

Abstract
We hypothesized that NO• is generated in isolated cardiac mitochondria as the source for ONOO− production during oxidative stress. We monitored generation of ONOO− from guinea pig isolated cardiac mitochondria subjected to excess Ca2+ uptake before adding succinate and determined if ONOO− production was dependent on a nitric oxide synthase (NOS) located in cardiac mitochondria (mtNOS). Mitochondria were suspended in experimental buffer at pH 7.15, and treated with CaCl2 and then the complex II substrate Na-succinate, followed by menadione, a quinone redox cycler, to generate O2•−. L-tyrosine was added to the mitochondrial suspension where it is oxidized by ONOO− to form dityrosine (diTyr) in proportion to the ONOO− present. We found that exposing mitochondria to excess CaCl2 before succinate resulted in an increase in diTyr and amplex red fluorescence (H2O2) signals, indicating that mitochondrial oxidant stress, induced by elevated mtCa2+ and succinate, increased mitochondrial ONOO− production via NO• and O2•−. Changes in mitochondrial ONOO− production dependent on NOS were evidenced by using NOS inhibitors L-NAME/L-NNA, TEMPOL, a superoxide dismutase (SOD) mimetic, and PTIO, a potent global NO• scavenger. L-NAME and L-NNA decreased succinate and menadione-mediated ONOO− production, PTIO decreased production of ONOO−, and TEMPOL decreased ONOO− levels by converting more O2•− to H2O2. Electron microscopy showed immuno-gold labeled iNOS and nNOS in mitochondria isolated from cardiomyocytes and heart tissue. Western blots demonstrated iNOS and nNOS bands in total heart tissue, bands for both iNOS and nNOS in β-tubulin-free non-purified (crude) mitochondrial preparations, and a prominent iNOS band, but no nNOS band, in purified (Golgi and ER-free) mitochondria. Prior treatment of guinea pigs with lipopolysacharride (LPS) enhanced expression of iNOS in liver mitochondria but not in heart mitochondria. Our results indicate that release of ONOO− into the buffer is dependent both on O2•− released from mitochondria and NO• derived from a mtCa2+-inducible nNOS isoform, possibly attached to mitochondria, and a mtNOS isoform like iNOS that is non-inducible.
Keywords: heart, liver mitochondria, mitochondrial oxidant stress, nitric oxide, nitric oxide synthase, peroxynitrite, superoxide
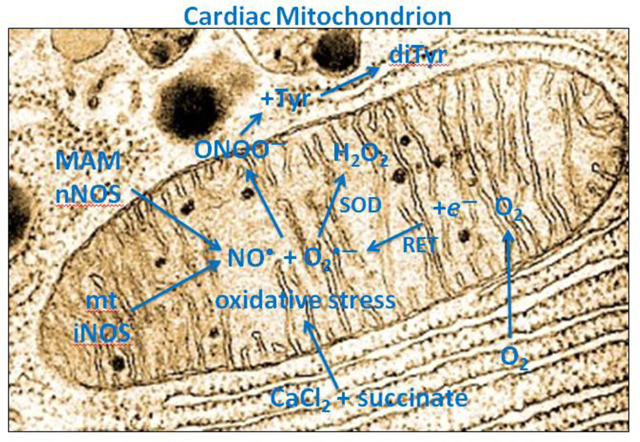
1. Introduction
Nitric oxide (NO•) [1], initially identified as the endothelium-derived relaxing factor, plays a crucial role in the gaseous regulation of multiple systems including cardiovascular, respiratory, endocrine, peripheral and central nervous, and immune systems. The synthesis of NO• is catalyzed by nitric oxide synthase (NOS), of which two isozymes are constitutive and called neuronal NOS (nNOS or NOS1) and endothelial NOS (eNOS or NOS3). The third isoform is called inducible NOS (iNOS or NOS2). These isoforms are ubiquitously expressed and found in all cell types. During NO• synthesis NOS converts L-arginine to NO• and L-citrulline. The activities of nNOS and eNOS depend on Ca2+ binding to calmodulin, so Ca2+ is a regulator of NO• generation [2,3,4,5]. In contrast, iNOS is largely upregulated by pro-inflammatory cytokines and hypoxia [6,7]. Much is known about the function of the isozymes of NOS by information obtained using specific NOS knockouts [5,8].
Initially, eNOS was thought to be the only constitutively expressed isoform in myocytes and was considered the major source of NO• in the autocrine regulation of myocardial contraction and Ca2+ homeostasis [2,9]. NO• plays a major role in modulating mitochondrial function, so the presence of any isoform of NOS in the inner mitochondrial membrane (IMM), outer mitochondrial membrane (OMM), the mitochondrial matrix, or in the immediate vicinity of mitochondria, would be important for assessing its function [7,10,11,12]. For example, findings have indicated that nNOS is present in cardiac sarcoplasmic reticulum (SR) [13,14,15] and that it may be a variant [16] of the canonical nNOS that targets the SR [16], a component of the so-called mitochondrial associated membranes (MAM) [13].
Under physiological conditions, mitochondria play a critical role in cellular energy production, gene expression, and metabolic regulation [17]. NO• is well known to block cytochrome c oxidase (complex IV) to stem electron transport at that site [18,19] and alter mitochondrial function. NO• has been implicated in regulating trans-mitochondrial (mt) Ca2+ flux and mitochondrial trans-membrane potential (ΔΨm) [20,21] and in modulating reactive oxygen species (ROS) and reactive nitrogen species (RNS) emission [22,23]. Nitrites, like peroxynitrite (ONOO−) [24], derived from NO• and O2•– (superoxide), can induce preconditioning protection against ischemic injury [25] but in high amounts they induce tissue and mitochondrial damage [24,26,27,28,29,30,31]. Pharmacologically blocking NOS results in coronary vasoconstriction [32], and RNS scavenging protects hearts against ischemia [26].
Despite the effects of stimulating or blocking NO• generation on mitochondrial function, it still remains controversial if mitochondria actually contain their own NOS (mtNOS) isoform [33] (review), either in the matrix or in the IMM [34]. Several studies have provided results suggesting the existence of a mtNOS based on western blots using antibodies to Ca2+-sensitive NOS [3,7,11,12,35,36]. mtNO• release was reported to be modulated by ΔΨm [37]. The generation of NO• was identified in rat liver mitochondrial homogenates using electron paramagnetic resonance and a spin trap [38]. Isolation of a NOS isoform in rat liver mitochondria suggests it differs from cytosolic NOS and is post-translationally modified to allow its transport through the IMM [39]. Conversely, other studies have reported that the mitochondrial electron transport chain (ETC) is a source of non-enzymatic NO• production via a nitrate reductase/ubiquinone cycle [40,41]. However, because this phenomenon was only observed in the absence of O2, its physiological relevance remains unclear [34]. It remains difficult to rule out contamination of isolated mitochondria with other organelle fragments that may contain NOS to release NO• that affects mitochondrial function.
Whether NO is generated by a NOS located in structures near mitochondria, or in the OMM, IMM, or matrix, NO• can diffuse easily across nearby membranes and physiologically modulate mitochondrial function. NO• can also negatively affect mitochondrial function via its conversion to ONOO−, a nonradical but highly toxic oxidant, or to other RNS [42]. ONOO− is a short-lived oxidant in an acidic, warm environment but it is stable in an alkaline, cold environment. It is produced by the reaction between the two radicals NO• and O2•– at diffusion-controlled rates (~1×1010 M–1 s–1) [42,43,44]. Peroxynitrite production is presumed to occur near mitochondrial sources of O2•– such as the ETC. Although NO• is relatively stable and freely diffusible through membranes, O2•– is short-lived, and as a charged species, demonstrates limited diffusion [45]. The induction of ONOO− production in vivo in specific compartments has been previously estimated to reach levels of 50–100 μM/min [46]. In a physiological buffer containing L-tyrosine (Tyr), Tyr is oxidized by ONOO− to form the fluorescent dimer dityrosine (diTyr). Thus, this diTyr assessment method has been used to monitor the formation of ONOO− indirectly, but in direct proportion to actual ONOO−, in extracellular fluid [24,29,47,48].
When hearts are subjected to ischemia reperfusion (IR) injury, or isolated cells are exposed to hypoxia or simulated ischemia, the presence of NO• with locally derived O2•– generates ONOO− that can be assessed either by diTyr production or by protein tyrosine nitration with 3-nitrotyrosine antibody [24,27,28,29]. When electron transport is blocked pharmacologically at complex I, III, or IV, mitochondria can produce very high levels of ROS, initially as O2•– and its dismutase product hydrogen peroxide (H2O2), that are dependent on Ca2+ loading and an acidic pH [22,48]. The presence of NO• largely determines if H2O2 or ONOO− is produced in greater amounts [26]. Menadione, a quinone redox cycler that induces one electron reduction of oxidants, has been shown to generate large amounts of O2•− and ONOO− [48,49].
Cardiac cell mitochondria have been reported to contain [10,23,50] or not to contain [51] a functional nNOS. Our goal was to induce oxidative stress in isolated, unpurified mitochondria by adding excess CaCl2 and then succinate to the mitochondrial buffer, and to examine for mtNOS activity assessed by di-Tyr production. We proposed that increased mtCa2+ loading activates a mtNOS to generate NO• [52] and that adding succinate, in the absence of pyruvate, reverses electron transfer at complex I to generate O2•– [53]. The result would be production of mtONOO–. Because superoxide dismutase (SOD) converts O2•– to H2O2, the SOD mimetic TEMPOL [54,55] and PTIO, a potent global NO• scavenger [55], would be expected to modulate the ONOO– levels. Ng-nitro-L-arginine-methyl ester (L-NAME), or another NOS inhibitor, Nω-nitro-L-arginine (L-NNA), would be expected to directly block any mtNOS activity and to stop ONOO– production. In vivo, mtCa2+ and succinate accumulate during cardiac IR injury [22,53]; the resulting accumulation of ONOO− can cause tyrosine nitration of proteins [24,26,48] and cause damage to mitochondrial proteins such as the voltage dependent anion channel (VDAC) and the ADP/ATP cotransporter adenine nucleotide translocase (ANT) [28].
Our first aim was to ascertain: a) if ONOO− production (diTyr levels) occurs in cardiac isolated mitochondria to indicate the presence of NOS, b) if ONOO− levels are altered during mitochondrial stress using modulators of O2•– or NO• generation, and c) if NOS inhibitors and ROS/RNS scavengers reduce the accumulation of mtONOO−. Our second aim was to assess if there is one or more traditional cytosolic NOS isoforms in isolated mitochondrial preparations from heart tissue and cardiomyocytes by western blotting and immuno-electron microscopy (IEM) using commercial anti-NOS antibodies.
2.0. Materials and Methods
2.1. Animals and treatments
All experiments were performed in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996) and were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. Albino guinea pigs were used in all experiments. In three additional experiments guinea pigs were first injected intraperitoneally (i.p.) with 40 mg/Kg lipopolysacharride (LPS, Escherichia coli 0111:B4, Sigma-Aldrich, Madrid, Spain), dissolved in 0.3 mL saline, to determine if this would induce expression of iNOS in cardiac and liver isolated mitochondria. Five hours after LPS injection, guinea pigs were anesthetized and euthanized by decapitation. Liver and heart were quickly extracted and mitochondrial preparations were prepared from both organs as described below.
2.2. Isolation of guinea pig cardiomyocytes
Hearts were isolated from ketamine-anesthetized (50 mg/kg ip) guinea pigs (250–350 g) and the coronary system was perfused via the aorta against a closed aortic valve as reported in detail previously [24,26,32,48,56]. Cardiomyocytes were isolated as we previously described using an isolation solution containing collagenase and protease [57,58]. Dissociated cardiomyocytes were stored in the Tyrode’s solution (in mM: 132 NaCl, 10 HEPES, 5 glucose, 5 KCl, 1 CaCl2, 1.2 MgCl2, adjusted to pH of 7.4) and used to isolate mitochondria.
2.3. Isolation of mitochondria and western blotting
Mitochondria were isolated from intact guinea pig heart [22,27,28,58], and liver, and from cardiomyocytes after isolating them first. In brief, hearts and livers were excised, minced into 1 mm3 pieces, and placed in a chilled isolation buffer A containing (in mM): 200 mannitol, 50 sucrose, 5 KH2PO4, 5 MOPS, 1 EGTA, 0.1% fatty acid free bovine serum albumin (BSA), and protease inhibitors with pH adjusted to 7.15 with KOH, and then homogenized. Fresh isolated cardiomyocytes were also suspended in buffer A and homogenized with a Dounce homogenizer. The homogenized slurries were centrifuged for 10 min at 8000 g at 4°C. After decanting the supernatant, the pellet was re-suspended in isolation buffer A and spun at 900 g for 10 min at 4°C; the supernatant was collected and re-centrifuged at 8000 g at 4°C. The final mitochondrial pellet following this centrifugation was re-suspended in isolation buffer. The mitochondria isolated from fresh cardiomyocytes were processed to identify mitochondrial NOS isoforms using immuno-gold electron microscopy. The mitochondria isolated from heart tissue were used to measure mitochondrial function or for further purification as described by Graham [59] and by us [58]. For purification, mitochondria were layered on 30% percoll in isolation buffer, and then centrifuged for 30 min at 95,000 g at 4°C. The resulting purified mitochondria were used for western blot analysis of NOS.
Mitochondrial membranes were also prepared as described previously by Giulivi et al. [38]. In brief, purified mitochondria from guinea pig heart and liver, obtained as described above, were homogenized by using a Dounce homogenizer with buffer B containing 1 mM EDTA, 5 mM β-mercaptoethanol, 50 mM Hepes and protease inhibitors (Roche, complete mini tablet) at pH 7.5. This homogenate was centrifuged at 4000 g for 10 min, and then the supernatant was further centrifuged at 150,000 g for 1 h at 4°C. The pellet containing the mitochondrial membranes was then washed with buffer C (buffer B plus 1 M KCl, 10% glycerol) and centrifuged again at 150,000 g for 30 min. The final pellet of mitochondrial membranes was then lysed with RIPA buffer containing 50 mM Tris-Cl pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS and protease inhibitors on ice for 30 min for western blot analysis.
Whole heart tissue, unpurified and purified isolated mitochondrial preparations, and isolated IMM preparations were lysed with RIPA buffer and then the lysates were boiled at 95°C for 5 min in 1x Laemmli buffer. The samples were resolved by SDS-PAGE and transferred onto PVDF membranes. The membranes were incubated with specific primary antibodies against iNOS (SC-7271 from Santa Cruz Biotechnology, and ab3523 and ab136918 from Abcam), nNOS (SC-5302 from Santa Cruz Biotechnology and C7D7 from Cell Signaling Technology, Cat. #4231), and eNOS (D9A5L from Cell Signaling Technology, Cat #32027). Washed membranes were incubated with the appropriate secondary antibody conjugated to horseradish peroxidase, then immersed in an enhanced chemiluminescence detection solution (Bio-Rad Laboratories). The band signals were obtained using ChemiDoc imaging system (Bio-Rad Laboratories). For monitoring the content of isolated mitochondria, blots were stripped with buffer (62.5 mM Tris-Cl, 2% SDS, and 100 mM β-mercaptoethanol at pH 6.8), and then probed with the mitochondrial specific antibody anti-VDAC (Cell Signaling Technology). To assess the purity of isolated mitochondria, western blots against the cytosol-specific antibody anti-β-tubulin (Cell Signaling Technology), anti-calnexin antibody (Cell Signaling Technology), anti-SERCA2 (sarcoplasmic/endoplasmic reticulum Ca2+-ATPase2, Santa Cruz Biotechnology), and anti-GM130 (cytosolic Golgi protein, Cell Signaling Technology) were performed using the same membranes as for the NOS antibodies.
2.4. Immuno-electron microscopy to identify and localize mitochondrial NOS isoforms
Immuno-electron microscopy (IEM) was used as previously described [58] to identify and localize isoforms of NOS in mitochondria isolated from guinea pig cardiomyocytes and in heart tissue. Briefly, the isolated mitochondrial pellet or the heart tissue samples were fixed with EM fixative and then processed using the protocols of Berryman and Rodewald [60]. Immuno-gold labeling was performed by floating grids on droplets of 0.1 M NaH2PO4 buffer containing 5% PB-BSA, then incubated with rabbit specific primary antibodies against nNOS (C7D7), eNOS (D9A5L), and iNOS (ab136918) diluted 1:50 for 90 min. Non-immune rabbit polyclonal serum was used as the negative control. This step was followed by washes in PB-BSA. The sections were then incubated with goat anti-rabbit IgG, or goat anti-mouse IgG, conjugated to 10 nM colloidal gold, rinsed in distilled water, and then stained with 2% aqueous uranyl acetate. Sections were examined in a JEOL JEM2100 transmission electron microscope at 75–80 kV for 30,000 to 60,000x magnification (conditions: ORCA 1792 ×2 gain, Bin 1; gamma: 0.95; normal contrast, no sharpening).
2.5. Mitochondrial function protocols
Mitochondrial functional experiments, described below, were conducted at room temperature (25°C), with unpurified (crude) mitochondria (0.5 mg protein/mL) suspended in experimental buffer D that contained (in mM) 130 KCl, 5 K2HPO4, 20 MOPS, 0.001 Na4P2O7, and 0.1% BSA. Protein content was determined by the Bradford method. The mitochondrial suspension protein concentration was adjusted to yield 12.5 mg protein/mL before initiating the mitochondrial functional protocols. This assured that only approximately 40 μM EGTA was carried over from the isolation buffer (buffer A) into the experimental buffer. In fully charged mitochondria, Ca2+ uptake after adding 75 μM CaCl2 is estimated to increase matrix mt[Ca2+] to approximately 15–20 μM [61].
2.6. Mitochondrial peroxynitrite accumulation assessed by di-tyrosine concentration
In a physiological buffer, L-tyrosine is oxidized by ONOO− to form the stable fluorescent dimer dityrosine (diTyr) [27,28,29,43,48]. This method is used to assess the proportional amount of ONOO− released into the extracellular fluid of isolated hearts or into the buffer of isolated mitochondria. Mitochondria were suspended in buffer E, which was buffer D plus 0.5 mM L-Tyrosine, in a 1 mL cuvette placed inside a spectrophotometer ((Horiba QM-8; Photon Technology International (PTI). ONOO− production was measured in Buffer E from mitochondria undergoing stress induced by adding to buffer E, 75 μM CaCl2 followed by 10 mM Na-succinate, with or without the redox cycler menadione (10 μM). Menadione, which generates ROS within the ETC and concomitantly decreases NADH consistent with redox cycling [62,63], was used to induce an additional mitochondrial oxidant stress as would occur during IR injury. DiTyr concentration was measured at excitation and emission wavelengths of λ322 nm and λ422 nm, respectively. To assess endogenous NO• generation induced by NOS and concomitant ONOO− production, NOS inhibitors L-NAME and L-NNA (10 mM), the SOD mimetic TEMPOL (2.5 and 10 mM), and the global NO• scavenger PTIO (100 μM and 500 μM), were added to buffer E before adding mitochondria.
ONOO− concentrations were estimated from a linear calibration curve in which eight authentic ONOO− standards (range 1–300 μM), frozen and stored in alkaline buffer [64], were thawed and added to buffer E containing the excess L-tyrosine plus CaCl2 (75 μM) (40 μM EGTA), succinate, and menadione, but no mitochondria. A linear calibration curve for diTyr (y), in arbitrary fluorescence units (AFU), as a function of (x), the [ONOO−] standards, gave: y = 92.3x −270 (R2 = 0.985), so that an increase of 10,000 AFUs would be equivalent to about 105 μM ONOO−. Buffer E, containing L-tyrosine, without added mitochondria, gave a background fluorescence of 10,000 AFU, indicating the baseline. Adding substrates with mitochondria to buffer E (induced oxidative stress only) increased fluorescence to approximately 25,000 AFUs, so an increase of 15,000 AFU approximates a basal concentration of 157 μM ONOO−.
2.7. Mitochondrial H2O2 production
Mitochondria were suspended in buffer D in a 1 mL cuvette inside the same cuvette-based spectrophotometer. The rate of H2O2 release was measured using amplex red (12.5 μM; Molecular Probes, Eugene, OR, USA) and horseradish peroxidase (0.1 U/mL), in the presence of CaCl2 (75 μM with 40 μM EGTA), Na-succinate (10 mM), and later, menadione (10 μM), at excitation and emission wavelengths of λ530 and λ583 nm, respectively. H2O2 is the direct product of O2•− when catalyzed by O2•− dismutase (SOD) in the absence of nitric oxide (NO•). To investigate a role of NO• in modulating H2O2 generation in isolated mitochondria, L-NAME and L-NNA (10 mM), TEMPOL (2.5 and 10 mM), PTIO (100 μM and 500 μM), and the complex I blocker rotenone (10 μM), were also added to buffer D (before the addition of mitochondria). H2O2 levels were calibrated over a range of 10–200 nM H2O2 added to buffer D in the absence of mitochondria but in the presence of amplex red and horseradish peroxidase as described previously [22].
2.8. Mitochondrial membrane potential
Mitochondrial membrane potential (ΔΨm) was assessed using rhodamine 123 (200 nM; Sigma) [62] inside the same cuvette-based spectrophotometer at excitation and emission wavelengths of λ503 nm and λ527 nm, respectively, in the presence of CaCl2 (75 μM with 40 μM EGTA) and Na-succinate (10 mM) with menadione (10 μM) added later for additional oxidative stress. The complex I blocker rotenone (10 μM) and/or the complex III blocker antimycin A (5 μM) were added to buffer D before mitochondria were added. The uncoupling agent FCCP (36 μM) was given at the end of each experiment to assess maximal ΔΨm depolarization.
2.9. Mitochondrial O2 consumption
Oxygen consumption rate (respiration) was measured polarographically using a Clark O2 electrode system (System S 200A; Strathkelvin Instruments). Respiration experiments using complex II substrate Na-succinate (10 mM) at pH 7.15 and without added CaCl2 were conducted initially to determine mitochondrial vitality. Respiration was measured before (state 2), after adding 250 μM ADP (state 3), and after complete phosphorylation of the added ADP (state 4). The respiratory control index (RCI), the ratio of states 3/4 respiration, had to be greater than 3 in the presence of 10 μM Na-succinate to continue experiments using the fluorescence probes. Mean RCI before initiating experiments was 4.4±0.3; after 4 h (approximate length of experiments) the RCI was 3.5±0.2 (data not shown graphically).
2.10. Statistical analysis
Functional data were collected and averaged at 60 s (after adding drugs and mitochondria), at 240 s (after adding CaCl2 and succinate but before menadione) and at 320–350 s (after adding menadione). All data points presented were expressed as average ±SEM. Repeated measure ANOVAs followed by a post hoc analyses using Student-Newman-Keuls’ test was performed to determine significant differences among groups. A P value < 0.05 (two-tailed) was considered significant. See figure legends for statistical notations.
3. Results
3.1. Effect of blocking ETC complexes I & III on altering ΔΨm induced by excess CaCl2 and succinate
In one set of experiments (Fig. 1) we assessed changes in ΔΨm and evaluated the effects of stress induced by a high mt[Ca2+] and succinate alone vs. pre-treatment with ETC complex I inhibitor rotenone, or complex III inhibitor antimycin A, and both together. An increase in rhodamine 123 fluorescence represents increasing ΔΨm depolarization. In the single case with succinate given before CaCl2 (purple trace), ΔΨm depolarization did not occur. Mitochondria exposed to CaCl2 loading first (90 s) exhibited depolarization (yellow trace), until the addition of succinate (140 s), which repolarized ΔΨm to baseline. Pre-treatment with rotenone before incubation resulted in greater ΔΨm depolarization from baseline with CaCl2 (blue trace) compared to untreated mitochondria (90 s); addition of succinate (150 s) repolarized ΔΨm to baseline; this indicates that membrane permeability transition (MPT) did not occur. Pre-treatment with antimycin A resulted in a greater ΔΨm depolarization after CaCl2 loading (90 s) compared to untreated stressed mitochondria or rotenone (green trace); addition of succinate did not repolarize ΔΨm to baseline. Pre-treatment with rotenone plus antimycin A caused the greatest ΔΨm depolarization after CaCl2 loading (red trace) and addition of succinate did not repolarize ΔΨm to baseline. For each treatment group, the latter addition of menadione (250 s) resulted in negligible changes in ΔΨm compared to baseline. Final addition of the proton gradient uncoupler FCCP demonstrated the maximal depolarization of ΔΨm for each treatment group. These results demonstrated that only CaCl2 loading followed by succinate effectively induces mitochondrial stress by causing ΔΨm depolarization and repolarization, respectively. The added effect of rotenone indicates that the changes in ΔΨm with CaCl2 and succinate are likely due to reverse electron transfer at complex I and not to inhibition of complex III.
Fig. 1.

Time-dependent changes in rhodamine 123 fluorescence (excitation λ503 nm; emission λ527 nm), as an indicator of membrane potential (ΔΨm) in guinea pig isolated mitochondria undergoing stress induced by: CaCl2 (75 μM) added at 90 s and Na-succinate (10 mM) added at 140 s (CaCl2 + succinate); Na-Succinate (10 mM) added first at 90 s and CaCl2 (75 μM) added at 150 s (succinate + CaCl2 ) did not depolarize ΔΨm. Pretreatments of vehicle, rotenone (10 μM), antimycin A (5 μM) and rotenone + antimycin A were added to the buffer before adding mitochondria and exposure to CaCl2 and succinate. Tracings shown were similar in mitochondria isolated from two other guinea pig hearts.
3.2. Effect of NOS inhibition, NO• scavenging, and a SOD mimetic, on mitochondrial H2O2 generation during stress induced by CaCl2 plus succinate
In a second set of experiments (Fig. 2) we measured H2O2 generation induced by adding CaCl2 and then succinate, before and after menadione, as in Fig. 1, and evaluated the effects of pre-treatments with NOS inhibitors L-NAME or L-NNA, SOD mimetic TEMPOL, global NO• scavenger PTIO, and complex I blocker rotenone on H2O2 generation. Adding succinate (150 s) after CaCl2 (90 s) caused the H2O2 generation rate (light blue) to increase. Pre-treatment with 2.5 mM TEMPOL (yellow) increased the basal H2O2 generation rate and delayed the succinate-mediated increase in H2O2 by 30 s. Pre-treatment with PTIO (green, purple) increased the basal rate of H2O2 generation and both concentrations delayed the succinate-mediated increase in H2O2 by approximately 30 s. Pre-treatment with L-NAME (brown) or L-NNA (dark blue) reduced that basal H2O2 generation rate and delayed the succinate-mediated increase in H2O2 by approximately 20 and 30 s, respectively. Pre-treatment with 10 μM rotenone (red) decreased the basal rate of H2O2 generated and abolished the succinate-mediated increase in H2O2 consistent with the repolarization of ΔΨm (Fig. 1). Following exposure to menadione, the rate of H2O2 generation decreased (270 s) compared to the succinate-mediated increase in H2O2 generation rate (180 s). Pre-treatment with L-NAME and L-NNA decreased the menadione-mediated H2O2 generation rate (270 s) compared to mitochondria that had undergone stress alone. Pre-treatment with 2.5 mM TEMPOL markedly increased the menadione-mediated H2O2 generation rate (270 s). Pre-treatment with PTIO (100 μM and 500 μM) also markedly increased the menadione-mediated H2O2 generation rate (270 s). Pre-treatment with 10 μM rotenone did not affect the menadione-mediated H2O2 production rate. These results indicated that excess CaCl2, preceding succinate addition, effectively simulates H2O2 generation (derived from O2, most likely via RET at complex I) in mitochondria; moreover, NOS inhibitors reduce, whereas PTIO and TEMPOL increase, the basal and menadione-induced H2O2 generation rates.
Fig. 2.

Time-dependent changes in amplex red fluorescence (excitation λ530 nm; emission λ583 nm) as an indicator of H2O2 production. Mitochondria were suspended in buffer D containing Amplex red (12.5 μM) and horseradish peroxidase (0.1 U/mL) in the presence of CaCl2 (75 μM added at 90 s with 40 μM EGTA), Na-succinate (10 mM added at 140 s), and menadione (10 μM added at 270 s). L-NAME and L-NNA (10 mM), TEMPOL (2.5 mM), PTIO (100 μM and 500 μM), and the complex I blocker rotenone (10 μM) were also added to buffer D before the addition of mitochondria. Tracings shown were similar in mitochondria isolated from two other guinea pig hearts.
3.3. Effect of NOS inhibition, NO• scavenging, and TEMPOL, on mitochondrial ONOO− production during stress induced by CaCl2 plus succinate
We then measured mitochondrial release of ONOO− indirectly via formation of diTyr in mitochondria undergoing oxidative stress as shown in Fig. 2, and again evaluated the effects of pre-treatment with NOS inhibitors L-NAME and L-NNA, the SOD mimetic TEMPOL, and the global NO• scavenger PTIO on altering ONOO− production (Fig. 3). Sample traces show changes over time (Fig. 3A). Starting fluorescence unit values reflect effects of the prior treatments on basal levels of ONOO−when mitochondria were added to the buffer. In summarized results, baseline (at 60 s) [ONOO−] after adding mitochondria, but before inducing stress, was highest in the control group (no pre-treatments), lowest in the L-NAME and L-NNA groups, and intermediate in the TEMPOL and PTIO groups (Fig. 3B).
Fig. 3.

A. Sample traces of time-dependent changes in dityrosine (diTyr) fluorescence (excitation λ322 nm; emission λ422 nm) as an indicator of ONOO− production in guinea pig isolated cardiac mitochondria stressed with CaCl2 + succinate. Mitochondria (0.5 mg/mL) were suspended in buffer E containing 0.5 mM L-tyrosine (buffer D) in the presence of CaCl2 (75 μM added at 90 s; with 40 μM EGTA), Na-succinate (10 mM added at 140 s), and the redox cycler menadione (10 μM added at 250 s). NOS inhibitors L-NAME and L-NNA (10 mM), the SOD mimetic TEMPOL (2.5 and 10 mM), and the global NO• scavenger PTIO (100 μM and 500 μM), were added to the buffer before the addition of mitochondria. B. Summary of the mean (±SEM) baseline ONOO− concentrations after treatments but before (at 60 s) CaCl2+succinate or addition of menadione. The presence of drugs added to the buffer before adding mitochondria reduced baseline [ONOO−] compared to Control (no treatment). [ONOO−] was calibrated over a range of 1–300 μM (y = 92.965x − 270.2; R² = 0.98). For all treatments n=6 guinea pig hearts. For treatments vs. no treatment (Control): * P < 0.05; ** P < 0.005; *** P < 0.001.
After adding CaCl2 and succinate, but before adding menadione (240 s) there was a moderate increase in ONOO− (+14.0 ±1.6 μM) compared to the 60 s baseline for each treatment group (Fig. 4A). Pre-treatment with NOS inhibitors L-NAME and L-NNA (both 10 mM) significantly decreased the amount of CaCl2 plus succinate-mediated ONOO− production (−15.6 ± 1.1μM, P <0.001) and (3.4 ±0.4 μM, P <0.001), respectively, compared to mitochondria only treated with CaCl2 and succinate. Pre-treatment with the SOD mimetic TEMPOL (2.5 mM and 10 mM) decreased CaCl2 plus succinate-mediated ONOO− production slightly, but the differences were not significant (+11.9 ±2.0 μM, P =0.96) and (+7.9 ±2.3 μM, P =0.12). Pre-treatment with the NO• scavenger PTIO (100 μM and 500 μM) also decreased CaCl2 plus succinate-mediated ONOO− production, and the difference was significant for 500 μM PTIO (+7.1 ± 1.9 μM, P <0.05), but not for 100 μM PTIO (+14.0 ± 0.7 μM, P =1.00).
Fig. 4.

A. Summary of the mean (±SEM) changes in ONOO− concentration (μM) from baseline (Δ[ONOO−]) after inducing stress (CaCl2 + succinate) but before (at 240 s) adding 10 μM menadione. B. Summary of the mean (±SEM) changes in ONOO− concentration (μM) from baseline (Δ[ONOO−]) after stress (CaCl2+succinate) and after adding (at 350 s) 10 μM menadione. For all treatments n=6 guinea pig hearts. For induced stress plus treatments vs. induced stress alone: * P<0.05; ** P <.005; *** P <0.001.
Mitochondria undergoing oxidative stress induced by excess CaCl2 plus with added menadione (at 350s) produced a greater increase in ONOO− (60.0 ±2.9 μM) (Fig. 4B) compared to CaCl2 plus succinate alone (P<0.001) in this treatment free group. Pre-treatment with L-NAME (−3.1 ±0.6 μM, P<0.001) and L-NNA (2.4 ±1.5 μM, P<0.02) significantly decreased menadione-mediated ONOO− production compared to that in the CaCl2 plus succinate treated mitochondria alone group (60.0 ±2.9 μM), respectively. Pre-treatment with 2.5 mM or 10 mM TEMPOL significantly decreased menadione-mediated ONOO− production (17.4 ±4.4 μM, P <0.03; −3.7 ±0.9 μM, P <0.001, respectively. Pre-treatment with 100 μM PTIO did not significantly decrease menadione-mediated ONOO− production (46.9 ±2.3 μM, P=0.99), but 500 μM PTIO significantly decreased ONOO− production (24.1 ±4.1 μM, P <0.05) compared to CaCl2 plus succinate treatment alone. These results indicated that mitochondrial ONOO− production becomes greatly attenuated during oxidative stress when mtNOS is inhibited or NO• is scavenged, or if more O2•– is converted to H2O2 by TEMPOL, and if menadione is present to enhance generation of O2•– within the ETC.
3.4. Western blotting to identify NOS isoforms in cardiac isolated mitochondrial preparations
Guinea pig heart tissue, isolated crude mitochondria, percoll-purified cardiac mitochondria, and enriched IMM samples were subjected to western blot analysis with eNOS, nNOS, or iNOS specific primary antibodies. To help determine the presence of a mtNOS, several antibodies against two NOS isoforms from different suppliers were used. β-tubulin, a cytosol-specific protein; calnexin and SERCA2, SR markers; and GM130, a Golgi marker were determined by western blots to assess the purity of the isolated mitochondrial samples. The β-tubulin band (51 kDa) was present in the whole-heart samples but was not present in crude (Figs. 5–7) or percoll-purified mitochondrial samples (Fig. 7). This indicated that the mitochondrial preparations, isolated using either of these procedures, were not likely contaminated by cytosolic proteins.
Fig. 5.

Western blots of guinea pig total cardiac tissue, purified cardiac and liver mitochondria, and mitochondrial membrane (mito memb) with anti-iNOS specific primary antibody ab3523 (abcam). β-tubulin was used to assess purity of isolated mitochondria; VDAC1 was used to monitor mitochondria content. iNOS was faintly detectable in IMM (A), undetectable in isolated cardiac mitochondria (B,C), but strongly detectable in total heart tissue (C). Prior treatment of guinea pigs with lipopolysaccharide (LPS) did not appear to enhance band density of iNOS in heart mitochondria (A,B). Mito indicates mitochondria isolated from two hearts.
Fig. 7.

Western blots of guinea pig total heart tissue, unpurified (crude), and percoll-gradient purified isolated cardiac mitochondria. Blots show levels of total heart iNOS and nNOS. However, purified isolated mitochondria (mito-β-tubulin, calnexin, SERCA2, and GM130 free), displayed only a low level of iNOS and absence of nNOS at approximately 150 kDa suggesting the unpurified, crude mitochondrial preparation could contain Golgi and SR attached MAM. VDAC1 was used to monitor mitochondria content.
The whole-heart homogenates displayed a strong iNOS band of approximately 130 kDa using anti-iNOS antibody ab3523 (Fig. 5C); isolated liver mitochondria (Fig. 5B,C) and liver mitochondrial membranes (Fig 5A) also showed similar iNOS bands using ab3523 (Fig. 5A-C); however, the iNOS band was absent in heart mitochondrial membranes, and also in isolated mitochondria, even after prior LPS treatment (Fig. 5A-C). Neither the anti-nNOS antibody C7D7 nor the anti-eNOS antibody D9A5L demonstrated antigen specificity in these preparations (western blots not shown).
Whole-heart homogenates again displayed strong iNOS bands at approximately 130 kDa using either anti-iNOS antibody ab136918 or SC-7271, whereas iNOS bands were present (unlike with ab3532 iNOS) but weak in crude mitochondrial preparations from two hearts (Fig. 6A,B).
Fig. 6.

Western blots of guinea pig total heart tissue and isolated cardiac mitochondria. The two blots show level of iNOS in total heart and isolated mitochondria (mito-β-tubulin free), respectively, using iNOS antibodies different from those used in Fig. 5. VDAC1 was used to monitor mitochondrial content.
Whole-heart homogenates displayed strong iNOS and nNOS bands, when using the anti-iNOS SC-7271 antibody and the anti-nNOS SC-5302 antibody (Fig. 7 lane 1). In the cardiac, crude isolated mitochondrial preparations, which did not express β-tubulin but moderately expressed GM130, the marker of Golgi, and less so SERCA2, both iNOS and nNOS were expressed (Fig. 7 lane 3). In the percoll-purified mitochondrial preparation (Fig. 7 lane 2), there were no antibody reactions to calnexin, SERCA2, or GM130, suggesting the absence of MAM in this mitochondrial preparation. Tested on the same samples, the same antibodies against iNOS (SC-7271) and nNOS (SC-5302) showed a positive band for iNOS but not for nNOS. However, a positive band of approximately 110 kDa was observed, which could be a fragment or variant of nNOS. Together, these results suggested that iNOS is within cardiac mitochondria and nNOS is associated closely with, but not within or on, cardiac mitochondria.
3.5. Immuno-electron microscopy to localize iNOS and nNOS isoforms in cardiac mitochondria
Anti-iNOS, nNOS, and eNOS antibodies, ab136918, C7D7, and D9A5L, respectively, were conjugated to immuno-gold and used to determine if these NOS isoforms were localized in guinea pig heart tissue and cardiomyocyte mitochondria. The IEM photomicrographs demonstrated the presence of nNOS in or near mitochondria of heart tissue cells (Fig. 8A) and iNOS in or near mitochondria of heart tissue cells (Fig. 8B,C) and for iNOS in an isolated myocyte mitochondrion (Fig. 8D). The anti-eNOS antibody revealed no immuno-gold granules in isolated mitochondria (IEM graphs not shown).
Fig. 8.

Immuno-electron microscopic (IEM) detection of NOS in mitochondrial preparations of guinea pig heart tissue and isolated cardiomyocytes. IEM graphs show presence of nNOS near or in mitochondria of heart tissue (A), presence of iNOS near or in heart tissue mitochondria (B,C), and presence of iNOS in one mitochondrion in a cardiomyocyte preparation (D).
4. Discussion
The main goals of this project were: 1) to assess the effects of inducing mitochondrial oxidant stress by adding excess CaCl2 before adding succinate to alter ΔΨm for inducing H2O2 generation in cardiac isolated mitochondria; 2) to measure ONOO− production as a marker of NO• release with O2•– in cardiac isolated mitochondria undergoing oxidative stress induced by adding CaCl2 and succinate in the presence and absence of drugs that modulate ROS/RNS levels or mitochondrial function; 3) to identify the location and isoforms of any cardiac mtNOS by western blotting and IEM; and 4) to compare guinea pig cardiac and liver mitochondria for iNOS expression. Here we report the functional activity of a mtNOS, which could be variant of cytosolic iNOS, and a Ca2+-inducible nNOS that may be a component of the MAM, that are individually or together responsible for the baseline presence and observed changes in levels of of the NO• + O2•– product ONOO− in isolated mitochondria. We found no evidence for the presence of eNOS in cardiomyocyte mitochondria.
4.1. Functional data supporting mitochondrial NOS
We investigated first the functional effects of inducing mitochondrial stress on changing ΔΨm and increasing H2O2 production, based on the mtCa2+ uptake and accumulation of succinate known to occur during authentic IR injury, and on restoration of ΔΨm, which can lead to increased ROS emission via complex I [53]. Indeed, RET leading to generation of O2•– at complex I is dependent on a strong ΔΨm [30]. We observed that ΔΨm depolarized during CaCl2 loading; this is due to the large influx of Ca2+ ions that overrides H+ pumping by the ETC Complexes during forward electron transfer. We previously reported that this is accompanied by oxidation of NADH [65] indicating decreased complex I activity. Afterward, addition of succinate repolarized ΔΨm to near baseline; this is likely due to RET to complex I. RET causes reduction of NAD+ to NADH and electron leak at complex I, which reduces O2 to O2•– [66,67]. Our protocol to induce mitochondrial ROS is in part based on our prior report [68], in which we found (reference Figs. 1,3) that succinate, but not pyruvate, when given after 80 μM CaCl2, enhanced the rate of H2O2 generation, repolarized ΔΨm, and restored the NADH level after they had been diminished by adding CaCl2. Importantly, in the present report we found that during the succinate-mediated repolarization phase, ONOO− levels increased along with the rate of H2O2 generation (derived from O2•–) (Figs. 2–4). This supports the notion that mtNOS activity, presumably induced by the high mtCa2+ coupled with the enhanced generation of O2 after succinate, is due to ΔΨm repolarization and RET as the cause for increased ONOO− production [20]. Inhibition of complex I by rotenone reduced H2O2 production dramatically, but rotenone-treated mitochondria were still able to maintain a polarized ΔΨm when energized with succinate (Fig. 1). However, pre-treatment with complex III blocker antimycin A prevented mitochondria from repolarizing with the addition of succinate. It is known that complex III inhibition with subsequent prevention of forward electron transfer via ETC causes electron leak and O2•– generation with loss of the H+ gradient and inhibition of ATP synthesis [69]. Thus, mitochondrial NOS activity and ROS/RNS production during induced mitochondrial stress, like authentic IR injury, is a ΔΨm-dependent processes.
We assessed the contribution of altering mtNOS activity on H2O2 generation (Fig. 2) using the same oxidative stress conditions and pre-treatments for assessing ONOO− production (Figs. 3,4). Mitochondria exposed to excess CaCl2 did not demonstrate increased rates of H2O2 generation until succinate was added, which supports our assertion that mitochondrial stress, like cardiac IR injury [22,53], enhances mitochondrial O2•– generation. Pre-treatment with NOS inhibitors L-NAME and L-NNA before adding mitochondria most-notably demonstrated, paradoxically, menadione-inhibited H2O2 generation. One might expect that decreased NO• synthesis and the subsequent decrease in ONOO− production would result in greater conversion of O2•– to H2O2. However, inhibition of NOS can also result in decreased O2•– generation, as all isoforms of NOS can alternatively generate O2•– rather than NO• under mostly pathological conditions [70]. Our data support this assertion as shown by a reduction in H2O2 generation during NOS inhibition. Additionally, the SOD mimetic TEMPOL [54] greatly increased H2O2 production during the induced stress; this verifies its SOD mimetic activity. Furthermore, pre-treatment with the complex I blocker rotenone demonstrated a markedly reduced rate of H2O2 production (Fig. 2), which suggests that ETC complex I plays a crucial role in ROS/RNS generation during mitochondrial stress [22]. Thus mitochondrial NOS plays a crucial role in the regulation of mitochondrial ROS/RNS production during induced stress by way of NO• synthesis, either within mitochondria or from nearby sources, and O2•– generation, primarily by mitochondrial ETC complex I in these experiments.
Assuming NO• is generated along with O2•– in isolated mitochondria, we assessed ONOO− production by measuring diTyr in the unpurified mitochondrial preparation as an indirect marker of NO• generation dependent on concomitant O2 generation. Mitochondrial ONOO− production is dependent on mitochondrial O2•– generation. We have shown that mitochondrial VDAC is nitrated by ONOO− after IR injury in isolated hearts by evidence for tyrosine nitration of VDAC using anti-tyrosine antibody in isolated mitochondria [27,71]. In our isolated mitochondria experiments described here, the NO• component, due to its high membrane solubility, could have been derived from a soluble iNOS or nNOS located within the MAM, the IMM, or in the mitochondrial matrix. Mitochondria placed in buffer not containing the NOS inhibitors or ROS/RNS scavengers had higher basal levels of ONOO−, indicating a greater presence of NO• (Fig. 3A, B). Mitochondria exposed to excess CaCl2 first and then succinate (Fig. 4A) produced a higher level of ONOO− than at baseline, and additional exposure to the redox cycler menadione (Fig. 4B) induced even greater levels of ONOO−. Mitochondria incubated with either NOS inhibitors, L-NAME or L-NNA, exhibited significant reductions in both CaCl2 + succinate-mediated (Fig. 4A) and menadione-mediated (Fig. 4B) ONOO− production. NOS inhibition by L-NAME or L-NNA was more effective at reducing ONOO− than by hastening the conversion of O2•– to H2O2 by TEMPOL or by scavenging NO• by PTIO. This may be due to the relatively high rate of reaction and short lifespan of NO• in the presence of O2•– [45].
Overall, these functional results indicate that mitochondria, per se, play a crucial role in the processing of NO• (assessed by ONOO−) during mitochondrial stress. Because L-NAME and L-NNA inhibited ONOO− production, this indicated that NOS was the source of NO• activity in the mitochondria. Chronic, oral administration of L-NAME to rats was reported to result in compensatory induction of iNOS in intestinal tissue [72]. The higher concentrations of TEMPOL and PTIO point to O2•– and NO• as the precursors of ONOO−.
4.2. Expression of mtNOS isoforms in cardiomyocyte mitochondria
Based on our functional studies indicating the presence of NO• in crude isolated mitochondrial preparations, we sought to localize NOS in cardiomyocytes and in cardiac isolated mitochondrial preparations via western blot analysis and IEM. In general, whole-heart tissue cross-reacted strongly with anti-iNOS specific primary antibodies, whereas isolated cardiac mitochondria samples clearly cross-reacted with two anti-iNOS antibodies, SC-7271 and ab136198 (Fig. 6), but cross-reacted very weakly with the anti-iNOS antibody ab3523 (Fig. 5). In non-purified mitochondria, there was no reactivity at all with the anti-eNOS antibody D9A5L and the anti-nNOS antibody C7D7, but there was reactivity with the anti-nNOS antibody SC-5302 (Fig. 7). In comparison, guinea pig liver mitochondria were strongly positive for the anti-iNOS antibody ab3523. This corroborates a prior study [39] reporting iNOS in liver mitochondria using ab3523, and supports the utility of using the various antibodies in the present study.
To assess for mitochondrial purity, mitochondrial samples were simultaneously probed with the cytosol-specific antibody anti-β-tubulin [2,28]. The cytosol-specific β-tubulin band was present in the whole-heart samples but was not present in the non-purified or purified mitochondrial samples, which indicates these mitochondrial preparations were largely free from contamination by cytosolic proteins. When we tested for iNOS in unpurified cardiac mitochondrial preparations using ab136198 and SC-7271 antibodies, we could detect iNOS bands. Our results indicate that total cardiac tissue as well as unpurified mitochondria expressed iNOS and nNOS isoforms; iNOS was also present in the cardiac IMM fraction. Purified mitochondria, however, only weakly expressed iNOS with questionable expression of an nNOS iso-form. This is in contrast to liver mitochondria which highly expressed iNOS in the IMM fraction.
Thus, despite fractionation and purification techniques to isolate only mitochondrial protein, it remained possible there was contamination by non-mitochondrial particles containing iNOS and nNOS. For example, nNOS has been shown to be expressed in the SR [15], a component of the MAM [13]. To assess mitochondrial purity, we repeated iNOS and nNOS western blots in percoll-purified mitochondrial fractions along with antibodies to calnexin, SERCA2, and GM130. Neither Golgi or SR proteins were evidenced but the iNOS band, although less dense, remained. It is possible that the antibodies we used for iNOS may not have been specific enough for the mitochondrial isoform variant. Even though a band for nNOS at approximately 150 kDa was not evident in purified mitochondria, a band of approximately 110 kDa was observed, which could be a mitochondrial variant. A 130 kDa nNOS variant has been found to associate with the cardiac myocyte SERCA [16], so we presume that the nNOS we observed in our mitochondrial preparations could be a MAM component.
IEM assessment of the location of immuno-gold particles tagged to anti-iNOS and anti-nNOS antibodies in whole-heart tissue and in mitochondria isolated from cardiomyocytes support the western blot data. Using IEM, there was firm evidence for iNOS in both cardiac tissue mitochondria and in the unpurified cardiomyocyte mitochondrial preparations; for nNOS, the evidence was not firm. This observation supports the presence of iNOS as a mtNOS in guinea pig cardiac mitochondria that may be neither well expressed or inducible and which may show weak specificity for several of the antibodies utilized.
In summary, this work provides new functional data supporting the activity of a mtNOS in cardiac mitochondria. We found that during mitochondrial oxidative stress, NOS plays a crucial role in regulating mitochondrial function via mitochondrial NO• synthesis, ONOO− production, O2•– generation, complex I and III activity, Ca2+ homeostasis, and maintenance of a polarized ΔΨm. Factors that might induce mtNOS in the heart are unclear, but mitochondrial oxidative stress, is accompanied by mitochondrial NO• release. The mtNOS found resembles iNOS, but there may also exist an nNOS variant located in the MAM of mitochondrial preparations from guinea pig hearts. These functional experiments, coupled with the anti-NOS antibody experiments, support other cited reports that suggest there are one or more functional mtNOS isoforms within or near mitochondria in isolated cardiac mitochondrial preparations. This may be the first report on the existence of a mitochondrial iNOS variant in cardiac muscle cells.
Highlights:
Cardiac isolated mitochondria produce and release ONOO−, a product of O2•− and NO•.
O2•− is generated by complex I during oxidative stress induced by excess CaCl2 and succinate.
NO• is generated within or near isolated mitochondria.
During oxidative stress mitochondrial ONOO− levels are modulated by O2•− and NO•.
Mitochondrial NOS is likely a variant of a non-inducible iNOS, but a nNOS may also contribute.
Acknowledgments
This project was funded by NIH 5T35 HL072483-34 and VA Merit Grant BX002539.
Footnotes
Conflicts of interest
The authors declare no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- [1].Palmer RM, Ferrige AG, Moncada S, Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor, Nature 327 (1987) 524–526. [DOI] [PubMed] [Google Scholar]
- [2].Balligand JL, Kobzik L, Han X, Kaye DM, Belhassen L, O’Hara DS, Kelly RA, Smith TW, Michel T, Nitric oxide-dependent parasympathetic signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes, J Biol Chem 270 (1995) 14582–14586. [DOI] [PubMed] [Google Scholar]
- [3].Kobzik L, Stringer B, Balligand JL, Reid MB, Stamler JS, Endothelial type nitric oxide synthase in skeletal muscle fibers: mitochondrial relationships, Biochem Biophys Res Commun 211 (1995) 375–381. [DOI] [PubMed] [Google Scholar]
- [4].Reiner M, Bloch W, Addicks K, Functional interaction of caveolin-1 and eNOS in myocardial capillary endothelium revealed by immunoelectron microscopy, J Histochem Cytochem 49 (2001) 1605–1610. [DOI] [PubMed] [Google Scholar]
- [5].Wei G, Dawson VL, Zweier JL, Role of neuronal and endothelial nitric oxide synthase in nitric oxide generation in the brain following cerebral ischemia, Biochim Biophys Acta 1455 (1999) 23–34. [DOI] [PubMed] [Google Scholar]
- [6].Robinson MA, Baumgardner JE, Otto CM, Oxygen-dependent regulation of nitric oxide production by inducible nitric oxide synthase, Free Radic Biol Med 51 (2011) 1952–1965. [DOI] [PubMed] [Google Scholar]
- [7].Lacza Z, Puskar M, Figueroa JP, Zhang J, Rajapakse N, Busija DW, Mitochondrial nitric oxide synthase is constitutively active and is functionally upregulated in hypoxia, Free Radic Biol Med 31 (2001) 1609–1615. [DOI] [PubMed] [Google Scholar]
- [8].Grunewald T, Beal MF, NOS knockouts and neuroprotection, Nat Med 5 (1999) 1354–1355. [DOI] [PubMed] [Google Scholar]
- [9].de Belder AJ, Radomski MW, Why HJ, Richardson PJ, Bucknall CA, Salas E, Martin JF, Moncada S, Nitric oxide synthase activities in human myocardium, Lancet 341 (1993) 84–85. [DOI] [PubMed] [Google Scholar]
- [10].Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM, Choi SY, de Groat WC, Peterson J, Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection, Proc Natl Acad Sci U S A 98 (2001) 14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bates TE, Loesch A, Burnstock G, Clark JB, Immunocytochemical evidence for a mitochondrially located nitric oxide synthase in brain and liver, Biochem Biophys Res Commun 213 (1995) 896–900. [DOI] [PubMed] [Google Scholar]
- [12].Bates TE, Loesch A, Burnstock G, Clark JB, Mitochondrial nitric oxide synthase: a ubiquitous regulator of oxidative phosphorylation?, Biochem Biophys Res Commun 218 (1996) 40–44. [DOI] [PubMed] [Google Scholar]
- [13].Csordas G, Thomas AP, Hajnoczky G, Calcium signal transmission between ryanodine receptors and mitochondria in cardiac muscle, Trends Cardiovasc Med 11 (2001) 269–275. [DOI] [PubMed] [Google Scholar]
- [14].Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC, Nitric oxide synthase in cardiac sarcoplasmic reticulum, Proc Natl Acad Sci U S A 96 (1999) 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hare JM, Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling, Proc Natl Acad Sci U S A 101 (2004) 15944–15948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Balke JE, Zhang L, Percival JM, Neuronal nitric oxide synthase (nNOS) splice variant function: Insights into nitric oxide signaling from skeletal muscle, Nitric Oxide 82 (2019) 35–47. [DOI] [PubMed] [Google Scholar]
- [17].Darley-Usmar V, The powerhouse takes control of the cell; the role of mitochondria in signal transduction, Free Radic Biol Med 37 (2004) 753–754. [DOI] [PubMed] [Google Scholar]
- [18].Sarti P, Arese M, Bacchi A, Barone MC, Forte E, Mastronicola D, Brunori M, Giuffre A, Nitric oxide and mitochondrial complex IV, IUBMB Life 55 (2003) 605–611. [DOI] [PubMed] [Google Scholar]
- [19].Sarti P, Arese M, Giuffre A, The molecular mechanisms by which nitric oxide controls mitochondrial complex IV, Ital J Biochem 52 (2003) 37–42. [PubMed] [Google Scholar]
- [20].Katakam PV, Wappler EA, Katz PS, Rutkai I, Institoris A, Domoki F, Gaspar T, Grovenburg SM, Snipes JA, Busija DW, Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase, Arterioscler Thromb Vasc Biol 33 (2013) 752–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ghafourifar P, Asbury ML, Joshi SS, Kincaid ED, Determination of mitochondrial nitric oxide synthase activity, Methods Enzymol 396 (2005) 424–444. [DOI] [PubMed] [Google Scholar]
- [22].Lindsay DP, Camara AK, Stowe DF, Lubbe R, Aldakkak M, Differential effects of buffer pH on Ca2+-induced ROS emission with inhibited mitochondrial complexes I and III, Front Physiol 6 (2015) 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dedkova EN, Blatter LA, Characteristics and function of cardiac mitochondrial nitric oxide synthase, J Physiol 587 (2009) 851–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Novalija E, Hogg N, Kevin LG, Camara AK, Stowe DF, Ischemic preconditioning: triggering role of nitric oxide-derived oxidants in isolated hearts, J Cardiovasc Pharmacol 42 (2003) 593–600. [DOI] [PubMed] [Google Scholar]
- [25].de Lima Portella R, Lynn Bickta J, Shiva S, Nitrite confers preconditioning and cytoprotection after ischemia/reperfusion injury through the modulation of mitochondrial function, Antioxid Redox Signal 23 (2015) 307–327. [DOI] [PubMed] [Google Scholar]
- [26].Camara AK, Aldakkak M, Heisner JS, Rhodes SS, Riess ML, An J, Heinen A, Stowe DF, ROS scavenging before 27 degrees C ischemia protects hearts and reduces mitochondrial ROS, Ca2+ overload, and changes in redox state, Am J Physiol Cell Physiol 292 (2007) C2021–2031. [DOI] [PubMed] [Google Scholar]
- [27].Yang M, Camara AK, Wakim BT, Zhou Y, Gadicherla AK, Kwok WM, Stowe DF, Tyrosine nitration of voltage-dependent anion channels in cardiac ischemia-reperfusion: reduction by peroxynitrite scavenging, Biochim Biophys Acta 1817 (2012) 2049–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yang M, Xu Y, Heisner JS, Sun J, Stowe DF, Kwok WM, Camara AKS, Peroxynitrite nitrates adenine nucleotide translocase and voltage-dependent anion channel 1 and alters their interactions and association with hexokinase II in mitochondria, Mitochondrion 46 (2019) 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yasmin W, Strynadka KD, Schulz R, Generation of peroxynitrite contributes to ischemia-reperfusion injury in isolated rat hearts, Cardiovasc Res 33 (1997) 422–432. [DOI] [PubMed] [Google Scholar]
- [30].Stowe DF, Camara AK, Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function, Antioxid Redox Signal 11 (2009) 1373–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Camara A, Stowe DF, Reactive Oxygen Species (ROS) and Cardiac Ischemia and Reperfusion Injury, in: Laher I (Ed.), Systems Biology of Free Radicals and Antioxidants, Springer-Verlag, Berlin, 2014, pp. 889–949. [Google Scholar]
- [32].Stowe DF, Boban M, Roerig DL, Chang D, Palmisano BW, Bosnjak ZJ, Effects of L-arginine and N omega-nitro-L-arginine methyl ester on cardiac perfusion and function after 1-day cold preservation of isolated hearts, Circulation 95 (1997) 1623–1634. [DOI] [PubMed] [Google Scholar]
- [33].Zaobornyj T, Ghafourifar P, Strategic localization of heart mitochondrial NOS: a review of the evidence, Am J Physiol Heart Circ Physiol 303 (2012) H1283–1293. [DOI] [PubMed] [Google Scholar]
- [34].Lacza Z, Pankotai E, Busija DW, Mitochondrial nitric oxide synthase: current concepts and controversies, Front Biosci (Landmark Ed) 14 (2009) 4436–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ghafourifar P, Cadenas E, Mitochondrial nitric oxide synthase, Trends Pharmacol Sci 26 (2005) 190–195. [DOI] [PubMed] [Google Scholar]
- [36].Zemojtel T, Kolanczyk M, Kossler N, Stricker S, Lurz R, Mikula I, Duchniewicz M, Schuelke M, Ghafourifar P, Martasek P, Vingron M, Mundlos S, Mammalian mitochondrial nitric oxide synthase: characterization of a novel candidate, FEBS Lett 580 (2006) 455–462. [DOI] [PubMed] [Google Scholar]
- [37].Valdez LB, Zaobornyj T, Boveris A, Mitochondrial metabolic states and membrane potential modulate mtNOS activity, Biochim Biophys Acta 1757 (2006) 166–172. [DOI] [PubMed] [Google Scholar]
- [38].Giulivi C, Poderoso JJ, Boveris A, Production of nitric oxide by mitochondria, J Biol Chem 273 (1998) 11038–11043. [DOI] [PubMed] [Google Scholar]
- [39].Tatoyan A, Giulivi C, Purification and characterization of a nitric-oxide synthase from rat liver mitochondria, J Biol Chem 273 (1998) 11044–11048. [DOI] [PubMed] [Google Scholar]
- [40].Kozlov AV, Staniek K, Nohl H, Nitrite reductase activity is a novel function of mammalian mitochondria, FEBS Lett 454 (1999) 127–130. [DOI] [PubMed] [Google Scholar]
- [41].Nohl H, Staniek K, Sobhian B, Bahrami S, Redl H, Kozlov AV, Mitochondria recycle nitrite back to the bioregulator nitric monoxide, Acta Biochim Pol 47 (2000) 913–921. [PubMed] [Google Scholar]
- [42].Gryglewski RJ, Palmer RM, Moncada S, Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor, Nature 320 (1986) 454–456. [DOI] [PubMed] [Google Scholar]
- [43].Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA, Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide, Proc Natl Acad Sci U S A 87 (1990) 1620–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A, Unraveling peroxynitrite formation in biological systems, Free Radic Biol Med 30 (2001) 463–488. [DOI] [PubMed] [Google Scholar]
- [45].Szabo C, Ischiropoulos H, Radi R, Peroxynitrite: biochemistry, pathophysiology and development of therapeutics, Nat Rev Drug Discov 6 (2007) 662–680. [DOI] [PubMed] [Google Scholar]
- [46].Alvarez MN, Piacenza L, Irigoin F, Peluffo G, Radi R, Macrophage-derived peroxynitrite diffusion and toxicity to Trypanosoma cruzi, Arch Biochem Biophys 432 (2004) 222–232. [DOI] [PubMed] [Google Scholar]
- [47].Ferdinandy P, Schulz R, Inhibition of peroxynitrite-induced dityrosine formation with oxidized and reduced thiols, nitric oxide donors, and purine derivatives, Antioxid Redox Signal 3 (2001) 165–171. [DOI] [PubMed] [Google Scholar]
- [48].Camara AK, Riess ML, Kevin LG, Novalija E, Stowe DF, Hypothermia augments reactive oxygen species detected in the guinea pig isolated perfused heart, Am J Physiol Heart Circ Physiol 286 (2004) H1289–1299. [DOI] [PubMed] [Google Scholar]
- [49].Klohn PC, Neumann HG, Impairment of respiration and oxidative phosphorylation by redox cyclers 2-nitrosofluorene and menadione, Chem Biol Interact 106 (1997) 15–28. [DOI] [PubMed] [Google Scholar]
- [50].Bombicino SS, Iglesias DE, Zaobornyj T, Boveris A, Valdez LB, Mitochondrial nitric oxide production supported by reverse electron transfer, Arch Biochem Biophys 607 (2016) 8–19. [DOI] [PubMed] [Google Scholar]
- [51].French S, Giulivi C, Balaban RS, Nitric oxide synthase in porcine heart mitochondria: evidence for low physiological activity, Am J Physiol Heart Circ Physiol 280 (2001) H2863–2867. [DOI] [PubMed] [Google Scholar]
- [52].Traaseth N, Elfering S, Solien J, Haynes V, Giulivi C, Role of calcium signaling in the activation of mitochondrial nitric oxide synthase and citric acid cycle, Biochim Biophys Acta 1658 (2004) 64–71. [DOI] [PubMed] [Google Scholar]
- [53].Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP, Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS, Nature 515 (2014) 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ahmed LA, Shehata NI, Abdelkader NF, Khattab MM, Tempol, a superoxide dismutase mimetic agent, ameliorates cisplatin-induced nephrotoxicity through alleviation of mitochondrial dysfunction in mice, PLoS One 9 (2014) e108889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Robin E, Derichard A, Vallet B, Hassoun SM, Neviere R, Nitric oxide scavenging modulates mitochondrial dysfunction induced by hypoxia/reoxygenation, Pharmacol Rep 63 (2011) 1189–1194. [DOI] [PubMed] [Google Scholar]
- [56].Aldakkak M, Camara AK, Heisner JS, Yang M, Stowe DF, Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts, Pharmacol Res 64 (2011) 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Camara AK, Begic Z, Kwok WM, Bosnjak ZJ, Differential modulation of the cardiac L- and T-type calcium channel currents by isoflurane, Anesthesiology 95 (2001) 515–524. [DOI] [PubMed] [Google Scholar]
- [58].Yang M, Camara AKS, Aldakkak M, Kwok WM, Stowe DF, Identity and function of a cardiac mitochondrial small conductance Ca2+-activated K+ channel splice variant, Biochim Biophys Acta Bioenerg 1858 (2017) 442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Graham JM, Purification of a crude mitochondrial fraction by density-gradient centrifugation, Curr Protoc Cell Biol Chapter 3 (2001) Unit 3 4. [DOI] [PubMed] [Google Scholar]
- [60].Berryman MA, Rodewald RD, An enhanced method for post-embedding immunocytochemical staining which preserves cell membranes, J Histochem Cytochem 38 (1990) 159–170. [DOI] [PubMed] [Google Scholar]
- [61].Blomeyer CA, Bazil JN, Stowe DF, Dash RK, Camara AK, Mg2+ differentially regulates two modes of mitochondrial Ca2+ uptake in isolated cardiac mitochondria: implications for mitochondrial Ca2+ sequestration, J Bioenerg Biomembr 48 (2016) 175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Huang M, Camara AK, Stowe DF, Qi F, Beard DA, Mitochondrial inner membrane electrophysiology assessed by rhodamine-123 transport and fluorescence, Ann Biomed Eng 35 (2007) 1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Loor G, Kondapalli J, Schriewer JM, Chandel NS, Vanden Hoek TL, Schumacker PT, Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis, Free Radic Biol Med 49 (2010) 1925–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pryor WA, Cueto R, Jin X, Koppenol WH, Ngu-Schwemlein M, Squadrito GL, Uppu PL, Uppu RM, A practical method for preparing peroxynitrite solutions of low ionic strength and free of hydrogen peroxide, Free Radic Biol Med 18 (1995) 75–83. [DOI] [PubMed] [Google Scholar]
- [65].Blomeyer CA, Bazil JN, Stowe DF, Pradhan RK, Dash RK, Camara AK, Dynamic buffering of mitochondrial Ca2+ during Ca2+ uptake and Na+-induced Ca2+ release, J Bioenerg Biomembr 45 (2013) 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chance B, Hollunger G, The interaction of energy and electron transfer reactions in mitochondria. IV. The pathway of electron transfer, J Biol Chem 236 (1961) 1562–1568. [PubMed] [Google Scholar]
- [67].Tretter L, Patocs A, Chinopoulos C, Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis, Biochim Biophys Acta 1857 (2016) 1086–1101. [DOI] [PubMed] [Google Scholar]
- [68].Aldakkak M, Stowe DF, Dash RK, Camara AK, Mitochondrial handling of excess Ca2+ is substrate-dependent with implications for reactive oxygen species generation, Free Radic Biol Med 56 (2013) 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Drose S, Brandt U, The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex, J Biol Chem 283 (2008) 21649–21654. [DOI] [PubMed] [Google Scholar]
- [70].Xia Y, Superoxide generation from nitric oxide synthases, Antioxid Redox Signal 9 (2007) 1773–1778. [DOI] [PubMed] [Google Scholar]
- [71].Dikalov SI, Harrison DG, Methods for detection of mitochondrial and cellular reactive oxygen species, Antioxid Redox Signal 20 (2014) 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Miller MJ, Thompson JH, Liu X, Eloby-Childress S, Sadowska-Krowicka H, Zhang XJ, Clark DA, Failure of L-NAME to cause inhibition of nitric oxide synthesis: role of inducible nitric oxide synthase, Inflamm Res 45 (1996) 272–276. [DOI] [PubMed] [Google Scholar]