

Abstract
Background:
The use of next-generation sequencing for fusion identification is being increasingly applied and aids our understanding of tumor biology. Some fusions are responsive to approved targeted agents, while others have future potential for therapeutic targeting. Although some pediatric central nervous tumors may be cured with surgery alone, many require adjuvant therapy associated with acute and long-term toxicities. Identification of targetable fusions can shift the treatment paradigm toward earlier integration of molecularly targeted agents.
Methods:
Patients diagnosed with glial, glioneuronal and ependymal tumors between 2002 and 2019 were retrospectively reviewed for fusion testing. Testing was done primarily using the ArcherDx FusionPlex Solid Tumor panel that assesses fusions in 53 genes. In contrast to many previously published series chronicling fusions in pediatric patients, we compared histological features and tumor classification subtype with the specific fusion identified.
Results:
We report 24 cases of glial, glioneuronal, or ependymal tumors from pediatric patients with identified fusions. With the exception of BRAF:KIAA1549 and pilocytic/pilomyxoid astrocytoma morphology, and possibly QKI-MYB and angiocentric glioma, there was not a strong correlation between histological features/tumor subtype and the specific fusion. We report the unusual fusions of PPP1CB-ALK, CIC-LEUTX, FGFR2-KIAA159, and MN1-CXXC5 and detail their morphological features.
Conclusions:
Fusion testing proved to be informative in a high percentage of cases. A large majority of fusion events in pediatric glial, glioneuronal and ependymal tumors can be identified by relatively small gene panels.
Keywords: CNS tumors, pediatric, Glioma, Glioneuronal, Ependymoma, molecular fusion
Introduction
Central nervous system (CNS) tumors are the second most common malignancy and leading cause of cancer-related death in children.1 For patients who achieve long-term remission, sequelae from intensive treatment, which can include chemotherapy, radiation therapy (RT), and stem cell transplantation, can result in lifelong physical and intellectual impairments. A substantial proportion of pediatric CNS tumors also pose a diagnostic challenge owing to their considerable diversity and the fact that histologically similar neoplasms vary widely in their biological behavior and response to therapy. While a few molecular alterations have come to be accepted as definitional, such as BRAF-KIAA1549 for pilocytic astrocytoma (PA)2, other alterations span a variety of different histologies and molecular grades3,4, limiting the ability to a priori ascertain the presence of fusions based on histology alone. A broad fusion panel is thus required for the identification of fusions with potential diagnostic or therapeutic utility.
Wider availability of next-generation sequencing (NGS) methods and the simultaneous emergence of targeted therapies have resulted in a paradigm shift in the diagnosis and treatment of pediatric CNS tumors. Information gained from molecular sequencing can result in 1) confirmation of histologic diagnosis, 2) refinement of histologic diagnosis, 3) consideration of an alternative diagnosis, and most importantly, 4) identification of therapeutic targets. The latter has the potential to reduce morbidity from more traditional therapies.
Many tertiary care centers currently utilize a broad, panel-based approach for assessment of potentially actionable genetic alterations in CNS tumors. Recently, a handful of studies have demonstrated the feasibility and utility of incorporating molecular diagnosis into the care of children with CNS tumors.4–8 These reports detail use of large 300–500-gene panels focused on mutations common in human cancers but not specific to pediatric tumors. They are also costly and often have limited reimbursement from insurance companies. Here, we report our experience using the ArcherDx FusionPlex Solid Tumor panel that assesses fusions in 53 genes. We show the remarkable efficacy of this approach in accurately diagnosing challenging cases and identifying therapeutically targetable aberrations in many of them. We describe several rare and previously unreported fusions, including PPP1CB-ALK, MN1-CXXC5, and CIC-LEUTX.
Materials and Methods
Cases were identified via a retrospective database review of all pediatric patients with a glial, glioneuronal, or ependymal tumor diagnosed between 2002 and 2019 that underwent fusion testing. For most cases, the initial surgery and subsequent management was performed at our institution. A minority of cases were biopsied at outside institutions; however, all histopathology was reviewed at the University of Colorado Denver (UCD). Fusion testing became available at our institution in 2016, after which most pediatric CNS tumor cases underwent prospective evaluation. In some cases, fusion testing was performed retrospectively on archived tissue to identify any relevant therapeutic targets. For all cases, clinical data were collected including age, sex, clinical presentation, imaging studies, tumor location, surgical extent, histology, treatment, and survival. This study was approved by the UCD Institutional Review Board (COMIRB 05–0149).
For ependymal tumors, fusion testing was performed predominantly to assist with diagnosis of RELA fusion-positive ependymomas, as stipulated in World Health Organization (WHO) 2016 classification guidelines. Embryonal tumors, sarcomas, and tumors presenting in fetal and immediate neonatal period (congenital gliomas) were excluded. A few cases in which molecular findings were helpful in distinguishing glial tumors with primitive neural differentiation and embryonal tumors are also included. Cases with the BRAF-KIAA1549 fusion, which defines a subset of low-grade gliomas (LGGs) and is the most common fusion in pediatric CNS tumors8–10 were excluded to focus on less common fusions in this patient population. A single challenging case including the BRAF-KIAA1549 fusion was included in which the pathologic diagnosis was revised following the discovery of a BRAF fusion years after initial diagnosis.
Genetic analysis
For most cases, fusion testing was performed using a CLIA-certified, RNA-based NGS assay performed in the Colorado Molecular Correlates (CMOCO) Laboratory in the Department of Pathology at UCD, using a commercially available assay of 53 genes (supplementary figure 1) from ArcherDx (Boulder, CO). This assay uses a proprietary Anchored Multiplex PCR (AMP™)-based enrichment to detect all fusions associated with the genes in a single sequencing assay, without prior knowledge of fusion partners or breakpoints. Nucleic acids were extracted and processed from FFPE material as reported previously.11 RNA libraries were sequenced on an Illumina MiSeq instrument using v3 chemistry (Illumina, San Diego, California). Raw sequence data were analyzed using the Archer Analysis software package (version 4.1.1.7; ArcherDx). Bioinformatically identified fusions were verified to be in-frame by manual inspection of the breakpoint. Sample processing, total nucleic acid extraction, library preparation, sequencing, and raw data analysis typically took 6–7 working days and final reports were typically issued in 10–14 working days. This panel of 53 genes has been used since the inception of the panel in 2016 and has not been updated.
The majority of samples tested by CMOCO were confirmed by research based RNAseq (Illumina HiSeq). In three patients, research RNAseq was done retrospectively without prior testing by CMOCO (see Table 1). RNA was isolated (Qiagen AllPrep) from tumor tissue snap frozen at the time of surgery (COMIRB 95–500). RNA libraries were synthesized from PolyA-selected total RNA using the Nugen Universal Plus mRNA-Seq kit and sequenced using either HiSeq4000 or NovaSeq6000 (Illumina). Reads were aligned to the human genome assembly GRCh38 using GSNAP (Genomic Short-read Nucleotide Alignment Program, version 2014–12-17).12 Fusion events were identified by two methods: 1) screening for candidate fusions (e.g. MN1-BEND2 in astroblastoma, case 18), by examining soft-clipped reads using the Integrative Genome Viewer (IGV)13, or 2) identification of other non-candidate fusions (e.g. CIC-LEUTX in anaplastic ganglioglioma, Case 16), using EricScript fusion finding software.14
TABLE 1.
Clinical, histological, and molecular characteristics of patients with CNS tumors with genomic rearrangements or fusions.
| ID | Age at Diagnosis (years) | Sex | Tumor Location | Histologic Diagnosis | WHO Grade | Genetic Alteration | Method of Testing | Therapy | Follow-up (months) | Follow-up Status |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.4 | F | Brainstem, thalamus, chiasm | Diffuse astrocytoma | II | FGFR1 TKD duplication | Archer | Biopsy → Carboplatin/ Etoposide | 10.5 | Progression |
| Vinblastine/ Avastin | 4.5 | Death | ||||||||
| 2 | 3 | M | Bilateral thalamus | Pediatric type oligodendroglioma | II | FGFR1 TKD duplication | Archer | Biopsy→ Obs | 1.5 | Progression |
| Frontal horn | PA | I | Carboplatin/ Vinblastine + RT | 25 | Death | |||||
| 3 | 2 | F | Hypo-thalamus | PMA | I | FGFR1 TKD duplication | Archer | Biopsy→ Carboplatin/ VCR /Gleevec/TMZ | 4 | Progression |
| Re-resection | 4 | Death | ||||||||
| 4 | 9 | M | Temporo-parietal | Extraventricular neurocytoma | II | FGFR1-TACC1 | Archer | STR → GTR + RT | 8 | Alive |
| 5 | 1.8 | M | Medullary/ Cervical spine | PA/PMA | FGFR1-TACC1 | Archer | STR→ Obs | 2 | Progression | |
| RT + Trametinib | 19 | Alive, on therapy | ||||||||
| 6 | 16 | F | Tectal plate | PA | I | FGFR-TACC1 | Archer | Biopsy→Obs | 15 | Alive |
| 7* | 6 | F | Peri-insular | Ganglioglioma | I | FGFR2-KIAA1598 fusion | Archer | New patient | N/A | N/A |
| 8^ | 5 | M | Thalamus | PA | II | MYB-QKI | Archer | Biopsy→ Carboplatin/ VCR | 3 | Progression |
| STR, RT→ MEK inhibitor → multi-kinase inhibitor + everolimus | 102 | Alive, progressive disease | ||||||||
| 9^ | 2 | M | Frontal | PMA (NF1-associated) | II | MYB-QKI | Archer | GTR | 15 | Progression |
| Frontal, Cervical spine | GTR→ Carboplatin → Trametinib | 42 | Alive, progressive disease | |||||||
| 10* | 8 | F | Cauda equina | EPN w/ myxo-papillary features | II | PPP1CB-ALK | Archer | GTR | 99 | Alive |
| 11 | 3 | F | Frontal | Epithelioid GBM | IV | ETV6-NTRK3 | Archer | STR + Carboplatin/ Etoposide | 2 | Progression |
| RT + NTRK inhibitor | 11 | Alive, on therapy | ||||||||
| 12 | 10 | F | Pons | DIPG | IV | ZKSCAN1-NTRK3 | Archer | Biopsy → RT + Avastin | 3 | Death |
| 13 | 6 | M | Thalamus | PA | I | GOPC-ROS1 | Archer | GTR | 13 | Alive |
| 14 | 7 | M | Cerebellum | PA | I | SRGAP3-RAF1 | Archer | GTR | 26 | Alive |
| 15 | 18 | F | Brainstem | Infiltrating glioma with equivocal ganglioglioma component | I-II | N/A | Archer | Biopsy→ RT + Avastin | 59 | Progression |
| Cerebellum | Anaplastic infiltrating glioma | PTPRZ1-MET | Re-Biopsy→ TMZ + RT | 3 | Alive, on therapy | |||||
| 16* | 19 | F | Fronto-parietal | Anaplastic ganglioglioma | III | CIC-LEUTX | Research RNA-Seq | GTR | 10 | Progression |
| RT + TMZ → STR + RT (CSI) + Carboplatin → Cytoxan/Doxo/Ifos/Etoposide | 56 | Alive, progressive disease | ||||||||
| 17* | 12 | F | Intra-ventricular | Metastatic anaplastic astrocytoma with epithelioid GBM features | III-IV | CIC-LEUTX | Foundation One | GTR + RT/TMZ | 3 | Alive, on therapy |
| 18* | 5 | F | Frontal | Astroblastoma | MN1-BEND2 | Research RNA-Seq | GTR | 85 | Alive | |
| 19# | 3 | M | Temporal | EPN | II | MN1-CXXC5 | Research RNA-Seq | GTR | 8 | Progression |
| 20 | 5 | M | Parietal | Anaplastic EPN | III | C11orf95-RELA | Archer | GTR + proton RT (elsewhere) | 13 | Progression |
| Metastatic anaplastic EPN | PD1 inhibitor + debulking of metastases x 2 | 3 | Alive, on therapy | |||||||
| 21 | 5 | F | Temporal | Metastatic anaplastic EPN | III | C11orf95-RELA | Archer | GTR + RT (elsewhere) | 10 | Progression |
| PD1 inhibitor | 5 | Death | ||||||||
| 22 | 11 | M | Parietal | Anaplastic EPN | III | C11orf95-RELA | St. Jude | NTR (elsewhere), re-resection +focal RT | 2 | Progression |
| Re-resection | 1 | Alive, on therapy | ||||||||
| 23 | 3 | F | Thalamus | PA | I-II | ETV-ITPR2 | Archer | STR + Carboplatin/ Vinblastine | 16 | Alive |
| 24# | 0.5 | M | 4th ventricle | Anaplastic EPN | III | BRAF-KIAA1549 | Archer | STR + Carboplatin/ Etoposide → GTR + RT | 77 | Alive |
indicates patients with a previously unreported or extremely rare fusion for a diagnosis.
indicates patients for whom histological and molecular findings were discordant and molecular findings led to a change in diagnosis.
indicates patients for whom a fusion result prompted diagnosis refinement.
TKD=tyrosine kinase domain, PA=pilocytic astrocytoma, STR=subtotal resection, GTR=gross total resection, PMA=pilomyxoid astrocytoma, VCR=vincristine, TMZ=temozolomide, Obs=observation, RT=radiation therapy, NF1=neurofibromatosis type 1, EPN=ependymoma, GBM=glioblastoma, DIPG=diffuse intrinsic pontine glioma, NTR=near-total resection, NTRK=neurotrophic receptor tyrosine kinase, Doxo=doxorubicin, Ifos=ifosfamide, PNET=primitive neuroectodermal tumor, autoSCT=autologous stem cell transplant, PD1=programmed cell death protein 1, Archer= Archer fusion panel conducted at CLIA-certified Colorado Molecular Correlates Laboratory (CMOCO)
An additional case, not tested by Archer, was evaluated using CLIA-certified Foundation One CDx Panel performed by Foundation Medicine, Inc (Cambridge, MA). A list of genes covered by this panel is available online at https://assets.ctfassets.net/vhribv12lmne/4ZHUEfEiI8iOCk2Q6saGcU/11dd3b532e30c34f56cb8e9b4a896783/F1CDx_TechSpecs_10-06_digital.pdf.
Results
Of all CNS tumors diagnosed between 2002 and 2019, 804 were classified as glioma, glioneuronal or ependymoma of which 86 underwent fusion analysis on any platform. Of 61 cases that underwent fusion analysis by CMOCO (Archer), 39 (64%) revealed a targetable genetic alteration, 50% of which involved a fusion or mutation in BRAF. Twenty-four illustrative fusion cases were included and are detailed in Table 1.
MYB-QKI fusion
Two cases of MYB-QKI fusion-positive CNS tumors were encountered. Patient 8 is a male, diagnosed at age 5 with a thalamic lesion. The initial pathologic diagnosis was LGG most consistent with PA (Fig. 1A–C). He was initially treated with chemotherapy but progressed quickly and subsequently underwent subtotal resection (STR) and RT. After stable disease for more than 6 years, his tumor again progressed, and he was treated with experimental targeted therapies including a RAS/RAF/MEK pathway inhibitor followed by a multi-kinase inhibitor. Unfortunately, he continues to have progressive disease more than 8 years from his original diagnosis. Patient 9 is a male with Neurofibromatosis Type 1 (NF1) who was diagnosed with a left frontal pilomyxoid astrocytoma (PMA) (Fig. 1D–F) at age 2 for which he underwent gross total resection (GTR) and was observed for 15 months before he progressed. He received chemotherapy but progressed within a year, including development of a new cervical spine lesion. He has been on trametinib for 2 years, but like patient 8, continues to have progressive disease.
Figure 1.
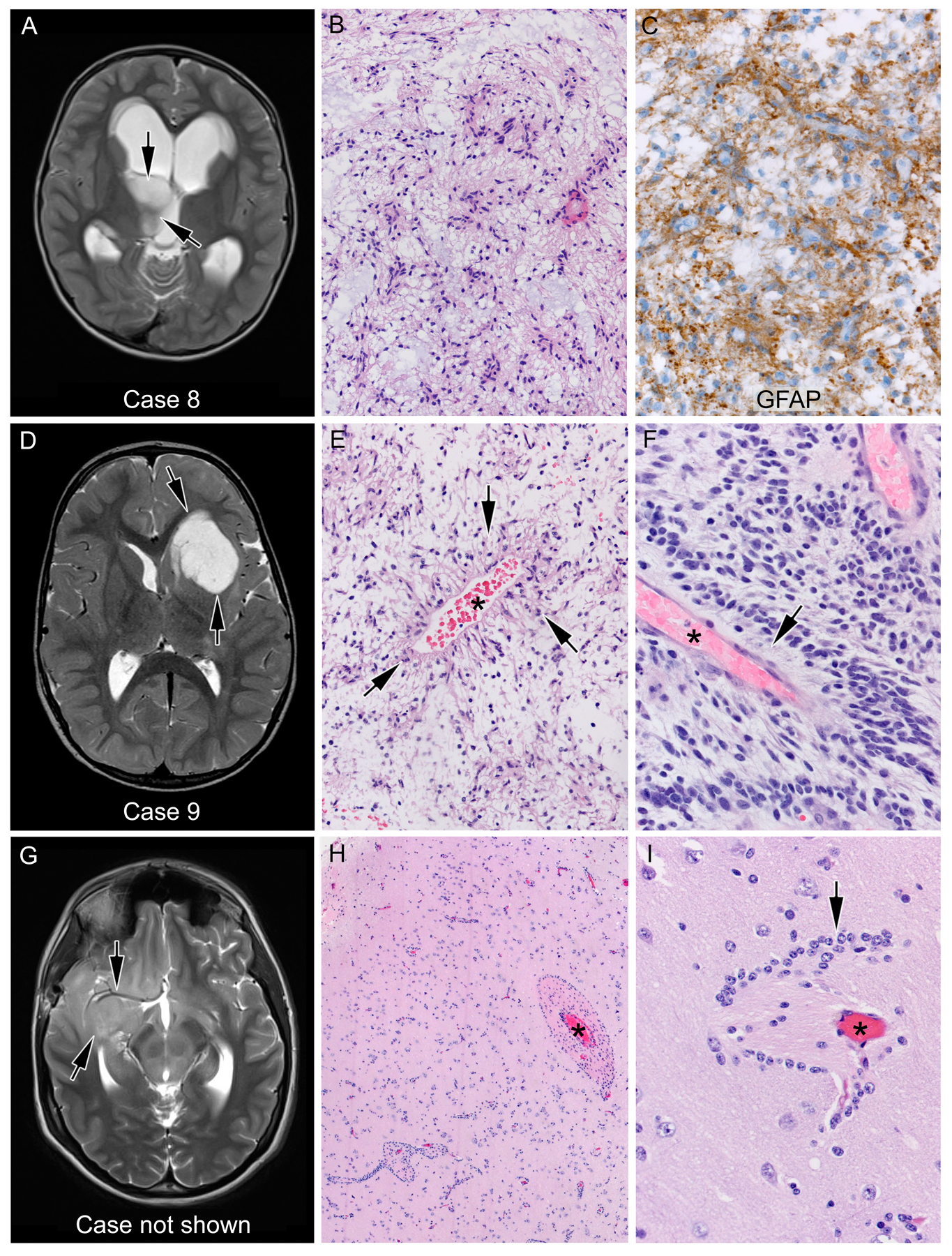
Angiocentric glioma; the presence of MYB-QKI alteration led to the refinement of histologic diagnosis. Two cases positive for MYB-QKI fusion were histologically more consistent with pilocytic astrocytoma (case 8, A-C), with a markedly piloid histology, or pilomyxoid astrocytoma (case 9, D-F), with distinct fibrillary processes (arrows) emanating from a blood vessel (asterisk). Conversely, a third case (G-I) showing low-grade glioma histology with prominent clustering of tumor cells (arrows) around blood vessels (asterisk), highly suggestive of angiocentric gliomas, failed to show MYB-QKI alteration, hence not meeting the current criteria for diagnosis of this entity. A, D, and G represent T2-weighted magnetic resonance images of the corresponding patients’ brains. Arrows indicate the location of the tumor.
In both cases, progression of disease and the availability of fusion testing led us to conduct fusion analysis, which revealed a MYB-QKI fusion. Although the initial diagnoses were defined as PA and PMA, retrospective review revealed rare regions of angiocentric histology in both. Of note, patient 9 is the first reported case of a MYB-QKI fusion in a patient with NF1. Conversely, MYB-QKI fusion was not always found in tumors with angiocentric histology: a 14-yr-old male diagnosed with high grade glioma involving the right frontotemporal cortex (Figure 1G) showed striking angiocentricity (Figure 1 H,I) mimicking angiocentric glioma pattern. This case was however negative for MYB-QK1 fusion.
PPP1CB-ALK fusion
Patient 10 was diagnosed with a PPP1CB-ALK fusion-positive ependymoma involving the cauda equina at age 8 years. She underwent GTR alone and has been disease-free for over 8 years. Anaplastic lymphoma kinase (ALK) gene fusions have been observed in numerous pediatric cancers including CNS tumors.15–17 The PPP1CB-ALK fusion, has only been reported in one case of congenital high-grade glioma (HGG); the patient did well following resection alone.18 Similarly, our patient required only resection for a sustainable remission.
CIC-LEUTX fusion
Patient 16 presented to an outside facility at age 19 with a mass in the corpus callosum (Fig. 2A). The tumor was resected, and the histologic diagnosis was anaplastic ganglioglioma (Fig. 2B–F). She required two additional surgeries in the first three years due to multiple local recurrences despite focal RT and temozolomide. Molecular analysis was performed on her third resection specimen, which demonstrated a CIC-LEUTX fusion by Archer. Following her third recurrence, she received craniospinal irradiation and multiple different intensive chemotherapy regimens, which were poorly tolerated. She ultimately stopped therapy 6 years after her initial diagnosis and is currently alive with disease.
Figure 2.
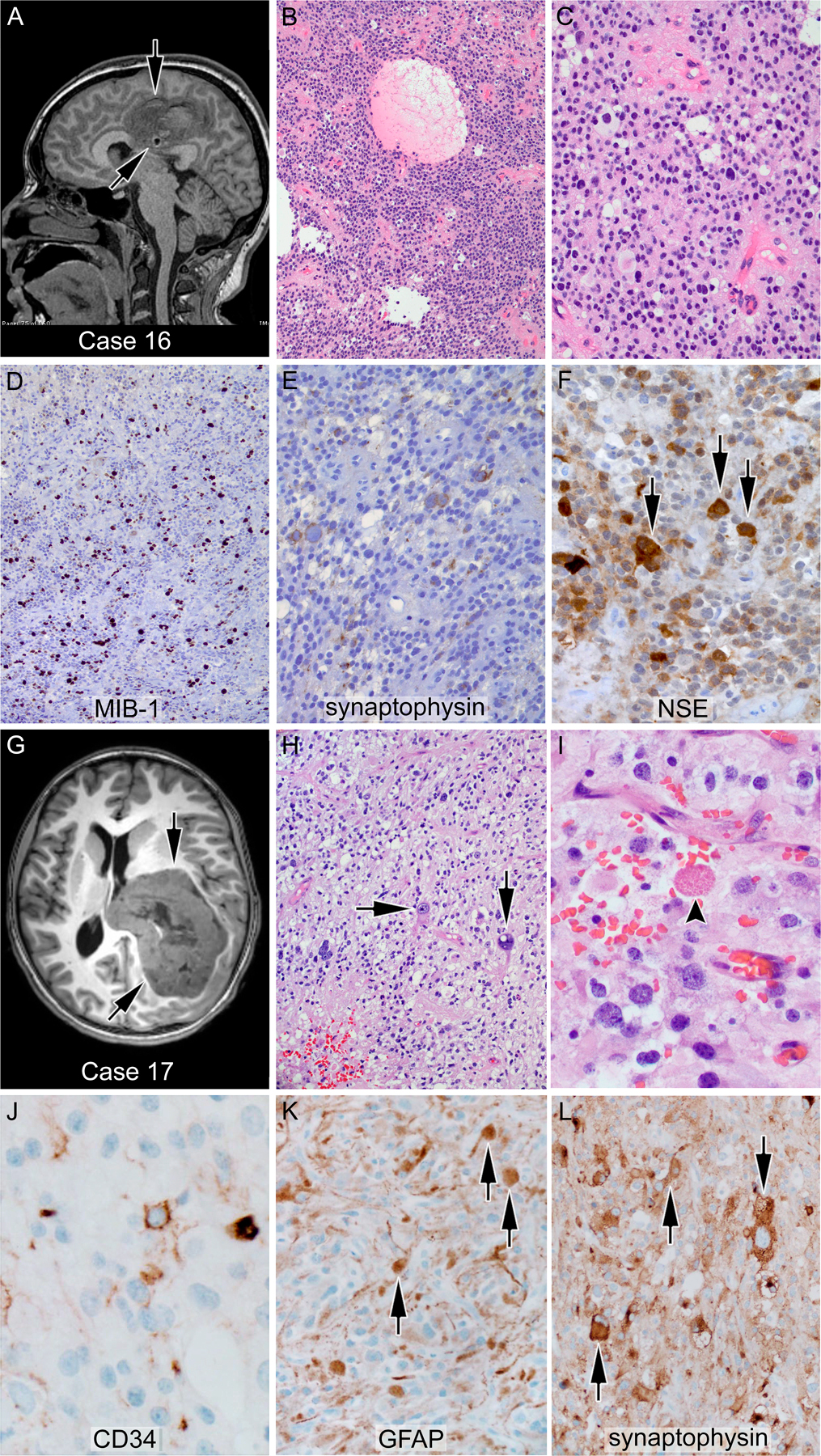
Glioneuronal tumors with CIC-LEUTX alteration; (A-F) case 16—anaplastic ganglioglioma: the tumor showed a population of monotonous cells with round nuclei and a second population of larger ganglionic cells (F, arrows) with positivity for neuronal markers, including synaptophysin and neuron-specific enolase. (G-L) Case 17—anaplastic pleomorphic xanthoastrocytoma: a markedly pleomorphic tumor with large bizarre cells (arrows), occasional xanthomatous cells, and rare eosinophilic granular bodies (I, arrowhead); an arborizing pattern of CD34 positivity was seen (J). The large cells (K, L arrows) are positive for both glial and neuronal markers such as GFAP and synaptophysin, respectively. More than 5/10 high power fields mitoses were found, sufficient for an anaplastic (grade III) diagnosis. A and G represent T2- and T1-weighted magnetic resonance images, respectively, of the corresponding patients’ brains. Arrows indicate the location of the tumor.
Patient 17 is a 12-year-old female who presented with a large intraventricular tumor (Fig. 2G). Pathology showed markedly pleomorphic tumor cells with large bizarre cells, occasional xanthomatous cells, and rare eosinophilic granular bodies (Fig. 2H and 2I). The large cells were simultaneously positive for glial markers including GFAP, and the neuronal marker synaptophysin (Fig. 2K and 2L). Additionally, an arborizing pattern of CD34 positivity was seen (Fig. 2J), as were multiple mitoses (>5/10 HPF). Taken together, these features were diagnostic for anaplastic pleomorphic astrocytoma, WHO grade III, with some features of epithelioid glioblastoma multiforme (GBM). Fusion testing revealed a CIC-LEUTX fusion. She is currently alive on therapy.
MN1-CXXC5 fusion
Patient 19 presented at age 3 with a left temporal mass. Histopathology was consistent with a supratentorial ependymoma for which he underwent GTR followed by observation. His tumor recurred 8 months later, at which time he underwent repeat surgery and focal RT. No high-grade markers were identified on repeat resection. Shortly after RT, a small nodule appeared in the prior radiation field. He underwent a third resection followed by radiosurgery and chemotherapy, and he remained disease-free for 5 years before another local recurrence. On re-resection, his tumor was consistent with anaplastic ependymoma histologically and tested positive for an MN1-CXXC5 fusion. Given the presence of this alteration, the diagnosis was amended to astroblastoma, which are rare glial tumors histologically defined by astrocytic cells with broad thick processes arranged around blood vessels.19 Astroblastoma can be difficult to differentiate from ependymoma and other tumors with perivascular pseudorosettes.
KIAA1549-BRAF fusion
Patient 24 presented at age 6 months with a large posterior fossa mass and marked hydrocephalus (Fig. 3D). He underwent STR, and pathology revealed a moderately cellular tumor with distinct perivascular arrangement of small monomorphic tumor cells with small nucleoli (Fig. 3E and 3F). The tumor was moderately positive for GFAP. EMA was negative. Several mitotic figures were seen, but there were no eosinophilic granular bodies or Rosenthal fibers. Based on these features, a diagnosis of anaplastic ependymoma was made. He received two cycles of chemotherapy followed by second look surgery, as complete resection is prognostically favorable for ependymoma. He continued chemotherapy until his first birthday, after which he received focal RT. At six years of age, this patient is disease-free but has significant physical and intellectual sequelae from his tumor and therapies. His tumor was retrospectively sent for molecular testing and was found to harbor the BRAF-KIAA1549 fusion. The diagnosis was amended to PA/PMA.
Figure 3.
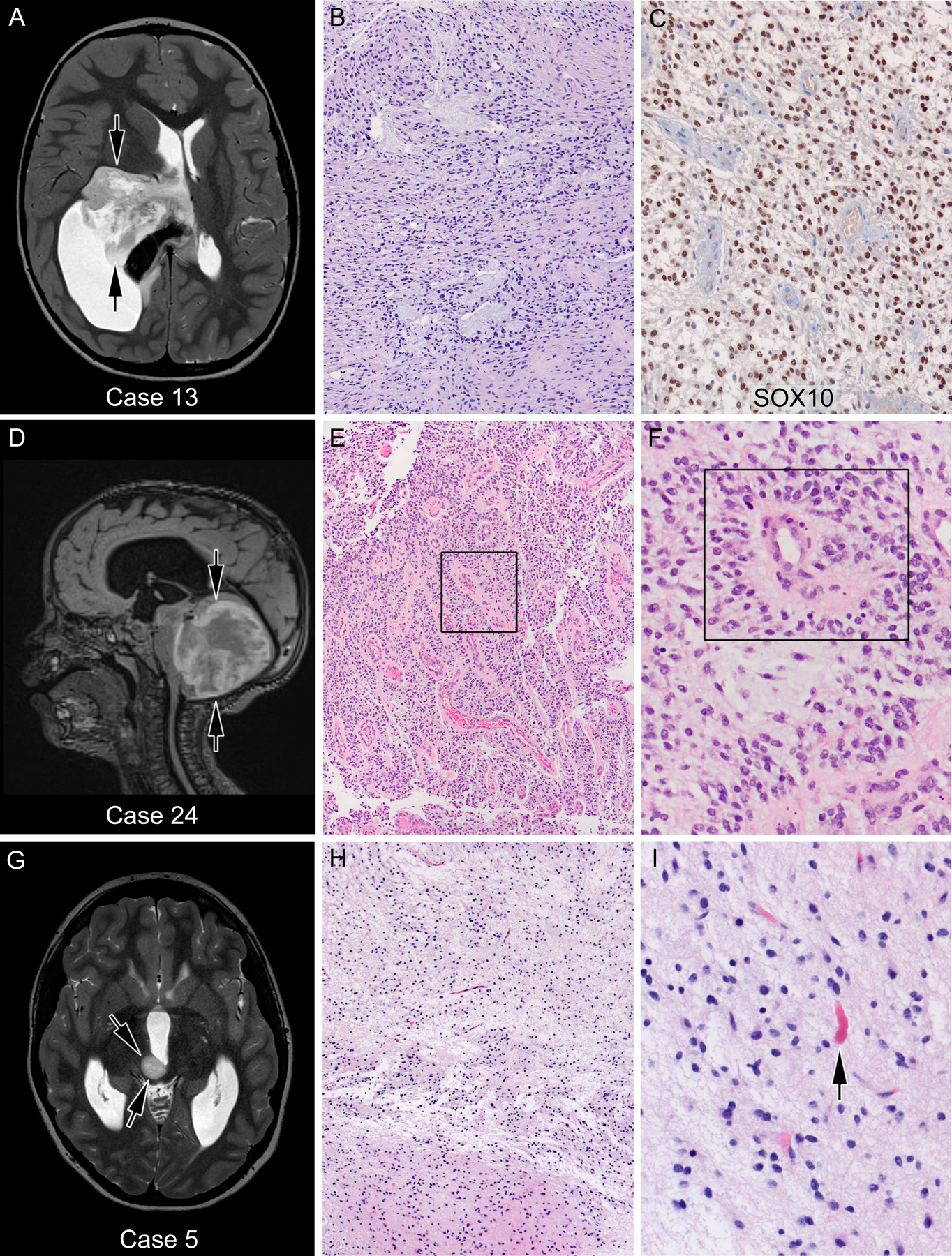
Gliomas with pilocytic astrocytoma histology showed various different fusions; (A-C) case 13—low-grade glioma with PA histology but without Rosenthal fibers or eosinophilic granular bodies, showed GOPC-ROS1 alteration. Conversely, a posterior fossa/fourth ventricular tumor (case 24, D-F) histologically diagnosed as anaplastic ependymoma because of prominent perivascular psuedorosettes (boxed) and treated as such showed BRAFKIAA fusion, proving to be pilocytic astrocytoma. In retrospect, this case showed strong and diffuse Sox10 positivity (not shown), also suggestive of PA as opposed to ependymoma. (G-I) Case 5—FGFR-TACC1 fusion was found in this tumor with classic PA histology with loosely arranged piloid astrocytic cells and scattered Rosenthal fibers (arrow). A and G represent T1-weighted magnetic resonance (MRI) images, while D represents a T2-weighted MRI image of the corresponding patients’ brains. Arrows indicate the location of the tumor
Discussion
Several recent publications have defined the utility of broad genomic screening in pediatric CNS tumors, including 1) University of California San Francisco using targeted sequencing of 510 genes5; 2) Dana-Farber Cancer Institute using the OncoPanel of 309 genes, OncoCopy, and array comparative genomic hybridization6; 3) University of Washington utilizing OncoPlex platform evaluating 262 cancer-related genes3; 4) University of Michigan utilizing transcriptome sequencing augmented by either whole exome sequencing or targeted sequencing of 1706 genes7 and 5) a study conducted at Foundation Medicine, Inc. reporting analysis of 315 cancer-related genes in 282 pediatric low- and high-grade gliomas.8 We report the utility of fusion analysis of pediatric CNS tumors using a smaller RNA-based panel consisting of 53 genes. Our results raise several important questions related to the diagnosis and treatment of pediatric CNS tumors, some of which are discussed below.
Novel and rare fusion events in pediatric gliomas
This study showed several fusion events that are novel or only rarely reported in CNS tumors, including CIC-LEUTX, PPP1CB-ALK and FGFR2-KIAA1598.
We report two cases of CIC-LEUTX fusion, one with anaplastic pleomorphic astrocytoma histology (case 17) and the other with features of anaplastic ganglioglioma (case 16). These cases constitute the first published report of CIC-LEUTX fusion in any CNS tumor. CIC abnormalities have been reported in CNS tumors as CIC-NUTM1 fusions20 but predominantly in sarcoma, with a single case of a CIC-LEUTX fusion seen in a 26-year-old female patient with angiosarcoma of her thigh.21 CIC alterations have also been reported in “Ewing-like tumors” with the most common fusion partner being DUX4.22 These tumors are characterized by a predominately small round blue cell morphology often with focal cell spindling, nuclear atypia, and stromal changes.23
ALK gene fusions were first identified in anaplastic large-cell lymphoma but now have been reported in many different pediatric cancers.15,16 Olsen et al. described ALK fusions in ependymoma-like gliomas, including KTN1-ALK and CCDC88A-ALK.17 Fusions involving PPP1CB are less common and have most frequently been reported in leukemias (fused with YPEL5) and recently reported as PPP1CB-ALK in a patient with a uterine leiomyosarcoma.24,25 The first case of PPP1CB-ALK fusion in a primary CNS tumor was in a 3 month-old girl with a supratentorial high-grade glioma (HGG).18 This patient underwent GTR followed by cyst fenestration at 9 months of age. At age 4, she experienced local recurrence that on re-resection showed a markedly different histology of LGG with mature ganglionic cells consistent with ganglioglioma.26
We identified one case of ganglioglioma with FGFR2-KIAA1598 fusion. This is a rare fusion and has reported in the literature only once in a CNS tumor.27 Similar to the case presented here, the previously reported case27 featured an oligodendroglial histology as the neoplastic glial component. Whether this histology is a general characteristic of tumors with FGFR2-KIAA1598 fusion remains to be determined.
Majority of fusion events are not definitional for pediatric tumor entities
While a few molecular fusions have come to define tumor entities with unique histologic characteristics and homogeneous clinical presentation, biological behavior, and treatment response (notable examples being KIAA1549-BRAF for PA and H3K27M for diffuse midline glioma), in most cases the same fusion can be seen in a variety of histologically disparate, clinically diverse tumors. As reported by others,8,28 FGFR alterations have been seen in a variety of glial and glioneuronal tumors, including PA and PMA, diffuse glioma, pediatric-type oligodendroglioma, ganglioglioma, and neurocytoma. PAs without BRAF alteration frequently show FGFR alterations.8 We found seven cases with FGFR fusions in our cohort (cases 1–7). Of these, FGFR-TACC1 fusion was seen in disparate tumor entities with PA, PMA, and extraventricular neurocytoma histologies, while one case featured FGFR1 alteration in the context of a diffuse histology, supporting the idea that these alterations are not specific to one type of LGG. Similarly, CIC-LEUTX fusions presented in the context of histologies compatible with anaplastic pleomorphic astrocytoma and anaplastic ganglioglioma. While MN-CXX5/MN1-BEND2 fusions have been proposed to be definitional for CNS HGNET tumors, recent data suggests that not all MN1 altered tumors behave aggressively. A recent study of MN1 altered tumors with astroblastoma histology show good prognosis unlike that for primitive neuroectodermal tumors (PNET)20. Whether a relatively benign clinical behavior also extends to MN1 altered tumors with PNET histology is unclear. Overall, these findings support the idea that few genetic alterations are definitional for a pediatric CNS tumor entity and that the diagnosis of the clear majority of pediatric CNS tumors requires integration of both traditional and molecular features of a tumor.2
Molecular findings lead to refinement of histologic diagnosis
The question of whether pediatric tumors should be defined by their histologic or molecular features remains open and controversial. We identified several cases in which the molecular features prompted re-review of histological features, including multiple resection specimens. Our 2 cases of MYB-QKI gliomas (patients 8 and 9) showed extremely rare and subtle features of angiocentric glioma that were only noted in retrospect with the knowledge of the molecular findings. In both cases, careful review specifically looking for characteristic angiocentric glioma features revealed very focal histological features compatible with this diagnosis. These cases, along with the previously reported cases29–32 suggest that MYB-QKI alteration present in LGGs may show only focal and rare angiocentric histology. Identification of MYB-QKI fusion should prompt careful re-review of all resection materials for this histologic feature. It should be noted, however, that neither LGG or angiocentric glioma behaves in an aggressive fashion, nor is further therapy usually required; thus, change in diagnosis based on this fusion may not have significant clinical or therapeutic impact. Though the MYB-QKI fusion has been shown to be a pathogenic event in angiocentric glioma29, whether the fusion will remain definitional for angiocentric glioma in the future is unclear. Although rare, cases of diffuse astrocytoma32 and ganglioglioma33 have also been reported to have the same alteration. The question of whether those case should be amended to angiocentric glioma remains.
Molecular findings leading to a change in histologic diagnosis
In three cases, molecular findings led to a change in histologic diagnosis. Gene fusions involving MN1 (22q12.3), usually with BEND2 (Xp22.13) but also with the alternative partner CXXC5 (5q31.2), have recently been reported in CNS tumors.20 These fusions were observed in a subset of tumors previously defined as PNET. Although many MN1-fused tumors showed a non-specific primitive neuroectodermal (PNET-like) morphology, a subset had classic features of astroblastoma, an entity previously histologically characterized by the presence of perivascular pseudorosettes composed of tumor cells with thick broad processes extending to a central blood vessel, often with hyalinized walls.19,34 These findings led to the review of molecular findings in histologically defined astroblastomas, a large proportion of which were shown to have MN1 alterations. Current understanding is that although astroblastoma histology is not specific for any particular tumor entity, a large number show MN1 alterations and cluster best with CNS-High grade neuroepithelial tumors.19,34 A recent study of MN1-altered tumors with astroblastoma histology show good prognosis unlike that expected for PNET. Whether a relatively benign clinical behavior also extends to MN1-altered tumors with PNET histology is unclear. Of note, of the two tumors with MN1 alterations in our cohort, one had classic astroblastoma histology (case 18) while the other was histologically diagnosed as anaplastic ependymoma (case 19), a known differential for astroblastoma.
BRAF-KIAA1549 is a common fusion seen in LGGs, particularly PA and PMA. PA presents with a wide variety of histologic findings that can mimic other benign and malignant neoplasms. The presence of BRAF-KIAA1549 alteration can therefore prevent misdiagnosis in cases of LGG with non-classic histology. Importantly, PA is usually not aggressive and can often be managed with resection alone. As exemplified by patient 24, in rare cases, PA can be confused with ependymoma which may lead to unnecessary systemic treatment. Following discovery of a BRAF fusion in case 24, histology was retrospectively reviewed and few additional features favoring pilocytic astrocytoma were found. Given the molecular finding, a differential of PMA was also raised which exists with PA in a histologic continuum.35 However, unlike PMA, prominent myxoid matrix and bipolar cells with thin wispy processes were not present in this tumor. Additional immunohistochemical studies were conducted, showing strong and diffuse Sox10 positivity in the tumor cells, a feature that we have recently reported to be more compatible with pilocytic astrocytoma than ependymoma.36 Notably the utility of SOX10 immunohistochemical staining in PA was not known at the time of the original diagnosis and routine broad panel fusion testing was not being done then. If this knowledge that been present at diagnosis, this patient would likely have been managed with surgery alone leading to a better long-term outcome. While SOX10 immunohistochemical staining proved useful in differentiating PA from ependymoma, it did not predict the presence of BRAF-fusion, as shown by case 13: a 6-year-old male with a thalamic tumor showing a low grade piloid histology with numerous myxoid areas suggestive of PA. SOX10 was strongly and diffusely positive, however fusion testing yielded the presence of a GOPC-ROS1 fusion.
Kline et al. reported5 a case that was histologically diagnosed as an ependymoma and subsequently amended as an HGG following the finding of an FGFR1 mutation. Somewhat analogous to case 24, their case showed strong Olig2 staining further supporting the diagnosis of an ependymal tumor.
Therapeutic implications of fusion testing
Pediatric CNS tumors are more genetically homogeneous and harbor fewer genetic drivers than their adult counterparts.37 This suggests the possibility of robust and more complete therapeutic response to targeted therapies. Additionally, targeted agents are particularly relevant in the pediatric population for which long-term sequelae from standard therapeutic modalities can be severe and may negatively affect quality of life. Durable responses to targeted inhibitors have been reported in NTRK-fusion positive and BRAF-altered gliomas.38,39 Active investigations of the therapeutic potential of other targeted inhibitors are underway (Table 2), including one trial integrating targeted therapy in the upfront setting (NCT02684058).
TABLE 2.
Targeted therapies that have undergone evaluation or are currently in clinical trials for pediatric CNS tumors
Designing a fusion panel
Recent data have suggested that pediatric CNS tumors, while histologically diverse, feature a limited set of driver fusion events37. We propose that a large majority of fusion events in pediatric glioma can be identified by relatively small gene panels. While other studies have utilized large several hundred genes panels, here we show that a smaller fusion-based assay comprised of carefully selected genes can identify a clear majority of fusion events and serve as the crucial first tier diagnostic tool for pediatric CNS tumors. We list the fusions found in our experience in pediatric gliomas, ependymomas and glioneuronal tumors (Table 1), which should be a part of fusion panels designed to identify alterations for therapeutic intent.
Conclusion
The recent rendition of the WHO classification of CNS tumors aims to define tumors as precisely and narrowly as possible to better prognosticate and treat. Our experience shows that small genomic platforms (around 50 genes) can identify key alterations in a large number of pediatric CNS tumors that may yield prognostically and therapeutically relevant information. Additionally, since few molecular alterations are definitional for tumor entities, diagnosis and classification of CNS tumors must rely upon integrating these features.
Supplementary Material
Acknowledgements
This work was supported, in part, by the UCD Molecular Pathology Shared Resource, The UCD Genomics and Microarray Core (National Cancer Institute Cancer Center Support Grant No. P30-CA046934), the Morgan Adams Foundation, and the Olivia Caldwell Foundation. We thank Lisa Litzenberger for assistance with histology figures.
Abbreviations
- ALK
Anaplastic lymphoma kinase
- CMOCO
Colorado Molecular Correlates
- CNS
central nervous system
- GBM
glioblastoma multiforme
- GTR
gross total resection
- HGG
high-grade glioma
- LGG
low-grade glioma
- NF1
Neurofibromatosis Type 1
- NGS
next-generation sequencing
- PA
pilocytic astrocytoma
- PMA
pilomyxoid astrocytoma
- PNET
primitive neuroectodermal tumors
- RT
radiation therapy
- STR
subtotal resection
- UCD
University of Colorado Denver
- WHO
World Health Organization
Footnotes
Conflict of Interest Statement
Kurt D. Davies has received sponsored travel from ArcherDx.
Contributor Information
Jessica A. Lake, Center for Cancer and Blood Disorders, Children’s Hospital Colorado, Aurora, CO, USA
Andrew M. Donson, The Morgan Adams Foundation Pediatric Brain Tumor Research Program, Department of Pediatrics, Center for Cancer and Blood Disorders, Children’s Hospital Colorado, Aurora, CO, USA
Eric Prince, Department of Neurosurgery, University of Colorado, Aurora, CO, USA.
Kurtis D. Davies, Department of Pathology, University of Colorado, Aurora, CO, USA
Anandani Nellan, The Morgan Adams Foundation Pediatric Brain Tumor Research Program, Department of Pediatrics, Center for Cancer and Blood Disorders, Children’s Hospital Colorado, Aurora, CO, USA.
Adam L. Green, The Morgan Adams Foundation Pediatric Brain Tumor Research Program, Department of Pediatrics, Center for Cancer and Blood Disorders, Children’s Hospital Colorado, Aurora, CO, USA
Jean Mulcahy Levy, The Morgan Adams Foundation Pediatric Brain Tumor Research Program, Department of Pediatrics, Center for Cancer and Blood Disorders, Children’s Hospital Colorado, Aurora, CO, USA.
Kathleen Dorris, The Morgan Adams Foundation Pediatric Brain Tumor Research Program, Department of Pediatrics, Center for Cancer and Blood Disorders, Children’s Hospital Colorado, Aurora, CO, USA.
Rajeev Vibhakar, The Morgan Adams Foundation Pediatric Brain Tumor Research Program, Department of Pediatrics, Center for Cancer and Blood Disorders, Children’s Hospital Colorado, Aurora, CO, USA.
Todd C. Hankinson, The Morgan Adams Foundation Pediatric Brain Tumor Research Program Departments of Neurosurgery and Pediatrics, Children’s Hospital Colorado, Aurora, CO, USA
Nicholas K. Foreman, The Morgan Adams Foundation Pediatric Brain Tumor Research Program, Department of Pediatrics, Center for Cancer and Blood Disorders, Children’s Hospital Colorado, Aurora, CO, USA
Mark D. Ewalt, Department of Pathology, University of Colorado, Aurora, CO, USA
Bette K. Kleinschmidt-DeMasters, Departments of Pathology, Neurology, Neurosurgery, University of Colorado, Aurora, CO, USA
Lindsey M. Hoffman, The Morgan Adams Foundation Pediatric Brain Tumor Research Program, Department of Pediatrics, Center for Cancer and Blood Disorders, Children’s Hospital Colorado, Aurora, CO, USA.
Ahmed Gilani, Department of Pathology, University of Colorado, Children’s Hospital Colorado, Aurora, CO, USA.
Data Availability Statement:
The data that support the findings of this study are available from the corresponding author upon request.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Ellison D, Hawkins C, Jones DTW, Onar-Thomas A, Pfister SM, Reifenberger G, Louis DN. cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol 2019;137:683–7. [DOI] [PubMed] [Google Scholar]
- 3.Cole BLLC, Stasi S, Stevens J, Lee A, Ojemann JG, Ellenbogen RG, and Leary SES Year 1 in the Molecular Era of Pediatric Brain Tumor Diagnosis: Application of Universal Clinical Targeted Sequencing in an Unselected Cohort of Children. JCO Precision Oncology 2018;2:1–13. [DOI] [PubMed] [Google Scholar]
- 4.Cole BLPC, Anderson M, Leary SE. Targeted Sequencing of Malignant Supratentorial Pediatric Brain Tumors Demonstrates a High Frequency of Clinically Relevant Mutations. Pediatr Dev Pathol 2018;21:380–8. [DOI] [PubMed] [Google Scholar]
- 5.Kline CN, Joseph NM, Grenert JP, et al. Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro Oncol 2017;19:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramkissoon SH, Bandopadhayay P, Hwang J, et al. Clinical targeted exome-based sequencing in combination with genome-wide copy number profiling: precision medicine analysis of 203 pediatric brain tumors. Neuro Oncol 2017;19:986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koschmann CWY, Kumar-Sinha C, Lonigro R, Vats P, Kasaian K, Cieslik M, Cao X, Anderson B, Frank K, Zhao L, Prensner JR, Zureick AH, Everett J, Mullan B, Marini B, Camelo-Piragua S, Venneti S, McKeever P, McFadden K, Lieberman AP, Leonard M, Maher CO, Garton H, Muraszko K, Robertson P, Robinson D, Chinnaiyan AM, and Mody R. Clinically Integrated Sequencing Alters Therapy in Children and Young Adults With High-Risk Glial Brain Tumors. JCO Precision Oncology 2018;2:1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson A, Severson E, Gay L, et al. Comprehensive Genomic Profiling of 282 Pediatric Low- and High-Grade Gliomas Reveals Genomic Drivers, Tumor Mutational Burden, and Hypermutation Signatures. Oncologist 2017;22:1478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lassaletta A, Zapotocky M, Bouffet E, Hawkins C, Tabori U. An integrative molecular and genomic analysis of pediatric hemispheric low-grade gliomas: an update. Childs Nerv Syst 2016;32:1789–97. [DOI] [PubMed] [Google Scholar]
- 10.Dimitriadis EAG, Tsotsou P, Simeonidi E, Stefanaki K, Patereli A, Prodromou N, Pandis N. BRAF alterations in pediatric low grade gliomas and mixed neuronal-glial tumors. J Neurooncol 2013;113:353–8. [DOI] [PubMed] [Google Scholar]
- 11.Davies KDLA, Sheren J, Nijmeh H, Gowan K, Jones KL, Varella-Garcia M, Aisner DL, Doebele RC. Comparison of Molecular Testing Modalities for Detection of ROS1 Rearrangements in a Cohort of Positive Patient Samples. J Thorac Oncol 2018;13:1474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010;26:873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benelli M, Pescucci C, Marseglia G, Severgnini M, Torricelli F, Magi A. Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics 2012;28:3232–9. [DOI] [PubMed] [Google Scholar]
- 15.Takita J The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci 2017;108:1913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosse YP. Anaplastic Lymphoma Kinase as a Cancer Target in Pediatric Malignancies. Clin Cancer Res 2016;22:546–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olsen TK, Panagopoulos I, Meling TR, et al. Fusion genes with ALK as recurrent partner in ependymoma-like gliomas: a new brain tumor entity? Neuro Oncol 2015;17:1365–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aghajan Y, Levy ML, Malicki DM, Crawford JR. Novel PPP1CB-ALK fusion protein in a high-grade glioma of infancy. BMJ Case Rep 2016;2016. [DOI] [PMC free article] [PubMed]
- 19.Lehman NL, Hattab EM, Mobley BC, et al. Morphological and molecular features of astroblastoma, including BRAFV600E mutations, suggest an ontological relationship to other cortical-based gliomas of children and young adults. Neuro Oncol 2017;19:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sturm D, Orr BA, Toprak UH, et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016;164:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang SC, Zhang L, Sung YS, et al. Recurrent CIC Gene Abnormalities in Angiosarcomas: A Molecular Study of 120 Cases With Concurrent Investigation of PLCG1, KDR, MYC, and FLT4 Gene Alterations. Am J Surg Pathol 2016;40:645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Specht K, Sung YS, Zhang L, Richter GH, Fletcher CD, Antonescu CR. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer 2014;53:622–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siegele B, Roberts J, Black JO, Rudzinski E, Vargas SO, Galambos C. DUX4 Immunohistochemistry Is a Highly Sensitive and Specific Marker for CIC-DUX4 Fusion-positive Round Cell Tumor. Am J Surg Pathol 2017;41:423–9. [DOI] [PubMed] [Google Scholar]
- 24.Velusamy T, Palanisamy N, Kalyana-Sundaram S, et al. Recurrent reciprocal RNA chimera involving YPEL5 and PPP1CB in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2013;110:3035–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ptakova N, Miesbauerova M, Kostun J, et al. Immunohistochemical and selected genetic reflex testing of all uterine leiomyosarcomas and STUMPs for ALK gene rearrangement may provide an effective screening tool in identifying uterine ALK-rearranged mesenchymal tumors. Virchows Arch 2018;473:583–90. [DOI] [PubMed] [Google Scholar]
- 26.Ng A, Levy ML, Malicki DM, Crawford JR. Unusual high-grade and low-grade glioma in an infant with PPP1CB-ALK gene fusion. BMJ Case Rep 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pekmezci M, Villanueva-Meyer JE, Goode B, et al. The genetic landscape of ganglioglioma. Acta Neuropathol Commun 2018;6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cole BLC, Paulson V, and Leary S. Pediatric brain tumors with FGFR1 mutations: A series of 14 cases assessing the morphologic spectrum and associated genetic alterations. Neuro Oncol 2019;21:ii85–6. [Google Scholar]
- 29.Bandopadhayay P, Ramkissoon LA, Jain P, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 2016;48:273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Aronco LRC, Gayden T, Crevier L, Decari JC, Perreault S, Jabado N, Bandopadhayay P, Ligon KL, Ellezam B,. Brainstem angiocentric gliomas with MYB-QKI rearrangements. Acta Neuropathol 2017;134:667–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan EBA, Siroh D, Van Ziffle J, Grenert JP, Kline CN, Tihan T, Perry A, Gupta N, Solomon DA. Angiocentric glioma with MYB-QKI fusion located in the brainstem, rather than cerebral cortex. Acta Neuropathol 2017;134:671–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qaddoumi I, Orisme W, Wen J, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 2016;131:833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roth JJ, Santi M, Rorke-Adams LB, et al. Diagnostic application of high resolution single nucleotide polymorphism array analysis for children with brain tumors. Cancer Genet 2014;207:111–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wood MD, Tihan T, Perry A, et al. Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities. Brain Pathol 2018;28:192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson MW, Eberhart CG, Perry A, et al. Spectrum of pilomyxoid astrocytomas: intermediate pilomyxoid tumors. Am J Surg Pathol 2010;34:1783–91. [DOI] [PubMed] [Google Scholar]
- 36.Kleinschmidt-DeMasters BK, Donson AM, Richmond AM, Pekmezci M, Tihan T, Foreman NK. SOX10 Distinguishes Pilocytic and Pilomyxoid Astrocytomas From Ependymomas but Shows No Differences in Expression Level in Ependymomas From Infants Versus Older Children or Among Molecular Subgroups. J Neuropathol Exp Neurol 2016;75:295–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rahal Z, Abdulhai F, Kadara H, Saab R. Genomics of adult and pediatric solid tumors. Am J Cancer Res 2018;8:1356–86. [PMC free article] [PubMed] [Google Scholar]
- 38.Toll SA, Tran HN, Cotter J, et al. Sustained response of three pediatric BRAF(V600E) mutated high-grade gliomas to combined BRAF and MEK inhibitor therapy. Oncotarget 2019;10:551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.AACR. Entrectinib Shows Pediatric Potential. Cancer Discov 2019. [DOI] [PubMed]
- 40.Geoerger BBF, DuBois SG, Fischer M, Geller JI, Gottardo NG, Marabelle A, Pearson ADJ, Modak S, Cash T, Robinson GW, Motta M, Matano A, Bhansali SG, Dobson JR, Parasuraman S, Chi SN. A Phase I Study of the CDK4/6 Inhibitor Ribociclib (LEE011) in Pediatric Patients with Malignant Rhabdoid Tumors, Neuroblastoma, and Other Solid Tumors. Clin Cancer Res 2017;23:2433–41. [DOI] [PubMed] [Google Scholar]
- 41.Banerjee A, Jakacki RI, Onar-Thomas A, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol 2017;19:1135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 2010;363:1801–11. [DOI] [PubMed] [Google Scholar]
- 43.Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 2018;19:705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.