

Abstract
Cancer-associated fibroblasts (CAFs) remain active even in the absence of cancer cells. However, the molecular mechanism underlying the sustained active status of CAFs is largely unrevealed. We found that in CAFs, DNMT3B was not only a target of miR-200b, miR-200c and miR-221, but was able to induce DNA methylation of miR-200s promoters. DNMT3B eventually reached a stably high level by the counteracting effect of decreasing miR-200b/c and increasing miR-221 in normal fibroblasts (NFs) with long-term exogenous TGF-β1 treatment, and DNMT3B further led to a low level of miR-200s which established CAF activation. Meanwhile, miR-200s/miR-221/DNMT3B signaling sustained autocrine TGF-β1 maintaining active CAF status. Destruction of the autocrine TGF-β1/miR-200s/miR-221/DNMT3B signaling led to demethylation of miR-200s promoters and further restored the NF phenotypes. Moreover, we confirmed that TCF12, the target of miR-141, stimulated c-Myc/Cyclin D1 axis in breast cancer cells to promote cancer growth by enhancing CXCL12 of CAFs. The current study reveals that the TGF-β1/miR-200s/miR-221/DNMT3B regulatory loop is responsible for the maintenance of CAFs status and is also necessary for CAF function in promoting malignance of breast cancer, which provides a potential target for CAF-driven therapeutic strategy.
Keywords: CAFs, Autocrine TGF-β1, DNMT3B, miR-200s, miR-221
1. Introduction
Cancer associated fibroblasts (CAFs) are the predominant stromal resident in tumor microenvironment. Previous studies highlighted that CAFs could promote cancer progression by encouraging tumor growth, angiogenesis, chemotherapy resistance and metastasis [1; 2]. Interestingly, other researches and our works found that primary CAFs isolated from human carcinomas can stay active even in vitro for a long term [3; 4], however, the nature of this phenomenon remains largely unknown. Due to lack of specific CAF markers or gene signatures and their multiple origins, investigators proposed that CAF is a “cell state” rather than a “cell type” [5], hypothesizing that CAFs can be maintained in a stable activation state to contribute to tumor malignance and CAFs can also be reversible dependent on the change of environmental factors. The CAF-driven therapeutic opportunities aimed at reversing the activated CAFs into an inactivated fibroblastic state are currently being tested [6]. Thus, understanding the molecular mechanisms underlying the maintenance of CAF “cell state” can optimize the strategy of finding out the CAF-driven cancer therapeutic opportunities.
Cancer researchers have a common consensus that CAFs undergo very rare somatic mutation, but more frequent occurrence of epigenetic events [7; 8; 9]. DNA methylation is one of the important routes of epigenetic changes. A series of evidence identified that paracrine cross-talk between normal fibroblasts (NFs) and cancer cells results in DNA methylation modification in NFs, which provides a central role in programming NFs into CAFs and functions of CAFs. For example, proinflammatory cytokine Leukemia Inhibitory Factor (LIF) reprograms and retains a pro-invasive phenotype of CAFs, which involved in DNA methyltransferase 3 beta (DNMT3B) mediated methylation in Src homology region 2 domain-containing phosphatase-1 (SHP-1) promoter and constitutive activation of Janus kinase/signal transducers and activators of transcription3 (JAK/STAT3) signaling [10]. Pancreatic ductal adenocarcinoma (PDAC) cells can induce DNA methylation of Suppressor Of Cytokine Signaling 1 (SOCS1) gene which further triggers insulin-like growth factor-1 expression in pancreatic CAFs to support PDAC growth [11]. Colon adenocarcinoma HT-29 cells instruct colon fibroblasts to acquire a CAF-like phenotype with global DNA methylation in a co-culture system [12]. These studies suggest that DNA methylation modification is a possible mechanism underpinning the emergence of the stable CAF phenotype.
Transforming growth factor β1 (TGF-β1) serves as a major cytokine essential for CAF activation. Our previous study have demonstrated that TGF-β1 derived from cancer cells is able to down-regulate microRNA-200s (miR-200s) expressions and lead to up-regulated expression of their targets, TCF12 and Fli-1, thus contributes to CAF activation [13]. DNA methylation associated inactivation of miR-200 members has been described in cancer. For example, CpG island hypermethylated or unmethylated status of miR-200s can be dynamically regulated corresponding to the epithelial to mesenchymal transition (EMT) and mesenchymal to epithelial transition (MET) phenotype [14; 15]. In this regard, the epigenetic silences of miR-200 families may be involved in TGF-β1-stimulated CAF stativity.
However, the involvement of TGF-β1 in epigenetic signature of CAF remains controversial. A few genes of TGF-β1-driven epigenetic changes have been described in CAFs, such as collagen type I alpha 1 chain (COL1A1), Cluster of Differentiation 90 (CD90) and Caveolin 1 (CAV1) which are identified to be highly important for CAF functions [16; 17; 18]. But one of studies, which demonstrated that TGF-β1 is necessary for reprogramming and maintenance of CAFs by interacting with C-X-C motif chemokine 12 (CXCL12) autocrine signaling network, appears to exclude the requirement of epigenetic mechanism in the maintenance of CAF state [19]. Further studies are needed for a better understanding of the molecular links between TGF-β1 signaling and durable CAF state.
Here, we showed that epigenetic silence of miR-200s coincides with elevated DNMT3B expression in CAFs. DNMT3B is not only a direct target of miR-221 which is up-regulated in CAFs, but also a target of miR-200b/c which are down-regulated in CAFs. The establishment and maintenance of autocrine TGF-β1/miR-200s/miR-221/DNMT3B positive feedback loop, mediated by exogenous TGF-β1 in a self-stimulating pattern, is essential for CAF state. We also revealed that epigenetic silencing of miR-141 directly activates TCF12 expression to facilitate breast cancer cell growth through mediating CXCL12 secretion in CAFs and further promoting c-Myc and Cyclin D1 expressions in breast cancer cells.
2. Materials and methods
2.1. Cell culture
Immortalized CAFs and NFs (immortalized using pBABE-hygro-hTERT), which have been described previously [3], and breast cancer MDA-MB-231, MCF-7 cells were cultured in Dulbecco’s Modified Eagle’s medium with 10% Fetal Bovine Serum (GIBCO-BRL) and 1% antibiotics.
For co-culture experiments, conditioned media from fibroblasts were collected every 48 h and cetrifuged at 500g for 5 min and was passed through a 0.22 um filter (Millipore). Cancer cells were cultured with the supernatant from fibroblasts. Exosomes were cleared by ultracentrifugation at 110,000g for 60 min at 4 °C.
For TGF-β1 treatment, recombinant human TGF-β1 (R&D systems) was used at 5 ng/ml. To block TGF-β1 signaling, the TGF-β R1 inhibitor SB431542 (Selleck) was used at a 10 μM final concentration. TGF-β neutralizing antibody (ab64715, Abcam) and control antibody (MAB002, R&D Systems) were used at 5 μg/mL. For epigenetic drug treatment, NFs were treated with 5-aza-2’-deoxycytidine (Sigma) at 3 μM or 5 μM for 3 or 5 days.
2.2. Plasmid construction, inhibitors and mimics
pLenti4.1-puro-pre-miR-200b/miR-200c, pLVX-shRNA-miR-200b and pLVX-shRNA-miR-200c were previously described [13]. Pre-miR-221 cDNA was amplified by PCR and cloned into the pLenti4.1-puro vector. pcDNA3/Myc-DNMT3B was purchased from Addgene (#35522) and reconstructed by sub-cloning into pLenti4.1-puro vector based on PCR approach. The synthetic shRNA oligonucleotides (Invitrogen) specifically targeting the miR-221 and DNMT3B gene were inserted into the pLVX-shRNA1 lentivector (Clontech, USA). To generate WT-DNMT3B 3′-UTR-Luc and Mut-DNMT3B 3′-UTR-Luc reporters, the synthetic oligonucleotides (Invitrogen) that correspond to the wild-type or the mutated binding sites of miR-200b/c in the 3′-UTR of DNMT3B were separately cloned into the pMIR-Reporter vector (Ambion, USA) at Spe1-HindIII sites. The binding sites were identified using the TargetScan database (MIT, www.targetscan.org). Mimics and inhibitors of miR-200s, miR-221 and the corresponding controls were purchased from GenePharma (Shanghai, China). All the shRNA and DNMT3B 3’-UTR-Luc sequences were listed in Table S1.
2.3. RNA preparation and qRT-PCR
Total RNA was isolated using Trizol (Invitrogen). PrimeScript RT Reagent Kit (RR047A, TaKaRa, Dalian, China) was used for reverse transcription of the purified RNA. The cDNA was subjected to qRT-PCR with SYBR Premix Ex Taq™ II (RR820A, TaKaRa, Dalian, China). The utilized primers to detect miR-200a, miR-200b, miR-200c and miR141 were previously shown [13]. The primers used for miR-221 and DNMT3B analysis by RT-PCR and qRT-PCR are listed in Table S2.
2.4. Western blot analysis
Western blot analysis was performed as described in an earlier study [3]. Briefly, 50 μg of total cell protein lysate were separated by 10% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. The following antibodies were used: DNMT3B (832121, mouse monoclonal antibody, 1:1000, NOVUS Biologicals), TCF12 (Sc-357, rabbit polyclonal antibody, 1:200, Santa Cruz), α-SMA (ab5694, rabbit polyclonal antibody, 1:1000, Abcam), c-Myc (D84C12, rabbit polyclonal antibody, 1:1000, #5605), Cyclin D1 (D86, rabbit polyclonal antibody, 1:500, BioWorld), p-ATM (2873, rabbit monoclonal antibody, 1:1000, Cell Signaling Technology Co), p-RB (BS6414, rabbit polyclonal antibody, 1:500, Bioworld), and α-actin (1:1000, Boster).
2.5. Luciferase reporter assay
1×105 CAFs were seeded in 24-well plates and co-transfected with total 800 ng of miR-200s expression plasmid and pMIR-DNMT3B 3′-UTR (wide type or mutant) as well as pRL-TK control plasmid (Promega, Madison, USA) using Lipofectamine 2000 reagent (Invitrogen). After culture for 48 hours, lysates were collected, and Renilla and firefly luciferase activities were measured with a Dual-Luciferase Reporter System (E1910, Promega).
2.6. Enzyme-linked immunosorbent assay (ELISA)
Conditioned media from 3×105 fibroblasts were harvested, and the concentration of TGF-β1 was measured by ELISA (ExCell Bio, Shanghai, China) according to the manufacturer’s instructions. The absorbance (450 nm) of each sample was analyzed using a standard ELISA plate reader.
2.7. Immunofluorescence (IF)
Cells were grown on prepared coverslips to 80% confluence and then fixed within 4% paraformaldehyde. After treatment with 0.1% Triton X-100, a 5% goat serum solution was used to block the non-specific binding. The sections were incubated with specific antibody against DNMT3B (832121, mouse monoclonal antibody, 1:500, NOVUS Biologicals), normal rabbit IgG was used as a negative control. The sections were then treated with a FITC-labeled goat anti-mouse secondary antibody (1:200, Sigma) and mounted in aqueous medium containing DAPI (Vector Labs). IF images were captured using a Nikon Eclipse 80i microscope (Eclipse 80i, Tokyo, Japan).
2.8. Flow cytometric analysis
Cells in the S-phase of the cell cycle were determined by flow cytometry as previously described [28]. Briefly, the co-cultured breast cancer cells were harvested after 24h of culture, and fixed with 70% ethanol at 4 °C for 1 h, resuspended in 1 ml of PBS containing propidium iodide (50 μg/ml) and RNase A (0.1 mg/ml) and incubated at 37 °C for 30 min. Cell cycle distribution was analyzed by flow cytometry (FACS Vantage SE, BD, Franklin Lakes, NJ, USA)
2.9. Bisulfite sequencing analysis
Genomic DNA was prepared using Genomic DNA mini kit (Qiagen) and subjected to bisulfite CT conversion reaction using EZ DNA methylation-Gold kit (ZYMO research). CpG islands of miR-200b-200a-429 and miR-200c-141 loci and primers were identified and designed using MethPrimer (http://www.urogene.org/methprimer). Primers are listed in Table S3. The amplified fragments were subcloned into pCR2.1-TOPO vector (Invitrogen), DNA methylation status was established by bisulfite genomic sequencing of multiple clones.
2.10. Statistical analysis
Statistical analysis was performed using SPSS standard version 17.0 software. The data are expressed as the mean ± standard deviations from at least three independent determinations. For single comparisons between two groups, a Student’s t test was used. Values of P<0.05 were considered significant.
3. Results
3.1. DNA methylation in miR-200s promoters is induced by DNMT3B
Epigenetic regulation of miR-200s has been known to mediate cell fate, like epithelial to mesenchymal transition (EMT) [14; 15]. However, whether methylation of miR-200s is related with cancer-associated fibroblasts (CAFs) activity is unknown. To this end, expressions of miR-200s in CAFs were examined upon 5-aza-2′-deoxycytidine (5′-AZA) treatment. Exposure of CAFs to 5′-AZA significantly enhanced miR-200a, miR-200b, miR-200c and miR-141 levels in CAFs (Fig 1A). MiR-200 family is composed of two clusters: miR-200b-200a-429 and miR-200c-141, CpG islands in the promoters of miR-200b-200a-429 and miR-200c-141 were mapped using the Methprimer platform [20] (Fig 1B).
Figure 1. DNA methylation in miR-200s promoters is induced by DNMT3B.
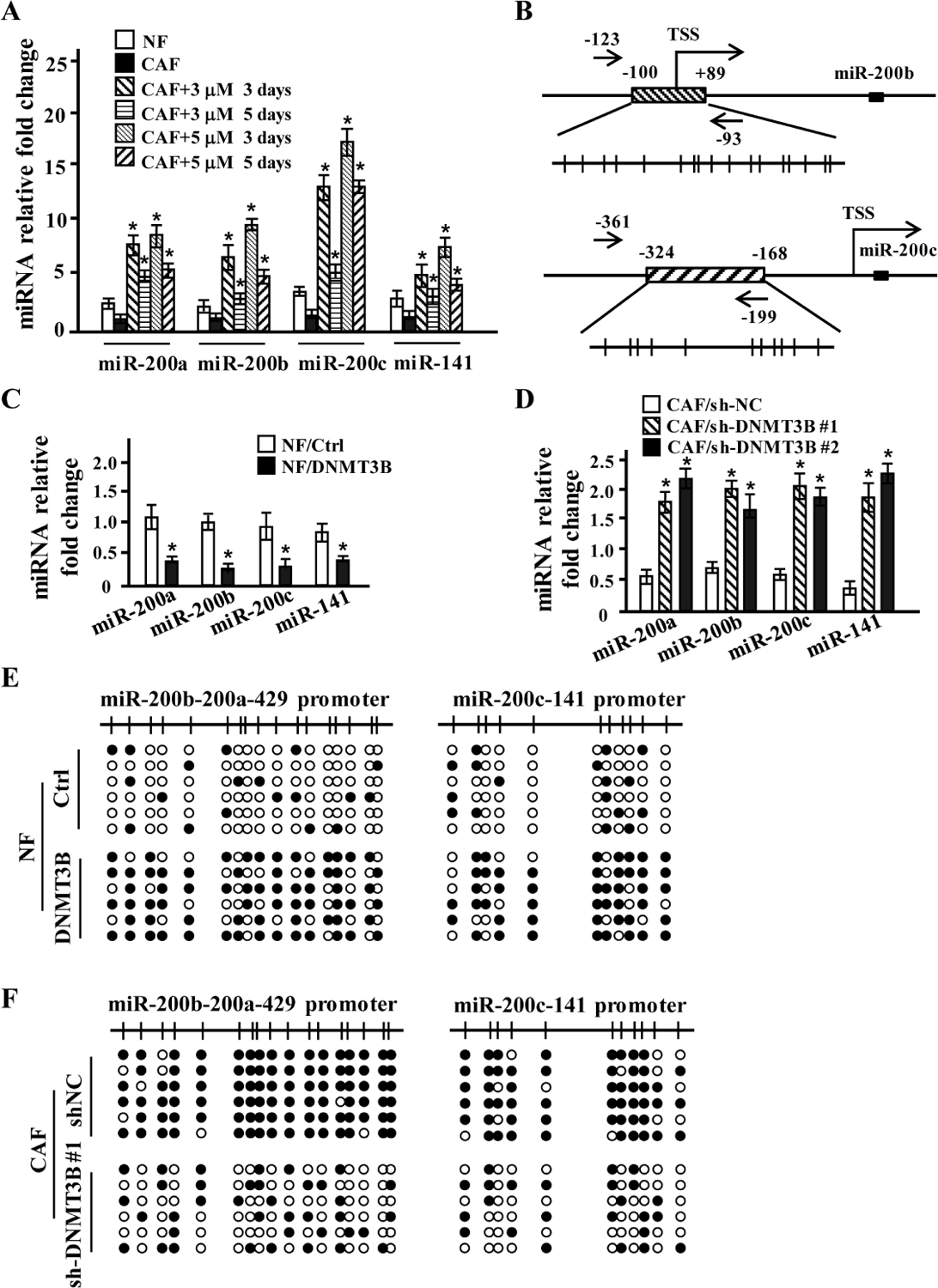
(A) The expression of miR-200s was confirmed using qRT-PCR in NFs treated by 3 or 5 μM of 5′-aza for 3 or 5 days. U6 was used as an internal control. Error bars, SD; t test, *p < 0.05. (n=3).
(B) Schematic illustrating DNA methylation in the promoters of miR-200s. Location of bisulfite genomic sequencing PCR primers (black arrows), CpG islands (boxes with diagonal), transcription start sites (TSS) and CpG dinucleotides (vertical lines) are shown.
(C) The expression of the miR-200s was examined by qRT-PCR in NFs overexpressing DNMT3B. U6 was used as an internal control. Error bars, SD; t test, *p < 0.05. (n=3).
(D) The expression of the miR-200s was evaluated by qRT-PCR in CAFs infected by two shRNAs against DNMT3B (CAF/shDMT3B #1 and CAF/shDNMT3B #2). U6 was used as an internal control. Error bars, SD; t test, *p < 0.05. (n=3).
(E-F) CpG island methylation in the promoters of miR-200b-a-429 and miR-200c-141 in NFs overexpressing DNMT3B (E) or in CAFs transfected with shRNA against DNMT3B was analyzed by bisulfite genomic sequencing (F).
To explore potential aberrant epigenetic modification in CAFs, we reviewed our previous mRNA and miRNA microarray data acquired from paired CAFs and NFs, and found that epigenetic modulators, those especially mediating DNA methylation, such as DNA methyltransferase (DNMT3A, DNMT3B and DNMT1), were dysregulated or potentially regulated by the changed miRNAs in CAFs (Fig S1A–S1B). The enhanced DNMT3B in CAFs was confirmed by qRT-PCR and western blotting analysis (Fig S1C–S1D), and DNMT3B was further revealed to be located in nucleus of CAFs (Fig S1E).
To examine the potential regulatory role of DNMT3B in miR-200 family expression, DNMT3B-overexpressed or depleted stable CAFs were established. We found that four of miR-200 members (miR-200b, miR-200a, miR-200c and miR-141) were decreased more than two folds in normal fibroblasts (NFs) transfected with a lentiviral vector encoding DNMT3B (NF/DNMT3B cells) (Fig 1C), whereas expressions of these miR-200 members in CAFs transfected with either lentiviral vector encoding shRNAs against DNMT3B (CAF/sh1-DNMT3B and CAF/sh2-DNMT3B) were more than 3 times in comparison with parental CAFs (Fig 1D). In agreement with the inhibitory effect of DNMT3B on miR-200s expressions in NFs, bisulfite sequencing showed increased CpG methylation in the promoters of miR-200b-200a-429 and miR-200c-141 in DNMT3B overexpressed NFs (Fig 1E). Conversely, efficient knockdown of DNMT3B in CAFs led to obvious demethylation of miR-200b-200a-429 and miR-200c-141 promoter (Fig 1F). These data indicate that DNMT3B promotes CpG methylation in miR-200s promoters, which results in silencing of miR-200s expressions in breast CAFs.
3.2. DNMT3B is a direct target of miR-200b/c and miR-221
As shown in Figure S1B, DNMT3B is predicted to be directly targeted by miR-200b/c or miR-221 as predicted. To confirm this prediction, qRT-PCR and western blotting were performed. The mRNA (Fig 2A) and protein (Fig 2B) levels of DNMT3B were attenuated in miR-200b-, miR-200c- and miR-200b/miR-200c-overexpressing CAFs, while were increased in miR-221-silenced CAFs. In contrast, stable knockdown of miR-200b, miR-200c or both of them in NFs using their specific shRNA led to increased DNMT3B in mRNA and protein levels, whereas overexpression of miR-221 resulted in decreased DNMT3B in both mRNA and protein levels (Fig 2C–2D). Based on the target sites of miR-200s and miR-221 in the 3′-UTR of DNMT3B (Fig 2E), luciferase assay was performed. The 3′-UTR of DNMT3B was suppressed by miR-200b/c and miR-221 in CAFs, and mutation of the binding sites in the 3′-UTRs canceled the response of DNMT3B to miR-200b/200c or miR-221 (Fig 2F). Taken together, these data show that DNMT3B is a direct target of miR-200b, miR-200c and miR-221.
Figure 2. DNMT3B is a direct target of miR-200b/c and miR-221.

(A and B) DNMT3B expression was examined by qRT-PCR (A) and Western blot (B) in CAFs expressing miR-200b, miR-200c and miR-200b/c or transfected with shRNA against miR-221. β-Actin was used as the loading control. Error bars, SD; t test, *p < 0.05. (n=3).
(C and D) DNMT3B expression was examined by qRT-PCR (C) and Western blot (D) in NFs expressing shRNA against miR-200b, miR-200c and miR-200b/c or overexpressing miR-221. β-Actin was used as an internal control or loading control. Error bars, SD; t test, *p < 0.05. (n=3).
(E) Schematic diagram of luciferase reporters with the wild-type DNMT3B 3′-UTR binding sites for miR-200b/c and miR-221.
(F) Luciferase activity of CAFs co-transfected with the indicated miRNAs and DNMT3B 3′-UTR reportor plasmids. miR-Ctrl, non-miRNA-expressing control; 3′-UTR wt, wild-type 3’-UTR of DNMT3B; 3’-UTR mut, mutant-binding sites for miR-200b/c or miR-221 in the target mRNA. All the luciferase reporter assay results were normalized to Renilla luciferase. Error bars, SD; t test, *p < 0.05. (n=3).
3.3. TGF-β1 activates miR-200b/c/miR-221/DNMT3B regulatory loop
TGF-β1 is a well-known inducer for CAF phenotype, and we prove that TGF-β1 derived from cancer cells can establish and sustain permanent activation of CAF through decreasing miR-200s expressions [13]. Investigation of the epigenetic-based mechanism controlling TGF-β1-induced miR-200s expressions revealed that higher levels of DNMT3B was acquired in NFs upon 20 days of TGF-β1 treatment. α-SMA, the marker of CAF activation, was also increased in those NFs, suggesting the role of DNMT3B in reprograming of NFs into CAFs (Fig S2A, left panel). Conversely, blockade of TGF-β1 signaling with the specific inhibitor, SB431542, blunted the ability of TGF-β1 to up-regulate α-SMA and DNMT3B expressions (Fig S2A, right panel). And SB431542 was also effective to counteract TGF-β1-reduced miR-200s expressions (Fig S2B). As expect, suppression of DNMT3B by siRNAs resulted in attenuating α-SMA induction in NFs incubated with TGF-β1 for 20 days in comparison with control NFs (Fig 3A). Similarly, the abandon of miR-200s expressions by TGF-β1 in activated CAFs were also efficiently rescued by siRNAs against DNMT3B (Fig 3B). Consistently, TGF-β1 could obviously trigger CpG methylation in promoter loci of miR-200b-a-429 and miR-200c-141 in NFs, however, CpG methylation in the promoter loci of these miR-200s was compromised by silencing DNMT3B (Fig 3C).
Figure 3. TGF-β1 activates miR-200b/c/miR-221/DNMT3B regulatory loop.

(A and B) Western blot analysis of α-SMA and DNMT3B expression (A) and qRT-PCR analysis of miR-200a, miR-200b, miR-200c and miR-141 (B) in TGF-β1-activated NFs which were transfected with siRNAs against DNMT3B. β-Actin and U6 were used as the loading control or an internal control. Error bars, SD; t test, *p < 0.05. (n=3).
(C) CpG island methylation of miR-200b-a-429 and miR-200c-141 was analyzed by bisulfite genomic sequencing in TGF-β1-activated NFs (TGF-β1-NFs) transfected with siRNAs against DNMT3B.
(D and E) qRT-PCR analysis of mRNA levels of miR-200s and miR-221 (D) and Western blot analysis of protein level of α-SMA and DNMT3B (E) in NFs treated with 5 ng/ml of TGF-β1 for 5, 8, or 11d, and cultured NFs for 5 days after TGF-β1 was removed at 11th day of treatment. U6 and β-Actin was used as an internal control or loading control. Error bars, SD; t test, *p < 0.05. (n=3).
(F) Western blot analysis of DNMT3B and α-SMA in TGF-β1-activated NFs transfected with miR-221 inhibitors. α-NC, inhibitors of control. α-miR-221, inhibitors of miR-221. β-Actin was used as the loading control.
(G and H) Western blot analysis of DNMT3B and α-SMA in miR-200b/c overexpressed-CAFs transfected with miR-221 inhibitors (G) and in miR-200b/c knocked down NFs transfected with miR-221 mimics (H). α-miR-221, inhibitors of miR-221. β-Actin is the loading control.
Given the reversed regulation role of miR-200s and miR-221 to DNMT3B expression in CAFs, we hypothesized that a balance of DNMT3B expression controlled by miR-200s and miR-221 may be essential to establish and maintain CAF status. To validate this hypothesis, we programmed NF into CAF with TGF-β1 and withdrew TGF-β1 after the treatment, then evaluated the expressions of miR-200s, miR-221, DNMT3B and CAF marker of α-SMA at different time points. As shown in Figure 3, after 5 days of TGF-β1 treatment, miR-200s levels began to decline, and miR-221 began to elevate (Fig 3D), but protein levels of DNMT3B and α-SMA were not affected at this stage (Fig 3E). With TGF-β1 treatment for 8 days, miR-200s were reduced about 50%, and levels of miR-221 and DNMT3B rather than α-SMA were increased compared with the control NFs (Fig 3D–3E). At 11th day of TGF-β1 treatment, miR-200s were obviously decreased (more than 70%) and miR-221 had a nearly two-fold up-regulation compared with control NFs (Fig 3D). Notably, expressions of miR-200s and miR-221 kept in stable at 5th day after TGF-β1 was withdrew at 11th day of treatment (Fig 3D). Coincidently with the change of miRNA, there was a significant increase of α-SMA (Fig 3E) which kept in stability for several months (data not shown). Interestingly, progressive reduction of miR-200b/c and increase of miR-221 were accompanied with progressively enhanced DNMT3B under TGF-β1 treatment from day 1 to day 8. The gradual up-expression of DNMT3B was halted since day 8, while expressions of miR-200b/c and miR-221 were kept decreasing and increasing respectively, suggesting a counteracting role between miR-200b/c and miR-221 might eventually reach a balance to sustain a relatively high level of DNMT3B in CAFs, ever after TGF-β1 was withdraw (Fig 3E). To further confirm the findings, we transiently inhibited miR-221 expression using siRNA in NFs treated with TGF-β1 for 11 days. DNMT3B expression was found to be up-regulated, and α-SMA was not changed (Fig 3F). MiR-221 inhibitors could restore the suppression of DNMT3B and α-SMA by miR-200b/c (Fig 3G). In contrast, ectopic miR-221 could inhibit DNMT3B and α-SMA expressions in miR-200b/c knocked down NFs (Fig 3H). Collectively, these data indicate that miR-200s and miR-221 can orchestrate to regulate DNMT3B expression in response to TGF-β1, and the dynamic stability of DNMT3B is crucial to maintain the activation of CAFs.
3.4. MiR-200b/c/miR-221/DNMT3B feedback loop influences TGF-β1 expression
TGF-β1 is a master cytokine secreted by both tumor cells and stromal CAFs [21; 22; 23]. To explore effects of miR-200b/c/miR-221/DNMT3B feedback loop on TGF-β1 expression and secretion, TGF-β1 protein level was measured following knockdown of DNMT3B in CAFs or ectopic expression in NFs. Lentivirus-mediated shRNA against DNMT3B caused decrease of TGF-β1 and p-Smad3 (a known downstream target of TGF-β1) (Fig 4A) and reduce of TGF-β1 secretion in CAFs (Fig 4B). In contrast, stable expression of DNMT3B in NFs increased TGF-β1 and p-Smad3 proteins (Fig 4C) as well as TGF-β1 secretion (Fig 4D). Next, we manipulated miR-200s and miR-221 expressions in stromal fibroblasts to evaluate TGF-β1 secretion. Knockdown of miR-200b or miR-200c, rather than miR-200a or miR-141, increased secreting TGF-β1 levels in NFs, however, miR-221 overexpression had no much effect on TGF-β1 production (Fig 4E, left panel). Otherwise, ectopic miR-200b or miR-200c decreased secreting TGF-β1 proteins in CAFs, while silence of miR-221 in CAFs resulted in enhanced secreting TGF-β1 levels (Fig 4E, right panel). These data indicate that miR-221 can antagonize miR-200b/c in regulating DNMT3B expression. And the antagonistic effect of miR-221 and miR-200b/c on DNMT3B thus maintains a relatively high level of autocrine TGF-β1 in CAFs.
Figure 4. MiR-200b/c/miR-221/DNMT3B feedback loop influences TGF-β1 expression.

(A) Western blot analysis of TGF-β1 and p-Smad3 expression in CAFs infected with two shRNAs against DNMT3B (CAF/sh-DMT3b #1 and CAF/sh-DNMT3B #2). β-Actin was used as the loading control.
(B) Enzyme-linked immunosorbent assay (ELISA) of TGF-β1 concentration in supernatant of CAF/sh-DMT3b #1 and CAF/sh-DNMT3B #2. Error bars, SD; t test, *p < 0.05, (n=3).
(C) Western blot analysis of TGF-β1 and p-Smad3 expression in NFs overexpressing DNMT3B. β-Actin is the loading control.
(D and E) ELISA of TGF-β1 concentration in supernatant of NFs overexpressing DNMT3B (D) and in supernatant of NFs respectively knocking down miR-200a, miR-200b, miR-200c and miR-141 or overespressing miR-221 (E, left panel) and CAFs respectively overexpressing miR-200a, miR-200b, miR-200c and miR-141 or knocking down miR-221 (E, right panel). Error bars, SD; t test, *p < 0.05 (n=3).
3.5. Autocrine TGF-β1 is necessary to maintain activity of miR-200s/miR-221/DNMT3B regulatory loop
DNMT3B is reported to be a de novo DNA methyltransferase and regulates gene expression in myogenic differentiation [24], indicating that DNMT3B dynamically regulates DNA methylation following cell differentiation. Thus, it raised a hypothesis that autocrine TGF-β1 might involve in sustaining the dynamic stability of miR-200s/miR-221/DNMT3B feedback loop to maintain CAF status. To confirm this hypothesis, a specific neutralized antibody against TGF-β1 or inhibitor SB431542 were employed. Blockage of TGF-β1 signaling using neutralized antibody or inhibitor SB431542 effectively reduced DNMT3B and α-SMA expressions (Fig 5A). Meanwhile, the increased miR-200s and decreased miR-221 were detected (Fig 5B). Notably, methylation of CpG islands in the promoter of miR-200s was impaired upon SB431542 treatment (Fig 5C). Our previous studies have identified that CAFs are characteristic of upregulated proliferation-associated gene [25], we further tested the effects of TGF-β1/miR-200s/miR-221/DNMT3B regulatory loop on their expressions. We found p-ATM, c-Myc and p-RB were increased in CAFs and TGF-β1-activated NFs, while SB431542 and 5’-AZA reduced their protein levels (Fig S3A). These data suggest that autocrine TGF-β1 is necessary to govern dynamic and reversible DNA methylation of miR-200 family loci and contributes to CAF activation.
Figure 5. Autocrine TGF-β1 is necessary to maintain activity of miR-200s/miR-221/DNMT3B regulatory loop.

(A and B) Western blot analysis of DNMT3B and α-SMA (A) and qRT-PCR analysis of miR-200s and miR-221 (B) in CAFs treated with TGF-β1 neutralized antibody (α-TGF-β1) or TGF-β1 signaling specific inhibitor, SB431542. Error bars, SD; t test, *p < 0.05 (n=3).
(C) CpG island methylation of miR-200b-a-429 and miR-200c-141 in CAF treated with SB431542 was analyzed using bisulfite genomic sequencing.
3.6. Autocrine TGF-β1/DNMT3B/miR-141 axis enhances TCF12 in CAFs to promote breast cancer cell proliferation
To understand the potential influences of autocrine TGF-β1/miR-200s/DNMT3B regulatory loop on progression of breast cancer cells, we analyzed tumor cell proliferation using co-culture system, in which cancer cells were co-cultured with supernatant from stromal fibroblasts. Indeed, the supernatant derived from NFs treated with TGF-β1 significantly increased the population of MCF-7 or MDA-MB-231 cells in S-phase, while SB431542, the TGF-β1 inhibitor, notably mitigated the promoting effect of TGF-β1-activated NF on cell proliferation of MCF-7 and MDA-MB-231 cells (Fig 6A–6B, column 1–3). In contrast, the supernatant from CAFs treated with 5′-AZA or CAFs with silenced DNMT3B dramatically decreased S-phase cell ratio of these cancer cells (Fig 6A–6B, column 5–6 and column 8–9), suggesting that interfering stability of CAF by manipulating autocrine TGF-β1, miR-200s or DNMT3B halts CAF effects on tumor cell proliferation under tumor microenvironment. Given the role of exosome and cytokines from CAF in cancer proliferation, we also tested cancer cell growth when exosomes were removed from supernatant of fibroblasts. The supernatant of NFs with or without exosomes had the same effect on proliferation of MCF-7 and MDA-MB-231 cells. Removel of exosomes from the supernatant of CAFs or TGF-β1-activated NFs reduced both MCF-7 and MDA-MB-231 cells in S-phase (Fig S4A). However, elimination of exosomes from the supernatant of TGF-β1-activated NFs with SB431542 or CAFs with 5’-AZA or DNMT3B knocked down CAFs didn’t endow difference in S-phase of MCF-7 and MDA-MB-231 cells compared to the supernatant of those fibroblasts with exosomes (Fig 6A–6B, colume 3–4, 6–7, 9–10). These results indicate cytokine may play a major role in TGF-β1/miR-200s/DNMT3B loop-mediated cancer cell growth.
Figure 6. Autocrine TGF-β1/DNMT3B/miR-141 axis enhances TCF12 in CAFs to promote breast cancer cell proliferation.

(A-B) The percentages of S-phase cells in cell cycle are shown by histogram in the MCF-7 (A) or MDA-MB-231 (B) cells co-cultured with supernatant from indicated fibroblasts. The labeled number represents the percent of cells in S-phase. Exo R, exosome were eliminated from the supernetant of fibroblasts by centrifugation. 5′-AZA, 5-aza-2′-deoxycytidine. Error bars, SD (n=3).
(C) Western blot analysis of TCF12 in TGF-β1 activated NFs and CAFs treated with TGF-β1 neutralized antibody (α-TGF-β1) or SB431542.
(D) Western blot analysis of DNMT3B and TCF12 in CAF/sh-DMT3b #1 and CAF/sh-DNMT3B #2, and NF/DNMT3B.
(E-F) The percentages of S-phase cells in cell cycle are shown by histogram in the indicated breast cancer cells co-cultured with supernatant from CAF/sh-TCF12 treated with miR-141 mimics (E) or co-cultured with supernatant from NF/TCF12 treated with miR-141 inhibitors (F). The labeled number represents the percent of cells in S-phase. Error bars, SD (n=3).
Our previous study confirmed that TCF12, the target of miR-141, can independently trigger the activation of breast CAF [13]. We wondered whether TCF12 contributes to TGF-β1/DNMT3B/miR-141 signaling-mediated cancer cell growth. Exposure of NFs to TGF-β1 for 9 days increased TCF12 expression, but blockage of autocrine TGF-β1 signaling using TGF-β1 neutralized antibody or SB431542 markedly reduced TCF12 levels in CAFs (Fig 6C). Similarly, knockdown of DNMT3B caused a marked drop in TCF12 levels in CAFs (Fig 6D, left panel), whereas ectopic DNMT3B induced higher level of TCF12 in NFs (Fig 6D, right panel). Furthermore, supernatant from NFs with ectopic TCF12 endowed cancer cells with a strong proliferation ability, which was compromised by miR-141 inhibitors (Fig 6E). Similarly, silencing TCF12 in CAFs decreased the population of MCF-7 and MDA-MB-231 cells in S phase, which was partially rescued by miR-141 mimics (Fig 6F). Collectively, these data support that autocrine TGF-β1/DNMT3B/miR-141/TCF12 axis in activated CAFs fuels tumor cell proliferation.
3.7. TCF12-driven CXCL12 secretion in CAFs stimulates c-Myc/Cyclin D1 signaling in proliferative breast cancer cells
CXCL12, CCL2 and VEGF are the known factors, secreted by CAFs, to induce progression of cancer cells [26; 27; 28], thus we tested whether these factors are responsible for TCF12-stimulated proliferation of breast cancer cells. We found CXCL12 was significantly increased in TCF12-overexpressing NFs and reduced in TCF12-knocked down CAFs (Fig 7A). Inhibition of TCF12 by miR-141 mimics blunted TGF-β1-stimulated CXCL12 expression in NFs (Fig 7B), suggesting TCF12 has an ability to promote CXCL12 expression. Next, we explored the contribution of TCF12-CXCL12 signaling to cancer cell proliferation, and found that neutralized antibody specifically against CXCL12 obviously attenuated S-phase cell ratio of cancer cells co-cultured with supernatant from TCF12-overexoressing NFs (Fig 7C). In contrast, supernatant from CAFs with TCF12 knocked-down mitigated cancer cell population in S-phase, and supplement of exogenous CXCL12 in these supernatant restored the ratio of S-phase cells (Fig 7D).
Figure 7. TCF12-driven CXCL12 secretion in CAFs stimulates c-Myc/Cyclin D1 signaling in proliferative breast cancer cells.

(A) Western blot analysis of CXCL12 in NFs overexpressing TCF12 and CAFs knocking down TCF12.
(B) Western blot analysis of CXCL12 in NF overexpressing miR-141 with or without TGF-β1 treatment.
(C-D) The percentages of S-phase cells in cell cycle are shown by histogram in the indicated breast cancer cells co-cultured with supernatant from NF/TCF12 treated with or without CXCL12 neutralized antibody (α-CXCL12) (C) and in the indicated breast cancer cells co-cultured with supernatant from CAF/sh-TCF12 treated with or without CXCL12 (D). The labeled number represents the percent of cells in S-phase. Error bars, SD; t test (n=3).
(E-F) Western blot analysis of c-Myc and Cyclin D1 in indicated breast cancer cells co-cultured with supernatant from NF/TCF12 treated with or without CXCL12 neutralized antibody (E) and in indicated breast cancer cells co-cultured with supernatant from CAF/sh-TCF12 treated with or without CXCL12 (F).
(G) Schematic depicting the role of autocrine TGF-β1/miR-200s/miR-221/DNMT3B regulatory loop in the maintainess of CAF active state and its promoting effect on proliferation of breast cancer cells through increasing TCF12 and CXCL12 expression in activated CAFs and stimulating c-Myc/Cyclin D1 axis in breast cancer cells.
C-Myc and Cyclin D1 are important in regulating proliferation in breast cancer cells [29; 30], thus assessment of c-Myc and Cyclin D1 expressions was carried. As expected, robust expressions of c-Myc and Cyclin D1 were observed in MCF-7 or MDA-MB-231 cells co-cultured with supernatant from TCF12-overexpressing NFs, and such increase of c-Myc and Cyclin D1 was severely blunted in tumor cells after removal of CXCL12 in the supernatant using CXCL12 neutralized antibody (Fig 7E). On the contrary, TCF12 expression was ablated by shRNA in CAFs notably repressed c-Myc and Cyclin D1 levels in MCF-7 or MDA-MB-231 cells, adding exogenous CXCL12 in the supernatant could restore c-Myc and Cyclin D1 expressions of tumor cells (Fig 7F). Taken together, TCF12-induced CXCL12 activates c-Myc and Cyclin D1 expression to facilitate tumor cell proliferation under cross-talk with CAFs.
4. Discussion
Tumor-promoting CAFs fuel cell malignancy [31] and tumor progression [26; 32], however, the biochemical alteration(s) underlying the active CAF state are poorly understood. In the present study, we reveal that autocrine TGF-β1 and miR-200s/miR-221/DNMT3B regulatory loop are interconnected in the induction and maintenance of CAF activity. TCF12, the target of miR-141 (a member of the miR-200s), enhances CXCL12 secretion, further promoting breast cancer growth through activation of c-Myc and Cyclin D1 (Fig 7G).
The major finding of this study is that DNMT3B-induced de novo DNA methylation of CpG islands in the miR-200s promoter depends on TGF-β1, which is reversible under blockage of TGF-β1 signaling. Changes in the degree of methylation of miR-200s are accompanied with miR-200s expression and reversible transformation of normal fibroblasts (NFs) into CAFs phenotype. Our finding is supported by a couple of studies showing DNA methylation-associated silencing of miRNAs plays a decisive role in determination of cell fate. For example, DNMT1 preserves stemness state of pancreatic ductal adenocarcinoma stem cells via epigenetic modulating miR-17–92 cluster [33]. CpG island hypermethylation of miR-9 fails to promote differentiation of neuronal progenitor and contributes to Medulloblastoma pathogenesis [34]. Hypermethylation of the miR-149 promoter and suppression of miR-149 promotes epithelial-to-mesenchymal transition (EMT) and stem-like traits in gastric cancer cells [35]. CAFs that undergo epigenetic silencing of miR-148 has a selective advantage to endow endometrial cancer cells with metastatic capacity [36]. Our study strengthens an insight on activity changes of CAFs, that is, the CAF state could be switched by altering these epigenetic factors.
Another important finding in this study is that DNMT3B is the target of both miR-200b/c and miR-221, fine-tuning the balance of DNMT3B by miR-200s and miR-221 controls CAF active state. This is consistent with the reported model where balance of key modulators determines a stable cell state. For example, the ratio of RANKL/OPG regulated by WNT signaling pathway determines the osteogenic differentiation of ligament stem cell [36]. The expression of core pluripotency genes regulated by mutual repression between Hoxa1 and Nanog determines the alternate state of pluripotency and differentiation [37].
The DNA methylation process is catalyzed by DNMTs (DNA methylation and DNA methyltransferases [38]. Therefore, high DNMTs expression is associated with DNA global hypermethylation [39]. Our finding indicates that bidirectional regulation of DNMT3B by miR-200s and miR-221 in CAFs seems to be a regulatory mechanism which may help to protect CAFs from DNA hypermethylation. As reported, global DNA hypomethylation in non-small cell lung cancer CAFs is only accompanied by some hypermethylated genes [40; 41].
We also show a reciprocal regulation between autocrine TGF-β1 and stability of miR-200s/miR-221/DNMT3B regulatory loop in CAFs. This is consistent with the notion that CAF active state is sustained by positive feedback signals. For example, YAP and matrix stiffening established a feed-forward self-reinforcing loop that helps to maintain the promoting effect of CAFs on tumor metastasis [42]. The IL-6/STAT3/NF-κB positive feedback loop is responsible for the sustained active status of cancer-associated fibroblasts [43]. Our results provide new evidence for TGF-β1 signaling in maintaining the durable CAF active state.
Accordantly, we provide evidence that either inhibition of autocrine TGF-β1 or suppression of DNA methylation by DNMT3B knockdown or 5-aza-2’-deoxycytidine (5′-AZA) is sufficient to attenuate breast cancer cell growth. Our findings are consistent with the recently published study, that is, demethylation agent 5′-AZA restored CAFs’ pro-tumoral capabilities [10]. Hence, epigenetic switch may provide a prospective therapeutic opportunity aimed to revert CAF state into a non-tumor-promoting fibroblastic state to inhibit cancer progression.
In the present study, we further uncover that TCF12 links the epigenetic mechanism which dictates CAF active status with their tumor-promoting behaviors. TCF12 expression is controlled by TGF-β1/miR-200s/DNMT3B regulatory loop, which plays a key role in promoting cancer cell growth through CXCL12-c-Myc axis. These data reinforce our previous finding that TCF12 is a direct target of miR-200s, and can drive CAF-induced tumor growth in null mice system [13].
Collectively, the present study significantly implicated the CAFs maintain their cancer-promoting state by establishing TGF-β1/miR-200s/miR-221/DNMT3B feedback loop.
Supplementary Material
Highlights.
In CAFs, DNMT3B is a direct target of miR-200b/c and miR-221, and DNMT3B has ability to induce DNA methylation of miR-200s promoters.
Increasing miR-221 counteracts decreasing miR-200b/c in regulating DNMT3B and autocrine TGF-β1 during the programming of NFs into CAFs by exogenous TGF-β1.
The autocrine TGF-β1/miR-200b/c/miR221/DNMT3B axis maintained in a self-stimulating pattern is necessary for persistently active CAFs.
Autocrine TGF-β1/DNMT3B/miR-141 axis enhances TCF12/CXCL12 signaling in active CAFs to promote breast cancer cell proliferation via c-Myc and Cyclin D1.
5. Acknowledgements
This work was supported by National Natural Science Foundation of China [grant numbers NSFC 81773078, NSFC 81402180 for Yixuan Hou; NSFC 81472476, NSFC 31671481 for Manran Liu; NSFC 81802653 for Xi Tang], and also partly supported the First Affiliated Hospital of Chongqing Medical University [PYJJ2018-04].
Abbreviations:
- CAFs
cancer-associated fibroblasts
- NFs
normal fibroblasts
- DNMT3B
DNA methyltransferase 3 beta
- TGF-β1
Transforming growth factor β1
- EMT
epithelial to mesenchymal transition
- MET
mesenchymal to epithelial transition
- 5′-AZA
5-aza-2′-deoxycytidine
Footnotes
Conflict of Interest
The authors declare no competing financial interests.
6. References
- [1].Kalluri R, The biology and function of fibroblasts in cancer. Nature reviews. Cancer 16 (2016) 582–598. [DOI] [PubMed] [Google Scholar]
- [2].La Porta CAM, Zapperi S, Explaining the dynamics of tumor aggressiveness: At the crossroads between biology, artificial intelligence and complex systems. Semin Cancer Biol (2018). [DOI] [PubMed] [Google Scholar]
- [3].Wang L, Hou Y, Sun Y, Zhao L, Tang X, Hu P, Yang J, Zeng Z, Yang G, Cui X, Liu M, c-Ski activates cancer-associated fibroblasts to regulate breast cancer cell invasion. Mol Oncol 7 (2013) 1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Karakasheva TA, Lin EW, Tang Q, Qiao E, Waldron TJ, Soni M, Klein-Szanto AJ, Sahu V, Basu D, Ohashi S, Baba K, Giaccone ZT, Walker SR, Frank DA, Wileyto EP, Long Q, Dunagin M, Raj A, Diehl JA, Wong KK, Bass AJ, Rustgi AK, IL-6 mediates cross-talk between activated fibroblasts and tumor cells in the tumor microenvironment. Cancer Res (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Madar S, Goldstein I, Rotter V, ‘Cancer associated fibroblasts’--more than meets the eye. Trends in molecular medicine 19 (2013) 447–453. [DOI] [PubMed] [Google Scholar]
- [6].Gascard P, Tlsty TD, Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev 30 (2016) 1002–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, Trivett M, Thompson E, Ramakrishna M, Gorringe K, Polyak K, Haviv I, Campbell I, No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat. Genet 40 (2008) 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jiang L, Gonda T, Gamble M, Salas M, Seshan V, Tu S, Twaddell W, Hegyi P, Lazar G, Steele I, Varro A, Wang T, Tycko B, Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 68 (2008) 9900–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hu M, Yao J, Cai L, Bachman K, van den Brûle F, Velculescu V, Polyak K, Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet 37 (2005) 899–905. [DOI] [PubMed] [Google Scholar]
- [10].Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, Philippe C, Herraiz Serrano C, Benamar S, Croce O, Sanz-Moreno V, Meneguzzi G, Feral CC, Cristofari G, Gaggioli C, Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun 6 (2015) 10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xiao Q, Zhou D, Rucki AA, Williams J, Zhou J, Mo G, Murphy A, Fujiwara K, Kleponis J, Salman B, Wolfgang CL, Anders RA, Zheng S, Jaffee EM, Zheng L, Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res 76 (2016) 5395–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ling E, Ringel A, Sigal-Batikoff I, Abu-Freha N, Vaknine H, Friah W, Reshef A, Pinsk I, Fich A, Lamprecht S, Human Colorectal Cancer Stage-dependent Global DNA Hypomethylation of Cancer-associated Fibroblasts. Anticancer Res 36 (2016) 4503–4507. [DOI] [PubMed] [Google Scholar]
- [13].Tang X, Hou Y, Yang G, Wang X, Tang S, Du YE, Yang L, Yu T, Zhang H, Zhou M, Wen S, Xu L, Liu M, Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ 23 (2016) 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cursons J, Pillman KA, Scheer KG, Gregory PA, Foroutan M, Hediyeh-Zadeh S, Toubia J, Crampin EJ, Goodall GJ, Bracken CP, Davis MJ, Combinatorial Targeting by MicroRNAs Co-ordinates Post-transcriptional Control of EMT. Cell Syst 7 (2018) 77–91 e77. [DOI] [PubMed] [Google Scholar]
- [15].Davalos V, Moutinho C, Villanueva A, Boque R, Silva P, Carneiro F, Esteller M, Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 31 (2012) 2062–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pan X, Chen Z, Huang R, Yao Y, Ma G, Transforming growth factor beta1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoS One 8 (2013) e60335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Neveu WA, Mills ST, Staitieh BS, Sueblinvong V, TGF-beta1 epigenetically modifies Thy-1 expression in primary lung fibroblasts. Am J Physiol Cell Physiol 309 (2015) C616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sanders YY, Liu H, Scruggs AM, Duncan SR, Huang SK, Thannickal VJ, Epigenetic Regulation of Caveolin-1 Gene Expression in Lung Fibroblasts. Am J Respir Cell Mol Biol 56 (2017) 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA, Orimo A, Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A 107 (2010) 20009–20014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li LC, Dahiya R, MethPrimer: designing primers for methylation PCRs. Bioinformatics 18 (2002) 1427–1431. [DOI] [PubMed] [Google Scholar]
- [21].Lamprecht S, Sigal-Batikoff I, Shany S, Abu-Freha N, Ling E, Delinasios GJ, Moyal-Atias K, Delinasios JG, Fich A, Teaming Up for Trouble: Cancer Cells, Transforming Growth Factor-beta1 Signaling and the Epigenetic Corruption of Stromal Naive Fibroblasts. Cancers (Basel) 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yu Y, Xiao C, Tan L, Wang Q, Li X, Feng Y, Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 110 (2014) 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tauriello D, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X, Byrom D, Matarin J, Calon A, Rivas E, Nebreda A, Riera A, Attolini C, Batlle E, TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554 (2018) 538–543. [DOI] [PubMed] [Google Scholar]
- [24].Megiorni F, Camero S, Ceccarelli S, McDowell HP, Mannarino O, Marampon F, Pizer B, Shukla R, Pizzuti A, Marchese C, Clerico A, Dominici C, DNMT3B in vitro knocking-down is able to reverse embryonal rhabdomyosarcoma cell phenotype through inhibition of proliferation and induction of myogenic differentiation. Oncotarget 7 (2016) 79342–79356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].S T, Y H, H Z, G T, L Y, Y S, L L, X T, YE D, M Z, T Y, L X, S W, C L, M L, Oxidized ATM promotes abnormal proliferation of breast CAFs through maintaining intracellular redox homeostasis and activating the PI3K-AKT, MEK-ERK, and Wnt-β-catenin signaling pathways. Cell cycle (Georgetown, Tex.) 14 (2015) 1908–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mezawa Y, Orimo A, The roles of tumor-and metastasis-promoting carcinoma-associated fibroblasts in human carcinomas. Cell Tissue Res 365 (2016) 675–689. [DOI] [PubMed] [Google Scholar]
- [27].De Palma M, Biziato D, Petrova TV, Microenvironmental regulation of tumour angiogenesis. Nature reviews. Cancer 17 (2017) 457–474. [DOI] [PubMed] [Google Scholar]
- [28].Shen H, Yu X, Yang F, Zhang Z, Shen J, Sun J, Choksi S, Jitkaew S, Shu Y, Reprogramming of Normal Fibroblasts into Cancer-Associated Fibroblasts by miRNAs-Mediated CCL2/VEGFA Signaling. PLoS Genet 12 (2016) e1006244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fallah Y, Brundage J, Allegakoen P, Shajahan-Haq AN, MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Musgrove E, Lee C, Buckley M, Sutherland R, Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc. Natl. Acad. Sci. U.S.A 91 (1994) 8022–8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sun Y, Yang D, Xi L, Chen Y, Fu L, Sun K, Yin J, Li X, Liu S, Qin Y, Liu M, Hou Y, Primed atypical ductal hyperplasia-associated fibroblasts promote cell growth and polarity changes of transformed epithelium-like breast cancer MCF-7 cells via miR-200b/c-IKKβ signaling. Cell Death Dis 9 (2018) 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Giordano C, Barone I, Vircillo V, Panza S, Malivindi R, Gelsomino L, Pellegrino M, Rago V, Mauro L, Lanzino M, Panno ML, Bonofiglio D, Catalano S, Ando S, Activated FXR Inhibits Leptin Signaling and Counteracts Tumor-promoting Activities of Cancer-Associated Fibroblasts in Breast Malignancy. Sci Rep 6 (2016) 21782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zagorac S, Alcala S, Fernandez Bayon G, Bou Kheir T, Schoenhals M, Gonzalez-Neira A, Fernandez Fraga M, Aicher A, Heeschen C, Sainz B Jr., DNMT1 Inhibition Reprograms Pancreatic Cancer Stem Cells via Upregulation of the miR-17–92 Cluster. Cancer Res 76 (2016) 4546–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fiaschetti G, Abela L, Nonoguchi N, Dubuc AM, Remke M, Boro A, Grunder E, Siler U, Ohgaki H, Taylor MD, Baumgartner M, Shalaby T, Grotzer MA, Epigenetic silencing of miRNA-9 is associated with HES1 oncogenic activity and poor prognosis of medulloblastoma. Br J Cancer 110 (2014) 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li P, Shan JX, Chen XH, Zhang D, Su LP, Huang XY, Yu BQ, Zhi QM, Li CL, Wang YQ, Tomei S, Cai Q, Ji J, Li JF, Chouchane L, Yu YY, Sun FZ, Xu ZH, Liu BY, Zhu ZG, Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res 25 (2015) 588–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Aprelikova O, Palla J, Hibler B, Yu X, Greer YE, Yi M, Stephens R, Maxwell GL, Jazaeri A, Risinger JI, Rubin JS, Niederhuber J, Silencing of miR-148a in cancer-associated fibroblasts results in WNT10B-mediated stimulation of tumor cell motility. Oncogene 32 (2013) 3246–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].De Kumar B, Parker HJ, Parrish ME, Lange JJ, Slaughter BD, Unruh JR, Paulson A, Krumlauf R, Dynamic regulation of Nanog and stem cell-signaling pathways by Hoxa1 during early neuro-ectodermal differentiation of ES cells. Proc Natl Acad Sci U S A 114 (2017) 5838–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang W, Xu J, DNA methyltransferases and their roles in tumorigenesis. Biomark Res 5 (2017) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sarabi M, Naghibalhossaini F, Association of DNA methyltransferases expression with global and gene-specific DNA methylation in colorectal cancer cells. Cell Biochem. Funct 33 (2015) 427–433. [DOI] [PubMed] [Google Scholar]
- [40].Zhang L, Liu W, Zhao J, Ma X, Shen L, Zhang Y, Jin F, Jin Y, Mechanical stress regulates osteogenic differentiation and RANKL/OPG ratio in periodontal ligament stem cells by the Wnt/beta-catenin pathway. Biochim Biophys Acta 1860 (2016) 2211–2219. [DOI] [PubMed] [Google Scholar]
- [41].Vizoso M, Puig M, Carmona FJ, Maqueda M, Velasquez A, Gomez A, Labernadie A, Lugo R, Gabasa M, Rigat-Brugarolas LG, Trepat X, Ramirez J, Moran S, Vidal E, Reguart N, Perera A, Esteller M, Alcaraz J, Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis 36 (2015) 1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E, Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 15 (2013) 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hendrayani SF, Al-Harbi B, Al-Ansari MM, Silva G, Aboussekhra A, The inflammatory/cancer-related IL-6/STAT3/NF-kappaB positive feedback loop includes AUF1 and maintains the active state of breast myofibroblasts. Oncotarget 7 (2016) 41974–41985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.