

Abstract
The successful use of PARP1 inhibitors like olaparib (Loparza®) in the treatment of BRCA1/2- deficient breast cancer has provided clinical proof of concept for applying personalized medicine based on synthetic lethality to the treatment of cancer. Unfortunately, all marketed PARP1 inhibitors act by competing with the cofactor NAD+ and resistance is already developing to this anti-cancer mechanism. Allosteric PARP1 inhibitors could provide a means of overcoming this resistance. A high throughput screen performed by Tulin et al. identified 5F02 as an allosteric PARP inhibitor that acts by preventing the enzymatic activation of PARP1 by histone H4. 5F02 demonstrated anti-cancer activity in several cancer cell lines and was more potent than olaparib and synergistic with olaparib in these assays. In the present study we explored the structure-activity relationship of 5F02 by preparing analogs that possessed structural variation in four regions of the chemical scaffold. Our efforts led to lead molecule 7, which demonstrated potent anti-clonogenic activity against BRCA-deficient NALM6 leukemia cells in culture and a therapeutic index for the BRCA-deficient cells over their BRCA-proficient isogenic counterparts.
Keywords: Allosteric, BRCA, Histone 4, PARP1, Synthetic Lethality
Introduction
Tumor cells accumulate spontaneous and drug-induced DNA damage, but they survive because of enhanced/altered DNA repair activities (Bartkova et al., 2005). The hypothesis that cancer cells are addicted to particular DNA repair pathways is supported by selective elimination of tumor cells by drugs/compounds targeting specific DNA repair mechanisms (Nickoloff et al., 2017). DNA double-strand breaks (DSBs), the most lethal DNA lesions, are usually repaired by BRCA1/2 -mediated homologous recombination (BRCA-HR) in proliferating cells (Karanam et al., 2012). The enzyme poly (ADP-ribose) polymerase 1 (PARP1) may prevent accumulation of DNA double-strand breaks (DSBs) by playing a key role in base excision repair (BER), single-strand break (SSB) repair, alternative non-homologous end-joining (Alt-NHEJ), and/or by facilitating MRE11-mediated recruitment of RAD51 to promote stalled replication fork restart (Metzger et al., 2013; Ying et al., 2012). The success of PARP inhibitor (PARPi) olaparib (Lynparza®, Figure 1) in treating BRCA1/2-deficient breast tumors has established a proof-of-concept of personalized cancer therapy utilizing synthetic lethality (Lord et al., 2015). Moreover, we demonstrated that PARPi’s trigger synthetic lethality also in proliferating and quiescent tumor cells deficient in DNA-PK mediated NHEJ (D-NHEJ) (Nieborowska-Skorska et al., 2017). Recently we showed that quiescent and proliferating cells from BRCA-HR and D-NHEJ -deficient hematopoietic malignancies are sensitive to synthetic lethality triggered by PARPi’s (Nieborowska-Skorska et al., 2017; Dasgupta et al., 2017; Meifrede et al., 2017).
Figure 1.

Structures of marketed PARP inhibitors and 5F02, an allosteric PARP1 inhibitor that acts by blocking histone H4=stimulated activation of the enzyme.
Three drug design strategies for inhibiting PARP1 have been pursued: competing with the cofactor NAD+, obstructing PARP1 binding with DNA and preventing enzymatic activation of PARP1 by histone H4 (Malyuchenko et al, 2015; Kirsanov et al., 2014; Jain and Patel, 2019). All of the marketed PARPi’s (Figure 1) act by competing with NAD+. Unfortunately, resistance to NAD+-competitive PARP1 inhibitors is rapidly developing. To overcome this problem, the Tulin group performed a high throughput screen on a 50,000 compound library to identify non-NAD-like PARP inhibitors (Thomas et al., 2016). In the course of that work, 5F02 (Figure 1) and related analogs were identified as a novel class of inhibitors of histone H4-mediated PARP1 activation. To our knowledge, this is the first report of compounds that inhibit PARP1 through this mechanism. In collaboration with the Tulin group we demonstrated that 5F02 demonstrated anti-tumor effects in a number of cell lines, exerted synergistic effect with olaparib in those assays and was more potent than olaparib in in vitro proliferation assays (Nieborowska-Skorska et al., 2019).
We wished to expand on these innovative findings by examining the structure activity relationship (SAR) of the interesting chemical scaffold presented by 5F02. To achieve a better understanding of the SAR and structure-property relationship (SPR) around the chemical scaffold inherent in 5F02 we explored four regions of the molecule (Figure 2), namely the piperazine moiety, the requirement for a positively charged quaternary amine group, the requirement for and ester functionality and the cyclododecyl substituent. To assess the role of physicochemical properties we simultaneously tested all target molecules for their maximum kinetic aqueous solubility in 2% DMSO/phosphate buffered saline and their stability in mouse and human liver microsomes in the presence and absence of NADPH. Our work led to the identification of an analog within this novel series that demonstrated selective anti-clonogenic activity against a leukemia cell line that is BRCA-deficient compared to BRCA-proficient isogenic counterparts.
Figure 2.

SAR Strategy for the 5F02 Chemical Scaffold.
Chemistry
Reagents were purchased from commercial suppliers (Sigma-Aldrich, Combiblocks, Enamine, Fisher Scientific) and used without further purification. Unless stated otherwise, reactions were run in the presence of normal atmospheric conditions. 1H NMR and 13C NMR data were collected on a Bruker 400 MHz Avance III spectrometer at ambient temperature in the identified solvent. 13C NMR were run at 100 MHz. Peak positions are given in parts per million downfield from tetramethylsilane as the internal standard. LC-MS analysis was performed on an Agilent Technologies 1200 series LC system coupled to a 6300 quadrapole MS. High resolution mass spectrometry analysis was performed by the Mass Spectrometry Lab, School of Chemical Sciences, University of Illinois at Urbana-Champaign on a Waters QTOF Ultima ESI instrument. Silica gel chromatography was performed using a Teledyne ISCO Combiflash Rf system with UV detection at 220 nM and 254 nM. Reversed phase chromatography was performed on a Gilson GX281 system using a C-18 column and a gradient of acetonitrile in water with 0.1% trifluoroacetic acid modifier. Detection was accomplished with UV at 220 nM and 254 nM. Purity (> 99.5% unless otherwise noted) of all final target compounds was confirmed by HPLC on two separate systems: 1) normal phase on a silica gel column using a gradient of ethyl acetate in hexane; 2) reversed phase on a C-18 column using a gradient of acetonitrile in water
Cyclododecyl 2-chloroacetate:
In a round bottom flask, cyclododecanol (20.8 gm, 113 mmol) was dissolved in benzene (225 mL), and then pyridine (11.4 mL, 141 mmol) was added. This mixture was cooled to 0°C and chloroacetyl chloride (12.7 gm, 113 mmol) was added dropwise. The stirred mixture was then heated at 78°C and allowed to stir for 3 hours. Upon completion, the reaction was allowed to cool to room temperature. The mixture was diluted with diethyl ether and washed with water, 10% aqueous HCl, and finally saturated aqueous sodium bicarbonate. The organic layer was dried over anhydrous magnesium sulfate and concentrated on a rotary evaporator. The residuals were purified by chromatography on silica gel (gradient of 0–10% hexanes in ethyl acetate). The desired product (19.1 gm, 65%) was obtained as a white solid. 1H NMR (400 MHz, CDCl3) δ ppm: 5.11–5.04 (m, 1H, O-CH), 4.00 (s, 2H, CH2-Cl), 1.78–1.67 (m, 2H, C2, C12 cyclododecyl –CH2), 1.57–1.46 (m, 2H, C2, C12 cyclododecyl –CH2), 1.44–1.26 (m, 18H, remaining cyclododecyl –CH2); ESIMS m/z (pos): 282.3 (100%)/284.3 (33%) [M+Na]+.
General Procedure for the synthesis of compounds 1a – 9a
In a reaction vial, cyclododecyl 2-chloroacetate (0.50 gm, 1.9 mmol) was combined with the appropriate cyclic secondary amine (2.5 mmol) and stirred at 100°C for 48 hours. The reaction was cooled to room temperature and the mixture were purified by silica gel chromatography (gradient of 0–40% hexanes in ethyl acetate) to provide the desired products 1a–9a.
Cyclododecyl 2-(piperidin-1-yl)acetate (1a):
This compound was obtained as a yellow oil; Yield = 38%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.05 (tt, J=7.15, 4.89 Hz, 1H, O-CH), 3.16 (m, 2H, N-CH2), 2.53 (m, 4H. C2, C6 of piperidine –CH2), 1.73–1.58 (m, 4H, C3, C5 of piperidine – CH2), 1.51–1.32 (m, 24H C4 of piperidine-CH2, cyclododecyl –CH2); ESIMS m/z (pos) 310.2 (100%) [M+H]+.
Cyclododecyl-2-(pyrrolidin-1-yl)acetate (2a):
This compound was obtained as a orange solid; Yield = 51%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.13–5.04 (m, 1H, O-CH), 3.34 (s, 2H, N-CH2), 2.72–2.64 (m, 4H, C2, C5 of pyrrolidine –CH2), 1.89–1.78 (m, 4H, C3, C4 of pyrrolidine – CH2), 1.78–1.67 (m, 2H, C2, C12 of cyclododecyl –CH2), 1.58–1.48 (m, 2H, C2, C12 of cyclododecyl –CH2), 1.48–1.24 (m, 18H, remaining cyclododecyl –CH2); ESIMS m/z (pos) 296.2 (100%) [M+H]+.
Cyclododecyl-2-(azepan-1-yl)acetate (3a):
This compound was obtained as a orange solid; Yield = 44%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.06–5.01 (m, 1H, O-CH), 3.36 (s, 2H, N-CH2), 2.68–2.63 (m, 4H, C2, C7 of azacycloheptyl ring –CH2), 1.85–1.76 (m, 4H, C3, C6 of azacycloheptyl ring –CH2), 1.74–1.64 (m, 2H, C3, C5 of azacycloheptyl ring –CH2), 1.55–1.43 (m, 2H, C3, C5 of azacycloheptyl ring –CH2), 1.42–1.19 (m, 22H, remaining cyclododecyl –CH2); ESIMS m/z (pos) 324.3 (100%) (M+H)+.
Cyclododecyl 2-(4,4-dimethylpiperidin-1-yl)acetate (4a):
This compound was obtained as a orange solid; Yield = 58%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.00–4.96 (m, 1H, O-CH), 3.33 (s, 2H, N-CH2), 2.61–2.55 (m, 4H, C2, C6 of piperidine –CH2), 1.83–1.72 (m, 4H, C3, C5 of piperidine –CH2), 1.70–1.61 (m, 2H, C2, C12 of cyclododecyl –CH2), 1.51–1.41 (m, 2H, C2, C12 of cyclododecyl –CH2), 1.42–1,19 (m, 18H, remaining cyclododecyl –CH2), 0.85 (s, 6H, piperidine –CH3); ESIMS m/z (pos) 338.3 (100%) [M+H]+.
Cyclododecyl 2-morpholinoacetate (5a):
This compound was obtained as a yellow solid; Yield = 22%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.04–4.96 (m, 1H, O-CH), 3.76–3.74 (m 4H, morpholino O-CH2), 3.13 (s, 2H, N-CH2), 2.62–2.60 (m, 4H, morpholino N-CH2), 1.68–1.61 (m, 2H, C2, C12 cyclododecyl –CH2), 1.48–1.41 (m, 2H, C2, C12 cyclododecyl –CH2), 1.36–1.23 (m, 18H, remaining cyclododecyl –CH2); ESIMS m/z (pos) 312.2 (100%) [M+H]+.
Cyclododecyl 2-thiomorphinoacetate (6a):
This compound was obtained as a yellow solid; Yield = 37%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.02–4.97 (m, 1H, O-CH), 3.34 (s, 2H, N-CH2), 2.68–2.61 (m, 4H, thiomorpholinyl N-CH2), 2.60–2.52 (m, 4H, thiomorpholinyl S-CH2), 1.73–1.64 (m, 2H, C2, C12 cyclododecyl –CH2), 1.55–1.47 (m, 2H C2, C12 cyclododecyl –CH2), 1.42–1.19 (m, 18H, remaining cyclododecyl –CH2); ESIMS m/z (pos) 328.2 (100%) [M+H]+.
Cyclododecyl 2-(piperazin-1-yl)acetate (7a):
This compound was obtained as a yellow solid; Yield = 8.5%,. 1H NMR (400 MHz, CDCl3) δ ppm: 5.04–4.96 (m, 1H, O-CH), 3.13 (s, 2H, N- CH2), 2.62 (m, 4H, piperazine N-CH2), 2.54–2.52 (m, 4H, piperazine N-CH2) 2.40–2.38 (m, 1H, piperazine NH), 1.68–1.61 (m, 2H, C2, C12 cyclododecyl –CH2), 1.48–1.41 (m, 2H, C2, C12 cyclododecyl –CH2), 1.36–1.23 (m, 18H, remaining cyclododecyl –CH2); ESIMS m/z (pos) 312.2 (100%) [M+H]+.
tert-Butyl 4-(2-(cyclododecyloxy)-2-oxoethyl)piperazine-1-carboxylate (8a):
This compound was obtained as a white solid; Yield = 26%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.06–4.97(m, 1H, O-CH), 3.48–3.32 (m, 4H, piperazine N-CH2), 3.13 (s, 2H, N-CH2), 2.48–2.46 (m, 4H, piperazine N-CH2), 167–1.62 (m, 2H, C2, C12 cyclododecyl –CH2), 1.47–1.42 (m, 2H, C2, C12 cyclododecyl –CH2), 1.43 (s, 9H, BOC-CH3)1.38–1.24 (m, 18H, remaining cyclododecyl –CH2); ESIMS m/z (pos) 411.3 (100%) [M+H]+.
Cyclododecyl-2-(4-benzylpiperidin-1-yl)acetate (9a):
This compound was obtained as a white solid; Yield = 26%. 1H NMR (400 MHz, CDCl3) δ ppm: 7.30–7.26 (m, 2H, Ar-H), 7.21 – 9.19 (m, 1H, Ar-H), 7.16–7.154 (m, 2H, Ar-H), 5.06–4.97(m, 1H, O-CH), 3.13 (s, 2H, N-CH2), 2.85–2.83 (m, 2H, benzyl –CH2), 2.48–2.46 (m, 4H, C2, C6 of piperiinel -CH2), 1.67–1.62 (m, 2H, C2, C12 cyclododecyl –CH2), 1.47–1.43 (m, 1H, C4 of piperidine –CH), 1.39–1.34 (m, 2H, C2, C12 cyclododecyl –CH2), 1.36–1.22 (m, 22H, C3, C5 of piperidine –CH2, remaining cyclododecyl –CH2); ESIMS m/z (pos) 411.3 (100%) (M+H)+.
General Procedure for the synthesis of compounds 1–6, 8–9
In a reaction vial, the appropriate amino-cyclododecyl acetate 1a–9a (0.65 mmol) was dissolved in methanol (2 mL), and then methyl iodide (0.72 mmol) was added. The vial was sealed and the resulting mixture was heat to 65°C and stirred at that temperature overnight. The reaction mixture was then cooled to room temperature and concentrated on a rotary evaporator and the resulting residue triturated with diethyl ether. The resulting precipitate was collected by vacuum filtration, washed with diethyl ether and dried in vacuo to provide the desired products 1–6, 8–9.
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1-methylpiperidin-1-ium iodide (1, 5F02):
This compound was obtained as a yellow solid; Yield = 75%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.14–5.07 (m, 1H, O-CH), 4.88 (s, 2H, N-CH2), 3.95–3.88 (m, 4H, C2, C6 of piperidinel –CH2), 3.41 (s, 3H, N-CH3), 2.41–2.29 (m, 4H, C3, C5 of piperidine –CH2), 2.29–2.16 (m, 2H, C4 of piperidine –CH2), 1.77–1.69 (m, 2H, C2, C12 cyclododecyl –CH2), 1.55–1.43 (m, 2H, C2, C12 cyclododecyl –CH2), 1.39–1.25 (m, 18H, remaining cyclododecyl –CH2); MS (ESI) (m/z) 324.3 (M)+.; 13C NMR (100 MHz, CDCl3) δ ppm 162.5 (C=O), 60.4 (cyclododecyl C1), 59.6 (N+–CH2), 51.9 (piperidinyl C2,6), 47.0 (N+–CH3), 27.6 (cyclododecyl C2,12), 22.5 (cyclododecyl C4,10), 22.3 (cyclododecyl C5,9), 21.9 (cyclododecyl C6,8), 21.7 (cyclododecyl C7), 19.5 (cyclododecyl C3,11), 19.1 (piperidinyl C4), 18.8 (piperidinyl C3,5); ESIMS m/z (pos) 324.3 [M]+; HRESIMS m/z (pos): 324.2896 C20H38NO2 (calcd. 324.2903).
1-(2-(Cyclododecyloxy)-2-(oxoethyl)-1-methylpyrrolidin-1-ium iodide (2):
This compound was obtained as an orange solid; Yield = 85%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.10–5.03 (m, 1H, O-CH), 4.85 (s, 2H, N-CH2), 4.08–4.01 (m, 4H, C2, C5 of pyrrolidine –CH2), 3,41 (s, 3H, N-CH3), 2.39–2.26 (m, 2H, C3, C4 of pyrrolidine, –CH2), 2.26–2.12 (m, 2H, C3, C4 of pyrrolidine – CH2), 1.76–1.65 (m, 2H, C2, C12 cyclododecyl –CH2), 1.54–1.44 (s, 2H, C2, C12 cyclododecyl – CH2), 1.38–1.21 (m, 18H, remaining cyclododecyl –CH2); 13C NMR (100 MHz, CDCl3) δ ppm 164.4 (C=O), 76.3 (cyclododecyl C1), 65.8 (pyrrolidinyl C2,5). 62.7 (N+-CH2). 49.7 (N+-CH3). 29.1 (cyclododecyl C3,11). 23.9 (cyclododecyl C4,10). 23.7 (cyclododecyl C5,9). 23.3 (cyclododecyl C6,8). 23.1 (cyclododecyl C7). 21.5 (cyclododecyl C3,11). 20.9 (pyrrolidinyl C3,4); ESIMS m/z (pos) 310.3 (100%) [M]+; HRESIMS m/z (pos): 310.2745 C19H36NO2 (calcd. 310.2746).
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1-methylazepan-1-ium iodide (3):
This compound was obtained as a yellow solid; Yield = 68%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.10–5.03 (m, 1H, O-CH), 4.71, (s, 2H, N-CH2), 4.07–3.98 (m, 2H, C2, C7 of azacycloheptyl ring, –CH2), 3.92–3.83 (m, 2H, C2, C7 of azacycloheptyl ring, –CH2), 3.59 (s, 3H, N-CH3), 2.06–1.86 (m, 4H, C3, C6 of azacycloheptyl ring –CH2), 1.83–1.62 (m, 6H, C3, C4 of azacycloheptyl ring and C2, C12 cyclododecyl –CH2), 1.55–1.43 (m, 2H, C2, C12 cyclodedecyl –CH2), 1.41–1.18 (m, 18H, remaining cyclododecyl –CH2); 13C NMR (100 MHz, CDCl3) δ ppm 163.1 (C=O), 76.06 (cyclododecyl C1), 65.9 (azacycloheptyl C2,7), 62.1 (N+-CH2), 51.4 (N+-CH3), 29.1 (cyclododecyl C2,12), 26.7 (azacycloheptyl C3,6), 23.9 (cyclododecyl C4,10), 23.7 (cyclododecyl C5,9), 23.3 (cyclododecyl C6,8), 23.1 (cyclododecyl C7), 22.2 (cyclododecyl C3,11), 20.9 (azacycloheptyl C4,5); ESIMS m/z (pos) 338.3 (100%) [M]+; HRESIMS m/z (pos): 338.3056 C21H40NO2 (calcd. 338.3059.).
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1,4,4-trimethylpiperidin-1-ium iodide (4):
This compound was obtained as a white solid; Yield = 71%. 1H NMR (400 MHz, DMSO) δ ppm: 5.11–5.02 (m, 1H, O-CH), 4.51 (s, 2H, N-CH2), 3.63–3.51 (m, 2H, C2, C6 of piperidine –CH2), 3.51–3.40 (m, 2H, C2, C6 of piperidine –CH2), 3.21 (s, 3H, N-CH3), 1.80–1.46 (m, 8H, C3, C5 of piperidine –CH2 and C2, C12 cyclododecyl –CH2), 1.42–1.22 (m, 18H, remaining cyclododecyl – CH2), 1.02 (s, 3H, piperidine –CH3), 1.01 (s, 3H, piperidine –CH3); 13C NMR (100 MHz, DMSO- d6) δ ppm 169.8 (C=O), 79.2 (cyclododecyl C1), 62.7 (N+-CH2), 60.1 (piperidinyl C2,6), 48.4 N+-CH3 (), 36.7 (piperidinyl C3,5), 34.2 (piperidinyl 4-CH3), 33.8 (cyclododecyl C2,12), 32.3 (piperidinyl C4), 28.7 (cyclododecyl C4,10), 28.5 (cyclododecyl C5,9), 28.1 (cyclododecyl C6,8), 27.9 (cyclododecyl C7), 25.6 (cyclododecyl C3,11); ESIMS m/z (pos) 352.3 (100%) [M]+; HRESIMS m/z (pos): 352.3209 C22H42NO2 (calcd. 352.3216).
4-(2-(Cyclododecyloxy)-2-oxoethyl)-4-methylmorpholin-4-ium iodide (5):
This compound was obtained as a yellow solid; Yield = 36%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.15–5.08 (m, 1H, O-CH), 4.47 (s, 2H, N-CH2), 4.02–3.88 (m, 4H, morpholinyl O-CH2), 3.74–3.57 (m, 4H, morpholinyl N-CH2), 3.38 (s, 3H, N-CH3), 1.79–1.68 (m, 2H, C2, C12 cyclododecyl –CH2), 1.57–1.46 (m, 2H, C2, C12 cyclododecyl –CH2), 1.41–1.19 (m, 18H, remaining cyclododecyl –CH2); 13C NMR (100 MHz, CD3OD) δ ppm 165.4 (C=O), 76.7 (cyclododecyl C1), 63.2 (morpholinyl O- CH2), 62.1 (morpholinyl N-CH2), 61.5 (N+-CH2), 49.0 (N+-CH3), 30.1 (cyclododecyl C2,12), 25.0 (cyclododecyl C4,10), 24.8, 24.6 (cyclododecyl C5,9), 24.5 (cyclododecyl C6,8), 24.3 (cyclododecyl C7), 22.1 (cyclododecyl C3,11); ESIMS m/z (pos) 326.2 (100%) [M]+; HRESIMS m/z (pos): 326.2696 C19H36NO3 (calcd. C).
4-(2-(Cyclododecyloxy)-2-oxoethyl)-4-methylthiomorphilin-4-ium iodide (6):
This compound was obtained as a yellow solid; Yield = 66%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.17–5.09 (m, 1H, O-CH), 4.51 (s, 2H, N-CH2), 4.35–4.14 (m, 4H, thiomorpholinyl N-CH2), 3.99–3.79 (m, 4H, thiomorpholinyl S-CH2), 3.56 (s, 3H, N-CH3), 1.81–1.67 (m, 2H, C2, C12 cyclododecyl –CH2), 1.59–1.47 (m, 2H, C2, C12 cyclododecyl –CH2), 1.43–1.23 (m, 18H, remaining cyclododecyl –CH2); 13C NMR (100 MHz, CDCl3) δ ppm 163.2 (C=O), 77.0 (cyclododecyl C1), 62.0 (thiomorpholinyl N-CH2), 57.7 (N+-CH2), 48.6 (N+-CH3), 37.9 (cyclododecyl C1), 28.7 (thiomorpholinyl S-CH2), 23.9 (cyclododecyl C4,10), 23.8 (cyclododecyl C5,9), 23.2 (cyclododecyl C6,8), 23.1 (cyclododecyl C7), 20.7 (cyclododecyl C3,11); ESIMS m/z (pos) 342.2 (100%) [M]+; HRESIMS m/z (pos) (M+) 342.2462 C19H36NO2S (calcd. 342.2467).
4-(tert-Butoxycarbonyl)-1-(2-(cyclodedecyloxy)-2-oxoethyl)-1-methylpiperazin-1-ium iodide (8):
This compound was obtained as a yellow solid; Yield = 30%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.13–5.09 (m, 1H, O-CH), 4.04–3.90 (m, 4H, piperazinyl –CH2), 3.75 (s, 3H, N-CH3), 3.72–3.65 (m, 4H, piperazinyl –CH2), 1.78–1.73 (m, 2H, C2, C12 cyclododecyl –CH2), 1.95–1.55 (m, 2H, C2, C12 cyclododecyl –CH2), 1.45 (s, 9H, BOC –CH3), 1.38–1.21 (m, 18H, remaining cyclododecyl –CH2); 13C NMR (100 MHz, CDCl3) δ ppm 163.7 (ester C=O), 153.5 (BOC C=O), 82.1 (BOC t-butyl quaternary C), 76.6 (cyclododecyl C1), 60.9 (piperizinyl N-CH2). 60.3 (piperizinyl N+-CH2), 47.9 (N+-CH3), 29.1 (cyclododecyl C1), 28.3 (BOC -CH3), 23.9 (cyclododecyl C4,10), 23.7 (cyclododecyl C5,9), 23.3 (cyclododecyl C6,8), 23,2 (cyclododecyl C7), 21.0 (cyclododecyl C3,11); ESIMS m/z (pos) 425.3 (100%) [M]+; HRESIMS m/z (pos): 425.3375 C24H45N2O4 (calcd. 425.3379).
4-(Benzyl)-1-(2-(cyclododecyloxy)-2-oxoethyl)-1-methylpiperidin-1ium iodide (9):
This compound was obtained as a yellow solid; Yield = 44%. 1H NMR (400 MHz, DMSO) δ ppm: 7.24–7.17 (m, 2H, Ar-H), 7.16–7.06 (m, 3H, Ar-H), 5.09–4.99 (m, 1H, O-CH), 4.97 (s, 2H, N-CH2), 4.03–3.93 (m, 4H, C2, C6 of piperidine –CH2), 3.44 (s, 3H, N-CH3), 2.68–2.55 (m, 2H, benzyl –CH2), 2.20–2.03 (m, 1H, C4 of piperidine -CH), 1.87–1.75 (m, 2H, C3, C5 of piperidine –CH2), 1.75–1.55 (m, 4H, C3, C5 of piperidine –CH2, C2, C12 cyclododecyl –CH2), 1.52–1.37 (m, 2H, C2, C12 cyclododecyl –CH2), 1.37–1.16 (m, 18H, remaining cyclododecyl –CH2); 13C NMR (100 MHz, CDCl3) δ ppm 164.0 (C=O), 138.2 (phenyl C1), 129.1 (phenyl C3,5), 128.6 (phenyl C2,6), 126.5 (phenyl C4), 75.8 (cyclododecyl C1), 64.0 (N+-CH2), 61.6 (piperidinyl C2,6), 45.3 (N+- CH3), 41.6 (benzyl –CH2), 34.2 (piperidinyl C4), 29.1 (cyclododecyl C2,12), 26.0 (piperidinyl C3,5), 23.8 (cyclododecyl C4,10), 23.6, (cyclododecyl C5,9), 23.3 (cyclododecyl C6,8), 23.2 (cyclododecyl C7), 21.0 (cyclododecyl C3,11); ESIMS m/z (pos) 414.4 (100%) [M]+; HRESIMS m/z (pos): 414.3369 C27H44NO2 (calcd, 414.3372),
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1-methylpiperazinium iodide (7):
4-(tert- Butoxycarbonyl)-1-(2-(cyclodedecyloxy)-2-oxoethyl)-1-methylpiperazin-1-ium iodide (8, 0.04 gm, 0.07 mmol) was dissolved in a 1:1 mixture of dichloromethane and trifluoroacetic acid (2 mL). The mixture was stirred at room temperature for 15 minutes and then concentrated on a rotary evaporator. The desires product (0.032 gm, 100 %) was obtained as a yellow solid by trituration with diethyl ether. 1H NMR (400 MHz, CDCl3) δ ppm: 5.11–5.04 (m, 1H, O-CH), 4.98 (s, 2H, N- CH2), 4.36–4.27 (m, 2H, C2, C6 of piperazine –CH2), 4.14–4.07 (m, 2H, C2, C6 of piperazine - CH2), 4.00–3.92 (m, 1H, piperazine NH), 3.66 (s, 3H, N-CH3), 3.15–3.01 (m, 2H, C3, C5 of piperazine –CH2), 2.98–2.88 (m 2H, C3, C5 of piperazine –CH2), 1.77–1.60 (m, 2H, C2, C12 cyclododecyl –CH2), 1.56–1.38 (m, 2H, C2, C12 cyclododecyl –CH2), 1.3.0, −1.20 (m, 18H, remaining cyclododecyl –CH2); 13C NMR (100 MHz, CDCl3) δ ppm 163.7 (C=O), 73.0 (cyclododecyl C1), 62.0 (N+-CH2), 61.0 (piperazinyl N+-CH2), 46.1 (N+-CH3), 37.5 (piperazinyl NH-CH2), 29.2 (cyclododecyl C2,12), 24.0 (cyclododecyl C4,10), 23.4 (cycyldodecyl C5,9), 23.3 (cyclododecyl C6,8), 21.5 (cyclododecyl C7), 21.0 (cyclododecyl C3,11); ESIMS m/z (pos) 325.3 (100%) [M]+; HRESIMS m/z (pos); 325.2850 C19H37N2O2 (calcd. 325.2855).
General procedure for the synthesis of compounds 10 – 13
In a reaction vial 1a (0.20 gm, 0.65 mmol) was dissolved in methanol (2 mL),and then the appropriate alkyl iodide (0.72 mmol) was added. The vial was sealed and the resulting mixture was heat to 65°C and stirred at that temperature overnight. The reaction was then cooled to room temperature, concentrated on a rotary evaporator and the resulting residue triturated with diethyl ether. The resulting precipitate was collected by vacuum filtration, washed with diethyl ether and dried in vacuo to obtain the desired products 10–13.
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1-ethylpiperidin-1-ium iodide (10):
This compound was obtained as an orange solid; Yield = 35%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.14–5.07 (m, 1H, O-CH), 4.48 (s, 2H, N-CH2 (to carbonyl)), 4.26–4.20 (q, J=8.0 Hz, 2H, ethyl –CH2), 3.92–3.81 (m, 4 H, C2, C6 of piperidine –CH2), 1.93–1.81 (m, 6H, C3,C4, C5 of piperidine –CH2), 1.79–1.72 (m, 2H, C2, C12 cyclododecyl –CH2), 1.56–1.49 (m, 2H, C2, C12 cyclododecyl –CH2), 1.40–1.28 (m, 18H, remaining cyclododecyl –CH2), 1.35 (t, J=8.0Hz, 3H, ethyl –CH3; 13C NMR (100 MHz, CDCl3) δ ppm 164.0 (C=O), 76.4 (cyclododecyl C1), 59.7 (N+-CH2), 57.0 (ethyl –CH2), 54.0 (piperidinyl C2,6), 29.2 (cyclododecyl C2,12), 24.0 (cyclododecyl C4,10), 23.8, (cyclododecyl C5,9), 23.5 (cyclododecyl C6,8), 23.3 (cyclododecyl C7), 21.1 (cyclododecyl C3,11), 20.5 (piperidinyl C4), 20.2 (piperidinyl C3,5), 8.3 (ethyl –CH3); ESIMS m/z (pos) 338.3 (100%) [M]+; HRESIMS m/z (pos): (M+): 338.3052 C21H40NO2 (calcd. 338.3059).
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1-(1-propyl)piperidin-1-ium iodide (11):
This compound was obtained as a yellow solid; Yield = 11%. 1H NMR (400 MHz, CDCl3) δ ppm: 5.09–5.03 (m, 1H, O-CH), 4.47 (s, 2H, N-CH2), 4.28–4.15 (q, J=7.4 Hz, 2H, propyl N-CH2), 3.85–3.74 (m, 2H, C2, C6 of piperidine –CH2), 3.73–3.58 (m, 2H, C2, C6 of piperidine –CH2), 1.93–1.66 (m, 10H, C3, C4, C5 piperidine –CH2, propyl –CH2, C2, C12 cyclodecyl –CH2), 1.53–1.38 (m, 2H, C2, C12 cyclododecyl –CH2), 1.32–1.27 (m, 18H, remaining cyclododecyl –CH2), 0.98 (t, J=7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ ppm 164.0 (C=O), 76.1 (cyclododecyl C1), 60.2 (N+-CH2), 60.0 (propyl N+-CH2), 57.4 (piperidinyl C2,6), 29.1 (cyclododecyl C2,12), 23.8 (cyclododecyl C4,10), 23.6 (cyclododecyl C5,8), 23.3 (cyclododecyl C6,8), 23.1 (cyclododecyl C7), 21.0 (cyclododecyl C3,11), 20.3 (piperidinyl C4), 20.1 (piperidinyl C3,5), 15.9 (propyl – CH2), 10.8 (propyl –CH3); ESIMS m/z (pos) 352.3 (100%) [M]+. HRESIMS m/z (pos): 352.3212 C22H42NO2 (calcd. 352.3216).
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1-(1-butyl)piperidin-1-ium iodide (12):
This compound was obtained as a yellow solid: Yield = 19%.1H NMR (400 MHz, CDCl3) δ ppm: 5.13–5.04 (m, 1H, O-CH), 4.47 (s, 2H, N-CH2), 4.21–4.16 (m, 2H, propyl N-CH2), 3.85–3.82 (m, 2H, C2, C6 of piperidine –CH2), 3.74–3.70 (m, 2H, C2, C6 of piperidine –CH2), 1.96–1.80 (m, 6H, C3, C5 of piperidine –CH2, butyl –CH2), 1.73–1.62 (m, 4H, C4 of piperidine, C2, C12 cyclododecyl –CH2), 1.54–1.45 (m, 2H, C2, C12 cyclododecyl –CH2), 1.43–1.16 (m, 20H, butyl –CH2, remaining cyclododecyl –CH2), 0.96 (t, J=7.28 Hz, 3H, butyl –CH3); 13C NMR (100 MHz, CDCl3) δ ppm 163.8 (C=O), 75.9 (cyclododecyl C1), 57.4 (butyl N+-CH2), 55.7 (N+-CH2), 52.8 (piperidinyl C2,6), 29.0 (cyclododecyl C2,12), 24.0 (cyclododecyl C4,10), 23.8 (cyclododecyl C5,9), 23.6 (cyclododecyl C6,8), 23.3 (cyclododecyl C7), 23.1 (cyclododecyl C3,11), 22.9 (butyl C2), 21.2 (piperidinyl C4), 21.0 (piperidinyl C3,5), 20.0 (butyl C3), 13.6 (butyl –CH3); MS (ESI) (m/z) 366.3 (M)+. ESIMS m/z (pos) 366.3 (100%) [M]+; HRESIMS m/z (pos): 366.2270 C23H44NO2 (calcd. 366.3372).
1-(2-(Cyclododecyloxy)-2-oxoethyl)-1-(1-pentyl)piperidin-1-ium iodide (13):
This compound was obtained as a yellow solid; Yield = 5%). 1H NMR (400 MHz, CDCl3) δ ppm: 5.14–5.07 (m, 1H, O-CH), 4.51 (s, 2H, N-CH2), 4.36–4.26 (m, 2H, pentyl N-CH2), 3.86–3.77 (m, 2H, C2, C6 of piperidine –CH2), 3.77–3.68 (m, 2H, C2, C6 of piperidine –CH2), 1.86–1.83 (m, 4H, C3, C5 of piperidine –CH2), 1.77–1.71 (m, 4H, C4 of piperidine –CH2, pentyl –CH2), 1.69–1.62 (m, 2H, C2, C12 cyclododecyl –CH2), 1.57–1.49 (m, 4H, pentyl –CH2, C2, C12 cyclododecyl –CH2), 1.40–1.29 (m, 20H, pentyl –CH2, remaining cyclododecyl –CH2), 0.92–0.89 (m, 3H, pentyl –CH3); 13C NMR (100 MHz, CDCl3) δ ppm 164.0 (C=O), 76.2 (cyclododecyl C1), 60.0 (butyl N+- CH2), 57.8 (N+-CH2), 59.1 (piperidinyl C2,6), 29.0, (cyclododecyl C2,12), 28.3 (pentyl C3), 23.9 (pentyl C2), 23.7 (cyclododecyl C4,10), 23.3 (cyclododecyl C5,9), 23.1 (cyclododecyl C6,8), 22.2 (cyclododecyl C7), 21.8 (cyclododecyl C3,11), 21.0 (piperidinyl C3,5), 20.3 (piperidinyl C4), 20.0 (butyl C3), 13.8 (butyl –CH3); ESIMS m/z (pos) 380.3 (100%) [M]+; HRESIMS m/z (pos): 380.3518 C24H46NO2 (calcd. 380.3529).
Synthesis of 1-(2-(cyclodecyloxy)-2-oxoethyl)-1-methylpiperidin-1-ium iodide (14)
Cyclodecyl 2-chloroacetate:
In a round bottom flask, cyclodecanol (Schenker and Gunthard, 1952) (3.0 gm, 19.2 mmol) was dissolved in benzene (30 mL), and then pyridine (1.9 mL, 24.0 mmol) was added. This mixture was cooled to 0°C and chloroacetyl chloride (1.53 mL, 19.2 mmol) was added dropwise. The stirred mixture was then heated at 78°C and allowed to stir for 3 hours. Upon completion, the reaction was allowed to cool to room temperature. The mixture was diluted with diethyl ether and washed with water, 10% aqueous HCl, and finally saturated aqueous sodium bicarbonate. The organic layer was dried over anhydrous magnesium sulfate and concentrated on a rotary evaporator. The residuals were purified by chromatography on silica gel (gradient of 0–10% ethyl acetate in hexane). The desired product (3.70 gm, 83%) was obtained as a white solid. 1H NMR (400 MHz, CDCl3) δ ppm: 5.15–5.09 (m, 1H, O-CH), 3.32 (s, 2H, CH2Cl), 2.12 – 2.04 (m, 2H, C2, C10 cyclodecyl –CH2), 1.75 – 1.70 (m, 2H, C2, C10 cyclosdecyl –CH2), 1.58 – 1.52 (m, 14H, remaining cyclododecyl –CH2); ESIMS m/z (pos) 255.1 (100%)/257.1 (33%) [M+Na]+.
Cyclodecyl 2-(piperidin-1-yl)acetate (14a):
In a reaction vial, cyclodecyl 2-chloroacetate (0.50 gm, 1.9 mmol) was combined with piperidine (0.25 mL, 2.5 mmol) and stirred at 100°C for 48 hours. The reaction was cooled to room temperature and the mixture were purified by silica gel chromatography (0–40% ethyl acetate in hexane) to provide the desired product 14a (0.24 gm, 38%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ ppm: 5.18–5.14 (m, 1H, O-CH), 3.22 (s, 2H, N-CH2), 2.53 – 2.47 (m, 4H, C2, C6 of piperidine –CH2), 1.88–1.79 (m, 2H, C3, C5 of piperidine-CH2), 1.69–1.40 (m, 22H, C3, C4, C5 of piperidine –CH2, remaining cyclodecyl –CH2); ESIMS m/z (pos) 282.2 (100%) [M+H]+.
1-(2-(Cyclodecyloxy)-2-oxoethyl)-1-methylpiperidin-1-ium iodide (14):
In a reaction vial, cyclodecyl 2-(piperidin-1-yl)acetate (0.20 gm, 0.7 mmol) was dissolved in methanol (2 mL), and then methyl iodide (97 μL, 0.77 mmol) was added. The vial was sealed and the resulting mixture was heat to 65°C and stirred at that temperature overnight. Upon return, the reaction mixture was concentrated on a rotary evaporator and the resulting residue triturated with diethyl ether. The resulting yellow solid was collected by vacuum filtration, washed with diethyl ether and dried in vacuo to provide the desired product (0.24 gm, 81 %) as an orange solid. 1H NMR (400 MHz, CDCl3) δ ppm: 5.21–5.15 (m, 1H, O-CH), 3.19 (s, 2H, N-CH2), 2.58 (s, 3H, N-CH3), 2.55 – 2.44 (m, 4H, C2, C6 of piperidine –CH2), 1.85–1.74 (m, 2H, C3, C5 of piperidine –CH2), 1.69–1.40 (m, 22H, C3, C4, C5 of piperidine –CH2, cyclodecyl –CH2); 13C NMR (100 MHz, CDCl3) δ ppm 163.8 (C=O), 77.6 (cyclodecyl C1), 61.8 (N+-CH2), 53.3 (piperidinyl C2,6), 48.5 (N+-CH3), 30.0 (cyclodecyl C2,10), 24.9 (cyclodecyl C4,8), 24.8 (cyclodecyl C5,7), 24.0 (cyclodecyl C6), 22.1 (cyclodecyl C3,9), 20.5 (piperidinyl C2), 20.2 (piperidinyl C3,5); ESIMS m/z (pos) 296.2 (100%) [M]+. HRESIMS m/z (pos): 296.2587 C18H34NO2 (calcd. 296.2590).
Synthesis of 1-(2-(Cyclooctyloxy)-2-oxoethyl)-1-methylpiperidin-1-ium iodide (15)
Cyclooctyl 2-chloroacetate:
In a round bottom flask, cyclooctanol (2.46 gm, 19.2 mmol) was dissolved in benzene (30 mL), and then pyridine (1.9 mL, 24.0 mmol) was added. This mixture was cooled to 0°C and chloroacetyl chloride (1.53 mL, 19.2 mmol) was added dropwise. The stirred mixture was then heated at 78°C and allowed to stir for 3 hours. Upon completion, the reaction was allowed to cool to room temperature. The mixture was diluted with diethyl ether and washed with water, 10% aqueous HCl, and finally saturated aqueous sodium bicarbonate. The organic layer was dried over anhydrous magnesium sulfate and concentrated on a rotary evaporator. The residuals were purified by chromatography on silica gel (gradient of 0–10% ethyl acetate in hexane). The desired product (2.46 gm, 63%) was isolated as a yellow oil. 1H NMR (400 MHz, CDCl3) δ ppm: 5.11 – 5.06 (m, 1H, O-CH), 3.35 (s, 2H, CH2Cl), 2.02 – 1.96 (M, 2H, C2, C10 cyclooctyl –CH2), 1.71 – 1.65 (m, 2H, C2, C10 cyclooctyl –CH2), 1.49 – 1.35 (m, 10H, remaining cyclooctyl –CH2); ESIMS m/z (pos) 227.1 (100%)/229.1 (33%) [M+Na]+.
Cyclooctyl 2-(piperidin-1-yl)acetate (15a):
In a reaction vial, cyclooctyl 2-chloroacetate (0.5 gm, 2.15 mmol) was combined with piperidine (0.28 mL, 2.79 mmol) and stirred at 100°C for 48 hours. The reaction was cooled to room temperature and the mixture were purified by silica gel chromatography (0–40% ethyl acetate in hexane) to provide the desired product 15a (0.28 gm, 46%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ ppm: 4.98–4.94 (m, 1H, O-CH), 3.19 (s, 2H, N-CH2), 2.50 – 2.44 (m, 4H, C2, C6 of piperidine –CH2), 1.75–1.58 (m, 2H, C3, C5 of piperidine –CH2), 1.50–1.40 (m, 18H, C3, C4, C5 of piperidine -CH2, cyclooctyl –CH2); ESIMS m/z (pos) 254.2 (100%) [M+H]+.
1-(2-(Cyclooctyloxy)-2-oxoethyl)-1-methylpiperidin-1-ium iodide (15):
In a reaction vial, cyclooctyl 2-(piperidin-1-yl)acetate (0.2 gm, 0.71 mmol) was dissolved in methanol (2 mL), and then methyl iodide (99 μL, 0.78 mmol) was added. The vial was sealed and the resulting mixture was heat to 65°C and stirred at that temperature overnight. Upon return, the reaction mixture was concentrated on a rotary evaporator and the resulting residue triturated with diethyl ether. The resulting yellow solid was collected by vacuum filtration, washed with diethyl ether and dried in vacuo to provide the desired product (0.22 gm, 74%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ ppm: 4.95–4.90 (m, 1H, O-CH), 3.16 (s, 2H, N-CH2), 2.6–2.60 (s, 3H, N-CH3), 2.52 (m, 4H, C2, C6 of piperidine –CH2), 1.77–1.57 (m, 2H, C3, C5 of piperidine –CH2), 1.51–1.39 (m, 18H, C3, C4, C5 of piperidine –CH2, cyclooctyl –CH2); 13C NMR (100 MHz, CDCl3) δ ppm 163.5 (C=O), 78.8 (cyclooctyl C1), 61.7 (N+-CH2), 61.1 (piperidinyl C2,6), 48.5 (N+-CH3), 31.5 (cyclooctyl C2,8), 26.8 (cyclooctyl C4,6), 25.3 (cyclooctyl C5), 22.8 (cyclooctyl C3,7), 22.7 (piperidinyl C4), 20.4 (piperidinyl C3,5); ESIMS m/z (pos) 268.2 (100%) [M]+; HRESIMS m/z (pos): 268.2271 C16H30NO2 (calcd. 268.2277).
Synthesis of 1-(2-(Cyclohexyloxy)-2-oxoethyl)-1-methylpiperidin-1-ium iodide (16) Cyclohexyl 2-(piperidin-1-yl)acetate (16a):
In a reaction vial, cyclohexyl 2-chloroacetate (Toronto Research Chemicals, Cat. # C992108) (0.5 gm, 2.83 mmol) was combined with piperidine (0.37 mL, 3.68 mmol) and stirred at 100°C for 48 hours. The reaction was cooled to room temperature and the mixture were purified by silica gel chromatography (0–40% ethyl acetate in hexane) to provide the desired product 16a (0.31 gm, 49%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ ppm 4.95–4.92 (m, 1H, O-CH), 3.17 (s, 2H, N-CH2), 2.51 – 2.46 (m, 4H, C2, C6 of piperidine –CH2), 1.81–1.70 (m, 2H, C3, C5 of piperidine –CH2), 1.51–1.42 (m, 14H, C3, C4, C5 of piperidine –CH2, cyclohexyl –CH2); ESIMS m/z (pos) 226.2 (100%) [M+H]+.
1-(2-(Cyclohexyloxy)-2-oxoethyl)-1-methylpiperidin-1-ium iodide (16):
In a reaction vial, cyclohexyl 2-(piperidin-1-yl)acetate (0.2 gm, 0.91 mmol) was dissolved in methanol (3 mL), and then methyl iodide (136 μL, 1.00 mmol) was added. The vial was sealed and the resulting mixture was heat to 65°C and stirred at that temperature overnight. Upon return, the reaction mixture was concentrated on a rotary evaporator and the resulting residue triturated with diethyl ether. The resulting yellow solid was collected by vacuum filtration, washed with diethyl ether and dried in vacuo to provide the desired product (0.19 gm, 58%) as an orange solid. 1H NMR (400 MHz, CDCl3) δ ppm: 4.82–4.75 (m, 1H, O-CH), 3.19 (s, 2H, N-CH2), 2.58 (s, 3H, N-CH3), 2.60–2.51 (m, 4H, C2, C6 of piperidine –CH2), 1.86–1.77 (m, 2H, C3, C5 of piperidine –CH2), 1.72–1.60 (m, 6H, C2, C6 cyclohexyl –CH2, C3, C4, C5 of piperidine –CH2), 1.56–1.20 (m, 8H, C2, C6 cyclohexyl –CH2, remaining cyclohexyl –CH2); 13C-NMR (100 MHz, CDCl3) δ ppm 163.6 (C=O), 76.1 (cyclohexyl C1), 61.7 (N+-CH2), 61.0 (piperidinyl C2,6), 48.5 (N+-CH3), 31.3 (cyclohexyl C2,6), 25.0 (cyclohexyl C5), 23.6 (cyclohexyl C3,5), 20.4 (piperidinyl C4), 20.2 (piperidinyl C3,5); ESIMS m/z (pos) 240.2 (100%) [M]+; HRESIMS m/z (pos): 240.1960 C14H26NO2 (calcd. 240.1964).
Synthesis of 1-(2-(Cyclopentyloxy)-2-oxoethyl)-1-methylpiperidin-1-ium iodide (17)
Cyclopentyl 2-chloroacetate:
In a round bottom flask, cyclopentanol (1.65 gm, 19.2 mmol) was dissolved in benzene (30 mL), and then pyridine (1.9 mL, 24.0 mmol) was added. This mixture was cooled to 0°C and chloroacetyl chloride (1.53 mL, 19.2 mmol) was added dropwise. The stirred mixture was then heated at 78°C and allowed to stir for 3 hours. Upon completion, the reaction was allowed to cool to room temperature. The mixture was diluted with diethyl ether and washed with water, 10% aqueous HCl, and finally saturated aqueous sodium bicarbonate. The organic layer was dried over anhydrous magnesium sulfate and concentrated on a rotary evaporator. The residuals were purified by chromatography on silica gel (gradient of 0–10% ethyl acetate in hexane). The desired product (1.72 gm, 55%) was isolated as a yellow liquid. 1H NMR (400 MHz, CDCl3) δ ppm: 5.11 – 5.04 (m, 1H, O-CH), 3.30 (s, 2H, CH2Cl), 1.99 – 1.93 9(m, 2H, C2, C5 cyclopentyl –CH2), 1.77 – 1.70 (m, 2H, C2, C5 cyclopentyl –CH2), 1.49 – 1.38 (m, 6H, remaining cyclopentyl –CH2); ESIMS m/z (pos) 185.0 (100%)/187.0 (33%) [M+Na]+.
Cyclopentyl 2-(piperidin-1-yl)acetate (17a):
In a reaction vial, cyclopentyl 2-chloroacetate (0.5 gm, 3.07 mmol) was combined with piperidine (0.40 mL, 4.0 mmol) and stirred at 100°C for 48 hours. The reaction was cooled to room temperature and the mixture were purified by silica gel chromatography (0–40% ethyl acetate in hexane) to provide the desired product 17a (0.33 gm, 51%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ ppm 5.10 – 5.04 (m, 1H, O-CH), 3.20 (s, 2H, N-CH2), 2.52 – 2.44 (m, 4H, C2, C6 of piperidine –CH2), 1.80 – 1.75 (m, 2H, C3, C5 of piperidine –CH2), 1.50 – 1.38 (m, 12H, C3, C4, C5 of piperidine –CH2, cyclopentyl –CH2); ESIMS m/z (pos) 212.2 (100%) (M+H)+.
1-(2-(Cyclopentyloxy)-2-oxoethyl)-1-methylpiperidin-1-ium iodide (17):
In a reaction vial, cyclopentyl 2-(piperidin-1-yl)acetate (0.2 gm, 0.94 mmol) was dissolved in methanol (3 mL), and then methyl iodide (140 uL, 1.03 mmol) was added. The vial was sealed and the resulting mixture was heat to 65°C and stirred at that temperature overnight. Upon return, the reaction mixture was concentrated on a rotary evaporator and the resulting residue triturated with diethyl ether. The resulting yellow solid was collected by vacuum filtration, washed with diethyl ether and dried in vacuo to provide the desired product (0.16 gm, 49%) as an yellow solid. 1H NMR (400 MHz, CDCl3) δ ppm: 5.20–5.15 (m, 1H, O-CH), 3.11 (s, 2H, N-CH2), 2.50–2.46 (m, 4H, C2, C6 of piperidine –CH2), 2.44 (s, 3H, N-CH3), 1.85–1.80 (m, 2H, C3, C5 of piperidine –CH2), 1.77–1.37 (m, 12H, C3, C4, C5 of piperidine –CH2, cyclopentyl –CH2); 13C NMR (100 MHz, CDCl3) δ ppm 164.7 (C=O), 80.3 (cyclopentyl C1), 61.8 (N+-CH2), 53.3 (piperidinyl C2,6), 47.9 (N+-CH3), 32.6 (cyclopentyl C2,5), 23.6 (cyclopentyl C3,4), 20.4 (piperidinyl C4), 20.2 (piperidinyl C3,5); ESIMS m/z (pos) 226.1 (100%) [M]+ HRESIMS m/z (pos): 226.1805 C13H24NO2 (calcd. 226.1807).
Synthesis of 1-(2-(cyclododecyl(methyl)amino)-2-oxoethoxy)-1-methylpiperidin-1ium iodide (18)
N-Methylcyclodedecanamine:
Cyclododecanone (3.0 gm, 16.5 mmol) and 2M methylamine in tetrahydrofuran (9.9 mL, 19.8 mmol) were dissolved in anhydrous tetrahydrofuran (40 mL). The mixture was cooled to 0°C and then treated with lithium aluminum hydride (1M in tetrahydrofuran, 8.2 mL, 8.2 mmol). The reaction mixture was allowed to come up to room temperature and then stirred overnight. The reaction was quenched by dropwise addition of saturated ammonium chloride and then extracted with dichloromethane. The organic layer was washed with water and brine, dried over anhydrous magnesium sulfate and concentrated on a rotary evaporator. The crude product (2.60 gm, 80%) was obtained as a white solid and was used in the subsequent reaction without further purification. ESIMS m/z (pos) 198.3 (100%) [M+H]+.
2-Chloro-N-cyclodedecyl-N-methylacetamide:
Crude N-methylcyclododecanamine (1.5 gm, 7.6 mmol) was dissolved in benzene (15 mL) and then pyridine (0.75 mL, 9.5 mmol) was added. The resulting stirred mixture was cooled to 0°C and treated dropwise with chloroacetyl chloride (0.61 mL, 7.6 mmol). Upon complete addition the reaction was heated to 78°C and stirred at that temperature for 3 hours. The reaction was then cooled to room temperature, diluted with diethyl ether and the organic layer washed with water, 10% aqueous HCl and saturated aqueous sodium bicarbonate. The organic layer was dried over anhydrous magnesium sulfate and concentrated on a rotary evaporator. The residue was purified by silica gel chromatography (gradient of 0 – 10% ethyl acetate in hexane) to provide the desired product (0.66 gm, 32% over two steps) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ ppm: 4.80–4.75 (m 1H, N-CH), 2.88 (s, 3H, N-CH3), 2.80 (s, 2H, CH2Cl), 1.72–1.61 (m, 2H, C2, C12 cyclododecyl –CH2), 1.58–1.20 (m, 20H, C2, C12 cyclododecyl –CH2, remaining cyclododecyl –CH2); ESIMS m/z (pos) 274.2 (100%)/276.2 (33%) [M+H]+.
N-Cyclododecyl-N-methyl-2-(piperidin-1-yl)acetamide:
In a reaction vial, 2-chloro-N- cyclodedecyl-N-methylacetamide (0.60 gm, 2.2 mmol) was combined with piperidine (0.28 mL, 2.8 mmol) and the sealed vial was stirred at 100°C for 48 hours. The reaction was cooled to room temperature and mixture was purified by silica gel chromatography (gradient of 0 – 40% ethyl acetate in hexane) to provide the desired product (0.085 gm, 12%) as a yellow solid. The 1H-NMR indicated the presence of rotational isomers. ESIMS m/z (pos) 323.3 (100%) [M+H]+. trans: 1H NMR (400 MHz, CDCl3) δ ppm: 4.81–4.74 (m, 1H, O-CH), 3.48 (br s, 2H, N-CH2), 3.17 (m, 2H, C2, C6 of piperidine –CH2), 2.80 (s, 3H, N-CH3), 1.68–1.48 (m, 10H, C2, C3, C4, C5, C6 of piperidine –CH2, C2, C12 cyclododecyl –CH2), 1.38–1.21 (m, 20H, C2, C12 cyclododecyl – CH2, remaining cyclododecyl –CH2); cis: 1H NMR (400 MHz, CDCl3) δ ppm: 4.28 (br s, 1H, O-CH), 3.95 (br s, 2H, N-CH2), 2.73 (s, 3H, N-CH3), 2.48–2.40 (m, 4H, C2, C6 of piperidine –CH2), 1.82–1.73 (m, 4H, C3, C5 of piperidine –CH2), 1.57–1.45 (m, 4H, C4 of piperidine –CH2, C12 of cyclododecyl –CH2), 1.38–1.21 (m, 20H, C2, C12 of cyclododecyl –CH2, remaining cyclododecyl –CH2).
1-(2-(Cyclododecyl(methyl)amino)-2-oxoethoxy)-1-methylpiperidin-1ium iodide (18):
In a reaction vial, N-cyclododecyl-N-methyl-2-(piperidin-1-yl)acetamide (0.20 gm, 0.62 mmol) was dissolved in methanol (2 mL), and then methyl iodide (93 μL, 0.68 mmol) was added. The vial was sealed and the resulting mixture was heat to 65°C and stirred at that temperature overnight. Upon return, the reaction mixture was concentrated on a rotary evaporator and the resulting residue triturated with diethyl ether. The resulting yellow solid was collected by vacuum filtration, washed with diethyl ether and dried in vacuo to provide the desired product (0.24 gm, 84 %) as an orange solid. 1H NMR (400 MHz, CDCl3) δ ppm; 1H NMR (400 MHz, CDCl3) δ ppm: 4.76–4.74 (m, 1H, O-CH), 4.68 (s, 2H, N-CH2), 4.09–4.03 (m, 2H, C2, C6 of piperidine –CH2), 3.79–3.74 (m, 2H, C2, C6 of piperidine), 3.44 (s, 3H, N-CH3), 2.97 (s, 3H, N-CH3), 1.97–1.85 (m, 4H, C3, C5 of piperidine –CH2), 1.82–1.74 (m, 2H, C2, C12 cyclododecyl –CH2), 1.69–1.60 (C4 of piperidine –CH2), 1.53–1.44 (m, 2H, C2, C12 cyclododecyl –CH2), 1.39–1.25 (m, 18H, remaining cyclododecyl –CH2); 13C NMR (100 MHz, CD3OD) δ ppm 167.3 (C=O), 66.1 (N+-CH2), 51.8 (piperidinyl C2,6), 51.5 (cyclododecyl C1), 48.3 (N+-CH3), 31.4 (cyclododecyl C2,12), 31.3 (amide N-CH3), 27.8 (cyclododecyl C4,10), 27.4 (cyclododecyl C5,9), 26.0 (cyclododecyl C6,8), 26.3 (cyclododecyl C7), 25.7 (piperidinyl C4), 24.8 (cyclododecyl C3,11), 23.6 (piperidinyl C3,5); ESIMS m/z (pos) 337.3 (100%) [M+]; HRESIMS m/z (pos): 337.3210 C21H41N2O (calcd. 338.3219).
In Vitro Physicochemical/ADME Assays
In order to assess the effects of aqueous solubility on the in vitro pharmacology of our compounds we assessed all final target molecules for their maximum kinetic aqueous solubility in 2% DMSO/phosphate buffered saline. As a further differentiator we also examined the stability of our final target molecules in mouse and human liver microsomes in the presence and absence of NADPH as an indicator of potential metabolic liability.
Experimental Procedure for Determining Maximum Kinetic Aqueous Solubility
Solubility assays were performed using Millipore MultiScreen®HTS-PFC Filter Plates designed for solubility assays (EMD Millipore, Billerica, MA). The 96-well plates consist of two chambers separated by a filter. Liquid handling was performed using JANUS® Verispan and MTD workstations (Perkin Elmer, Waltham, MA). 4 μL of drug solutions (10 mM in DMSO) are added to 196 μL of the appropriate medium in the top chamber to give a final DMSO concentration of 2% and a theoretical drug concentration of 200 μM. Plates are gently shaken for 90 min and then subjected to vacuum. Insoluble drug is captured on the filter. 160 μL of the filtrate is transferred to 96-well Griener UV Star® analysis plates (Sigma–Aldrich, St. Louis, MO) containing 40 μL of acetonitrile. The drug concentration in the filtrate is measured by UV absorbance on a Spectromax® Plus microplate reader (Molecular Devices, Sunnyvale, CA) using Softmax Pro software v. 5.4.5. Absorbances at 5 wavelengths (280, 300, 320, 340, and 360 nM) were summed to generate the UV signal. Assays were performed in triplicate. Standard curves were generated by adding 4 μL of 50× of five concentrations of test compounds in DMSO to 40 μL of acetonitrile in UV Star plates followed by 156 μL of the appropriate solubility medium. Analysis and statistics were performed using GraphPad® Prism v. 5.04. Data are reported as the maximum concentration observed in the filtrate. The following solubility medium was used in the assays.
Phosphate buffer (pH 7.4):
Millipore-defined universal buffer—45 mM potassium phosphate, 45 mM sodium acetate, 45 mM ethanolamine, pH = 7.4.
Experimental Procedure for Determining Stability in Mouse and Human Liver Microsomes
The clearance of test compounds in mouse or human liver microsomes was determined at 37 °C. Assays were conducted in 96-deep well polypropylene plates. Test compounds (1 μM) were incubated in 0.5 mL of 100 mM potassium phosphate buffer (pH 7.4) with 0.5 mg/mL pooled liver microsomes from male CD-1 mice (Life Technologies, Grand Island, NY) or pooled liver microsomes from humans (ThermoFisher, Waltham, MA), 2 mM tetra-sodium NADPH and 3 mM magnesium chloride for 60 min at 37 °C with gentle shaking. At five time points, 75 μL of reaction mixture was transferred to 96-shallow well stop plates on ice containing 225 mL of acetonitrile with 0.1 μM propafenone. Control reactions (lacking NADPH) were performed in a similar manner to demonstrate NADPH dependency of compound loss. Standard curves for test compounds were generated using 5 concentrations in duplicate that were processed as above but with zero incubation time. Stop plates were centrifuged at 2000g for 10 min and then 170 μL of the supernatants were transferred to a Waters Aquity® UPLC 700 μL 96-well sample plate with cap mat (Waters, Milford, MA). The amount of compound remaining in the supernatant was quantified by LC/MS/MS using a Waters TQ MS (electrospray positive mode) coupled to a Waters Aquity® UPLC (BEH column, C18 1.7 μm, 2.1 × 50 cm, gradient of acetonitrile/water/0.1% formic acid). Propafenone was used as the internal standard. GraphPad® Prism v 5.04 was used for nonlinear fitting of time course data to generate Clint values.
General Biology
In order to assess the ability of compounds to inhibit H4-stimulated PARP1 activity we developed a benchtop variation of the high throughput screening assay used by the Tulin group to identify 5F02 (Thomas et al. 2016). In this assay, H4 stimulates PARP1 to poly-ADP- ribosylate the histone. Compounds are assessed for their ability to inhibit this activity. Simultaneous with this assay, compounds were assessed for two drug-like properties using plate-based high throughput assays, namely maximum solubility in 2% DMSO/phosphate buffered saline and potential liability to oxidative and hydrolytic metabolism as assessed by stability in mouse and human liver microsomes. Compounds that demonstrated reasonable potency and drug-like properties in these assays were then assessed for anti-tumor effects and BRCA-selectivity. To test the effect of our various 5F02 analogs against BRCA-deficient and BRCA-proficient tumors, BRCA-pathway proficient NALM6 acute lymphoblastic leukemia cell line cells and BRCA-pathway deficient RAD54−/− isogenic counterparts were employed as described before.
Experimental Procedure for Determining Inhibition of H4-Stimulated PARP1 activation
Nunc® MaxiSorp™ 96 well plates were coated with 10 ng/well (50 uL) of recombinant human histone H4 in phosphate-buffered saline (PBS) overnight at room temperature. All subsequent procedures at room temperature. Plates were washed (200 uL; 5 min/wash) three times and then blocked for one hour with PBS-T (PBS + 0.05 % Tween 20) with 5 % non-fat milk. Plates were then washed twice with PBS-T and once with assay buffer (AB: 50 mM Tris/HCl, pH 8.0; 25 mM MgCl2; 0.1 % Triton® X-100). 20 uL/well of multiple concentrations of test compounds in assay buffer were then preincubated with 20 uL/well of 0.625 U/mL human Parp-1 (Trevigen #4668–500-01) for fifteen minutes. The twenty minute reactions were initiated by the addition of 10 uL/well of 50 uM NAD+ and stopped by washing three times with PBS-T.
Plates were then incubated with 50 uL/well (1:4000 in PBS-T/5% milk) of primary antibody (Tulip #1020; anti-poly(ADP-Ribose) polymer; clone 10H) for one hour. Plates were washed three times and then incubated with 50 uL/well (1: 1500 in PBS-T/milk) of secondary HRP- conjugated goat anti-mouse antibody for one hour. Plates were washed three times with PBS-T and once with PBS and then 50 uL/well of SureBlue reagent was added. After ten minutes, 50 ul/well of 1N hydrochloric acid stop solution was added. Absorbance (A450-A650) was measured with a Molecular Devices SpectraMax Plus® plate reader. IC50’s were determined using the Graph Pad® Prism v 5.04 nonlinear curve fitting program.
Experimental Procedure for Measuring the Effect of Drugs on RAD54−/− Nalm-6 Human Leukemia Cells and RAD54+/+ Nalm-6 Parental Cells
The protocol for the Nalm-6 cellular assays have been described elsewhere (Karanam et al., 2012; Sullivan-Reed et al., 2018). Briefly, test compounds were added to Nalm-6 isogenic RAD54−/− human leukemia cells or Nalm-6 RAD54+/+ parental cells (Horizon Discovery Group, Cambridge, UK) for 3–5 days followed by plating in methylcellulose. Clonogenic activity was assessed 7 days after plating. Data were plotted and IC50 values determined using the GraphPad® Prism v 5.04 nonlinear curve fitting program.
Results and Discussion
Compounds 1–17 were prepared based on the method of Koslov et al. (Koslov et al., 1989) and using the general method depicted in Scheme 1 and experimental details are provided in the Supplementary Information section. The required cycloalkyl alcohols were acylated with chloroacetylchloride. The resulting intermediates was treated with the desired cyclic secondary amine and those intermediates (1a – 6a, 8a – 17a) were finally treated with the required alkyl iodide in refluxing methanol to provide final target compounds 1 – 6 and 8 – 17. Methylation of intermediate 7a proved problematic so the desired product 7 was ultimately obtained by deprotection of the BOC-derivative 8 with trifluoroacetic acid. To prepare compound 18 the method depicted in Scheme 2 was employed. Cyclododecylmethylamine was prepared from cyclododecanone via reductive amination using a two-step sequence that involved imine formation followed by reduction with lithium aluminum hydride. The secondary amine was then converted to 18 using the methodology depicted in Scheme 1.
Scheme 1.

Synthesis of compounds 2–17. Reagents and Conditions: (a) chloroacetyl chloride, pyridine, benzene, reflux 3 hr; (b) cyclic secondary amine, 100°C; (c) alkyl iodide, methanol, reflux, (d) 18 hr; trifluoroacetic acid, r.t., 15 minutes.
Scheme 2.

Synthesis of compound 18. Reagents and Conditions: (a) 66% aq, methylamine, methanol, reflux, 2 hr; (b) lithijm aluminum hydride, tetrahydrofuran; (c) chloroacetyl chloride, pyridine, benzene, reflux 3 hr; (d) cyclic amine, 100°C; (e) iodomethane, methanol, reflux, 18 hr.
The results of the SAR study on the piperidine moiety of 5F02 are shown in Table 1. 5F01 (1) displayed an IC50 value for inhibiting H4-stimulated PARP1 activity of around 100 nM. Changes in the size of the piperidinyl ring (compounds 2 and 4) induced an increase in H4/PARP1 inhibitory potency. Likewise, addition of additional lipophilic bulk in the 4-position of the piperidine ring (e,g., the gem-di-methyl of compound 4, the tert-butyl group of compound 8 and the benzyl moiety of compound 9) enhanced potency somewhat, suggesting that the lipophilic binding pocket that accommodates the 6-membered ring of 5F02 may be somewhat longer and wider than the volume occupied by the chair comformation of that 6-membered ring. The greatest enhancement in potency seen with structural changes in this region of the molecule came with the introduction of a hydrogen bond accepting oxygen atom in the morpholine derivative (compound 5). This effect was not mimicked with the thiomorpholine derivative (compound 6), although without a structural model it is not possible to ascertain if this is caused by the electronic nature of the sulfur atom or the change in the ring shape and size induced by the sulfur atom. All of the analogs within this group demonstrated similar maximum aqueous solubility (100 – 200 uM range in 2% DMSO/PBS) that were well within the range tested in our assays. All of the analogs tested, including 5F02, were found to be labile to metabolism in both mouse and human liver microsomes in the presence of NADPH, something that is not totally unexpected given the presence of the cycloalkyl group. However, all of the analogs within this group were stable in microsomes in the absence of NADPH, suggesting that the ester group is surprisingly stable to enzymatic hydrolysis. To address the question of the necessity of the positive charge on the quaternary nitrogen we tested the penultimate intermediates (lacking the quaternary methyl group) of each final target molecule. We had previously shown that the des-methyl, non-quaternary intermediate leading to 5F02 was essentially inactive in the H4/PARP1 assay established in the Tulin lab (Karpova et al., 2019).
Table 1.
In vitro H4/PARP1 inhibitory activity, maximum kinetic aqueous solubility and mouse and human liver microsomal stability of compounds 1–9.
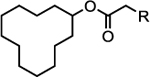 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cd # | R | logP | H4/PARP Inhibition IC50, nM | Liver Microsomal Stability (t1/2, min) | Max. Aq. Solubility (uM) | |||
| Mouse | Human | |||||||
| + NADPH | - NADPH* | + NADPH* | - NADPH* | |||||
| 1 |  |
5.11 | 104 | 4.9 | 92% | 3.4 | 88% | 177 |
| 2 |  |
4.66 | 52.4 | 3.8 | 98% | < 2 | 92% | 125 |
| 3 | 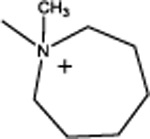 |
5.56 | 54 | 1.8 | 87% | < 2 | 95% | 140 |
| 4 | 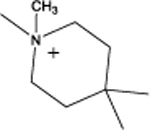 |
5.52 | 45.6 | 2.6 | 84% | < 2 | 92% | 95 |
| 5 |  |
3.65 | 34.5 | 5.1 | 100% | < 2 | 87% | 200 |
| 6 |  |
4.32 | 156 | 4 | 88% | < 2 | 85% | 110 |
| 7 | 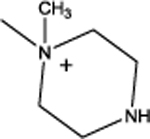 |
3.34 | 466 | 13.3 | 100 | 4 | 100 | 185 |
| 8 | 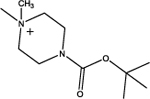 |
5.08 | 55.8 | 4.1 | 100 | < 2 | 83 | 200 |
| 9 | 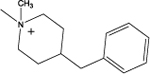 |
6.79 | 61.8 | 5 | 100 | < 2 | 87 | 113 |
Percent compound remaining after 1 hour @ 37°C.
A representative group of non-charged derivatives (Intermediates 1a – 9a) showed no inhibition of H4-stimulated PARP1 activity in our hands at concentrations up to 10 uM (Table S1, Supplementary Information section). Thus, either the charge or the presence of the quaternary alkyl group is necessary for potent inhibitory activity.
Since the initial SAR results indicated that the quaternary nitrogen played some role in binding, we next explored the size of the alkyl group on that quaternary nitrogen. The results are shown in Table 2. The ethyl analog (compound 10) was less potent than 5F02 (compound 1). Interestingly, the more flexible n-propyl (compound 11) and n-butyl (compound 12) derivatives, while not quite as potent as 5F02, were more potent than the ethyl analog. A size limitation was reached with the n-pentyl group (compound 13), which demonstrated a significant loss in potency compared to the other derivatives in this group. Maximum aqueous solubility for these compounds was similar to that seen with 5F02. Compounds 12 and 13, with the longer alkyl chains, demonstrated increased stability in mouse liver microsomes in the presence of NADPH compared to compounds 1–11. However, both compounds showed some loss of material in liver microsomes in the absence of NADPH, suggesting a potential liability to hydrolytic metabolism.
Table 2.
In vitro H4/PARP1 inhibitory activity, maximum kinetic aqueous solubility and mouse and human liver microsomal stability of compounds 10–13.
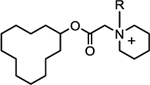 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cd # | R | logP | H4/PARP Inhibition IC50, nM | Liver Microsomal Stability (t1/2, min) | Max. Aq. Solubility (uM) | |||
| Mouse | Human | |||||||
| + NADPH | - NADPH* | + NADPH* | - NADPH* | |||||
| 1 | Me | 5.11 | 104 | 4.9 | 92% | 3.4 | 88% | 177 |
| 10 | Et | 5.52 | 174 | 4.7 | 96% | < 2 | 87% | 200 |
| 11 | n-Pr | 5.97 | 142 | 8.9 | 100% | < 2 | 100% | 194 |
| 12 | n-Bu | 6.42 | 136 | 16.2 | 89% | < 2 | 100% | 186 |
| 13 | n-Pentyl | 6.87 | 258 | 16.0 | 70% | < 2 | 100% | 196 |
Percent compound remaining after 1 hour @ 37°C.
The next region of the 5F02 scaffold that we explored was the cyclododecyl group. To accomplish this task we systematically reduced the ring size in the order: 12 (1):10 (14):8 (15):6 (16):5 (17). The results are presented in Table 3.
Table 3.
In vitro H4/PARP1 inhibitory activity, maximum kinetic aqueous solubility and mouse and human liver microsomal stability of compounds 14–17.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cd # | R | logP | H4/PARP Inhibition IC50, nM | Liver Microsomal Stability (t1/2, min) | Max. Aq. Solubility (uM) | |||
| Mouse | Human | |||||||
| + NADPH | - NADPH* | + NADPH* | - NADPH* | |||||
| 1 |  |
5.11 | 104 | 4.9 | 92% | 3.4 | 88% | 177 |
| 14 |  |
4.21 | 53.7 | 6.2 | 99% | 7.7 | 100% | 181 |
| 15 | 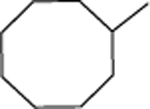 |
3.31 | 80.0 | 2.2 | 97% | > 60 | 95% | 182 |
| 16 |  |
2.41 | 77.1 | 2.9 | 100% | > 60 | 100% | 187 |
| 17 |  |
1.96 | 57.1 | 21.2 | 97% | > 60 | 90% | 200 |
Percent compound remaining after 1 hour @ 37°C.
Reducing the ring size, on the whole, resulted in derivatives with increased H4/PARP1 inhibitory potency, although as with the other structural changes the increase in potency was not dramatic. This limited sensitivity to structural changes is not uncommon for small molecule inhibitors of protein-protein interactions (Jin et al., 2014). The greatest effect was seen with the cyclodecyl derivative 14 and the cyclopentyl analog 17, which both demonstrated approximately 2-fold greater inhibitory potency than 5F02. Reducing the size of the cycloalkyl ring, however, had a beneficial effect on stability to oxidative metabolism. While the cyclodecyl analog was still very labile to oxidative metabolism in mouse and human liver microsomes, compounds 15 – 17 were found to be stable in human liver microsomes for up to 60 minutes (the longest time tested) and the cyclopentyl derivative 17 also demonstrated increased stability in mouse liver microsomes (t1.2 = 21.2 minutes). All of these derivatives were stable in mouse and human liver microsomes in the absence of NADPH, and these results were not influenced by solubility limitations. Thus, the cyclocodecyl group of 5F02 appears to be a weak spot for oxidative metabolism.
The last region of the molecule we explored was the ester group. We had previously reported that substitution of the ester with a secondary amide significantly reduced the H4/PARP1 inhibitory activity of the 5F02 scaffold significantly (Karpova et al., 2019). Hypothesizing that either a hydrogen bond donor in this position might be detrimental to binding or the presence of the secondary amide might be stabilizing an unfavorable conformation compared to the ester group (Lewin and Frucht, 1975), we prepared the tertiary amide derivative 18. Screening results are summarized in Figure 2.
While not as potent as the homologous ester analogs 1–9, compound 18 did inhibit H4/PARP1 activity to a greater extent than the analogous secondary amide (Karpova et al., 2019), giving an IC50 value in our hands of 191 nM. Unfortunately, the compound was still labile to oxidative metabolism in liver microsomes.
Because no dramatic SAR trends were realized in our SAR studies we elected to test most of the analogs in our in vitro clonogenic cancer cell model. The model employs the human isogenic leukemia cell line NALM6 wherein the cells have been made BRCA-deficient by eliminating RAD54 activity (RAD54−/−) (Fujita et al., 2013). Results in this cell line are compared to results obtained with RAD54+/+ parental cells which possess RAD54 activity and are BRCA- proficient to ascertain the selectivity of compounds for BRCA-deficient cancers. Results are presented in Figure 3.
Figure 4. The effect of 5F02 allosteric analogs on the clonogenic activity of RAD54 deficient and proficient leukemia cells.

NALM6 parental cells and NALM6 RAD54−/− isogenic counterparts (103/100 μL) were treated for 72 hours with the indicated compounds followed by plating in methylcellulose as described before (Nieborowska-Skorska et al., 2017).7 Results represent mean ± SD percentage of colonies in comparison to untreated cells from triplicate experiments.
5F02 (1) demonstrated a therapeutic window for killing BRCA-deficient cells (open circles) compared to BRCA-proficient cells (closed circles). In general, compounds that demonstrated a lower IC50 value in the in vitro H4-stimulated PARP1 assay tended to be more potent in the NALM6 model, although most of the compounds tested showed little or no selectivity for BRCA- deficient cells over the BRCA-proficient parental cells. The two exceptions to this trend toward lack of selectivity were compounds 5 and 7, which were both more potent at killing BRCA- deficient NALM6 RAD54−/− leukemia cells than the RAD54+/+ BRCA-proficient parental counterparts. The reasons for this are not clear at present, but the results emphasize the limitations of relying only on biochemical assays for profiling anti-cancer compounds and the need to support biochemical results with data generated in live tissue.
The structural changes made to date resulted in only limited improvements in metabolic stability and the trend was seen more in mouse liver microsomes than in human liver microsomes. Those changes that imparted the greatest improvement in liver microsomal stability resulted in compounds that lacked BRCA-deficient selectivity. That being said, 5F02 possessed a longer than expected in vivo half-life, which might be the result of its relatively high plasma protein binding and/or saturation of metabolism at higher doses (Karpova et a., 2019). Surprisingly, the ester group appears relatively stable to hydrolytic metabolism, which could indicate that it is shielded from enzymatic hydrolysis by the cycloalkyl group and/or the quaternary amine moiety.
Conclusions
In conclusion, we have expanded our previous findings on non-NAD like inhibitors of H4- stimulated PARP1 activity to obtain a better understanding of the SAR and SPR within this chemical scaffold. Compound 7 displayed potent, BRCA-selective anti-cancer activity in the NALM6 human leukemia cell line and a greater stability in mouse liver microsomes compared to 5F02 (1) while compound 17 gave indication that manipulating the cycloalkyl group may lead to compounds with improved metabolic stability. Synergy studies with compound 7 and olaparib are currently in progress, as are mechanistic studies to better understand the mechanism for the selectivity seen with 5F02 and our lead molecule. These results will be reported in the future.
Supplementary Material
Figure 3.

Structure and screening data for compound 18.
Acknowledgements
The authors would like to thank Dr. Furong Sun and the staff at the Mass Spectrometry Lab, School of Chemical Sciences, University of Illinois at Urbana-Champaign for providing the high resolution mass spectrometry data.
Funding
This work was funded by the National Institutes of Health/National Cancer Institute under R01 CA186238 to TS. MT was supported by an Etiuda6 scholarship awarded to her by the Polish National Science Centre.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
All experiments carried out in the course of this work were performed in compliance with OSHA regulations and under protocols that were approved by the Temple University Institutional Biosafety Committee.
References and Notes
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zeigler K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lkas J, Bartek J (2005) DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434:864–870 [DOI] [PubMed] [Google Scholar]
- Dasgupta Y, Golovine K, Nieborowska-Skorska M, Drugging DNA repair to target T-ALL cells. Leuk Lymphoma 59:1746–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, Sasanuma H, Yamamoto KN, Harada H, Kurosawa A, Adachi N, Omura M, Hiraoka M, Takeda S, Hirota K (2013) Inerference in DNA replication can cause mitotic chromosomal breakage unassociated with double-strand breaks. PLoSOne 8: e60043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain PG, Patel BD (2019) Medicinal chemistry approaches of poly ADP-ribose polymerase 1 (PARP1) inhibiors as anticancer agents – a recent update. Eur J Med Chem 165:198–215 [DOI] [PubMed] [Google Scholar]
- Jin L, Wang W, Fang G (2014) Targeting protein-protein interaction by small molecules. Ann Rev Pharmacol Toxicol 54:435–456. [DOI] [PubMed] [Google Scholar]
- Karanam K, Kafri R, Loewer A, Layay G (2012) Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell 47:320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova Y, Wu C, Divan A, McDonnell ME, Hewlett E, Makhov P, Gordon J, Ye M, Reitz AB, Childers WE, Skorski T, Kolenko V, Tulin AV (2019) Non-NAD-like PARP-1 inhibitors in prostate cancer treatment. Biochem Pharmacol 167:149–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsanov KI, Kotova E, Makhov P, Golovine K, Lesovaya EA, Kolenko VM, Yakubovskaya MG, Tulin AV (2014) Minor groove binding ligands disrupt PARP-1 activation pathways. Oncotarget 5:428–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslov NS, Kovaleva VN, Vecher EI, Boreiko EI, Vladyko GV, Korobchenko LV (1989) Synthesis and antiviral properties of cyclohexanol amino esters. khimichnykh navuk 2:66–68 [Google Scholar]
- Lewin AH, Frucht M (1975) Restricted rotationin amides VII. Methods of resonance assignment in tertiary amides – an evaluation. Org Mag Res 7:26–225 [Google Scholar]
- Lord CJ, Tutt AN, Ashworth A (2015) Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu Rev Med 66:455–470 [DOI] [PubMed] [Google Scholar]
- Maifrede S, Martin K, Podszywalow-Bartnicka P, Sullivan-Reed K, Langer SK, Nejati R, Dasgupta Y, Hulse M, Gritsyuk D, Nieborowska-Skorska M, Lupey-Green LN, Zhao H, Piwocka K, Wasik MA, Tempera I, Skorski T (2017) Mol Cancer Res 15:967–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malyuchenko NV, Lotova EY, Kulaeva OI, Kirpichnikov MP, Studitskiy VM (2015) PARP1 inhibitors: antitumor drug design. Acta Naturae 7:27–37 [PMC free article] [PubMed] [Google Scholar]
- Metzger MJ, Stoddard BL, Monnat RJ Jr (2013) PARP-mediated repair, homologous recombination, and back-up no-homologous end joining-like repair of single-strand breaks. DNA Repair (Amst) 12:529–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieborowska-Skorska M, Sullivan K, Dasgupta Y, Podszywalow-Bartnicka P, Hosner G, Maifrede S, Martinez E, Di Marcantonio D, Bolton-Gillespie E, Cramer-Morales K, Lee J, Li M, Slupianek A, Gritsyuk D, Cerny-Reiterer S, Seferynska I, Stoklosa T, Bullinger L, Zhao H, Gorbunova V, Piwocka K, Valent P, Civin CI, Muschen M, Dick JE, Want JC, Bhatia S, Bhatia R, Eppert K, Minden MD, Sykes SM, Skorski T (2017) Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J Clin Invest 127:2392–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieborowska-Skorska M, Maifrede S, Ye M, Toma M, Hewlett E, Gordon J, Le BV, Sliwinski T, Zhao H, Piwocka K, Valent P, Tulin AV, Childers W, Skorski T (2019) Non-NAD-like PARP1 inhibitors enhanced synthetic lethal effect of NAD-like PARP inhibitors against BRCA1-deficient leukemia. Leuk Lymphoma 60:1098–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickoloff JA, Jones D, Lee S-K, Williamson EA, Hromas R (2017) Drugging the cancers addicted to DNA repair. J Natl Can Inst 109:djx059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenker PK, Gunthard HH (1952) Carbon rings LX. The ten-membered ring. V. Helv. Chem. Acta 35:1598–1615 [Google Scholar]
- Sullivan-Reed K, Bolton-Gillespie E, Dasgupta Y, Langer S, Siciliano M, Nieborowska-Skorska M, Hanamshet K, Belyaeva EA, Bernhardy AJ, Lee J, Moore M, Zhao H, Valent P, Metlawska-Wasowska K, Muschen M, Bhatia S, Bhatia R, Johnson N, Wasik MA, Mazin AV, Skorski T (2018) Simultaneous targeting of PARP1 and RAD52 triggers dual synthetic lethality in BRCA-deficient tumor cells. Cell Rep 23:3127–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Ji Y, Lodhi N, Kotova E, Pinnola AD, Golovine K, Makhov P, Pechenkina K, Kolenko V, Tulin AV (2016) Non-NAD-like poly(ADP-ribose) polymerase-1 inhibitors effectively eliminate cancer in vivo. EBioMedicine 13:90–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hamdy FC, Helleday T (2012) Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 72:2814–2821 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.