

Abstract
Fibrotic disease is a major cause of morbidity and mortality and is characterized by the transition of resident fibroblast cells into active myofibroblasts, identified by their expression of alpha smooth muscle actin. Myofibroblast differentiation is regulated by growth factor signaling and mechanical signals transduced through integrins, which converge at focal adhesion proteins (Src and FAK) and MAPK signaling, but lead to divergent outcomes. While details are known about individual pathways, little is known about their interactions. To this end, an ODE-based model of this cell signaling network was developed in parallel with in vitro experiments to analyze potential mechanisms of crosstalk and regulation of αSMA production. We found that cells lacking Src or FAK produce significantly less or more αSMA than wild type cells, respectively. Transforming growth factor beta 1 and fibroblast growth factor signal through ERK and MAPK p38 with different dynamic profiles to increase or decrease αSMA expression, respectively. Our model effectively recreated αSMA expression levels across a set of 22 experimental conditions and matched some features of transient phosphorylation of ERK and p38. These results support a potential mechanism for regulation of fibroblast differentiation: αSMA production is promoted by active p38 and Src and opposed by ERK.
Keywords: Alpha-smooth muscle actin (αSMA), Transforming growth factor beta (TGF-β1), Fibroblast growth factor (FGF), Src, Focal adhesion kinase (FAK), p38, Extracellular signaling-related kinase (ERK), Integrin
INTRODUCTION
Fibroblast cells play a key role in producing and maintaining connective tissue throughout the body. The ability of these cells to differentiate into active myofibroblasts is important during development and wound healing, but prolonged myofibroblast activation can lead to overproduction of extracellular matrix proteins and stiffening of the surrounding tissue. This stiffening can cause heightened differentiation of neighboring fibroblasts through force transduction pathways and can lead to detrimental fibrotic pathologies in many organ systems.42 One hallmark of the myofibroblast phenotype is the production of alpha smooth muscle actin (αSMA; all acroynms are shown in Table 1) stress fibers, which transmit intracellular forces and increase the contractility of the cells and surrounding tissue.42,47 Clarifying the inputs and intracellular mechanisms which govern myofibroblast differentiation will provide insights into the pathophysiology of many fibrotic diseases.
TABLE 1.
Acronyms and abbreviations.
| Term | Description |
|---|---|
| αSMA | Alpha smooth muscle actin: myofibroblast marker |
| TGF-β1 | Transforming growth factor beta 1 |
| TβR2 | Type II TGF-β1 receptor |
| FGF | Fibroblast growth factor |
| FAK | Focal adhesion kinase |
| ERK | Extracellular signaling-related kinase |
| Src | Tyrosine kinase found in focal adhesions |
| MAPK | Mitogen activated protein kinase |
| p38 | MAPK involved in non-canonical TGF-β1 signaling |
| MEF+/+ | Wild type mouse embryonic fibroblasts (MEFs) |
| FAK−/− | FAK null MEFs |
| SYF−/− | MEFs lacking Src, Yes, and Fyn, three Src family kinases |
Mechanical stress and transforming growth factor beta 1 (TGF-β1) are known to promote the myofibroblast phenotype42 and fibroblast growth factor (FGF) has been shown to promote the quiescent fibroblast phenotype,29 but the intracellular effectors of these environmental cues have significant cross-talk,27,37 as seen in Fig. 1. Cells can experience mechanical tension and substrate rigidity through integrins, which are transmembrane proteins that transduce forces from the ECM to intracellular structures like focal adhesions and stress fibers. Different isoforms of integrin subunits are activated to transmit mechanical signals by specific ECM proteins. Integrins with β3 subunits are activated by fibronectin and transmit mechanical signals through the tyrosine kinase Src.35,38 Src and β3 integrin together enhance TGF-β1 non-canonical signaling to p38.9,14,15 β1 integrin subunits activate focal adhesion kinase (FAK) in a stiffness dependent manner.13 Src and FAK are important in the formation and maintenance of focal adhesions and are known to form a complex and activate each other’s kinase activity to enhance downstream signaling.35,41
FIGURE 1.

Relevant signaling network. (a) The cartoon depicts the major pathways of myofibroblast regulation and convergence on p38 and ERK kinases. Degradation and interactions details are not shown for the sake of clarity. (b) The schematic shows the model protein species and reactions. Proximity to the yellow node indicates that the rate of the reaction depends on the activation of the upstream protein species. The dashed line represents the contribution of Src which is described in Eq. (2).
Signaling through integrins and growth factors appears to converge at the intracellular level on two particular kinases, p38 and ERK. TGF-β1 signaling and β1 integrin signaling both activate p38, which has been shown to promote the myofibroblast phenotype.24,33 Conversely, ERK is required for FGF induction of the quiescent fibroblast phenotype.11 FAK serves as a docking site for Src and enhances Src activation and signaling to p38,48 while transducing signals from integrins and FGF to enhance ERK activation and limit αSMA production.8,17 FGF and TGF-β1 stimulate both p38 and ERK; however, they are known to lead to divergent outcomes.12,29 The complex and dynamic interactions of these signaling pathways complicates the regulation of fibroblast differentiation.
Computational models of cell signaling networks have been developed and used in many biological systems to clarify complex interactions, especially when intracellular activation states are difficult to quantify.2 Some models have been developed for subsets of this system to clarify specific mechanisms, but do little to address network effects and responses.7,44,49 Modeling biological networks is challenging due to the high number of interactions, the range of relevant time scales, and the difficulty of acquiring quantitative data of intracellular kinetics. Despite these hurdles, many model strategies have been developed and successfully implemented in similar network settings.2 A model developed by Janes et al.26 integrates complex cytokine signals to predict apoptosis, and they countered these difficulties by focusing on data-derived models. Further analysis of the same model indicated that the dynamic range of a given intracellular signaling event is more important for system function than the signal strength, which lessens the need to define system components with absolute protein numbers or concentrations.25
The goals of this work are to clarify the roles of FAK and Src in linking integrin and cytokine signaling, characterize the signaling profiles of TGF-β1 and FGF through p38 and ERK in the regulation of αSMA, and develop a quantitative model to evaluate and compare potential mechanisms for protein interactions in the regulation of myofibroblast differentiation.
In this study, we developed a computational network model to clarify the complex crosstalk between TGF-β1, FGF, and integrin signaling regulating the differentiation of fibroblasts. Known TGF-β1, FGF, and integrin signaling to p38 and ERK through Src and FAK from previously reported literature informed the development of an ODE-based model of fibroblast differentiation in different chemical and mechanical environments. The model was refined by fitting to experimental results for αSMA production and dynamic phosphorylation events. Sensitivity analysis and two model comparison techniques were developed to evaluate different model hypotheses and delineate potential mechanisms.
MATERIALS AND METHODS
Cell Culture
Wild type mouse embryonic fibroblasts (MEF+/+), MEFs lacking Src, Yes and Fyn (SYF−/−),31 and MEFs lacking FAK (FAK−/−)23 were used in this study. These cell lines were provided by Steven Hanks (Vanderbilt University), and were originally isolated by Phillipe Soriano (Fred Hutchinson Cancer Research Center)31 and Dusko Ilic (Kumamoto University School of Medicine),23 respectively. Cells were cultured in DMEM supplemented with 10% FBS, 1% antibiotic-antimicrobial and 1% non-essential amino acids. Unless otherwise noted, cells were plated at a density of 8000 cells/cm2 on tissue culture plastic (TCP) and kept in serum-free conditions during treatment with 1 ng/mL TGF-β1 or 10 ng/mL FGF.
PDMS for Stiffness Studies
Polydimethylsiloxane (PDMS) (Sylgard 184 from Dow Corning) culture surfaces were made with 10:1:0, 10:1:5, and 20:1:2 ratios of silicone-elastomer base to elastomer curing reagent to silicone oil as previously described.4,39 Bulk stiffness of these formulations was measured to be 2.1, 0.9 and 0.24 MPa, respectively.39 The dishes were then sterilized under UV light for 40 min and coated with human full-length fibronectin in filtered carbonate-bicarbonate buffer overnight to ensure proper cell adhesion.
Immunohistochemistry
Coverslips were coated in fibronectin by overnight incubation in a 50 μg/mL solution in sterile carbonate-bicarbonate buffer and rinsed in PBS before addition of cells. After 24 h of treatment, cells were fixed with 4% paraformaldehyde and permeabilized in 0.4% triton, blocked in 1% BSA, and stained with a Cy3 conjugated monoclonal αSMA antibody (Sigma). Slides were mounted in Prolong gold with DAPI mounting media to stain the nuclei and imaged at 20× magnification.
Quantification of αSMA Production by Indirect ELISA
Indirect ELISA assays with αSMA polyclonal antibody (Abcam) were used to quantify αSMA expression after 24 h in serum-free conditions as previously described.21
Quantification of MAPK Activation by Western Blot
Western blots were used to study dynamics of MAPK activation in MEF+/+ and FAK−/− cells as previously described.21 Briefly, cells were serum-starved for 3–4 h before treatment, and were lysed and diluted to equal protein concentration in RIPA buffer supplemented with protease and phosphatase inhibitors. Relative p38 and ERK phosphorylation was quantified by densitometry analysis and normalized to a loading control (β-actin, total ERK, or total p38) (cell signaling antibodies) and then to the average MEF+/+ no treatment case within each time point for each experiment.
Statistical Analysis
For all experiments measuring outputs across a range of cell types and treatments, a two-way ANOVA was run within each time point to determine significant effects of cell type and treatment, and interactions between the two. The Holms–Sidak method and individual student t tests with an overall significance level of 0.05 were used for multiple comparisons within cell-type and treatment groups. One-way ANOVA was used for dose response experiments which were limited to one cell type. Non-parametric tests (ANOVA on ranks or rank sum tests) were used if the samples failed the Shapiro–Wilks normality test or had unequal variance (p < 0.05).
Model Development
The model is a system of ODEs describing the dynamics of relative protein activation and αSMA production in fibroblasts. To simplify the model, a normalized closed system was assumed, wherein the total amount of each protein species in the signaling pathway is conserved at a value of 1. While many of these proteins have multiple phosphorylation states and conformations which affect their enzyme activity, most protein species were simplified to 2 activation states, “on” or “off”. Integrin adhesion, activation, and clustering was represented by a dimerization step similar to the strategy used in the integrin signaling model by Hammer et al.49 Src, FAK, and TβR2 were given three activation states to capture more system interactions. In total, the model contains nine active variables (Table 2), 27 kinetic rate coefficients (Table 3), and 12 inputs and boundary conditions which can be varied experimentally and in silico. Figure 1b shows a general descriptive schematic of the interactions and protein species represented in the model. First order activation rates proportional to the relative activation of the upstream species were used to model signaling cascades, unless more specific interactions were known. Substrate mechanics were incorporated into the model at the level of signaling from activated integrins to intracellular kinases via a scaling factor proportional to the log of substrate stiffness.13 Michaelis–Menten kinetics was used in cases of direct phosphorylation, as with Src activation of FAK tyrosines in the 400–900 range and Src phosphorylation of TGFβ receptor 2 (TβR2). A more detailed description of model formulation can be found in online Appendix.
TABLE 2.
List of active variables.
| Variable name | Description |
|---|---|
| B1on | Amount of activated β1 integrin as a fraction of total |
| B3on | Amount of activated β3 integrin as a fraction of total |
| TBRT | TGF-β1 receptor (TβR2) with TGF-β1 ligand attached |
| pTBR2 | Phosphorylated and ligand bound TGF-β1 receptor (TβR2) |
| pS | Activated Src kinase |
| pFAK | FAK phosphorylated on tyrosine 397 |
| 3pF | FAK phosphorylated on tyrosines in 400–900 range with active kinase activity |
| pP | Active p38 |
| pE | Active ERK |
The total amount of each protein is conserved and given a value of 1, so the inactive species fraction is calculated at each time point as 1 − Σ (active protein species).
TABLE 3.
List of parameter values.
| Parameter | Description | Value (h−1) | Source |
|---|---|---|---|
| k1f | Rate of β1 integrin adhesion and activation | 23 | Estimated from Ref. 49 |
| k1r | Rate of β1 integrin deactivation | 0.567 | Estimated from Ref. 49 |
| k2f | Rate of β3 integrin adhesion and activation | 23 | Estimated from Ref. 49 |
| k2f | Rate of β3 integrin deactivation | 0.567 | Estimated from Ref. 49 |
| k3f | Rate of TGF-β1 ligand attachment to TBR2 receptor | 60 | Ref. 44 |
| k3r | Rate of TGF-β1 disassociation | 15 | Estimated from Ref. 44 |
| kIpF | Rate of β1 integrin activation of FAK | 0.454 | Estimated from Refs. 13,49 |
| kIpS | Rate of β3 integrin activation of Src | 20.15 | Estimated from Refs. 1,49 |
| kTpS | Rate of TGFβ receptor activation of Src | 120 | Estimated from Refs. 14,15,21 |
| kSpF | Rate of Src association with FAK and activation of secondary phosphorylation sites | 29 | Estimated from Refs. 7,41,49 |
| KmSF | Michaelis–Menten constant for Src activation of FAK | 0.1 | Estimated from Ref. 7 |
| kFAKpE | Rate of FAK activation of ERK | 48 | Estimated from Refs. 46,49 |
| kFGFpFAK | Rate of FGF activation of FAK | [2.2–8.4] | Estimated from Refs. 17,29 and optimized |
| kFGFpERK* | Rate of FGF activation of ERK | [5.5–16.6] | Estimated from Refs. 17,29 and optimized |
| kSpT | Rate of Src phosphorylation of TBR2 | 40 | Estimated from Refs. 14,15 |
| KmST | Michaelis–Menten constant for Src activation of TBR2 | 0.1 | Estimated from Refs. 14,15 |
| kSpP | Rate of Src phosphorylation of p38 | 10 | Estimated from Refs. 7,41,49 |
| kTpP* | Rate of TGF-β1 activation of pp38 | [13.3–371] | Estimated from Refs. 14,15 |
| kIpP* | Rate of β1 integrin activation of p38 | 5 | Estimated from Refs. 24,33 |
| kPr | Rate of p38 dephosphorylation | 580 | Ref. 30 |
| kEr | Rate of ERK dephosphorylation | 210 | Estimated from Ref. 49 |
| kSr | Rate of Src dephosphorylation | 432 | Estimated from Ref. 49 |
| kFr | Rate of FAK dephosphorylation | 48 | Estimated from Ref. 49 |
| kTr | Rate of ligand induced TBR2 deactivation | 15 | Ref. 44 |
| kE | Intrinsic rate of ERK activation | 2 | Estimated and optimized |
| kαSMAf* | Rate of p38 promotion of αSMA | 0.5 | Estimated and optimized for each model |
| kαSMAr | Rate of αSMA degradation | 0.515 | Estimated from Ref. 20 |
Initial estimates of values were calculated from literature and varied to find the optimal parameter set.
Parameters that were optimized to calibration data set indicated by *.
Regulation of αSMA Production
In the simplification of this system, we focused on p38 and ERK as the primary regulators of αSMA production. Phosphorylated p38 (pp38) promotes the production of αSMA, while pERK inhibits αSMA accumulation by slowing the rate of production. According to the model proposed by Kawai-Kowase et al.,29 ERK activated by FGF signaling prevents smooth muscle gene expression by interfering with serum response factor (SRF) function via an unknown mechanism. We represented this in the model with the following equation:
| (1) |
After initial experimental results showed a dramatically lower amount of αSMA in SYF−/− cells despite comparable levels of p38 phosphorylation, a modified equation for αSMA production was devised:
| (2) |
Parameter Estimation
Parameters were estimated by comparison with previously published models and by calculating the maximum relative activation changes in relevant experimental contexts. Both p38 and ERK are activated via cascades of signaling events downstream of growth factor receptors, Src, and FAK19,49 but these cascades are approximated as a single step with a lumped parameter for the sake of model simplicity. To estimate values for these lump parameters, we measured αSMA expression and relative ERK and p38 phosphorylation in MEFs after 24 h of treatment with 1 or 10 ng/mL TGF-β1 and FGF. We also tuned our model’s sensitivity to changes in mechanical stiffness by measuring αSMA in cells plated on PDMS of stiffnesses ranging from 230 kPa to 2.14 MPa and on TCP (stiffness ~3 GPa16). The MATLAB optimization function lsqnonlin was used to vary up to three parameters at once to find the set of parameters which minimized the mean squared error (MSE) of the model fit to the growth factor sensitivity curves or the stiffness curve. While comparing candidate models, two parameters (kTpP and kFGFpERK) were optimized to fit the growth factor calibration data set for each model.
Sensitivity Analysis
Both the initial conditions and rate constants were varied over 2 decades around the primary estimation, and the relative change in output (αSMA concentration) and sensitivity coefficients S [relative change in output per relative change in parameter (P)] were calculated and ordered. This analysis provides insight into the bottlenecks and critical junctions where the system is more or less sensitive to perturbations:
| (3) |
Candidate Model Development and Statistical Comparison
We developed a set of candidate models (described in Table 4 and in Online Appendix) which contain modified signaling mechanisms, reflecting different hypotheses. Two of the hypotheses posited were that a negative feedback loop in the regulation of Src or ERK phosphorylation would improve model fit to experimental data. We used a data set independent from the calibration curves used to refine the model to evaluate model fit and quantitatively assess the likelihood of certain interaction mechanisms. After simulating the set of eight experiments with each candidate model, we calculated the χ2 statistic for the set of experimental results. The χ2 statistic is a metric for measuring model fit while accounting for variability in experimental measurements:
| (4) |
TABLE 4.
Model comparison and statistical analysis.
| Model | Features | MSE | χ2 | ΔAIC |
|---|---|---|---|---|
| 01 | No Src dependency or Src feedback or ERK feedback | 3.92 | 1700 | 22.6 |
| 02 | No Src dependency term, but Src and ERK feedback | 0.53 | 234 | 5.97 |
| 03* | Src dependency and Src feedback | 0.056 | 16.5 | 0 |
| 04 | Src dependency, Src and ERK feedback | 0.173 | 42.2 | 1.68 |
| 05 | Model 04 without stiffness dependence of IpP | 0.179 | 60.2 | 1.76 |
| 06 | Model 04 with β3 integrin positive feedback | 0.169 | 42.4 | 3.63 |
| 07* | Model 03 with calpain negative feedback to ERK | 0.0363 | 21.9 | 3.7 |
| 08 | Model 04 with no p3F activation of p38 | 0.190 | 45.3 | 1.68 |
Indicates that the χ2 value for the given model has a p < 0.05 for a χ2 distribution with 15 degrees of freedom, indicating that the model predictions are not significantly different from the experimental data set.
When χ2 is minimized, the agreement between the model prediction and the data is optimized.
Model Evaluation Using the Akaike Information Criterion (AIC)
The Akaike information criterion (AIC) is a metric for comparing models with different numbers of independent parameters (K), to attempt to optimize both the accuracy and the model simplicity, or parsimony, of different models variants:5,40
| (5) |
N reflects the number of experimental data points and MSE is the mean squared error. This criterion can be used to great effect in determining the relative likelihood of multiple models. Some of the candidate models considered in this study contain additional feedback loops and mechanisms which increase the number of relevant parameters while improving model fit.
RESULTS
Src and FAK Significantly Regulate αSMA Production via Integrin Signaling
MEFs with genetically deleted focal adhesion proteins express significantly different levels of αSMA in serum-free conditions than wild type cells (Fig. 2). A two-way ANOVA shows significant interaction (p < 0.001) between cell type and treatment. SYF−/− cells expressed significantly less αSMA than MEF+/+ cells in all treatment groups (Fig. 2a) and have noticeably altered morphology, with a rounded cell shape and few well-defined actin bundles or stress fibers (Fig. 2b). Furthermore, there is no significant difference in αSMA expression between treatment groups within the SYF−/− cells. FAK−/− cells express significantly more αSMA than MEF+/+ cells regardless of treatment, and both growth factors cause a significant effect relative to untreated FAK−/− cells (Fig. 2a). MEF+/+ and FAK−/− both have a fibroblast-like morphology, but in both treated and non-treated groups, a higher percentage of FAK−/− cells express αSMA (Fig. 2b). TGF-β1 causes an increase in intensity and frequency of αSMA expression and stress fiber formation in both cell types (Fig. 2b). FGF has the opposite effect, causing a loss of stress fibers and αSMA expression (Fig. 2b). The inverted roles that FAK and Src play in regulating myofibroblast differentiation prompted a more detailed look at downstream signals.
FIGURE 2.

Focal adhesion proteins FAK and Src regulate myofibroblast differentiation. (a) αSMA measured by ELISA after 24 h of culture in serum free media, 1 ng/mL TGF-β1, or 10 ng/mL FGF. All FAK−/− and SYF−/− groups were significantly different from the wild type MEF+/+ cells (p < 0.001). * denotes a significant difference between the NT and treated MEFs (p < 0.05). # denotes significant difference from the FAK−/− NT group. ^ denotes significant difference from the MEF+/+ sample under a given treatment. (b) Representative images of αSMA stress fiber assembly in cells grown on fibronectin-coated coverslips and stained for αSMA (red) and dapi nuclear stain (blue). The scale bar represents 50 μM.
TGF-β1, FGF, and Stiffness Modulate αSMA in a Predictable Manner
We conducted calibration experiments to correlate growth factor concentration and stiffness to internal signaling and regulation of αSMA and refine our initial estimates of lump parameters. Our first calibration experiment (Figs. 3a, 3b, and 3c) clarified the relationship between growth factor concentration, equilibrium p38 and ERK phosphorylation, and αSMA expression in MEF+/+ cells. At 24 h, there is no significant change in pp38 with treatment with 1 or 10 ng/mL FGF, but there is a significant log-linear increase proportional to TGF-β1 concentration (Fig. 3a). There is significant ERK phosphorylation after 24 h treatment with TGF-β1 that is independent of TGF-β1 concentration. There is also a significant increase in ERK activation with FGF treatment, which is highly dependent on FGF concentration (Fig. 3b). These data were used to refine estimates of kTpP, kFGFpERK, and kαSMAf (Table 3). The variability of the experimental measurements was considered in the optimization protocol; we selected the set of parameters which gave the minimum χ2 for each candidate model. By this technique, we achieved good agreement with our calibration curves, with χ2 values as low as 17.6 (p = 0.128) for the set of 15 growth factor measurements, indicating that the residuals between the model predictions and the set of experimental data are not significantly different from the expected value, given the variability of the data.
FIGURE 3.

Calibration curves for reaction to growth factors and stiffness. (a–c) Concentration dependent changes to p38 and ERK activation and αSMA expression in response to TGF-β1 and FGF. (d) Sensitivity to stiffness in production of αSMA in MEF+/+ and FAK−/− cells. * indicates significant difference from the no treatment/TCP condition within each cell type. ^ indicates significant difference from MEF+/+ sample within substrate. Active p38 and pERK data from densitometry of western blots (a, b) and αSMA determined from indirect ELISA (c, d). Average results are presented (n = 4–12). These data were used to refine model parameters.
We next measured αSMA production over a range of substrate stiffness (Fig. 3d) and found a statistically significant interaction between cell type and substrate stiffness (p = 0.009). αSMA production was significantly reduced when cells were cultured on PDMS, with the lowest αSMA expression corresponding to a PDMS stiffness of 900 kPa. There was no significant difference between αSMA expression on fibronectin-coated TCP and uncoated TCP in either cell type.
αSMA in FAK−/− cells is significantly higher than in MEF+/+ (p < 0.001) on TCP (stiffness = 3E6 kPa) but is not statistically different at lower stiffness, indicating that FAK−/− are more sensitive to changes in stiffness than MEF+/+ cells. These data were also used to refine model fit and parameter estimation, especially in determining kIpP. χ2 values as low as 4.5 (p = 0.87) were calculated for the set of 11 substrate measurements. After quantifying MAPK phosphorylation and αSMA expression at 24 h, we directed our focus to the details of dynamic signaling.
TGF-β1 and FGF Induce MAPK Phosphorylation with Different Dynamic Profiles
Western blot data show significant and sustained p38 phosphorylation in response to TGF-β1, which peaks at 1 h in both MEF+/+ and FAK−/− cells and remains significantly enhanced (p = 0.017) after 24 h of treatment (Figs. 4a, and 4c). While the shape of this activation is consistent between cell types, the peak magnitude in the FAK−/− is significantly lower (p = 0.007). Steady state p38 phosphorylation at 24 h is also significantly lower in FAK−/− cells relative to MEF+/+ (p = 0.046). In the same set of experiments, FGF induces a rapid increase in p38 phosphorylation, which attenuates to less than 1.5-fold of the non-treated group within 1 h. FGF induced a dramatic increase in ERK phosphorylation in both cell types in 5 min that persisted for at least 3 h (Figs. 4d, and 4e) and was still significantly elevated at 24 h (Fig. 4f). TGF-β1 induced a slight significant increase at 30 min, which faded to insignificance within 1 h. However, both cell types show significantly enhanced ERK phosphorylation at 24 h after treatment with TGF-β1 (Fig. 4f). The dynamic ERK and p38 trends produced by model simulations share the same general shape as the experimental results, but the relative values of short term phosphorylation are lower (Figs. 4g, 4h, 4i, 4j, 4k, and 4l). Model predictions from both models 03 and 04 are shown in Figs. 4g, 4h, 4i, 4j, 4k, and 4l to illustrate the difference between the models with (model 04) or without (model 03) a feedback term for ERK. In both models, pp38 is predicted to be lower in FAK−/− cells relative to MEF+/+ cells (Figs. 4g, 4h, and 4i), which is consistent with experimental trends (Figs. 4a, 4b, and 4c). However, the p38 activation curves predicted by the model peak sooner (20 min vs. 1 h) and at a lower relative size (~1.8 vs. ~6) than the experiment data showed. In model 04, short term ERK phosphorylation in FAK−/− cells is comparable to MEF+/+, which matches the experimental data more closely than model 3. Furthermore, model 04 predicts a more substantial increase in pERK in response to TGF-β1 than model 03. In both of these models, FGF does not directly activate p38, since the short duration and relatively low level of activation would not have a significant enough effect on αSMA content to justify the addition of model complexity. The models do predict a slight rise in p38 activation in MEFs following FGF stimulation that is transduced through FAK enhanced Src activation (Fig. 4g).
FIGURE 4.
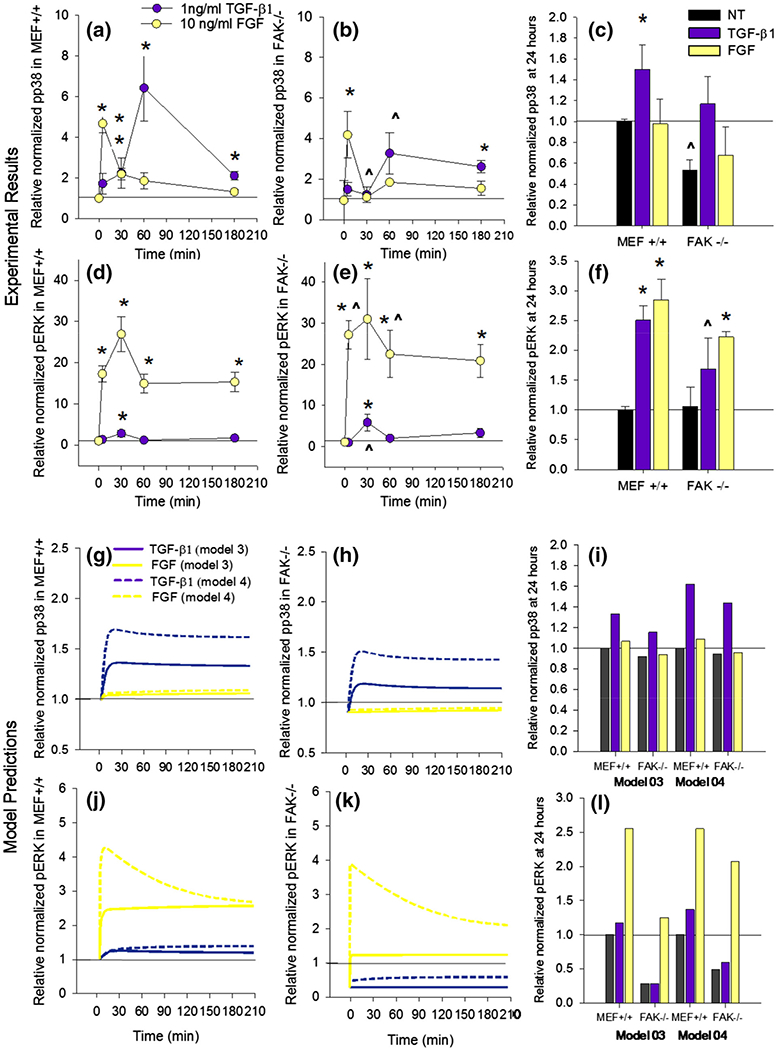
Different dynamic activation profiles for activation of ERK and p38. Averaged results of western blot densitometry analysis for pp38 (a–c) and ERK (d–f) activation over a 3 h time course in MEF+/+ (a, d) and FAK−/− (b, e) cells treated with 1 ng/mL TGF-β1 or 10 ng/mL FGF. Average p38 and ERK activity after 24 h of treatment (c, f). * indicates significant difference (p < 0.05) from average no treatment within cell type and time course. ^ indicates significant difference (p < 0.05) from the MEF+/+ sample within treatment and time point. Relative p38 and pERK activation predicted by models 03 and 04 at corresponding time points (g–l).
Sensitivity Analysis
Sensitivity analysis of the models with optimized parameters predicted that αSMA production in FAK−/− cells would be more sensitive to TGF-β1, FGF, and stiffness relative to MEF+/+ cells (Table SC1). Analysis of basal αSMA production revealed that of the boundary constraints and initial conditions, the total amount of ERK has the largest effect on relative αSMA in MEF+/+ and FAK−/− models, with sensitivity coefficients of −0.94 and −0.71 respectively. Basal αSMA is also sensitive to the total amounts of p38, β1 and β3 integrin across all models, cell types, and treatment groups. The different candidate models considered had variable sensitivities to Src and FAK, due to their differing model structure (Table SC2). Model 01, which contained the original equation for αSMA regulation (Eq. 1), was inversely related to Src, while models 03 and 04, with the modified equation, had positive S values for total Src. Between models 03 and 04, model 03 had higher predicted sensitivity to changes in both total FAK and total Src. The model’s response to TGF-β1 stimulation is most sensitive to changes in rate constants controlling the activation and deactivation of p38, Src, and TβR2. While the rank and sign of sensitivities is conserved between MEF+/+ and FAK−/− models, the magnitude of the parameters is often higher in FAK−/− models. One interesting exception is kTpP, the rate of p38 phosphorylation by TGF-β1, which is slightly lower in FAK−/− models (0.12 vs. 0.18). In MEF+/+ simulations, the response to FGF is most sensitive to the rate of FGF activation of ERK, the deactivation rate of FAK, and the rate of FAK-based activation by integrins. SYF−/− models have no reaction to TGF-β1, and their reaction to FGF is more sensitive to FGF and the rate of ERK activation by FGF than the MEF+/+ model. Sensitivity to both TGF-β1 and FGF was predicted to increase on fibronectin-coated PDMS (stiffness 900 kPa) relative to TCP. A subset of key sensitivity parameters can be seen in Table SC1.
Model can Predict Results Across Substrate and Cell Type
With optimized model parameters, we tested our model’s ability to predict the effect of growth factor treatment on cells lacking Src and FAK that we observed in vitro. Figure 5a shows the model results (models 3 and 4) plotted over the experimental results (same as Fig. 2). We also measured the combined effects of treatment and substrate stiffness by treating cells plated on fibronectin-coated PDMS (Fig. 5b) and found a statistically significant interaction between substrate and treatment (p = 0.004). Further, there is a significant difference (p < 0.05) between TCP and PDMS for non-treated and TGF-β1-treated cells, but not for FGF-treated cells. Within each substrate, both FGF and TGF-β1 treatment cause a significant (p < 0.05) change in αSMA expression. The model predictions for models 03 and 04 are plotted over the experimental results, and were within a standard deviation for all but the TGF-β1-treated sample on PDMS, the FAK−/− cells treated with TGF-β1, and the SYF−/− cells (marked by *).
FIGURE 5.
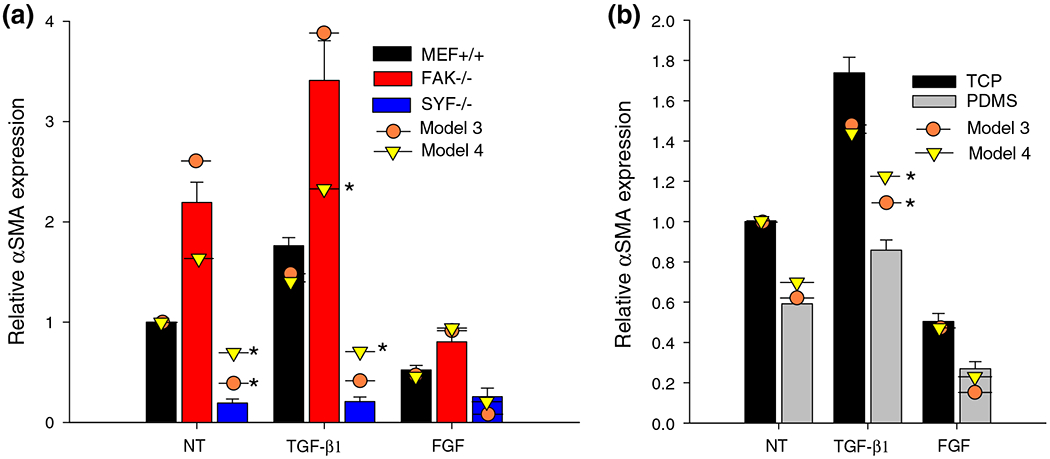
Model fit to TGF-β1 and FGF treatment across cell types and substrates. Average values of αSMA in MEF+/+ and FAK−/− cells (a) after 24 h treatments and (b) in MEFs on TCP or fibronectin coated PDMS. Model predictions from model 03 and model 04 with optimized parameters are plotted as circles and triangles, respectively. Model predictions which are more than one standard deviation away from experimental values are marked by *.
Model Comparisons
Eight candidate models (described in Table 4) were developed and evaluated to find the ideal fit to both steady state protein activation and dynamic protein phosphorylation events. The relative AIC (calculated as the difference between a given model’s AIC and the minimum AIC) provides a useful criterion for eliminating inferior models and improving model parsimony. After parameter optimization of kTpP and kFGFpERK for each model, simulated αSMA outputs were compared against the validation data set (Fig. 5) and the MSE, χ2 statistic, AIC, and ΔAIC were calculated (Table 4). The relative strength of evidence for any model (in comparison) can be estimated as e−ΔAIC/2. In other words, a model with ΔAIC > 10 is 148.4 times less likely than the best model.5,40 We found that model 03 had the lowest AIC and also had a very low MSE and χ2 for the set of 24 h data. Model 03 contained the modified αSMA production equation (Eq. 2) which has a Src-dependent term and negative feedback to Src but did not contain negative feedback to ERK. ΔAIC for the equivalent model (02) with the unmodified equation for αSMA production (Eq. 1) was 5.95, giving strong evidence that the Src-dependent term is supported by the data. Model 03 did not have a negative feedback loop for ERK, which means that the observed change in relative ERK phosphorylation from 3 to 24 h (Figs. 4d, 4e, and 4f) could not be replicated by this model (Figs. 4j, 4k, and 4l). The equivalent model with negative feedback to ERK (model 04) had an ΔAIC of 1.68, so it is reasonably close to the optimal model. Furthermore, after optimization model 04 was able to achieve lower χ2 values and better matching to the calibration data (see Table SB1). Models 03 and 04 simulations are presented in Figs. 4 and 5. Model 07 had the lowest MSE and best fit to the experimental data set via the addition of a calpain feedback loop which degrades β3 integrin and FAK, but this addition of model complexity increased the AIC score above the simpler models 03 and 04. These data indicate that the features of the model presented in this study have reasonable support from the data.
DISCUSSION
Using genetically modified MEFs, we have highlighted the importance of Src family kinases and FAK in the regulation of myofibroblast differentiation. Our results demonstrate a profound inhibitory effect of removing Src on αSMA production and stress fiber assembly. Densitometry revealed comparable levels of p38 and ERK phosphorylation in SYF−/− cells relative to MEF+/+ cells (data not shown), so the effect is likely operating through a different mechanism. This prompted the addition of a Src-dependent term to the αSMA production equation to capture the significant αSMA reduction in SYF−/− cells (Eq. 2). Without the addition of that term, SYF−/− cells in silico behave similarly to FAK−/−, since the absence of Src prevents the activation of FAK kinase ability. Even with a modified Src equation, model 04 was unable to fully replicate the significant effect of Src’s absence. It is likely that signaling downstream of Cas and other Src substrates is necessary for proper αSMA synthesis. Src kinase activity is required for activation of the transcription factor SRF, which is necessary for production of αSMA.28,43 This result is consistent with recent reports of Src’s prominent role in non-canonical TGF-β1 signaling in the context of myofibroblast differentiation.21
Interestingly, the absence of FAK, a protein which is known to enhance Src activation and signal to p38, caused a significant increase in αSMA. Given its complex role in multiple signaling cascades, it is not surprising that reports of the effect of FAK on myofibroblast differentiation vary. Blocking FAK expression in cardiac fibroblasts with siRNA has been shown to decrease force induced αSMA promoter activity.8 Furthermore, Ding et al.10 have reported that αSMA production in serum-free conditions and after TGF-β1 treatment is higher in FAK expressing MEFs compared with FAK−/− counterparts. They found that FAK-related non-kinase blocked TGF-β1-induced FAK activation, p38 and pERK phosphorylation, and αSMA upregulation in primary fibroblasts and in FAK−/− cells.10 Alternatively, others have reported that FAK is involved in FGF signaling to ERK in response to FGF cells, and FAK−/− cells exhibit enhanced αSMA accumulation and persistence after treatment with FGF.17,29 They also report reduced basal ERK phosphorylation in FAK−/− cells, and proposed a model for FGF signaling to ERK requiring FAK.17 This informed the development of our model and is consistent with the decreased initial ERK phosphorylation and increased αSMA that our model predicted. Additionally, we found that FGF was able to induce significant ERK phosphorylation and lower αSMA in the absence of FAK, which indicates that FAK is not required for FGF and ERK-based inhibition of αSMA.
One of the major goals of this project was to clarify the interactions between growth factor and integrin signaling in the regulation of myofibroblast differentiation. We first showed that decreasing substrate stiffness can significantly lower the expression of αSMA in MEFs through a fibronectin-integrin interaction that is significantly altered in FAK−/− cells (Fig. 3d). Sensitivity analysis of the model predicted that FAK−/− cells would be more sensitive to changes in stiffness, which is observed in the experimental data (Fig. 3d). The relationship between αSMA and stiffness has been shown previously and is correlated with changes in p38 activation.33 Both β1 and β3 integrins have been shown to have mechanosensitive capabilities and are involved in outside-in signaling to intracellular kinases like FAK and Src.13,38 Our model uses an activation function proportional to the log of stiffness to simulate integrin activation of FAK, Src, and p38, which gives good agreement with experimental results (Fig. 3d). We further showed the combinatorial effect of substrate changes and growth factor treatments and found a significant interaction, justifying the development of an integrated signaling model (Fig. 5b). All the data in Fig. 5 were well matched by a models whose parameters had been optimized to an independent data set (Fig. 3), which strengthens our proposed model on the roles on p38 and pERK. Model 04 had a closer fit to the dynamic phosphorylation of p38 and pERK, while model 03 better matched the validation data set of basal αSMA in different cell types. The largest discrepancy between model prediction and experimental results, response to TGF-β1 in cells on PDMS, highlights an area needing more detailed investigation: the effect of stiffness and integrin signaling on TGF-β1 pathways. β3 integrins are known to interact with TGF-β1 signaling to Src and p383 and are a likely target for further insights.
Surprisingly, our time course results show that ERK phosphorylation in non-treated FAK−/− cells is not significantly different from MEF+/+ at 24 h and is significantly higher at 30 min. Sensitivity analysis of the model predicted that FAK−/− cells would be more sensitive to changes in TGF-β1 and FGF. Our experimental results seem to confirm that FAK−/− cells have a higher sensitivity to environmental perturbations, such as when the media is changed at the start of the time course. Like FAK, ERK’s role in αSMA regulation has been presented from multiple perspectives. Some have argued that ERK is necessary for TGF-β1 induced activation,6,10 while others proposed a largely inhibitory role.10,17,18,29,45 Several groups have shown that MEK1/2 inhibition significantly increases αSMA expression in fibroblast-like cells.22,32 ERK is a major player in a large set of signaling pathways, which interacts with several other MAPKs to transduce a variety of signals. For instance, ERK is necessary for TGF-β1 induced upregulation of collagen-1 and cadherin-11, two other markers of myofibroblast differentiation.22,34 One of the goals of this project was to investigate the possibility of matching the observed upregulation of ERK by TGF-β1 and FGF in a model with a relatively straightforward αSMA regulatory scheme.
We developed a computational model of these overlapping signaling pathways and a set of tools for network analysis and hypothesis generation. Sensitivity analysis of the model predicted higher sensitivity to TGF-β1 and FGF and stiffness in FAK−/− cells relative to MEF+/+. Experimentally, FAK−/− cells demonstrated a larger relative change in response to FGF than in wild type cells (63 or 49% decrease, respectively), but a smaller relative response to TGF-β1 (55 or 73% increase, respectively) (Fig. 4a). According to the constitutive equation for αSMA activation, the sensitivity of equilibrium αSMA to both p38 and pERK is inversely proportional to ERK activation, so lower basal ERK activation, as found in FAK−/− cells, should cause higher sensitivity to all parameters which affect ERK and p38 activation. Functionally, Eqs. (1) and (2) mean that the presence of active ERK dampens the sensitivity of the system to changes in MAPK activation. Since the growth factors present in serum can cause a significant increase in ERK activation, we performed all of our ELISA and western blot experiments in serum-free conditions. Sensitivity to both TGF-β1 and FGF was predicted to increase with decreasing stiffness according to the model prediction, and was observed in the case of FGF (54.5 vs. 49.5% decrease) though the reverse is true for TGF-β1 (45 vs. 73% increase) (Fig. 5b). Further investigation into altered signaling on softer substrates could help clarify this discrepancy. Our model comparison revealed that a direct dependency on active Src greatly enhanced the quality of model fit. It also indicated that including more complex network interactions, like calpain-based negative feedback, can improve model accuracy, but not enough to justify additional model complexity. Future analysis of the model using larger calibration and validation data sets will give more insight into the dynamics of myofibroblast regulation. Overall, this study demonstrates the feasibility of p38/ERK/Src-based regulation of αSMA production during fibroblast differentiation.
The decision to limit active model variables and fit lump parameters to 24 h time points limits the predictive power of the model, especially for quantitatively matching the short term dynamic changes. We decided that this would be the best technique for modeling changes in αSMA expression, which occurs at a slower rate than dynamic phosphorylation changes. The model was developed from a variety of previously published models and experimental studies, so the relative rate constants presented here are not necessarily relatable to quantitative reaction kinetic experiments. Furthermore, the decision to hold the total value of the protein species constant at 1 limits the model accuracy. It has been shown that TGF-β1 promotes expression of β3 integrin,36 which could create feedback loops and have a large impact on model predictions. Our sensitivity analysis indicated that αSMA production is very sensitive to changes in total β1 and β3 integrin values (see Table SC1). As techniques for quantifying protein expression and activation improve, this model can be expanded and refined. The work presented here in parallel with experimental results is intended to give insight into the interactions between growth factor and integrin signaling in myofibroblast differentiation.
In this study we have demonstrated that an ODE-based computational model of relative protein expression can capture a subset of the dynamic and steady-state events observed during fibroblast differentiation. We have further shown that the mechanism of Src/p38/ERK based regulation of αSMA as described herein is a feasible model for regulation of myofibroblast differentiation. Model simulations were able to replicate some of the dynamic features of TGF-β1 and FGF signaling to p38 and pERK and show that despite the fact that ERK increases with TGF-β1 treatment, it may still be acting primarily as a negative regulator of αSMA. Our experimental results indicate that Src family kinases are crucial for αSMA synthesis in fibroblasts and demonstrate that FAK plays an important role in integrating signals for the regulation of αSMA production and myofibroblast differentiation.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the National Science Foundation (CAREER Award to WDM (1055384) and Graduate Research Fellowship to AKS (DGE-0909667)) and by the National Institutes of Health (HL094707 and HL115103, both to WDM).
Biography
W. David Merryman PhD is an Assistant Professor in the Departments of Biomedical Engineering, Pharmacology, Medicine, and Pediatrics at Vanderbilt University. His research interests are cardiovascular mechanobiology, cell and soft tissue biomechanics, tissue engineering, and bioengineering ethics. Prior to his arrival at Vanderbilt, Dave was an Assistant Professor of Biomedical Engineering at the University of Alabama at Birmingham and prior to that, a Research Associate of the McGowan Institute for Regenerative Medicine and Bioengineering at the University of Pittsburgh, where he was an American Heart Association Pre-doctoral Fellow. Dave completed his BS and MS in Engineering Science at the University of Tennessee and was awarded the Alumni Promise award from UT in 2011 which recognizes outstanding alumni under 40 years of age. Dave has been awarded the Early Career Award from the Wallace H. Coulter Foundation, the Scientist Development Grant from the American Heart Association, the NSF CAREER Award, the K Award from the National Institutes of Health (NHLBI), and the Y.C. Fung Young Investigator Award from the American Society of Mechanical Engineers. An internationally known expert in heart valve mechanobiology with over 40 peer-reviewed publications in the field, Dave has given over 20 invited talks at various conferences and at the most prestigious universities. In 2014, he delivered an invited talk on “Technologies for the Heart” at the National Academy of Engineering’s Frontiers of Engineering annual meeting for a select group of the nation’s outstanding young engineers between the ages of 30 and 45.

Footnotes
This paper is part of the 2014Young Innovators Issue.
ELECTRONIC SUPPLEMENTARY MATERIAL
The online version of this article (doi: 10.1007/s12195-014-0344-9) contains supplementary material, which is available to authorized users.
CONFLICT OF INTEREST
Alison K. Schroer, Larisa M. Ryzhova, and W. David Merryman declare that they have no conflicts of interest.
ETHICAL STANDARDS
No human or animal studies were carried out by the authors for this article.
REFERENCES
- 1.Arias-Salgado EG, et al. Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc. Natl. Acad. Sci. USA 100(23):13298–13302, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bajikar SS, and Janes KA. Multiscale models of cell signaling. Ann. Biomed. Eng 40(11):2319–2327, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blobe GC, Schiemann WP, and Lodish HF. Role of transforming growth factor beta in human disease. N. Engl. J. Med 342(18):1350–1358, 2000. [DOI] [PubMed] [Google Scholar]
- 4.Brown XQ, Ookawa K, and Wong JY. Evaluation of polydimethylsiloxane scaffolds with physiologically-relevant elastic moduli: interplay of substrate mechanics and surface chemistry effects on vascular smooth muscle cell response. Biomaterials 26(16):3123–3129, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Burnham KP, and Anderson DR. Model Selection and Multi-Model Inference: A Practical Information-Theoretic Approach. New York: Springer, 2002. [Google Scholar]
- 6.Caraci F, et al. TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol. Res 57(4):274–282, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Caron-Lormier G, and Berry H. Amplification and oscillations in the FAK/Src kinase system during integrin signaling. J. Theor. Biol 232(2):235–248, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Chan MW, et al. FAK, PIP5KIgamma and gelsolin cooperatively mediate force-induced expression of alpha-smooth muscle actin. J. Cell Sci 122(Pt 15):2769–2781, 2009. [DOI] [PubMed] [Google Scholar]
- 9.Merryam WD, and Sewell-Loftin MK. The role of Src in strain- and ligand-dependent phenotypic modulation of mouse embryonic fibroblasts In: Proceedings in Bioengineering (ASME). Farmington, PA, 2011. [Google Scholar]
- 10.Ding Q, et al. Focal adhesion kinase (FAK)-related non-kinase inhibits myofibroblast differentiation through differential MAPK activation in a FAK-dependent manner. J. Biol. Chem 283(40):26839–26849, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eubanks TR, et al. The effects of different corticosteroids on the healing colon anastomosis and cecum in a rat model. Am. Surg 63(3):266–269, 1997. [PubMed] [Google Scholar]
- 12.Faust D, et al. Differential p38-dependent signalling in response to cellular stress and mitogenic stimulation in fibroblasts. Cell Commun. Signal 10:6, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedland JC, Lee MH, and Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science 323(5914):642–644, 2009. [DOI] [PubMed] [Google Scholar]
- 14.Galliher AJ, and Schiemann WP. Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 8(4):R42, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galliher AJ, and Schiemann WP. Src phosphorylates Tyr284 in TGF-beta type II receptor and regulates TGF-beta stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 67(8):3752–3758, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Gilbert PM, et al. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 329(5995):1078–1081, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenberg RS, et al. FAK-dependent regulation of myofibroblast differentiation. FASEB J. 20(7):1006–1008, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Gu X, and Masters KS. Role of the MAPK/ERK pathway in valvular interstitial cell calcification. Am. J. Physiol. Heart Circ. Physiol 296(6):H1748–H1757, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hendriks BS, Hua F, and Chabot JR. Analysis of mechanistic pathway models in drug discovery: p38 pathway. Biotechnol. Prog 24(1):96–109, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Hsu HJ, et al. Stretch-induced stress fiber remodeling and the activations of JNK and ERK depend on mechanical strain rate, but not FAK. PLoS ONE 5(8):e12470, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutcheson JD, et al. 5-HT(2B) antagonism arrests non-canonical TGF-beta1-induced valvular myofibroblast differentiation. J. Mol. Cell. Cardiol 53(5):707–714, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutcheson JD, et al. Cadherin-11 regulates cell–cell tension necessary for calcific nodule formation by valvular myofibroblasts. Arterioscler. Thromb. Vasc. Biol 33(1):114–120, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ilic D, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377(6549):539–544, 1995. [DOI] [PubMed] [Google Scholar]
- 24.Ivaska J, et al. Integrin alpha2beta1 mediates isoform-specific activation of p38 and upregulation of collagen gene transcription by a mechanism involving the alpha2 cytoplasmic tail. J. Cell Biol 147(2):401–416, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janes KA, Reinhardt HC, and Yaffe MB. Cytokine-induced signaling networks prioritize dynamic range over signal strength. Cell 135(2):343–354, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janes KA, et al. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science 310(5754):1646–1653, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Junttila MR, Li SP, and Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 22(4):954–965, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Katsch K, et al. Actin-dependent activation of serum response factor in T cells by the viral oncoprotein tip. Cell Commun. Signal 10:5, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawai-Kowase K, et al. Basic fibroblast growth factor antagonizes transforming growth factor-beta1-induced smooth muscle gene expression through extracellular signal-regulated kinase 1/2 signaling pathway activation. Arterioscler. Thromb. Vasc. Biol 24(8):1384–1390, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Khanna P, et al. Model simulations reveal VCAM-1 augment PAK activation rates to amplify p38 MAPK and VE-cadherin phosphorylation. Cell. Mol. Bioeng 4(4):656–669, 2011. [Google Scholar]
- 31.Klinghoffer RA, et al. Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 18(9):2459–2471, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu S, et al. FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix-remodeling phenotype. Mol. Biol. Cell 18(6):2169–2178, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer-ter-Vehn T, et al. Extracellular matrix elasticity modulates TGF-beta-induced p38 activation and myofibroblast transdifferentiation in human tenon fibroblasts. Invest. Ophthalmol. Vis. Sci 52(12):9149–9155, 2011. [DOI] [PubMed] [Google Scholar]
- 34.Mishra R, et al. TGF-beta-regulated collagen type I accumulation: role of Src-based signals. Am. J. Physiol. Cell Physiol 292(4):C1361–C1369, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Mitra SK, and Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol 18(5):516–523, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Pechkovsky DV, et al. Transforming growth factor beta1 induces alphavbeta3 integrin expression in human lung fibroblasts via a beta3 integrin-, c-Src-, and p38 MAPK-dependent pathway. J. Biol. Chem 283(19):12898–12908, 2008. [DOI] [PubMed] [Google Scholar]
- 37.Plopper GE, et al. Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol. Biol. Cell 6(10):1349–1365, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roca-Cusachs P, et al. Clustering of alpha(5)beta(1) integrins determines adhesion strength whereas alpha(v)beta(3) and talin enable mechanotransduction. Proc. Natl. Acad. Sci. USA 106(38):16245–16250, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sewell-Loftin MK, et al. A novel technique for quantifying mouse heart valve leaflet stiffness with atomic force microscopy. J. Heart Valve Dis. 21(4):513–520, 2012. [PMC free article] [PubMed] [Google Scholar]
- 40.Shankaran H, et al. Integrated experimental and model-based analysis reveals the spatial aspects of EGFR activation dynamics. Mol. BioSyst 8(11):2868–2882, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas JW, et al. SH2- and SH3-mediated interactions between focal adhesion kinase and Src. J. Biol. Chem 273(1):577–583, 1998. [DOI] [PubMed] [Google Scholar]
- 42.Tomasek JJ, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol 3(5):349–363, 2002. [DOI] [PubMed] [Google Scholar]
- 43.Tominaga T, et al. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell 5(1):13–25, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Vilar JM, Jansen R, and Sander C. Signal processing in the TGF-beta superfamily ligand-receptor network. PLoS Comput. Biol 2(1):e3, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walker GA, et al. Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res 95(3):253–260, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Wang JG, et al. Uniaxial cyclic stretch induces focal adhesion kinase (FAK) tyrosine phosphorylation followed by mitogen-activated protein kinase (MAPK) activation. Biochem. Biophys. Res. Commun 288(2):356–361, 2001. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, et al. Mechanical force regulation of myofibroblast differentiation in cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol 285(5):H1871–H1881, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe T, et al. Adaptor protein Crk induces Src-dependent activation of p38 MAPK in regulation of synovial sarcoma cell proliferation. Mol. Cancer Res 7(9):1582–1592, 2009. [DOI] [PubMed] [Google Scholar]
- 49.Yee KL, Weaver VM, and Hammer DA. Integrin-mediated signalling through the MAP-kinase pathway. IET Syst. Biol 2(1):8–15, 2008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.