

Abstract
Heat shock protein 90 (Hsp90) was the focus of a recent meeting in the Swiss Alps, where the Hsp90 community met to discuss the operation and functions of this ubiquitous and essential molecular chaperone.
Molecular chaperones of the Hsp90 family execute the late-stage folding of a significant fraction of the eukaryotic cell proteome. So-called ‘client’ proteins of Hsp90 have diverse cellular functions, but an overarching functional theme for many of them is an involvement in signal transduction. This causes several biological processes to be affected when the activity of Hsp90 becomes limiting. Here we summarize some of the new insights presented at the 5th International Conference on the Hsp90 Chaperone Machine, held recently in Les Diablerets, Switzerland—most notably, in regard to structural and mechanistic aspects of the chaperone cycle and how Hsp90 can empower evolution. The meeting also covered the continued development and emergent medical applications of highly selective inhibitors of Hsp90.
Asymmetry in Hsp90–cochaperone complexes
Hsp90 acts in cooperation with a number of accessory proteins, or cochaperones. Different cochaperones sequentially associate with Hsp90 as the chaperone cycle progresses; their roles include the regulation of Hsp90 ATPase activity and the bridging of Hsp90 to Hsp70 or client proteins. Not only do the different cochaperones often show preferences for different conformational states of Hsp90, but by binding at discrete stages of the Hsp90 cycle, they also exert temporal control over the conformational changes within the Hsp90–client complex and the residence time of the client on Hsp90. Evidence is now accumulating that many of these complexes are asymmetric. That is, Hsp90, a dimeric molecule (Fig. 1), sometimes associates with just a single cochaperone molecule, as when a single Aha1 molecule bridges the two subunits simultaneously to stimulate ATPase activity1, and at other times associates with several different cochaperones.
Figure 1.
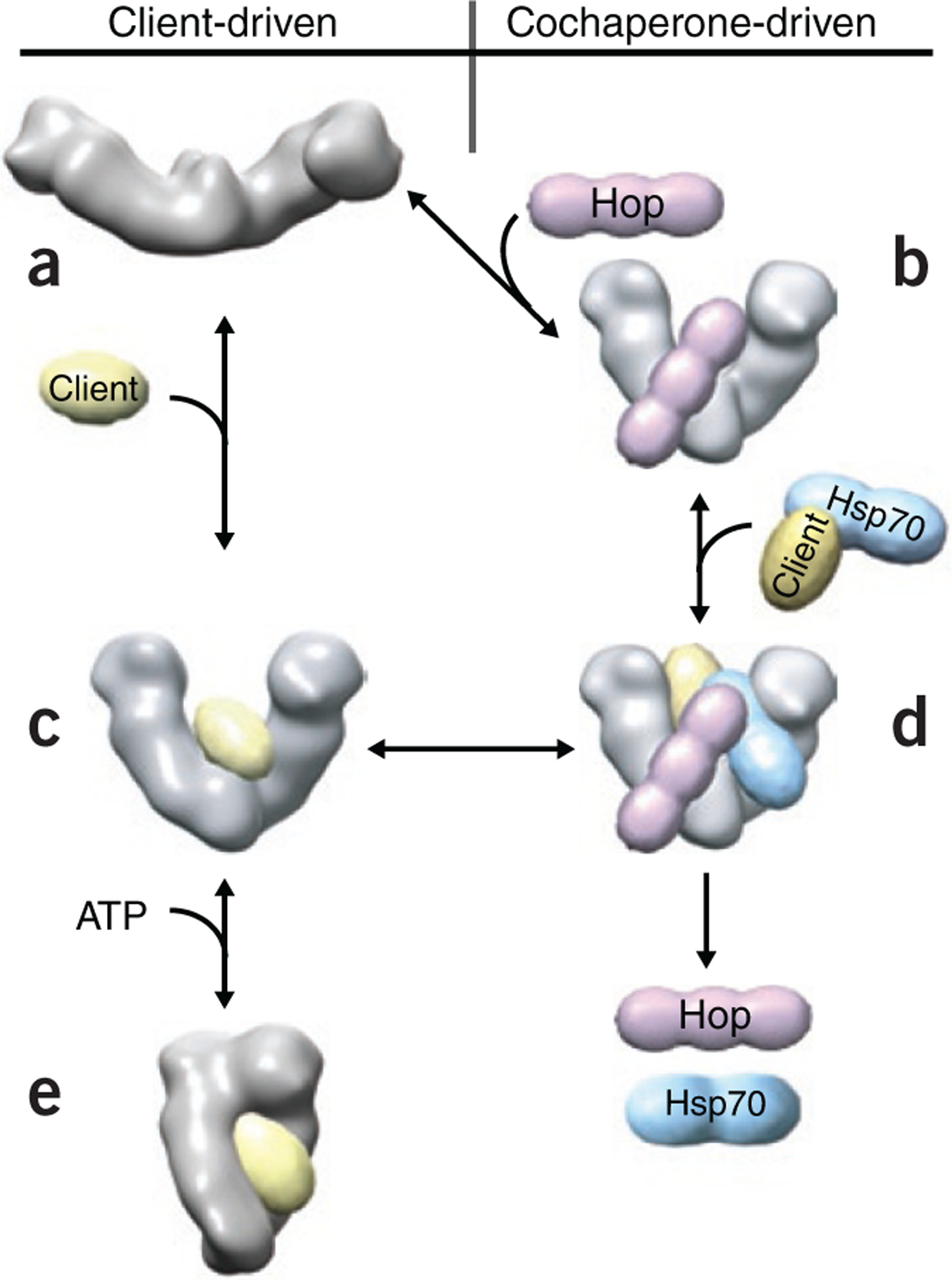
A model of Hsp90 client loading.
(a) EM structure of the apo-state. (b) EM structure of the Hsp90–Hop complex. (c) The NMR, SAXS and FRET data for the staphylococcal nuclease Δ131Δ-loaded Hsp90. (d) A hypothetical model of client loading on Hsp90 via Hsp70 and Hop. (e) Final closed ATP-bound conformation.
Structures a, b and c suggest a common structural pathway for both client-driven and cochaperone-driven loading of client proteins to the Hsp90 dimer via a V-shaped structure (b and c); the latter being intermediate between the apo form a and the final closed ATP-bound conformation e. Figure courtesy of D. Southworth, T. Street and D. Agard, University of California, San Francisco.
Johannes Buchner (Technische Universität München, Garching, Germany) described how fluorescence resonance energy transfer (FRET), when used in combination with analytical ultracentrifugation (AUC), can monitor these cochaperone exchanges during the progression from one Hsp90 complex to another. Cpr6 can bind simultaneously with Sti1, indicating that the two C-terminal MEEVD motifs in the Hsp90 dimer are capable of interacting with separate TPR domain–containing cochaperones. Addition of p23 and AMPPNP to the Hsp90–Sti1 complex resulted in a partial displacement of Sti1, with further displacement occurring on addition of Cpr6.
The cochaperone Sgt1 links Hsp90 function to nucleotide-binding leucine-rich repeat (NLR) receptors of innate immunity. In plants, Sgt1 acts together with the disease resistance protein Rar1, a cochaperone with tandem cysteine- and histidine-rich domains (CHORDs). Chris Prodromou (University of Sussex, Brighton, UK) presented the crystal structure of the symmetrical complex formed by the Hsp90 N-terminal domain (NTD), the CHORD II domain of Rar1 and the CS domain of Sgt1 (ref. 2). This symmetrical structure is believed to convert to an asymmetric structure, as the CHORD I and CHORD II domains of Rar1 can both bind the Hsp90 NTD, but only the CHORD II domain can associate with Sgt1. An exciting finding from this work is the unusual mechanism whereby Rar1 binding stimulates the Hsp90 ATPase activity. Rar1 displaces the ATP-lid from Hsp90’s ATP binding site and, by physically inserting itself between each NTD of the Hsp90 dimer, prevents the NTD domain dimerization that had previously been considered a prerequisite for ATP hydrolysis. Other cochaperones may also be found to stimulate the Hsp90 ATPase in this way.
Addressing the conformational flexibility of Hsp90
Matthias Mayer (Zentrum für Molekular Biologie der Universität Heidelberg) presented investigations into the conformational flexibility of Hsp90 by amide hydrogen-deuterium exchange and mass spectrometry (HX-MS). These experiments reveal that the eukaryotic Hsp90s are considerably more flexible than their Escherichia coli counterpart HtpG, and this difference may allow cochaperones (which are absent from E. coli) to mould Hsp90 for interaction with its myriad of different client proteins. Sti1 binding was found to restrict Hsp90’s flexibility throughout the entire length of the protein. The single molecule FRET analysis by Thorsten Hugel (Technische Universität München, Garching, Germany) also highlighted this conformational flexibility of Hsp90. It has revealed not only that the C-terminal domains (CTDs) within the Hsp90 dimer undergo rapid association and dissociation3, but also that there is an anti-correlation between this dimerization and the dimerization of the NTDs, such that the CTDs tend to be nonassociated when the NTDs are dimerized and vice versa. The functional significance of this observation is still unclear.
Hsp90 and cochaperone interaction networks
Didier Picard (University of Geneva) described a comprehensive in silico protein-protein interaction network for Hsp90 based on existing protein interaction databases, with GO term annotation clustering the proteins according to specific pathways. A prediction of this network has been experimentally validated in his laboratory, suggesting that the network will be an indispensible resource for the Hsp90 community. Picard maintains the Hsp90 interactor database (http://www.picard.ch/downloads/downloads.htm). Brian Freeman (University of Illinois, Urbana) described the protein interaction network of the cochaperone p23/Sba1, established partly from a synthetic growth analysis screen in yeast, by crossing a sba1Δ mutant with ~4,500 single-gene deletion strains. Interestingly, less than one-third of the identified p23 interactors overlap with known interactors of Hsp90. A holistic view, however, showed that these p23 and Hsp90 interactors could often be traced to the same complex or pathway, indicating that although p23 can act independently of Hsp90, it does so in a manner that is frequently complementary to Hsp90 function. The study highlighted the importance of p23 in Golgi transport and nuclear functions, including RNA processing, DNA repair and replication, chromatin remodeling, telomere maintenance and transcription.
Hsp90 is controlled by regulatory phosphorylation
There is now substantial evidence that Hsp90 is the target of regulatory phosphorylation events. Len Neckers (National Cancer Institute (NCI), US National Institutes of Health (NIH), Bethesda, Maryland) outlined the discovery of two topologically related phosphorylation sites in the Hsp90 NTD. Phosphorylation at these sites in yeast Hsp82 and human Hsp90α is mediated by homologous kinases, affects both client and cochaperone interactions with Hsp90, and influences the sensitivity of yeast and cancer cells to Hsp90 inhibitors4. Several other sites of phosphorylation in the middle and CTD regions of yeast Hsp90 were described by Joanna Soroka and Johannes Buchner (Technische Universität München, Garching, Germany); the analyses of unphosphorylated and phosphomimetic mutant forms of Hsp90s in vivo and in vitro reveal the functional importance of these phosphorylation events.
Client protein recognition and stabilization
How Hsp90 recognizes the partly folded state of its client proteins and facilitates structural transitions within these clients are important, yet still poorly understood, aspects of Hsp90 function. David Agard (University of California, San Francisco) described use of the staphylococcal nuclease deletion mutant Δ131Δ as a model client. Results from small-angle X-ray scattering (SAXS) and NMR show that a single Δ131Δ molecule binds the Hsp90 dimer, stabilizing a V-shaped Hsp90 conformation (Fig. 1). FRET experiments confirm that ATP binding accelerates the closure of this Hsp90 dimer. In addition, a 15-Å cryo-EM structure of the Hsp90–Hop complex reveals how Hop blocks the Hsp90 ATPase yet promotes client binding via a newly discovered V-shaped Hsp90 conformation, which is intermediate between the apo and ATP-bound conformations (Fig. 1).
Stefan Rüdiger (Utrecht University) described the use of selective methyl labeling of isoleucine residues to map the binding of the client protein Tau by NMR. Tau is a microtubule-associated protein whose aggregation in neurons has been implicated in a number of neurodegenerative diseases. Binding of Tau was found to produce chemical shift perturbations in a region of the Hsp90 NTD not involved in known cochaperone interactions. Nucleotide addition generated significant chemical shifts in other parts of the molecule, suggesting the intriguing possibility that the client binding site may change throughout the ATPase cycle in a manner that is synchronous with the conformational changes of Hsp90.
Hsp90 is thought to promote the stability of its clients, because these are generally destabilized when cells are treated with Hsp90 inhibitors. The meeting revealed some exciting new twists as to how Hsp90 can influence protein stability in vivo. Walid Houry (University of Toronto) reported that Hsp90 facilitates the processing of pre-ribosomal RNA by means of a protein stabilization ‘cascade’. In association with the small TPR cochaperone Tah1, Hsp90 first stabilizes a factor, Pih1, thereby enabling the formation of R2TP, a complex comprising two essential helicases, Rvb1 and Rvb2, together with Tah1 and Pih1 (ref. 5). R2TP then in turn stabilizes Nop58, a component essential for assembly of the box C/D small nucleolar ribonucleoproteins (snoRNPs) that function to methylate pre-rRNA.
The p53 tumor suppressor remains one of the most tantalizing of Hsp90 clients. Mutation of p53, generating dysfunctional states of this largely unstructured transcription factor, is a major causative element in the development of human cancer. Maciej Zylicz (International Institute of Molecular and Cell Biology, Warsaw) outlined how wild-type p53 undergoes a dynamic cycling with Hsp90. Experiments in which WT p53 was used in conjunction with mutant forms of Hsp90 revealed that it is binding of ATP by Hsp90, not the ATPase activity of this chaperone, that causes the Hsp90–p53 complex to dissociate and that allows binding of the native p53 to the WAF1 promoter. Hsp90–p53 association triggers a conformational change, possibly a partial p53 unfolding, and the subsequent ATP-dependent dissociation of this Hsp90–p53 complex enables p53 to attain a conformation where it is capable of binding the promoter sequence6.
A new dimension to Hsp90 downregulation of the stress response
Hsp90 negatively regulates heat shock transcription factor (HSF). Patricija van Oosten-Hawle (Northwestern University, Evanston, Illinois) described the cellular and organismal effects of overexpressing Hsp90 in different tissues of Caenorhabditis elegans. Within individual cell types, elevated Hsp90 suppressed the HSF-directed heat shock response. Unexpectedly, though, Hsp90 overexpression in one specific tissue seems to have an effect on the heat shock response in the organism as a whole. Although the C. elegans heat shock response is known to be controlled by thermosensory neurons7, it is still unknown how this alteration to Hsp90 levels in one tissue induces the stress response of an adjacent tissue.
Hsp90 deficiency and metazoan sterility
It has long been unclear why vertebrate species have two isoforms of cytosolic Hsp90, a heat shock–inducible isoform, Hsp90α, and a constitutively expressed isoform, Hsp90β (ref. 8). Hsp90α is expressed primarily in primordial germ cells and has been identified as an extracellular component associated with tumor progression9. Ahmed Chadli (Medical College of Georgia, Augusta) described how UNC45A (also called GCUNC45) is the only cochaperone so far found to distinguish between human Hsp90α and Hsp90β, having 7–10 times greater affinity for Hsp90α than for Hsp90β (ref. 10). UNC45A is required for optimal transcriptional activity of the progesterone and glucocorticoid receptors. Didier Picard investigated why the loss of Hsp90β in Hsp90β−/− (Hsp90ab1−/−) mice causes embryonic lethality, whereas Hsp90α seems dispensable for normal development, and found that Hsp90α−/− (Hsp90aa1−/−) male mice are sterile despite having an active Hsp90β, as their germ cells show evidence of a normal meiosis I until prophase but lack any subsequent spermatogenesis.
C. elegans has only a single cytosolic Hsp90, expressed primarily in germline cells and during oocyte development. It too is important in fertility. Klaus Richter (Technische Universität München, Garching, Germany) described RNA interference (RNAi) experiments that showed depletion of Sti1 caused sterility with impaired gonad development, whereas depletion of Hsp90 resulted in the protruding vulva (pvl) phenotypes11 previously observed for depletion of Cdc37.
Defects in Hsp90 mimic caloric restriction effects
Caloric restriction, defined as a reduction in energy intake without malnutrition, extends lifespan in species as diverse as yeast, worms, flies and rodents. In metazoans it acts by downregulating insulin-like growth factor (IGF) signaling12. Jill Johnson (University of Idaho, Moscow) revealed that a point mutation in the yeast Hsp90 is alone sufficient to recreate in glucose-grown cells the transcriptional responses that are normally only seen when cells are grown on a non-optimal carbon source (akin to caloric restriction) or starved for nitrogen. Yair Argon (University of Pennsylvania, Philadelphia) described how Grp94, the single Hsp90 family protein of the endoplasmic reticulum (ER), is essential for the folding of pro-IGF in a pre-Golgi compartment. Grp94−/− cells lack IGF secretion capacity and have a stress sensitivity rescued by the addition of exogenous IGF13,14. By influencing the levels of circulating IGF, Grp94 chaperone activity might prove to be a significant influence over the IGF-mediated signaling that drives the events of senescence and aging in diverse species12. Naturally occurring polymorphisms in the human Grp94 gene affecting the functionality of Grp94 might also be a significant cause of variation in circulating levels of IGF in humans.
Hsp90 empowering evolutionary change
Susan Lindquist (Whitehead Institute for Biomedical Research, Cambridge, Massachusetts) tackled the issue of how the protein homeostasis reservoir created by Hsp90 acts to promote not just phenotypic stability but also—when Hsp90 activity becomes limiting in development—the expression of cryptic mutations as Hsp90-contingent complex traits, with the latter leading to a rapid diversification of phenotypes15. She described how, by crossing ecologically and genetically diverse yeast strains and then analyzing their meiotic progeny, it is possible to correlate quantitative trait loci with high-resolution genome maps, thereby allowing one to ask the question of how phenotypes undergo alteration when Hsp90 activity levels become limiting. Remarkably, the correlations between phenotype and genotype improve with inhibition of Hsp90 or with stress, revealing that Hsp90 determines the adaptive value of preexisting genetic variation15.
Renato Paro (ETH Zürich, Basel) outlined how environmental cues and Hsp90 might influence epigenetic controls over gene expression. In Drosophila, Hsp90 stabilizes trithorax (trx), a protein involved in maintaining the gene expression crucial for development. Chromatin immunoprecipitation followed by deep sequencing reveals that these two proteins colocalize at the chromatin regions that correlate with gene expression. Hsp90 and trx also colocalize at interbands and at the major heat shock puffs on polytene chromosomes after heat shock16. Hsp90 appears therefore to be intimately involved in relaying environmental signals to the network that supervises cellular identities.
Arabidopsis thaliana provides another amenable system for studying how compromised Hsp90 activity results in an increased phenotypic variation17 and how Hsp90 may integrate growth and development with environmental cues. Jennifer Lachowiec (University of Washington, Seattle) described how brassinosteroids (BR) regulate essential processes in the growth and development of A. thaliana, binding to membrane-associated receptors that in turn signal to a set of downstream transcription factors. She revealed that, whereas Hsp90 stabilizes the inactive, phosphorylated form of one of the BR-responsive transcription factors, closely related paralogs presumed to act redundantly with this transcription factor appear to be Hsp90-independent. BR pathway genes are polymorphic in divergent A. thaliana strains, and the diversity of altered developmental phenotypes among these strains appearing with compromised Hsp90 exposes these polymorphisms to natural selection.
A different approach to this theme of how Hsp90 drives evolution is to address how chaperones shape the evolution of protein sequence space. RNA viruses show the highest known rates of mutation in nature, and the essential role of Hsp90 in picornavirus replication is the requirement for this chaperone in capsid protein maturation18. Judith Frydman (Stanford University, Palo Alto, California) described how compromised Hsp90 activity affects the evolutionary trajectory whereby mutations appear in this model client. She emphasized how the frequency and the nature of mutations will tend to reflect both influences that are intrinsic to the protein (protein robustness, notably a preference for neutral changes or ‘designable’ protein folds) as well as those that are extrinsic (chaperones, protein quality control machinery). Frydman then described how, when Hsp90 activity is compromised, amino acid substitutions decrease in frequency and become much more random in nature, whereas the frequency of silent mutations that slow translation increases. Full Hsp90 activity therefore facilitates diversity by helping to ensure that proteins evolve only through specific types of mutation and by disfavoring (‘buffering against’) mutations that are detrimental.
Disease states associated with defects in Hsp90-mediated protein homeostasis
Evidence is increasing that certain diseases reflect a defective folding or clearance of client proteins by the Hsp90 system. William Balch (The Scripps Research Institute, La Jolla, California) described a comparative interactome analysis by MS of normal and ΔF508 mutant forms of the cystic fibrosis (CF) transmembrane conductance regulator (CFTR) chloride channel, both in the absence and presence of proteostasis regulators (PRs), small molecules that regulate Hsp90 function. Normally ΔF508, the CFTR mutation associated with the most common form of CF, causes the retention of this channel protein in the ER, though the exit of this mutant form from the ER to the cell surface is rescued by growth at low temperature. Although the interactome data revealed that the ΔF508 CFTR protein underwent several aberrant protein interactions, these were largely corrected upon treatment with PRs, in a manner that resembles the rescue of CFTR function at low temperature. Appropriate pharmacological manipulation of the Hsp90 chaperone system might therefore be able to rescue the dysfunctional folding of the ΔF508 CFTR protein, thereby providing a potentially new therapy for CF.
Attention also focused on tau, a protein that normally binds and stabilizes microtubules, but that, in a number of neurodegenerative diseases (‘tauopathies’) that include Alzheimer’s and Parkinson’s, forms aggregates in neurons. Chad Dickey (University of South Florida, Tampa) described how the Hsp90 system can facilitate either a degradation or a stabilization of tau after microtubule disassembly. Either inhibition of Hsp90 or small interfering RNA (siRNA) downregulation of two cochaperones (p23 or FKBP51) facilitates the clearance of tau. Conversely, an overexpression of FKBP51 stabilizes tau, possibly by affecting tau kinase activity19. Full understanding of this chaperone processing of tau will clearly be important to the development of therapies designed to counteract the aggregation in neurodegenerative disease.
Hsp90 inhibitors in cancer
The ongoing development and clinical evaluation of novel Hsp90 inhibitor cancer drugs attests to the high interest of the pharmaceutical industry in this drug target. Both Jane Trepel (NCI, NIH) and Paul Workman (Institute of Cancer Research, London, UK) reviewed progress in this area and the challenges that confront the development of Hsp90 inhibitors to treat cancer. Ever since 17-allylaminodemethoxygeldanamycin (17-AAG) first entered the clinic 12 years ago, 16 other Hsp90 inhibitors have been, or are currently being, clinically evaluated. The effects of Hsp90 inhibition on both normal and tumor cell physiology are complex because of the wide variety of cellular processes affected by Hsp90, necessitating the development of new systems approaches to better evaluate and predict anti-cancer efficacy20. Nonetheless, several recent clinical trials have demonstrated promising activity in selected malignancies21. It may become necessary to develop inhibitors of various cochaperones and of Hsp70 family proteins that can either be administered alone or used synergistically with inhibitors of Hsp9022,23. Alternatively, drugs could be designed against a specific client. Marc Cox (University of Texas, El Paso) described a unique class of nuclear receptor alternate site modulators (NRAMs) that target the FKBP52 cochaperone dependence of the androgen receptor (AR). AR is frequently mutated to impart an enhanced FKBP52 dependence in late-stage prostate cancer. Cox described a lead compound that interacts with the AR ligand-binding domain but that does not compete with either hormone or coactivator binding. It thereby prevents hormone-induced AR dissociation from the Hsp90–FKBP52 chaperone complex and AR nuclear translocation and transcriptional activity.
The development of inhibitors binding within the Hsp90 NTD continues unabated. Zhenhai Gao (Novartis) reviewed development of the fully synthetic, orally available NVPHSP990. This, unlike most other Hsp90 inhibitors, can penetrate into the brain. It is now in phase 1 clinical trials in solid and hematological tumors. Gao also described new secreted biomarkers identified by an in silico data-mining approach. Several of these have been validated as useful pharmacodynamic readouts of Hsp90 inhibitor activity in vivo and may therefore prove useful in the ongoing clinical development of Hsp90 inhibitors. Weiwen Ying (Synta Pharmaceuticals) described an Hsp90 inhibitor (STA-9090) that is up to 100-fold more potent than 17-AAG. Active in vitro and in vivo against a wide range of tumor types, STA-9090 shows potent activity in cancers that have developed resistance to tyrosine kinase inhibitors. Based on encouraging signs of activity, a phase 2 study in non–small cell lung cancer has been expanded.
Ultimately the effectiveness of cancer drugs is often compromised by the emergence of drug resistance. As the ATP pocket of Hsp90 is highly conserved, part of the compelling case for developing NTD inhibitors that bind within this pocket rests on the belief that Hsp90 mutations that disrupt drug binding should severely compromise chaperone function and therefore not be selected under drug pressure. Peter Piper (University of Sheffield, Sheffield, UK) described Hsp90 mutations that provide partial resistance to either radicicol24 or 17-AAG in vivo in yeast engineered to express the human Hsp90s25. Importantly, binding of either drug is affected by distinct amino acid substitutions, suggesting that should resistance to a particular Hsp90 inhibitor develop in vivo, it is likely that sensitivity to other Hsp90 inhibitors will be maintained.
One of the drawbacks of NTD inhibitors is that they tend to induce the cytoprotective heat shock response and therefore increase levels of major chaperones such as Hsp70 and Hsp90 (ref. 26). Ultimately this may limit their clinical usefulness. Other drugs that target the Hsp90 CTD may show promise in this regard. Brian Blagg (University of Kansas, Lawrence) described the development of two distinct classes of CTD inhibitor, starting from the structure of the first CTD inhibitor, novobiocin. One class induces a robust heat shock response at nanomolar concentrations, protects neurons against toxic protein (Aβ)-induced apoptosis, and counteracts diabetic peripheral neuropathy27,28. A second class of CTD inhibitors fails to induce a heat shock response and is highly cytotoxic to tumor cells in vitro and in vivo29. Giorgio Colombo (Instituto di Chimica del Riconoscimento Moleculare, Milan, Italy) outlined the first in silico discovery of Hsp90 inhibitors that also target the CTD30. Importantly, by this completely independent approach, they identified a drug binding site similar to that of Blagg and colleagues.
Targeting Hsp90 to treat infectious diseases
Finally, it is now apparent that Hsp90 inhibitor drugs will likely have a clinical utility beyond cancer. A number of parasitic diseases seem very amenable to these agents. Leah Cowen (University of Toronto) described how abrogating Hsp90 function is a promising strategy to counteract both the virulence and the emergence of drug resistance by fungal pathogens. In the major human pathogen Candida albicans, Hsp90 can promote resistance to two widely deployed classes of antifungal drugs, the azoles and the echinocandins31,32, via its influence on calcineurin or protein kinase C signaling pathways. The morphogenetic switching of C. albicans from its yeast to its hyphal state (filamentation), a key determinant of virulence, can be triggered by combined heat and serum treatment. Hsp90 inhibitors can bypass the temperature requirement for filamentation that relies on the Ras–protein kinase A pathway33. Selective downregulation of C. albicans Hsp90, using a tetracycline (Tet) promoter–regulated Hsp90 gene, led to a clearance of the fungal burden in systemically infected mice33.
Hsp90 is also a promising target for antimalarial drug development. Both Utpal Tatu (Indian Institute of Science, Bangalore, India) and Dylan Pillai (University of Toronto) described the antimalarial activity of Hsp90 inhibitors. 17-AAG, the Hsp90 inhibitor that has undergone extensive clinical trials in cancer patients, has proven effective in preventing parasite growth in human erythrocytes and in a mouse model34. PU-H71, another inhibitor in clinical evaluation in cancer patients, is also an effective inhibitor of malaria Hsp90. Using high-throughput screening, Pillai identified three malaria-specific Hsp90 inhibitors that kill parasites at nanomolar concentrations in vitro and synergize with the anti-malarial drug chloroquine35. Importantly, these compounds were able to kill a multi-drug-resistant clinical isolate of malaria that demonstrated reduced responsiveness to artemisinin, the anti-malarial currently recommended by the World Health Organization.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Contributor Information
Cara K Vaughan, Institute of Structural Molecular Biology, Birkbeck College, London, UK..
Len Neckers, National Cancer Institute, Bethesda, Maryland, USA..
Peter W Piper, University of Sheffield, Sheffield, UK..
References
- 1.Retzlaff M et al. Mol. Cell 37, 344–354 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Zhang M, Kadota Y, Prodromou C, Shirasu K & Pearl LH Mol. Cell 39, 269–281 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ratzke C, Mickler M, Hellenkamp B, Buchner J & Hugel T Proc. Natl. Acad. Sci. USA 107, 16101–16106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mollapour M et al. Mol. Cell 37, 333–343 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao R et al. J. Cell Biol 180, 563–578 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walerych D et al. J. Biol. Chem 285, 32020–32028 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prahlad V, Cornelius T & Morimoto RI Science 320, 811–814 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sreedhar AS, Kalmar E, Csermely P & Shen YF FEBS Lett 562, 11–15 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Eustace BK & Jay DG Cell Cycle 3, 1098–1100 (2004). [PubMed] [Google Scholar]
- 10.Chadli A, Felts SJ & Toft DO J. Biol. Chem 283, 9509–9512 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaiser AM, Brandt F & Richter KJ Mol. Biol 391, 621–634 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Fontana L, Partridge L & Longo VD Science 328, 321–326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostrovsky O, Ahmed NT & Argon Y Mol. Biol. Cell 20, 1855–1864 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ostrovsky O, Makarewich CA, Snapp EL & Argon Y Proc. Natl. Acad. Sci. USA 106, 11600–11605 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taipale M, Jarosz DF & Lindquist S Nat. Rev. Mol. Cell Biol 11, 515–528 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Tariq M, Nussbaumer U, Chen Y, Beisel C & Paro R Proc. Natl. Acad. Sci. USA 106, 1157–1162 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Queitsch C, Sangster TA & Lindquist S Nature 417, 618–624 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Geller R, Vignuzzi M, Andino R & Frydman J Genes Dev 21, 195–205 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jinwal UK et al. J. Neurosci 30, 591–599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trepel J, Mollapour M, Giaccone G & Neckers L Nat. Rev. Cancer 10, 537–549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim YS et al. Curr. Top. Med. Chem 9, 1479–1492 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davenport EL et al. Targeting heat shock protein 72 enhances Hsp90 inhibitor-induced apoptosis in myeloma. Leukemia (2010). [DOI] [PubMed] [Google Scholar]
- 23.Powers MV et al. Cell Cycle 9, 1542–1550 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Prodromou C et al. ACS Chem. Biol 4, 289–297 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Millson SH, Prodromou C & Piper PW Biochem. Pharmacol 79, 1581–1588 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Morimoto RI & Santoro MG Nat. Biotechnol 16, 833–838 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Lu Y, Ansar S, Michaelis ML & Blagg BS Bioorg. Med. Chem 17, 1709–1715 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Urban MJ et al. ASN Neuro 2, e00040 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donnelly AC et al. Med. Chem. Commun 1, 165–170 (2010). [Google Scholar]
- 30.Morra G, Genoni A, Neves MA, Merz KM Jr. & Colombo G Curr. Med. Chem 17, 25–41 (2010). [DOI] [PubMed] [Google Scholar]
- 31.LaFayette SL et al. PLoS Pathog 6, e1001069 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh SD et al. PLoS Pathog 5, e1000532 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shapiro RS et al. Curr. Biol 19, 621–629 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pallavi R et al. J. Biol. Chem published online, doi: 10.1074/jbc.M110.155317 (13 September 2010). [DOI] [Google Scholar]
- 35.Shahinas D, Liang M, Datti A & Pillai DR J. Med. Chem 53, 3552–3557 (2010). [DOI] [PubMed] [Google Scholar]