1 promotes proliferation and inhibits apoptosis in non-small cell lung cancer. Oncol. Lett. 2018;15:731–740. doi: 10.3892/ol.2017.7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang D, et al. PYCR1 promotes the progression of non-small-cell lung cancer under the negative regulation of miR-488. Biomed. Pharmacother. 2019;111:588–595. doi: 10.1016/j.biopha.2018.12.089. [DOI] [PubMed] [Google Scholar]
- 99.Sang S, Zhang C, Shan J. Pyrroline-5-Carboxylate Reductase 1 Accelerates the Migration and Invasion of Nonsmall Cell Lung Cancer In Vitro. Cancer Biother Radiopharm. 2019;34:380–387. doi: 10.1089/cbr.2019.2782. [DOI] [PubMed] [Google Scholar]
- 100.Weijin F, et al. The clinical significance of PYCR1 expression in renal cell carcinoma. Medicine. 2019;98:e16384. doi: 10.1097/MD.0000000000016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ding J, et al. Human mitochondrial pyrroline-5-carboxylate reductase 1 promotes invasiveness and impacts survival in breast cancers. Carcinogenesis. 2017;38:519–531. doi: 10.1093/carcin/bgx022. [DOI] [PubMed] [Google Scholar]
- 102.Zhuang J, et al. PYCR1 interference inhibits cell growth and survival via c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathway in hepatocellular cancer. J. Transl. Med. 2019;17:343. doi: 10.1186/s12967-019-2091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang QL, Liu L. PYCR1 is associated with papillary renal cell carcinoma progression. Open Med. (Wars.) 2019;14:586–592. doi: 10.1515/med-2019-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yan K, et al. Knockdown of PYCR1 inhibits proliferation, drug resistance and EMT in colorectal cancer cells by regulating STAT3-Mediated p38 MAPK and NF-kappaB signalling pathway. Biochem. Biophys. Res. Commun. 2019;520:486–491. doi: 10.1016/j.bbrc.2019.10.059. [DOI] [PubMed] [Google Scholar]
- 105.Zeng T, et al. Knockdown of PYCR1 inhibits cell proliferation and colony formation via cell cycle arrest and apoptosis in prostate cancer. Med Oncol. 2017;34:27. doi: 10.1007/s12032-016-0870-5. [DOI] [PubMed] [Google Scholar]
- 106.Hollinshead KER, et al. Oncogenic IDH1 mutations promote enhanced proline synthesis through PYCR1 to support the maintenance of mitochondrial redox homeostasis. Cell Rep. 2018;22:3107–3114. doi: 10.1016/j.celrep.2018.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kardos GR, Wastyk HC, Robertson GP. Disruption of proline synthesis in melanoma inhibits protein production mediated by the GCN2 pathway. Mol. Cancer Res. 2015;13:1408–1420. doi: 10.1158/1541-7786.MCR-15-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Norton JA, Gorschboth CM, Wesley RA, Burt ME, Brennan MF. Fasting plasma amino acid levels in cancer patients. Cancer. 1985;56:1181–1186. doi: 10.1002/1097-0142(19850901)56:5<1181::AID-CNCR2820560535>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 109.Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020;21:183–203. doi: 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015;517:302–310. doi: 10.1038/nature14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kilberg MS, Shan J, Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009;20:436–443. doi: 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Han J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Deval C, et al. Amino acid limitation regulates the expression of genes involved in several specific biological processes through GCN2-dependent and GCN2-independent pathways. FEBS J. 2009;276:707–718. doi: 10.1111/j.1742-4658.2008.06818.x. [DOI] [PubMed] [Google Scholar]
- 115.Ou R, et al. Downregulation of pyrroline-5-carboxylate reductase-2 induces the autophagy of melanoma cells via AMPK/mTOR pathway. Tumour Biol. 2016;37:6485–6491. doi: 10.1007/s13277-015-3927-8. [DOI] [PubMed] [Google Scholar]
- 116.Larjava H, Plow EF, Wu C. Kindlins: essential regulators of integrin signalling and cell-matrix adhesion. EMBO Rep. 2008;9:1203–1208. doi: 10.1038/embor.2008.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Burridge K. Focal adhesions: a personal perspective on a half century of progress. FEBS J. 2017;284:3355–3361. doi: 10.1111/febs.14195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu W, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu, M. et al. Inhibiting both proline biosynthesis and lipogenesis synergistically suppresses tumor growth. J. Exp. Med.217.10.1084/jem.20191226 (2020). [DOI] [PMC free article] [PubMed]
- 123.Haider S, et al. Genomic alterations underlie a pan-cancer metabolic shift associated with tumour hypoxia. Genome Biol. 2016;17:140. doi: 10.1186/s13059-016-0999-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schworer S, et al. Proline biosynthesis is a vent for TGFbeta-induced mitochondrial redox stress. EMBO J. 2020;39:e103334. doi: 10.15252/embj.2019103334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kuo CL, et al. Mitochondrial oxidative stress by Lon-PYCR1 maintains an immunosuppressive tumor microenvironment that promotes cancer progression and metastasis. Cancer Lett. 2020;474:138–150. doi: 10.1016/j.canlet.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 126.Liu Y, et al. Ornithine aminotransferase promoted the proliferation and metastasis of non-small cell lung cancer via upregulation of miR-21. J. Cell Physiol. 2019;234:12828–12838. doi: 10.1002/jcp.27939. [DOI] [PubMed] [Google Scholar]
- 127.Yoon KA, Nakamura Y, Arakawa H. Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. J. Hum. Genet. 2004;49:134–140. doi: 10.1007/s10038-003-0122-3. [DOI] [PubMed] [Google Scholar]
- 128.Liu J, et al. Alternative splicing events implicated in carcinogenesis and prognosis of colorectal cancer. J. Cancer. 2018;9:1754–1764. doi: 10.7150/jca.24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vassalli G. Aldehyde dehydrogenases: not just markers, but functional regulators of stem cells. Stem Cells Int. 2019;2019:3904645. doi: 10.1155/2019/3904645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mo D, et al. miR-1290 is a potential prognostic biomarker in non-small cell lung cancer. J. Thorac. Dis. 2015;7:1570–1579. doi: 10.3978/j.issn.2072-1439.2015.09.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Marcato P, et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 2011;29:32–45. doi: 10.1002/stem.563. [DOI] [PubMed] [Google Scholar]
- 132.Sahu N, et al. Proline starvation induces unresolved ER stress and hinders mTORC1-dependent tumorigenesis. Cell Metab. 2016;24:753–761. doi: 10.1016/j.cmet.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 133.Verbruggen N, Hermans C. Proline accumulation in plants: a review. Amino Acids. 2008;35:753–759. doi: 10.1007/s00726-008-0061-6. [DOI] [PubMed] [Google Scholar]
- 134.Matysik J, Alia, Bhalu B, Mohanty P. Molecular mechanisms of quenching of reactive oxygen species by proline under stress in plants. Curr. Sci. 2002;82:8. [Google Scholar]
- 135.Wondrak GT, Jacobson MK, Jacobson EL. Identification of quenchers of photoexcited States as novel agents for skin photoprotection. J. Pharm. Exp. Ther. 2005;312:482–491. doi: 10.1124/jpet.104.075101. [DOI] [PubMed] [Google Scholar]
- 136.Krishnan N, Dickman MB, Becker DF. Proline modulates the intracellular redox environment and protects mammalian cells against oxidative stress. Free Radic. Biol. Med. 2008;44:671–681. doi: 10.1016/j.freeradbiomed.2007.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kuo ML, et al. RRM2B suppresses activation of the oxidative stress pathway and is up-regulated by p53 during senescence. Sci. Rep. 2012;2:822. doi: 10.1038/srep00822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu X, Xue L, Yen Y. Redox property of ribonucleotide reductase small subunit M2 and p53R2. Methods Mol. Biol. 2008;477:195–206. doi: 10.1007/978-1-60327-517-0_15. [DOI] [PubMed] [Google Scholar]
- 139.Yasuda T, et al. DJ-1 cooperates with PYCR1 in cell protection against oxidative stress. Biochem Biophys. Res. Commun. 2013;436:289–294. doi: 10.1016/j.bbrc.2013.05.095. [DOI] [PubMed] [Google Scholar]
- 140.Kawate T, Tsuchiya B, Iwaya K. Expression of DJ-1 in cancer cells: its correlation with clinical significance. Adv. Exp. Med. Biol. 2017;1037:45–59. doi: 10.1007/978-981-10-6583-5_4. [DOI] [PubMed] [Google Scholar]
- 141.Lin Y, et al. High expression of DJ-1 promotes growth and invasion via the PTEN-AKT pathway and predicts a poor prognosis in colorectal cancer. Cancer Med. 2018;7:809–819. doi: 10.1002/cam4.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou J, et al. DJ-1 promotes colorectal cancer progression through activating PLAGL2/Wnt/BMP4 axis. Cell Death Dis. 2018;9:865. doi: 10.1038/s41419-018-0883-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Quiros PM, Barcena C, Lopez-Otin C. Lon protease: a key enzyme controlling mitochondrial bioenergetics in cancer. Mol. Cell Oncol. 2014;1:e968505. doi: 10.4161/23723548.2014.968505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gibellini L, et al. Silencing of mitochondrial Lon protease deeply impairs mitochondrial proteome and function in colon cancer cells. FASEB J. 2014;28:5122–5135. doi: 10.1096/fj.14-255869. [DOI] [PubMed] [Google Scholar]
- 145.Cheng CW, et al. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013;4:e681. doi: 10.1038/cddis.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fukuda R, et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 147.Hagedorn CH, Phang JM. Transfer of reducing equivalents into mitochondria by the interconversions of proline and delta 1-pyrroline-5-carboxylate. Arch. Biochem. Biophys. 1983;225:95–101. doi: 10.1016/0003-9861(83)90010-3. [DOI] [PubMed] [Google Scholar]
- 148.Hagedorn CH, Phang JM. Catalytic transfer of hydride ions from NADPH to oxygen by the interconversions of proline and delta 1-pyrroline-5-carboxylate. Arch. Biochem. Biophys. 1986;248:166–174. doi: 10.1016/0003-9861(86)90413-3. [DOI] [PubMed] [Google Scholar]
- 149.Eggleston LV, Krebs HA. Regulation of the pentose phosphate cycle. Biochem J. 1974;138:425–435. doi: 10.1042/bj1380425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yeh GC, Phang JM. Stimulation of phosphoribosyl pyrophosphate and purine nucleotide production by pyrroline 5-carboxylate in human erythrocytes. J. Biol. Chem. 1988;263:13083–13089. [PubMed] [Google Scholar]
- 151.Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342:1234850. doi: 10.1126/science.1234850. [DOI] [PubMed] [Google Scholar]
- 152.Comes S, et al. L-Proline induces a mesenchymal-like invasive program in embryonic stem cells by remodeling H3K9 and H3K36 methylation. Stem Cell Rep. 2013;1:307–321. doi: 10.1016/j.stemcr.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.D’Aniello C, et al. A novel autoregulatory loop between the Gcn2-Atf4 pathway and (L)-Proline [corrected] metabolism controls stem cell identity. Cell Death Differ. 2015;22:1094–1105. doi: 10.1038/cdd.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Clevers H. The cancer stem cell: premises, promises and challenges. Nat. Med. 2011;17:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 155.Sharif T, Martell E, Dai C, Singh SK, Gujar S. Regulation of the proline regulatory axis and autophagy modulates stemness in TP73/p73 deficient cancer stem-like cells. Autophagy. 2019;15:934–936. doi: 10.1080/15548627.2019.1586321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tomasini R, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677–2691. doi: 10.1101/gad.1695308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rufini A, et al. p73 in cancer. Genes Cancer. 2011;2:491–502. doi: 10.1177/1947601911408890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Agostini M, Annicchiarico-Petruzzelli M, Melino G, Rufini A. Metabolic pathways regulated by TAp73 in response to oxidative stress. Oncotarget. 2016;7:29881–29900. doi: 10.18632/oncotarget.8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Agostini M, et al. TAp73 promotes anti-senescence-anabolism not proliferation. Aging. 2014;6:921–930. doi: 10.18632/aging.100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Amelio I, et al. TAp73 promotes anabolism. Oncotarget. 2014;5:12820–12934. doi: 10.18632/oncotarget.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rufini A, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev. 2012;26:2009–2014. doi: 10.1101/gad.197640.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Du W, et al. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat. Cell Biol. 2013;15:991–1000. doi: 10.1038/ncb2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jiang P, Du W, Yang X. A critical role of glucose-6-phosphate dehydrogenase in TAp73-mediated cell proliferation. Cell Cycle. 2013;12:3720–3726. doi: 10.4161/cc.27267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Abuawad A, Mbadugha C, Ghaemmaghami AM, Kim DH. Metabolic characterisation of THP-1 macrophage polarisation using LC-MS-based metabolite profiling. Metabolomics. 2020;16:33. doi: 10.1007/s11306-020-01656-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang R, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Najafi M, et al. Macrophage polarity in cancer: a review. J. Cell Biochem. 2019;120:2756–2765. doi: 10.1002/jcb.27646. [DOI] [PubMed] [Google Scholar]
- 168.Yan Y, et al. 1-Pyrroline-5-carboxylate released by prostate Cancer cell inhibit T cell proliferation and function by targeting SHP1/cytochrome c oxidoreductase/ROS Axis. J. Immunother. Cancer. 2018;6:148. doi: 10.1186/s40425-018-0466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Reversade B, et al. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat. Genet. 2009;41:1016–1021. doi: 10.1038/ng.413. [DOI] [PubMed] [Google Scholar]
- 170.Nakayama T, et al. Mutations in PYCR2, encoding pyrroline-5-carboxylate reductase 2, cause microcephaly and hypomyelination. Am. J. Hum. Genet. 2015;96:709–719. doi: 10.1016/j.ajhg.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zaki MS, et al. PYCR2 Mutations cause a lethal syndrome of microcephaly and failure to thrive. Ann. Neurol. 2016;80:59–70. doi: 10.1002/ana.24678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Magini P, et al. P5CS expression study in a new family with ALDH18A1-associated hereditary spastic paraplegia SPG9. Ann. Clin. Transl. Neurol. 2019;6:1533–1540. doi: 10.1002/acn3.50821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Panza E, Martinelli D, Magini P, Dionisi Vici C, Seri M. Hereditary spastic paraplegia is a common phenotypic finding in ARG1 deficiency, P5CS deficiency and HHH syndrome: three inborn errors of metabolism caused by alteration of an interconnected pathway of glutamate and urea cycle metabolism. Front Neurol. 2019;10:131. doi: 10.3389/fneur.2019.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bender HU, et al. Functional consequences of PRODH missense mutations. Am. J. Hum. Genet. 2005;76:409–420. doi: 10.1086/428142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- 176.Mitsubuchi H, Nakamura K, Matsumoto S, Endo F. Inborn errors of proline metabolism. J. Nutr. 2008;138:2016S–2020S. doi: 10.1093/jn/138.10.2016S. [DOI] [PubMed] [Google Scholar]
- 177.Pandhare J, Cooper SK, Phang JM. Proline oxidase, a proapoptotic gene, is induced by troglitazone: evidence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J. Biol. Chem. 2006;281:2044–2052. doi: 10.1074/jbc.M507867200. [DOI] [PubMed] [Google Scholar]
- 178.Chen S, et al. SIRT3 regulates cancer cell proliferation through deacetylation of PYCR1 in proline metabolism. Neoplasia. 2019;21:665–675. doi: 10.1016/j.neo.2019.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]