

Abstract
Many genetic diseases are caused by T-to-C point mutations. Hence, editing of mutated genes represents a promising strategy for treating these disorders. We engineered an artificial RNA editase by combining the deaminase domain of APOBEC1 (apolipoprotein B mRNA editing catalytic polypeptide 1) with a guideRNA (gRNA) which is complementary to target mRNA. In this artificial enzyme system, gRNA is bound to MS2 stem-loop, and deaminase domain, which has the ability to convert mutated target nucleotide C-to-U, is fused to MS2 coat protein. As a target RNA, we used RNA encoding blue fluorescent protein (BFP) which was derived from the gene encoding GFP by 199 T > C mutation. Upon transient expression of both components (deaminase and gRNA), we observed GFP by confocal microscopy, indicating that mutated 199C in BFP had been converted to U, restoring original sequence of GFP. This result was confirmed by PCR–RFLP and Sanger’s sequencing using cDNA from transfected cells, revealing an editing efficiency of approximately 21%. Although deep RNA sequencing result showed some off-target editing events in this system, we successfully developed an artificial RNA editing system using artificial deaminase (APOBEC1) in combination with MS2 system could lead to therapies that treat genetic disease by restoring wild-type sequence at the mRNA level.
Subject terms: RNA editing, Gene therapy
Introduction
Genetic engineering, a set of techniques for regulating the expression and functions of intracellular target genes, has been widely used in basic research as well as medicinal and therapeutic applications1. Nowadays, several other genome editing techniques have made it possible to manipulate genomic information in a targeted manner1–5. These methods include zinc-finger nucleases (ZFNs), transcription activator–like effector (TALE) nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9)6,7. These enzymes can be introduced into cells for a technique known as genome editing with engineered nucleases. The latest and currently popular enzyme for the genome editing is CRISPR/ Cas9. The CRISPR/Cas9 system originally evolved as an adaptive immune system that protects bacteria and archaea from viral (phage) and plasmid infection. But in recent years, CRISPR/Cas9 has been adapted for genome editing, representing an important scientific breakthrough. CRISPR/Cas9 can mediate gene modifications at particular locations, allowing scientists to rapidly and efficiently edit the genomes of a wide range of organisms. Accordingly, this system also has the potential to be used in cell therapy. The CRISPR/Cas9 system consists of several components of which the Cas9 protein itself and the sgRNA (synthetic single guideRNA) are essential for genome editing8, which is also considered to be very effective system for controlling off target effect during genome editing9.
Base editing is another form of genome editing that enables direct, irreversible conversion of one base pair to another at a target genomic locus without requiring double-stranded DNA breaks, The most commonly used base editors are third-generation designs (BE3) comprising (i) a catalytically impaired CRISPR-Cas9 mutant10,11. On the other hand, these existing base editors also sometimes create unwanted C-to-T alterations when more than one C is present in the enzyme’s five-base-pair editing window which the Gehrke et al., group has tried to reduce bystander mutations using an engineered human APOBEC3A (eA3A) domain with Cas912.
Although these techniques are expected to find applications in the treatment of diseases, it remains difficult to achieve accurate genome editing in all affected cells. Moreover, incorrect genome editing has the potential to cause cancers and other diseases.
Therefore, we think genome editing is a suitable method for treatment of fertilized eggs or ex vivo. For the treatment of patients, trillions of cells and various tissues must be delivered with the gene editing tools, so genome editing is not a suitable technique for gene therapy for the treatment of patients as genome editing can potentially cause mutations and delivery process is also not convenient. By contrast, RNA is expressed in all tissues and cells, and if RNA repair errors occur due to incorrect RNA editing, the mutated RNAs are quickly degraded and do not affect the genome sequence13,14. Therefore, from the standpoint of patient treatment, RNA editing is preferable than genome editing. Accordingly, we developed an artificial RNA editing system, based on the APOBEC1 deaminase, for restoration of the wild-type genetic code in genetic diseases caused by T-to-C mutations. Some examples of such disorders include: ADA deficiency, cystic fibrosis, elliptocytosis, antithrombin III deficiency, and others15–18. A search of the NCBI disease mutation database ClinVar revealed 762 registered cases of T-to-C mutations (Supplementary Data S1).
In previous studies, artificial A-to-I RNA editing was done by using ADAR family enzymes tethered to gRNAs complementary to the target sequence19,20. For tethering, Stafforst and colleagues used the SNAP-tag21–23, Montiel-Gonzalez and colleagues used Lambda-N protein and box B element RNA24, and our group used the MS2 system25,26.
To date, however, no study has been published involving MS2 system for C-to-U RNA editing. We postulated that an artificial RNA editase for C-to-U conversion could be designed similarly to the A-to-I editases reported previously. For the purpose apolipoprotein B-mRNA editing family member was used.
Members of the apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC) and activation-induced cytidine deaminase (AICDA/AID) families are single strand specific cytidine deaminases, expressed in multiple cells and tissues, which catalyze cytidine-to-uridine (C-to-U) base substitutions in RNA, viral DNA, and genomic DNA27. APOBEC1, a member of the APOBEC family can also perform editing on its own in vitro, this has been confirmed by overexpression in cell lines, as well as in vivo in the absence of cofactors28,29. Previously, it was thought that cis-acting elements and trans-acting factors are necessary and sufficient to carry out C-to-U RNA editing in vitro. Consistent with this, one group of researchers reported that pure GST-APOBEC1 can edit apoB mRNA (having an optimal amount) without any auxiliary factors, moreover, this apoenzyme is thermostable30.
Zenpei and colleagues (2017)31, reported a fusion of CRISPR-Cas9 and activation-induced cytidine deaminase (Target-AID) for site-directed mutagenesis at genomic regions specified by single guide RNAs (sgRNAs); their system could be used for genetic engineering in crop plants such as rice and tomato31. Mali and colleagues (2013) focused on human genome editing with the use of the Cas932. But these studies were basically considering the genome editing not RNA editing, which is our concern. So in this study, we have focused on an artificial C-to-U RNA editing system as a tool for de novo gene therapy.
Using an MS2-tagged system, we previously restored the original sequence of a G-to-A mutation via A-to-I editing with the ADAR1 deaminase25,26,33. Here, by contrast, we sought to restore C-to-U in the context of a T-to-C mutation using our artificial enzyme system along with a specific gRNA. Tagging with MS2 is based on the natural interaction between the MS2 bacteriophage coat protein and a stem-loop structure from the phage genome34, which has been used for biochemical purification of RNA–protein complexes and combined with green fluorescent protein (GFP) expression to enable detection of RNA in living cells35. By using the phenomena in bacteriophage regarding the coat protein and stem loop, APOBEC 1 was bound to the MS2 coat protein and the gRNA bound to MS2 stem loop with a view to initiate the binding of the coat protein and stem loop thus allowing the gRNA to guide the deaminase at the specific location/site and perform editing at the targeted nucleotide sequence. This is the first study in which the MS2 system has been engineered to create an RNA editing enzyme complex that is capable of targeting a specific point mutation. Using the GFP point mutant blue fluorescence protein (BFP) as a model target RNA, our artificial RNA editing system could successfully convert C-to-U at the mRNA level, restoring the wild-type sequence.
Results
Restoration of RNA using an artificial enzyme system
For the purpose of confirming the editing activity of our developed artificial enzymatic RNA editing system, we used human embryonic kidney (HEK) 293 cells stably expressing BFP, a point-mutant derivative of GFP. In these cells, restoration of the original GFP sequence could be readily visualized as a change from blue to green fluorescence. To actually restore the sequence, we needed two more factors: the gRNA and the APOBEC1 deaminase, both the constructs were prepared under the control of the pol II CMV IE-94 promoter in the pCS2-MT and pCS2 + only expressing vectors, respectively.
The bacteriophage has the characteristics that its coat protein can tightly bind with the stem loop and this tight interaction aids in specific recognition of the nucleotides within the mRNA. We took advantage of this phenomenon, regarding tight junction between the coat protein and stem loop, by combining the MS2 coat protein with the APOBEC 1 and MS2 stem loop with the gRNA (Fig. 1a, Supplementary Data 8), which in turn guided the deaminase to reach the specific target nucleotide and the whole system showed the activity by editing C-to-U.
Figure 1.

(a) Schematic diagram of the MS2 system, The MS2 coat protein is attached to the APOBEC 1 catalytic domain and the MS2 stem loop is attached to the gRNA under the control of the CMV promoter. The MS2 coat protein and stem loop can bind to each other, enabling detection of a specific nucleotide sequence within the mRNA. (b) Stably BFP expressing HEK 293 cells were transfected with wild type of GFP, one factor (e.g., either APOBEC1or only guide RNA) or two factors (APOBEC1 + guide RNA). Green fluorescence expression was observed only when two factors were present, implying that both factors were necessary for C-to-U editing. Imaging was performed by LSM confocal microscopy.
LSM confocal microscopy
For the confirmation of the editing of our developed artificial enzymatic system, we observed the expression of the fluorescence in the BFP expressed HEK 293 cells which we observed via fluorescence microscopy. To obtain more clear images, we observed by using an LSM confocal microscope (Fig. 1b). When only one factor was transfected (e.g., either only APOBEC1 or only gRNA) into the BFP stably transformed cells, only blue fluorescence was observed but not green fluorescence, which proved that no restoration had taken place. When both the factors were transfected together (APOBEC1 + gRNA) into the same BFP stably transformed HEK 293 cells, green fluorescence was detected. More precisely, expression of BFP fluorescence to GFP was restored in cells, where the genetic code was restored because of the editing of C-to-U, and most of the fluorescence was in the cytosols (Fig. 1b).
Confirmation of sequence restoration by PCR–RFLP
Target specificity is an important consideration in the context of genetic code restoration. Therefore, to verify specificity at the sequence level, we conducted reverse-transcriptase PCR followed by restriction fragment length polymorphism (RFLP) analysis of the BFP and GFP genes using the BtgI restriction enzyme. BtgI can only cut BFP, but neither the wild type GFP nor restored GFP (Fig. 2a). The total length of the PCR amplified fragment was 324 bp; digestion with BtgI yielded two bands at 201 bp and 123 bp. According to the sequence preference of the enzyme, only the BFP was cut, whereas the wild type GFP and restored GFP remained undigested at 324 bp. When GFP was restored, the two shorter bands were still present at 201 bp and 123 bp because the whole transfection process was done into the BFP stably transformed HEK 293 cells (Fig. 2b). And, 100% of editing is near about impossible as a result there were still remaining part of BFP.
Figure 2.

(a) Schematic illustration of RFLP. The BtgI restriction enzyme can cut the BFP but not wild type of GFP or restored GFP (C to U converted) (b) PCR–RFLP of cDNA extracted from transfected cells (HEK 293 stably expressing BFP), restriction-digested with BtgI. BFP was cleaved into two fragments, 201 and 123 bp, whereas restored GFP was not cleaved and remained at 324 bp. (c) For all data the statistical analysis (mean ± SEM) was calculated, where n = 5, Image J software was used for measuring the band intensity (https://imagej.net/Citin).
When only one factor (either APOBEC1 or gRNA) was transfected into the BFP-expressing HEK 293 cells, the PCR fragment was completely digested by the restriction enzyme, and there was no remaining band at 324 bp. Consistent with the confocal microscopy observations, neither of the two factors on its own could restore BFP to GFP; consequently, almost 100% of the PCR fragment of BFP was cut by the BtgI enzyme.
For the uncut bands of only BFP, both the bands at 123 and 201 bp were strong but when BFP was transfected with only one factor either APOBEC1 or guideRNA the band intensity decreased. Although in case of the restored sample where both the editing factors were transfected together, remained uncut band was found at 324 bp (Fig. 2b), if the intensity is compared with the two other bands at 123 bp and 201 bp of the same lane of sample, we could see overall the intensity was decreased.
In case of BFP alone (without any editing factor such as APOBEC 1 or gRNA), the fragment was completely digested by the restriction enzyme. The band at 324 bp was only present when the genetic code was restored to GFP (conversion from BFP to GFP, C-to-U), i.e., when the cells were co-transfected with gRNA and APOBEC1, only then the genetic code was restored from BFP to GFP, but still two small bands were there at 201 bp and 123 bp, as within a single cell 100% editing is not possible and our data also proves that.
We did the densitometry analysis of the band intensity from the PCR–RFLP PAGE electrophoresis bands. Calculation of the PCR–RFLP band intensity confirmed that BFP had been restored to GFP. To standardize the data, the PCR–RFLP experiment was repeated several times (N = 5), the intensity was measured by ImageJ software (NIH, https://imagej.net/Citing), and editing efficiency was calculated individually in each replicate. Ultimately, the mean editing efficiency was 20.4% (0.204 ± 0.05), (Fig. 2c).
Confirmation by Sanger sequencing
For more evidence of restoration the sample we also performed the Sanger’s sequencing, which we did by using both the sense and antisense primers (Fig. 3). Dual peaks were only observed when cells were transfected with both the editing factors (edited T and unedited C: sense; edited A and unedited G: antisense) (Fig. 3a and b). In case of both the sense and antisense primers, sequencing of PCR-amplified cDNA extracted from the transfected cells, confirmed that editing was successfully done and no off-target editing had occurred in the surrounding cytosines, 21 bp upstream and downstream of the target.
Figure 3.
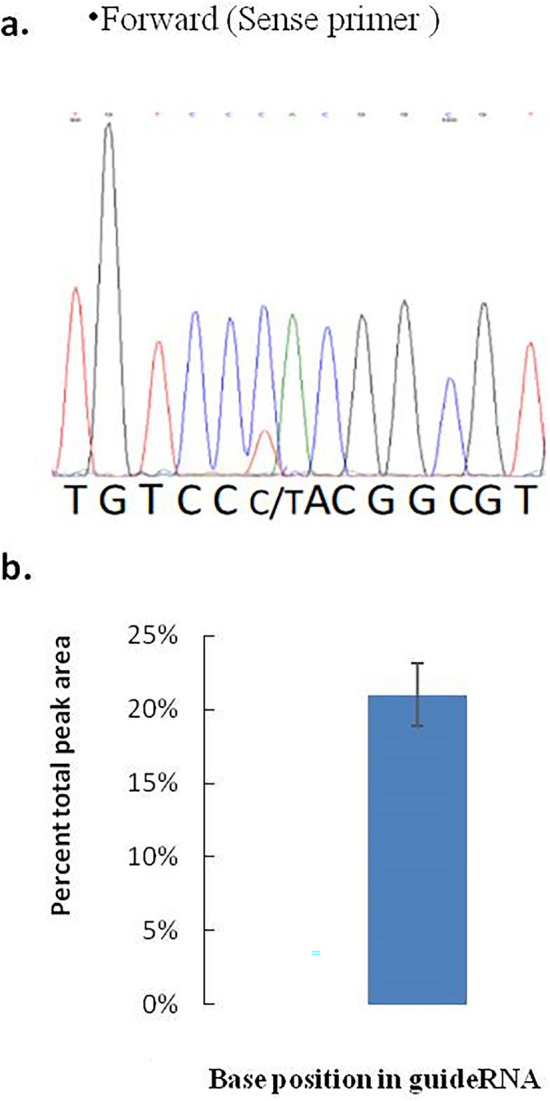
Confirmation of the restoration of BFP to GFP (C to U) by Sanger’s sequencing. (a) Forward or Sense primer (CCA to CTA), the dual peaks were observed, which were due to the restoration of the genetic code from the C to U (BFP to GFP), after the application of the two editing factors (APOBEC1 deaminase and gRNA). (b) Edit-R analysis of the Sense and antisense chromatogram height of the edited part of the peak and the statistical analysis (mean ± SEM) has been done, where the n = 5. Edit R Version 10 (https://moriaritylab.shinyapps.io/editr_v10/) analysis has been done by using the Sanger’s sequencing Ab.1 file.
Editing efficiencies, calculated based on peak area and peak height were 21.5% and 21%, respectively (Supplementary Data S4), consistent with the value obtained by PCR–RFLP result. This experiment and the whole analysis were performed five times (N = 5) under identical conditions, and similar results were obtained each time.
The peak chromatogram has also been analyzed by Edit-R software (Version 10) through which we have also obtained 21% editing which is the identical result that we calculated via the peak height and peak area calculation from the Sanger’s sequencing peak analysis (3c, 3d).
Confirmation of sequence restoration and observation of off-target effects by total RNA-sequencing (RNA-seq)
Next, we performed whole RNA sequencing (RNA-seq) to evaluate off-target effects (Supplementary Data S2 and S3, S6 and S7). The total number of reads before trimming were 30,554,972 for BFP_1 HEK 293 (Control: BFP-expressing HEK 293, targeted mRNA) and 28,523,210 for HEK_293 (Tested: in which GFP was restored after transfection with both APOBEC1 and gRNA); 30,482,237 and 28,459,029 reads, respectively, remained after trimming (Supplementary Data S2 and S3). The total number of reads sequenced did not differ between cell lines, so we can assume that any difference in the amount of expressed mRNA over the total length of the BFP gene was correlated with the number of mapping reads.
Each sequence was compared with the human genome database to identify gene nucleotide substitution. Since human genome sequence did not including GFP gene, so we actually used the human data base + GFP.
In the BFP_1 (Control) mapped reads, a mutation with two or fewer reads that support a mutation, was identical to the mutation detected in tested HEK 293 cells (restored GFP). All the detected mutations that were found in both the control and tested samples were presented in Supplementary Data S6 and S7. For proper identification of the off target effects of the developed system the RNA-seq result was analyzed through several ways (Fig. 4). In the restored sample, about 6.7% of all SNVs showed C-to-U alterations except the targeted position (Fig. 4a), which is quite low. In the box plot, the median value of the restored C-to-U alterations or the complementary G-to-A alterations is approximately 1 compared to the efficiency of editing-negative control (BFP stable transformed HEK 293 cells-target) (Fig. 4b). These results indicated some off-target effects. However, at more than 10 coverage of comparable C-to-U or G-to-A sites, the jitter plots showed that there are hundreds of specific off-target sites (Fig. 4c). Of these, more than 5% of C-to-U change occurred at 369 sites. Overall we can predict from the above mentioned analysis of the RNA-seq result is that the APOBEC 1-MS2 system along with sgRNA is quite specific and the off target effect is not so high.
Figure 4.

APOBEC 1(DD)-MS2 system induces some off-target C-to-U RNA editing in HEK293 cells. (a) Percentages of expressed genes with at least one edited cytosine (C-to-U or G-to-A) in total SNVs. (b) Box plots showing rate of cytosines edited by APOBEC1(DD)-MS2 compared to control (mutated BFP target in HEK 293 cells), yellow line is median. (c) Jitter plots showing transcriptome-wide efficiencies of C-to-U or G-to-A edits (y-axis) identified from RNA-seq experiments in HEK293 cells
modified by APOBEC1 (DD)-MS2 vs editing- negative control (BFP target, stably transformed in HEK 293 cells). n: total number of modified cytosines identified.
In HEK_293 derived reads, the number of reads that mapped, among them very small amount of reads were not present in the BFP_1 (control). Among the mutations detected in HEK 293 (tested, restored GFP), 9053 was not present in BFP_1, including 396 C-to-T mutations and one CC-to-TT mutation. The frequency of each base change is shown in the Supplementary Data S6, S7.
On the other hand, 5910 of the reads obtained from BFP_1 (target mRNA, control) cells mapped to BFP. For 199C, in HEK_293 (restored GFP, tested) derived reads, a C-to-T change occurred in one of the two reads mapped to base 199. On the other hand, in reads derived from BFP_1 (control) cells, we detected one C > A substitution, one C > G substitution, and 1608 unedited C nucleotides.
In addition, in the BFP_1 (control), we observed T > C mutations at 199th (Targeted site) position which was not found in case of the HEK_293 derived reads as this sample was the restored one, BFP to GFP. As a precaution, we removed the human genome sequence and mapped the reads of each cell line to the BFP gene sequence only, but the situation did not change.
Further, we looked for off-target effects. A total of 18,567 mutations were present in BFP_1 (control, Supplementary Data S6), whereas only 9053 mutations were present in HEK_293 (tested, Supplementary Data S7), suggesting that off-target effects are not a major problem in our system. The off target effects are not much significant (Fig. 4).
Discussion
In this study, we successfully established an artificial deaminase system based on APOBEC1 and showed that it functions as designed in C-to-U RNA editing. We also showed that the MS2 system is compatible with the design of an RNA editing enzyme model. The experimental data suggest that the deaminase domain of APOBEC1, which lacks the double-stranded RNA-binding domain (dsRBD), can be made competent for RNA editing by addition of an artificial gRNA. Because the deaminase domain of APOBEC1 lacks both a nuclear localization signal and a nuclear export signal, the engineered enzyme system should be localized to the cytoplasm, where it is likely to encounter the target mRNA. However, it remains to be determined whether small amounts of the MS2-fused deaminase chimera also localize to the nucleus.
Montiel-Gonzalez et al., (2016)36 used the Lambda N system for site-directed RNA editing along with the ADAR adenosine deaminase. In this study, we used the MS2 system, which is more efficient and has been used more frequently than the Lambda N system in RNA studies; in respect of the editing efficiency MS2 is stronger than Lambda-N system and efficient. The binding affinity towards the hairpin is 10–8 M for the Lambda N system, whereas the affinity towards the AUUA stem loop sequence is 10–9 M for MS2, which improves to 10–10 M when the AUCA stem loop sequence is present. This explains why MS2 is stronger than the Lambda N system37.
Concerning the restoration of genetic code with the developed artificial deaminase enzyme system only when BFP-expressing cells were transfected with two factors, the catalytic domain and the gRNA, we could detect conversion of (mutant) BFP to GFP. No GFP fluorescence was observed when one of the factors either APOBEC1 or gRNA was transfected alone, indicating that the other cellular factors are not involved in the editing. Further optimization of the system, including the gRNA sequence, the usability freedom of the MS2 moiety with the enzyme, and the relative proportions of the two factors, should be performed in future work. Appropriate concentrations of enzyme and reporter substrate can eliminate minor auto-editing38,39.
Our PCR–RFLP analysis confirmed that after the application of both the deaminase and gRNA, the restored GFP was not cleaved by BtgI, whereas the BFP stably transformed cells which were transfected with either APOBEC1 or gRNA, these were cleaved into two fragments of 201 bp and 123 bp. This is consistent with the sequence preference of the restriction enzyme BtgI, and completely supports the results of Luyen et al., (2015, 2017)40,41. These observations prove that APOBEC1 and gRNA together can restore the GFP sequence by RNA editing from the BFP transcript.
Non-restored BFP was completely cleaved into the 201 and 123 bp fragments, and no 324 bp fragment remained. By contrast, the restored GFP was not cleaved by the enzyme. A remained 324 bp band was observed due to the editing where both the enzyme and gRNA were applied together, however, still we could see the cleaved bands at 201 bp and 123 bp in the same sample of same lane where restored GFP was expressed (Fig. 2b), because 100% restoration of the genetic code is not possible which we have also observed from our confocal images. We calculated the editing rate by densitometry analysis from PAGE band intensities, which we analyzed using the ImageJ software (NIH, https://imagej.net/Citing), and found that the mean editing efficiency was 20.4%. In this regard it is mentionable that the intensity of the larger band is stronger than that of the smaller bands. So visually the band at 324 bp might look a bit stronger than that of the bands at 201 and 123 bp. But we have done the densitometry analysis of the band intensity and then calculated the restoration percentage which is 20.4%, but really the abundant is not so weak.
Further confirmation of ability of the developed system was obtained by Sanger’s sequencing. The sequencing analysis revealed that the restored GFP gave a peak corresponding to wild-type GFP: TCCTACGG instead of TCCCACGG with the sense primer, and CCGTAGGAC instead of CCGTGGGAC with the antisense primer. The dual peak could be observed only when the restoration of the genetic code happened due to editing. We calculated the editing efficiency based on peak height for both the sense and antisense primers. This calculation indicated that 21.5% of BFP was edited using our deaminase approach.
We also used the Edit-R software (version 10) to analyze the sequence result following Kluesner group (42). We measured the peak chromatogram and calculated the editing percentage of the particular nucleotide. The analysis showed that average 21% of the C has been restored to U which is identical with the measurement that we have done by peak height and peak area. We analyzed both the sense and antisense sequence data and in both the cases have got the same average 21% editing rate.
BFP contains a single mutation, so we detected only a single peak at that specific site. We found no off-target editing in the 21 bp upstream or downstream of the mutated site. We believe that the frequency of off-target editing is almost zero as the read through of the targeted BFP and restored GFP (after transfection with the two factors) was very similar.
Interestingly, our total RNA sequencing (RNA-seq) analysis revealed that, in HEK cells, expression of both GFP expressing mRNA and BFP protein were potentially reduced. It is possible that the gRNA may have mediated some sort of RNA interference (RNAi) activity, although, we have no corroborating data to this effect. Although when an expression vector is introduced into the cell, an endogenous transcription factor binds to the vector promoter. Therefore, transcription of endogenous genes is totally suppressed. Necessary transcription factors are deprived. Moreover BFP and GFP both are protein and they have half-life and the fluorescence intensity of BFP is weaker than that of GFP. These could be probable reasons that at such particular time the BFP intensity was reduced which was difficult to observe. Although in future studies the underlying assumptions should be validated.
In terms of editing efficiency, several factors should be considered; in the future, improvement of these factors could increase the efficiency. For example, we used the MS2-6X stem loop, although previous experiments suggested that the MS2-12X stem loop is much more active. Hence, a gRNA constructed using MS2-12X could increase editing efficiency. Even the 1X MS2 on the either side of the guideRNA may also have a positive impact on the editing efficiency, which has already been observed by the Katreakar et al., but for A to I editing with the ADAR1 enzyme and CRISPR approach20.
Another approach might be optimization of the length of the gRNA, which also determines the degree of off-target editing. In previous experiments, we found that the best length of a guide sequence for site-specific editing is 21 ntd: if we increase the length of a guide, efficiency may increase, but the level of off-target editing increases simultaneously (Supplementary data S5-unpublished). As our system is same with the previous one, only the difference is editing enzyme and the target mRNA that is why we have used that knowledge for designing the guideRNA for this study as well.
Transfection with two factors has a greater effect on cellular conditions than transfection with a single factor. Hence, if we can make a single construct in which the deaminase and the gRNA are encoded on the same construct, this should make transfection easier and likely also increase efficiency.
The developers of the CRISPR-Cas9–AID approach to sequence restoration reported editing efficiencies of 26.2% for rice and 53.8% for tomato31,42, though it was a genome editing technique. This is significantly higher than the value we achieved in the present study (~ 21%), which was based on combining MS2 and APOBEC1 to edit the mutated GFP/BFP mRNA in vitro. Notably, however, this is the first time that the MS2–APOBEC1 combination has been used for C-to-U editing to correct T-to-C mutations.
In our system, off-target effects were not so high whether the system was completely based on the RNA editing and MS2 system along with the sgRNA and APOBEC 1 deaminase was used. The developers of Cas9-AID also reported a very low rate of off-target editing, 0.14–0.38%, implying that off-target effects are very rare with systems of this kind, although their system was of genome editing whereas our system is for RNA editing31. Even the group who worked with the Adenosine base editors along with the CRISPR system they have also found low rate of indels which is only 0.1%11, but their system convert targeted A•T base pairs efficiently to G•C approximately with 50% efficiency in human cellular system, although their system was of genome editing and applied with CRISPR-ABEs11. Afterwards the Zuo group has done the Cytosine base editing in vivo and they have also found only zero to four indels in embryos and none of them overlapped with the predicted off-target sites43. Here they used the base editors/cas9 and the GOTI (genome-wide off-target analysis by two-cell embryo injection) technique for detecting the off target events in vivo.
Moreover, to prepare the CRISPR-Cas9–AID restoration tool, Zenpei et al., (2017)31 used pmCDA1, which has a backbone of ~ 10 kb, while the length of the Cas9 gene is 3156 bp. Thus, in total their construct is ~ 13 kb in length, which may be too large to deliver in a therapeutic context. Both MS2 and APOBEC1 are smaller than the components of CRISPR-Cas9, and as vectors we used pCS2-only and pCS2-MT, which have significantly smaller backbones, ~ 4.5 kb. Therefore, our system is smaller, so it is easier to manipulate and deliver, and has a better editing efficacy than the Cas9-AID combination tool, although that Cas9-AID system was used for the genome editing whereas our developed system is for RNA editing. Moreover, according to the RNA-seq result off-target C > U/G > A RNA editing occurred at higher levels at the restored sample in 931 sites (Fig. 4a and 4c) in comparison to the control sample. Frequency of editing at some of these off-target sites appears to be > 50% (Fig. 4c). These are not trivial numbers for off-target editing sites and their editing levels. Perhaps this is not surprising since after the blast analysis of the complementary sequence of the 21 ntd of the target BFP sequence (guideRNA) with the human genome sequence we found that some homologous sequences are there in some of the genes of human genome sequence. Moreover, in Fig. 4C particularly several sites which are in the same chromosome are having an editing frequency higher than 50%, which is quite different from the other published papers on the Base editing44. In those papers the editing frequency is quite lower. We think this could be because of the MS2-based approach. We also think because the sites are prone to form secondary structure that is why the editing frequency is higher.
Our findings indicated that our system could successfully restore gene expression by editing point mutations (U-to-C) in the RNA of patients with various diseases caused by such mutations, thereby alleviating the symptoms of their disorders by performing C-to-U editing. The ability of MS2-APOBEC1 and gRNA to edit mutated RNA sequences could be used to overcome genetic diseases caused by T-to-C mutation. Future studies should seek to optimize editing efficiency, e.g., by using gRNAs of different lengths and altering the MS2 stem loop. If possible, the practical applications of this system should be tested in animal models.
Methods
Target plasmid construction
The target BFP gene was prepared by a point mutation at 199th position of GFP by following the protocols of Luyen et al., (2012, 2015)40,45. The BFP construct was transfected into HEK 293 cells and transfectants were selected with G418 @ 500 ng/mL. Positive colonies were picked and confirmed by Sanger sequencing.
APOBEC1 deaminase plasmid construction
To target the enzyme to the BFP codon of interest, we cloned the deaminase domain of APOBEC1 downstream of MS2 in pCS2 + MT vector under the control of the pol II CMV IE-94 promoter, using the XhoI and XbaI (Takara, Shiga, Japan) restriction sites. The resultant plasmid was designated as pCS2 + MT-MS2HB-APOBEC1. APOBEC1 was PCR-amplified from HEK 293 cell line using primers with the appropriate restriction sites: XhoI catalytic APOBEC1 Fw: tccactcgagatgccctgggagtttgacgtctt; XbaI catalytic APOBEC1 Rv: acggtctagattaagggtgccgactcagaaactc; (red color font indicates restriction sites, blue color font highlighting the tri-nucleotide atg/tta is a leader sequence allowing proper recognition by the restriction enzyme). Positive colonies were picked and confirmed by Sanger sequencing. The open reading frame and catalytic domain were confirmed using the ExPASY Bioinformatics resource portal and NCBI-BLAST.
Preparation of the gRNA to direct the deaminase to target
To prepare the gRNA, a 21 ntd sequence complementary to the target mRNA having a mismatch A at the target C position (Supplementary data 8), was inserted upstream of MS2-RNA by adding the guide sequence with the forward primer of PSL-MS2-6X (pCS2 + guide-MS2-RNA) and was ligated with the pCS2 + Only vector plasmid under the control of the CMV IE-94 promoter. MS2 6X means the repetition of the sequence of MS2 stem loop part for six times. There are also 12 X and 24 X MS2 stem loop available, but for this research we have used the 6X MS2 stem loop.
The sequence used for this purpose was as follows: atcaGAATTCCACTGCACGCCGTAGGACAGGGAATGGCCATG. The atca tetra-nucleotide is a leader sequence allowing proper recognition by the restriction enzyme; bold font indicates the restriction site; italic font represents the 21 ntd guide; and the underlined portion indicates the forward primer for MS2-6X. Positive colonies were picked and confirmed by Sanger sequencing. The gRNA was chosen based on previous works done by our group with ADAR1.
Previously, we used ADARs to restore the wild-type sequence at various mutant stop codons. In that work, we designed guides of different lengths: 19, 21, or 23 ntd (upstream and downstream). We found that a 21 ntd guide was more efficient than 19 or 23 ntd versions, and that the 23 ntd guide created some off-target effects that were not detectable when the shorter sequences were used (Supplementary Data 5-still unpublished). In light of its optimal efficiency and minimal off-target effects, we used the 21 ntd gRNA in this study2,4. We have used this knowledge of the previous experiment for designing the guideRNA as we have used the same MS2 system in this case as well but the deaminase has been changed because of the change of the mutation type. Here we have used APOBEC1 deaminase instead of ADAR1 as we have chosen T-to-C mutation instead of G to A mutation. Regarding the location of the target whether it should be in the 5′ (as we have done) or middle or 3′ position of the guideRNA, we have tried with different positions and we have found that the best efficiency we have got from the 5′ position (7th position in 21 bp guideRNA) of the target in the guideRNA (Supplementary datda 5). Katrekar et al. (2019) also applied the guideRNA of 20 bp length and they placed the mismatch (at the site of target) at 6th position in 20 bp guideRNA20,46.
The gRNA expression was driven by the RNA Pol II promoter (CMV-IE94), which is also expressed in the human cells.
Culturing cells and transfection
Stably BFP-expressing HEK 293 cells (50–70% confluent) were transfected by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA); 3 × 105 cells were seeded in each well of a 12-well plate. Each well contained 1 mL Opti-MEM (Gibco) and 4 µL of Lipofectamine 2000, and was transfected with 800 ng of APOBEC1 deaminase and 700 ng of gRNA into each well. The samples were then cultured in an incubator at 37 °C for 6 h. After 6 h, the Opti-MEM was replaced with D-MEM (Gibco) to optimize cell growth, and the cells were incubated at 37 °C for 48 h prior to observation.
Observation of fluorescence by confocal microscopy
Cells were observed on an FV1000D confocal laser-scanning microscope (Olympus, Shinjuku-ku, Tokyo, Japan) under optimized conditions. To obtain very clear images, our conditions were designed to increase the effective resolution, dye selection, determination of the exposure time as well as the adjusted magnification, which enabled us to determine the exact location of the fluorescence in our cell samples, and particularly within the single cells.
RNA extraction and cDNA synthesis from transfected cells
Following microscopic observation, cells were harvested from the dish, and total RNA was extracted using the TRIzol reagent (Invitrogen). cDNA was synthesized from the extracted RNA using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen).
Confirmation of sequence restoration by PCR–RFLP
To confirm successful restoration of the wild-type sequence, PCR products amplified using GoTaq polymerase (Promega, Madison, WI, USA) on a GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA, USA) were digested with a restriction enzyme that differentiated between the edited and unedited DNA sequences. The amplicons were subjected to run in 6% polyacrylamide gel electrophoresis followed by staining with SYBR Green dye (Invitrogen). 100 ng of cDNA was used for each PCR reaction, the total reaction volume was 20 ul. After the PCR amplification 10 ul of PCR product was taken for restriction digestion, where incubation was done at 37 °C for 2 h with BtgI (New England BioLabs) restriction enzyme, which cleaved the BFP sequence into two fragments of 201 and 123 bp but left the restored GFP sequence undigested. During the gel electrophoresis equal volume (2 ul) of digested product was loaded into the 14 well comb. Imaging was done using the LAS 3000 imager.
The presence of the intact 324 bp sequence confirmed restoration of C to U in the mRNA (i.e., conversion of BFP to GFP). Band intensity was measured using the Image J software (NIH, https://imagej.net/Citing) and editing efficiency was calculated from the band intensities using the following equation:
Sanger sequencing
After doing PCR with equal amount of cDNA (100 ng) in each reaction of 20 ul volume, the PCR products were resolved on 1% agarose gels, the bands were cut out and frozen. DNA was purified using the QIAquick Gel Extraction kit, and concentration was measured on an ND-1000 spectrophotometer. Sequencing of the purified DNA was performed using the Big Dye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Technologies, Waltham, MA, USA) using the forward and reverse primers for GFP. The raw sequencing data were analyzed using the Sequence Scanner software, version 2 (Applied Biosystems). When the edited and unedited products were presented together, a dual peak (C [unedited] and T [edited]) was observed at the target site. Following the previous works on calculation of editing efficiency from peak area and peak height, we also calculated by measuring the area26 and peak height47,48 using the ImageJ software (NIH, https://imagej.net/Citing).
Editing efficiency (sense) =
The chromatogram height has also been measured and thus the editing rate has been analyzed using the Edit-R software, version 10 according to the instruction given by the Kluesner group49.
Total RNA-sequencing (RNA-seq)
RNA-seq analysis was performed by Filgen (Nagoya, Japan). Read trimming to eliminate low-quality reads from the analysis was performed using the CLC Genomics Server 11 (Qiagen). Reads with the following properties were removed: bases with Phred score < 13; more than three ‘N’ nucleotides; length below 100 bp. Mapping was performed using the RNA-seq analysis function.
RNA-seq mapping parameters were as follows:
Reference type = Genome annotated with genes and transcripts
Reference sequence = Homo sapiens (hg38) sequence + BFP1 sequence
Gene track = Homo sapiens (hg38) (Gene) + BFP1 gene
mRNA track = Homo sapiens (hg38) (mRNA) + BFP1 mRNA
Mismatch cost = 2
Deletion cost = 3
Length fraction = 0.8
Similarity fraction = 0.8
Global alignment = No
Auto-detect paired distances = No
Strand specific = Both
Maximum number of hits for a read = 10
The average coverage of the coding region in the mapping results was 82.6 for HEK_293T3F (edited GFP sample after application of two factors in BFP stable cells) and 7.5 for BFP_1 (targeted mRNA of BFP stable HEK 293 cells), and 85% of sequence reads were mapped onto exons.
Basic Variant Detection in CLC Genomics Server 11 was used for variant detection:
-
Minimum coverage = 10 (lower limit for the number of reads mapped to the mutation location).
- Minimum count = 5 (lower limit for the number of reads actually calling the mutation).
- Minimum frequency (%) = 1.0 (lower limit for the percentage of reads that mapped with mutations, calculated as counts / coverage).
- Mutations that met all three conditions were detected.
For analyzing the RNA-seq result several illustration was done such as: percentages of expressed genes with at least one edited cytosine (C-to-U or G-to-A) in total SNVs, Box plots showing rate of cytosines edited by APOBEC1(DD)-MS2 compared to editing-negative control detecting the off target events, and Jitter plots showing transcriptome wide efficiencies of C-to-U or G-to-A edits (y-axis) identified from RNA-seq experiments in HEK 293 cells modified by APOBEC1(DD)-MS2 following the process of Grunewald et al., group29,50.
Supplementary information
Acknowledgments
The authors gratefully acknowledge scholarships from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan. This research was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (26670167, 17H02204 and 18K19288 to T.T.).
Author contributions
Experiments, writing, and editing: S.B.; guidance in experiments: M.S.; concept and editing: T.T.*.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
The original online version of this Article was revised: The original version of this Article contained an error in the sequence of the macular mutation. Full information regarding the correction made can be found in the correction for this Article.
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41598-025-89490-3
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
9/9/2024
A Correction to this paper has been published: 10.1038/s41598-024-71985-0
Change history
2/11/2025
This article has been retracted. Please see the Retraction Notice for more detail: 10.1038/s41598-025-89490-3
Supplementary information
is available for this paper at 10.1038/s41598-020-74374-5.
References
- 1.Heidenreich, M. & Zhang, F. Applications of CRISPR-Cas systems in neuroscience. Nat. Rev. Neurosci.17(1), 36–44. 10.1038/nrn.2015.2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katrekar, D. & Mali, P. In vivo RNA targeting of point mutations via suppressor tRNAs and adenosine deaminases. Cold Spring Harbour (BioRxiv)10.1101/210278 (2017). [Google Scholar]
- 3.Kim, Y. G., Cha, J. & Chandrasegaran, S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci.93(3), 1156–1160. 10.1073/pnas.93.3.1156 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porteus, M. H. & Carroll, D. Gene targeting using zinc finger nucleases. Nat. Biotechnol.23(8), 967–973. 10.1038/nbt1125 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Zhang, F. et al. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol.29(2), 149–153. 10.1038/nbt1775 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsu, P. D., Lander, E. S. & Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell157(6), 1262–1278. 10.1016/j.cell.2014.05.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas, G., Shannon, J. S., Sai-lan, S. & Jia, L. Genome editing technologies: Principles and applications. Cold Spring Harbor Perspect. Biol.8, a023754. 10.1101/cshperspect.a023754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marx, V. Base editing a CRISPR way. Nat. Methods15, 767–770. 10.1038/s41592-018-0146-4 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Zhang, X. H., Tee, L. Y., Wang, X. G., Huang, Q. S. & Yang, S. H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Therapy-Nucl. Acids4, e264. 10.1038/mtna.2015.37 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature533(7603), 420–424. 10.1038/nature17946 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaudelli, N. M. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature551, 464–471. 10.1038/nature24644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gehrke, J. M. et al. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol.36, 977–982. 10.1038/nbt.4199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gott, J. M. & Emeson, R. B. Functions and mechanisms of RNA editing. Annu. Rev. Genet.34, 499–531 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Keegan, L. P., Gallo, A. & O’Connell, M. A. The many roles of an RNA editor. Nat. Rev. Genet.2(11), 869–878. 10.1038/35098584 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Aisling, M. F. & Andrew, R. G. Adenosine deaminase deficiency: A review. Orphanet: J. Rare Dis.13, 65. 10.1186/s13023-018-0807-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lane, D. A., Kunz, G., Olds, R. J. & Thein, S. L. Molecular genetics of antithrombin deficiency. Blood Rev.10(2), 59–74. 10.1016/S0268-960X(96)90034-X (1996). [DOI] [PubMed] [Google Scholar]
- 17.Olds, R. J. et al. A common point mutation producing type 1A antithrombin III deficiency: AT129 CGA to TGA (Arg to Stop). Thromb. Res.64(5), 621–625. 10.1016/S0049-3848(05)80011-8 (1991). [PubMed] [Google Scholar]
- 18.Hirschhorn, R., Tzall, S., Ellenbogen, A. & Orkin, S. H. Identification of a point mutation resulting in a heat-labile adenosine deaminase (ADA) in two unrelated children with partial ADA deficiency. J. Clin. Invest.83(2), 497–501 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maas, S., Rich, A. & Nishikura, K. A-to-I RNA editing: Recent news and residual mysteries. J. Biol. Chem.278, 1391–1394. 10.1074/jbc.R200025200 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Katrekar, D. et al. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods16(3), 239–242. 10.1038/s41592-019-0323-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stafforst, T. & Schneider, M. F. An RNA Deaminase conjugates electively repairs point mutations. Angew. Chem. Int. Ed51(44), 11166–11169. 10.1002/anie.201206489 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Vogel, P., Hanswollemenke, A. & Stafforst, T. Switching protein localization by site directed RNA editing under control of Light. ACS Synth. Biol.6, 1642–1649. 10.1021/acssynbio.7b00113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogel, P. & Stafforst, T. Site-directed RNA editing with antagomir deaminases-A tool to study protein and RNA function. Chem. Med. Chem.9(9), 2021–2025. 10.1002/cmdc.201402139 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Hiroki, S. et al. Introduction of pathogenic mutations into the mouse Psen1 gene by Base Editor and Target-AID. Nat. Commun.9, 2892. 10.1038/s41467-018-05262-w(1-8) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azad, M. T. A., Bhakta, S. & Tsukahara, T. Site-directed RNA editing by adenosine deaminase acting on RNA (ADAR1) for correction of the genetic code in gene therapy. Gene Ther.24(12), 779–786. 10.1038/gt.2017.90 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Bhakta, S., Azad, M. T. A. & Tsukahara, T. Genetic code restoration by artificial RNA editing of Ochre stop codon with ADAR1deaminase. Protein Eng. Des. Select.31(12), 1–8. 10.1093/protein/gzz005 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Liqun, L. et al. APOBEC3 induces mutations during repair of CRISPR–Cas9-generated DNA breaks. Nat. Struct. Mol. Biol.25, 45–52. 10.1038/s41594-017-0004-6 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Blanc, V. et al. APOBEC1 complementation factor (A1CF) and RBM47 interact in tissue-specific regulation of C to U RNA editing in mouse intestine and liver. RNA25(1), 70–81. 10.1261/rna.068395.118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grunewald, J. et al. Transcriptome-wide off-target RNA editing induced by CRISPR guided DNA base editors. Nature569, 433–437. 10.1038/s41586-019-1161-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chester, A. N. N., Weinreb, V., Chrles, W., Carter, J. R. & Navaratnam, N. Optimization of apolipoprotein B mRNA editing by APOBEC1 apoenzyme and the role of its auxiliary factor ACF. RNA10, 1399–1411. 10.1261/rna.7490704 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zenpei, S. et al. Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat. Biotechnol.35, 441–443. 10.1038/nbt.3833 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Mali, P. et al. RNA-guided human genome engineering via Cas9. Science339(6121), 823–826. 10.1126/science.1232033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanswillemenke, A., Kuzdere, T., Vogel, P., Jékely, G. & Stafforst, T. Site-directed RNA editing in vivo can be triggered by the light-driven assembly of an artificial riboprotein. J. Am. Chem. Soc.137(50), 15875–15881. 10.1021/jacs.5b10216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johansson, H. E., Liljas, L. & Uhlenbeck, O. C. RNA recognition by the MS2 phage coat protein. Sem. Virol.8(3), 176–185. 10.1006/smvy.1997.0120 (1997). [Google Scholar]
- 35.Bertrand, E. et al. Localization of ASH1 mRNA particles in living yeast. Mol. Cell2(4), 437–445. 10.1016/S1097-2765(00)80143-4 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Montiel-González, M. F., Vallecillo-Viejo, I. C. & Rosenthal, J. J. An efficient system for selectively altering genetic information within mRNAs. Nucl. Acids Res.44(21), 157–168. 10.1093/nar/gkw738 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montiel-Gonzalez, M. F., Quiroz, J. F. D. & Rosenthal, J. J. C. Current strategies for site-directed RNA editing using ADARs. Sci. Direct Methods156, 16–24. 10.1016/j.ymeth.2018.11.016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fukuda, M. et al. Construction of a guide-RNA for site-directed RNA mutagenesis utilizing intracellular A-to-I RNA editing. Sci. Rep.7(41478), 1–13. 10.1038/srep41478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider, M. F., Wettengel, J., Hoffmann, P. C. & Stafforst, T. Optimal guideRNAs for re-directing deaminase activity of hADAR1 and hADAR2 in trans. Nucl. Acids Res42(10), e87. 10.1093/nar/gku272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luyen, T. V. et al. Changing blue fluorescent protein to green fluorescent protein using chemical RNA editing as a novel strategy in genetic restoration. Chem. Biol. Drug Des.86, 1242–1252. 10.1111/cbdd.12592 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Luyen, T. V. & Toshifumi, T. C-to-U editing and site-directed RNA editing for the correction of genetic mutations. BioScience Trends Adv. Publ.11(3), 243–253. 10.5582/bst.2017.01049 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Keiji, N. et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Nat. Commun.353(6305), 8729. 10.1126/science.aaf8729 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Zuo, E. et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science364(6437), 289–292. 10.1126/science.aav9973 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin, A. S. et al. A panel of eGFP reporters for single base editing by APOBEC-Cas9 editosome complexes. Sci. Rep.9, 497. 10.1038/s41598-018-36739-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luyen, V. T. et al. Chemical RNA editing as a possibility novel therapy for genetic disorders. Int. J. Adv. Comput. Sci.2(6), 237–241 (2012). [Google Scholar]
- 46.Chen, G., Katrekar, D. & Mali, P. RNA guided Adenosine deaminases: Advances and challeneges for therapeutic RNA editing. Biochemistry58, 1947–1957. 10.1021/acs.biochem.9b00046 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Eggington, J. M., Greene, T. & Bass, B. L. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun.2(319), 1–9. 10.1038/ncomms1324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rinkevich, F. D., Schweitzer, P. A. & Scott, J. G. Antisense sequencing improves the accuracy and precision of A-to-I editing measurements using the peak height ratio method. BMC Res. Notes5(63), 1–6. 10.1186/1756-0500-5-63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kluesner, M. G. et al. EditR: a method to quantify base editing via Sanger sequencing. CRISPR J.1(3), 239–250. 10.1089/crispr.2018.0014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grunewald, J. et al. CRISPR adenine and cytosine base editors with reduced RNA off-target activities. Nat. Biotechnol.37, 1041–1048. 10.1038/s41587-019-0236-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.