

Abstract
Thrombosis is a principle cause of various life-threatening cardiovascular diseases. However, current antithrombotic treatments using drugs only offer limited efficacy due to short half-life, low targeting ability to the thrombus site, and unexpected bleeding complications. Taking into account of the biological characteristics of thrombus including upregulation of hydrogen peroxide (H2O2) and abundance of fibrin, we engineered a H2O2-responsive nanocarrier for thrombus-targeting delivery of an antithrombotic agent (i.e., tirofiban). The nanocarrier was composed of a drug-conjugated dextran nanocore and a red blood cell (RBC) membrane shell, and its surface was functionalized with a fibrin-targeting peptide, CREKA. Tirofiban was conjugated to dextran through a H2O2-cleavable phenylboronic ester linkage. The fibrin-targeting RBC membrane-cloaked dextran-tirofiban conjugate nanoparticles (i.e., T-RBC-DTC NPs) can scavenge H2O2 and provide controlled release of tirofiban to achieve site-specific antithrombotic effects. In RAW 264.7 cells and HUVECs, the T-RBC-DTC NPs effectively scavenged H2O2 and protected cells from H2O2-induced cytotoxicity. In the ferric chloride-induced carotid thrombosis mouse model, the T-RBC-DTC NPs efficiently accumulated at the injured carotid artery and exhibited significantly enhanced antithrombotic activity compared to free drug. The T-RBC-DTC NPs also exhibited good biocompatibility according to histology analysis. Overall, our results indicated that this bioengineered nanocarrier offers a promising therapeutic strategy for thrombotic disorders.
1. Introduction
Thrombosis is the major pathogenesis of various life-threatening cardiovascular diseases such as myocardial infarction [1], ischemic stroke [2], and pulmonary embolism [3]. Acute blood vessel obstruction caused by thrombosis can dramatically reduce the blood flow, leading to extensive cell death, organ dysfunction, and even death. For thrombus therapy, rapid removal of clots is a key requirement to recanalize the occluded vessels. Both surgical intervention and antithrombotic therapy are effective treatment methods clinically. However, due to the high risk, high cost, and invasiveness of surgical procedures, antithrombotic drugs remain a preferable choice [2,3]. Nevertheless, these drugs are unable to target the thrombus and may impair normal hemostatic capabilities, and subsequently generates a high risk of undesirable bleeding complications. Moreover, the short half-life of most antithrombotic drugs requires repeated administration, leading to additional cost, inconvenience, and risks. Therefore, innovative strategies for targeted delivery of antithrombotic drugs to the thrombus site are urgently needed to reduce side effects and extend the half-life of drugs for better therapeutic outcomes.
Reactive oxygen species (ROS) are upregulated at the thrombus site, which are generated by the injured endothelium and activated platelets [4,5]. The elevated ROS levels in turn lead to further endothelial dysfunction and platelet activation, thereby facilitating the propagation of thrombus [6,7]. ROS also mediate endothelial expression of inflammatory cytokines and promotes platelet-endothelium interactions and vessel occlusion [8,9]. Therefore, scavenging excessive ROS would be a promising therapeutic strategy for thrombus [10]. Moreover, for thrombus-targeting drug delivery nanosystems, elevated ROS levels in the thrombus microenvironment can be utilized as a smart trigger for controlled drug release. The incorporation of targeting ligands in drug delivery nanosystems can improve localization and accumulation of drug to the desired thrombus site. Biological factors in the thrombus microenvironment, such as glycoprotein [11–15], P-selectin [16–18], collagen [19], fibrin network [20–22], and thrombin [23], have been widely explored as targets for the design of clot-targeting therapeutics. Among these biological factors, fibrin has been described as a significant hallmark of thrombus with high specificity [24,25]. During thrombus formation, fibrins can be generated locally from the circulating fibrinogens catalyzed by thrombin and coagulation factors. Fibrins can crosslink and form a dense biopolymeric mesh that protects the clot from mechanical stress and proteolytic attack and promotes the recruitment of activated platelets. Given that fibrins are only abundantly present at the thrombus site, fibrin-targeting drug delivery systems can enable thrombus-targeting therapy. A peptide, CREKA (Cys-Arg-Glu-Lys-Ala), identified through a phage display, has been reported as a clot-binding peptide, which showed favorable targeting ability to fibrin [21,26,27]. CREKA is a desirable targeting ligand as it is linear and contains only 5 amino acid residues. In addition, cysteine, the terminal amino acid in CREKA, allows for convenient bioconjugation via a thiol-maleimide reaction [28].
A number of nanoplatforms have been developed for the delivery of antithrombotic drugs, including liposomes [15], polymers [17,29], prodrugs [13], and mesoporous silica nanoparticles [30]. However, it remains challenging to simultaneously achieve thrombus targeted drug delivery, stimulus-controlled drug release, ROS scavenging and long circulation time. To address these challenges, we sought to develop a polymer-drug conjugate that can take advantage of the physiological and biological characteristics of thrombus. Considering hydrogen peroxide (H2O2) is involved in the signaling pathways in the vasculature [7], H2O2-responsive thrombus-targeting red blood cell (RBC) membrane-cloaked dextran-tirofiban conjugate nanoparticles (T-RBC-DTC NPs) were developed for antithrombotic therapy. As a proof-of-concept, tirofiban was chosen as the model antithrombotic drug. Tirofiban is a synthetic, non-peptide antagonist of the platelet glycoprotein IIb/IIIa receptor with a half-life about 2 h, which specifically inhibits fibrinogen-dependent platelet aggregation and has been described as an effective antiplatelet agent in the management of acute coronary syndromes [31,32]. As shown in Fig. 1, the dextran-tirofiban conjugate (DTC) was first synthesized by conjugating tirofiban to dextran through phenylboronic ester linkage. This linkage can be rapidly oxidized and cleaved by H2O2, and thus can scavenge H2O2 during the oxidation reaction. Moreover, the H2O2-responsive linker can inhibit drug leakage during circulation, and provide controlled drug release at the thrombus site. Once the polymer-drug linkage is cleaved at the thrombus site, tirofiban can be restored to its original chemical structure, thereby maintaining its drug efficacy. Thereafter, the T-RBC-DTC NPs were fabricated by coating CREKA-functionalized RBC membrane on the surface of the self-assembled dextran-tirofiban conjugate nanoparticles (DTC NPs). CREKA-functionalized RBC membrane provides thrombus-targeting capability and can enhance the biocompatibility and circulation time of the nanosystem [33–36]. Notably, platelet membrane may be more suitable than RBC membrane for thrombus targeting, due to its thrombus-homing property in nature and the role of platelet in the thrombotic processes. However, tirofiban, an antiplatelet agent, is likely to compromise the receptor on the platelet membrane and thus may impact the targeting capability. Instead, we used RBC membrane coating and incorporated fibrin-targeting peptides to achieve thrombus targeting. In this work, the H2O2 scavenging capability of the T-RBC-DTC NPs was first demonstrated by cell culture models. In vivo imaging study demonstrated robust homing of the T-RBC-DTC NPs at the thrombus site. Furthermore, the T-RBC-DTC NPs significantly suppressed thrombus formation in a ferric chloride (FeCl3)-induced carotid arterial thrombosis mouse model.
Fig. 1.
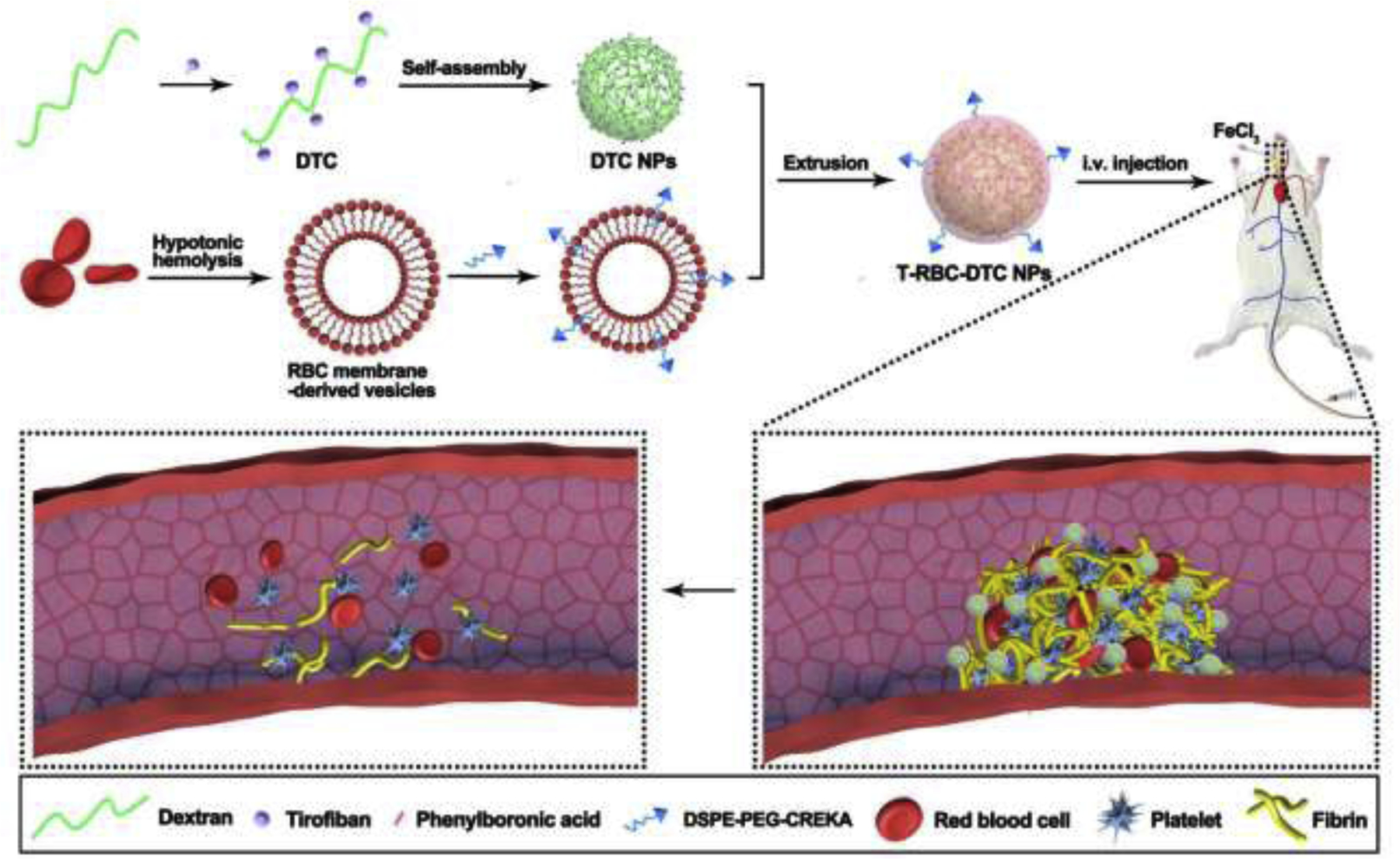
A schematic diagram of the T-RBC-DTC NPs as potent thrombus-targeting antithrombotic agents.
2. Materials and methods
2.1. Materials
2-(4-(bromomethyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane and diethanolamine were purchased from TCI (Tokyo, Japan) and Alfa Aesar (Tewksbury, MA, USA), respectively. 2-distearoyl-sn-glycero-3-phospho-ethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG2000-Mal), CREKA (Cys-Arg-Glu-Lys-Ala), and 4’,6-diamidino-2-phenylindole (DAPI) were obtained from NOF Corp (Tokyo, Japan), ABI Scientific Inc. (Sterling, VA, USA), and Abcam (Cambridge, MA, USA), respectively. Dextran (molecular weight ~ 20 kDa), poloxamer 188, 2,7-dichlorofluorescein diacetate, and dihydroethidium were purchased from Sigma-Aldrich (St. Louis, MO, USA). The whole blood from female ICR mice was purchased from BioIVT elevating science (Winchester, VA, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from VWR (Radnor, PA, USA). Other reagents were purchased from ThermoFisher Scientific (Fitchburg, WI, USA) and used as received unless otherwise stated.
2.2. Cell culture
Human umbilical vein endothelial cells (HUVECs) were cultured in endothelial basal medium supplemented with growth supplements at 37 °C in 5% CO2 atmosphere. Mouse macrophage cell line RAW 264.7 cells were maintained with RPMI-1640 cell culture medium (Gibco, USA) supplemented with 10% (v/v) fetal bovine serum (Gibco, USA) and 100 U/mL penicillin-streptomycin (Gibco, USA), in a humidified atmosphere containing 5% CO2 at 37 °C.
2.3. Synthesis and characterization of the dextran-tirofiban conjugate
2.3.1. Synthesis of the phenylboronic acid-tirofiban conjugate
Tirofiban (516.3 mg), 2-(4-(bromomethyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (504.6 mg), K2CO3 (647.8 mg), and KI (194.5 mg) were dissolved in 30 mL of anhydrous DMF and stirred at room temperature for 16 h. The product was purified and obtained by repeated precipitation in diethyl ether for 4 times and dried under vacuum. 1H-NMR (Fig. S2, 400 MHz, DMSO-d6, ppm): 7.6 (d, 2H), 7.3 (d, 2H), 7.1 (d, 2H), 6.7 (d, 2H), 3.9 (t, 2H), 3.6 (m, 1H), 3.4 (s, 2H), 2.9–3.2 (d, 2H), 2.6–2.8 (m, 4H), 1.9 (m, 2H), 1.1–1.7 (m, 13H), 1.3 (s, 12H), 0.8 (t, 3H).
2.3.2. Deprotection of the phenylboronic acid-tirofiban conjugate
Phenylboronic acid-tirofiban conjugate (306 mg) and diethanolamine (245.1 mg) were dissolved in 9 mL of anhydrous methanol. The reaction was stirred at room temperature for 16 h. The product was precipitated in diethyl ether and collected by centrifugation. The precipitate was dissolved in methanol. The pH of the resulting solution was adjusted to ~3 by adding HCl solution. Thereafter, the reaction was carried out at room temperature for 3 h. After precipitation by diethyl ether, the supernatant was collected and concentrated under vacuum to obtain the deprotected product. 1H-NMR (Fig. S3, 400 MHz, DMSO-d6, ppm): 7.6 (d, 2H), 7.3 (d, 2H), 7.1 (d, 2H), 6.7 (d, 2H), 4.3 (t, 2H), 3.9 (m, 3H), 3.3 (m, 4H), 2.7–3.1 (m, 4H), 1.0–1.9 (m, 15H), 0.8 (t, 3H).
2.3.3. Synthesis of the dextran-tirofiban conjugate
Deprotected phenylboronic acid-tirofiban conjugate (55 mg) and dextran (Mw~20 kDa, 15.5 mg) were dissolved in 1 mL of anhydrous DMSO in a dry round bottom flask equipped with molecular sieves. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (17.4 μL) was added to the solution and stirred overnight at room temperature. The polymer was purified by dialysis using a dialysis bag (molecular weight cut off, 3.5 kDa) against DMSO and 1 % DBU. Then, the product was precipitated into ethyl acetate and dried overnight under vacuum. 1H-NMR (Fig. S4, 400 MHz, DMSO-d6, ppm): 6.5–8.0 (m, 3.87H), 3.0–4.0 (m, 10.2H), 1.0–2.0 (m, 10.51H), 0.8 (m, 1.8 H).
2.3.4. Synthesis of the Cy 5.5-tagged dextran-tirofiban conjugate
Dextran-tirofiban conjugate (30 mg), disuccinimidyl carbonate (DSC, 237.2 mg), and pyridine (74.6 uL) were dissolved in 2 mL of anhydrous DMSO. The reaction was stirred at room temperature for 24 h. The solution was dialyzed against DMSO. Then, the product was precipitated into ethyl acetate and dried overnight under vacuum. Subsequently, the DSC-conjugated polymer and Cy 5.5-amine at the same mole ratio were dissolved in 2 mL of anhydrous DMSO under nitrogen. The reaction was carried out in the dark overnight. After dialysis against DMSO, the product was precipitated into ethyl acetate and dried overnight under vacuum.
2.4. Synthesis of the DSPE-PEG-CREKA
DSPE-PEG-CREKA was synthesized via a Michael addition reaction. CREKA bearing a thiol group terminal and DSPE-PEG-Mal were dissolved in DMSO/chloroform solution (1:1, v/v) at the molar ratio of 1.2:1. The resulting solution was gently stirred at room temperature overnight. Thereafter, the DMSO/chloroform solution was dried by a rotary evaporator. The product was purified by dialysis with a dialysis tubing (MWCO = 3.5 kDa) against DI water to remove residual CREKA followed by lyophilization.
2.5. Preparation and characterization of the T-RBC-DTC NPs
2.5.1. Preparation of RBC membrane-derived vesicles
The RBC membrane-derived vesicles were prepared using the hypotonic hemolysis method [33–35]. Briefly, whole blood collected from female ICR mice was centrifuged (3000 rpm, 5 min) at 4 °C to separate erythrocytes from serum and leukocytic cream. The resultant erythrocytes were washed with 1 × PBS (pH 7.4) 3 times and then suspended in 0.25 × PBS at 4 °C for 1 h. After this hypotonic buffer treatment, the solution was centrifuged (12000 rpm, 10 min) to remove the released hemoglobin. The process was repeated until the hemoglobin was completely removed. The pellet was then resuspended in water and sonicated. The resulting vesicles were subsequently extruded 11 times through 400 nm polycarbonate porous membranes using an Avanti mini extruder. The membrane protein concentration was determined by the BCA assay.
2.5.2. Preparation of the DTC, RBC-DTC and T-RBC-DTC NPs
The DTC NPs were prepared by self-assembly of the dextran-tirofiban conjugate, by dissolving 4 mg of dextran-tirofiban conjugate in 1 mL of poloxamer 188 solution (0.1%, w/v). To prepare RBC membrane-coated dextran-tirofiban conjugate nanoparticles (RBC-DTC NPs), the RBC membrane-derived vesicles were mixed with freshly made DTC NPs at a membrane protein to polymer weight ratio of 1:1. The mixture was then extruded through a 400 nm and a 200 nm polycarbonate porous membrane sequentially using an Avanti mini extruder.
To prepare CREKA-functionalized RBC membrane-derived vesicles, the RBC membrane-derived vesicles containing 5 mg of membrane protein were first incubated with 1 mg of DSPE-PEG-CREKA at 37 °C for 30 min to ensure the complete insertion of DSPE-PEG-CREKA into the lipid bilayer of the vesicles. The resulting DSPE-PEG-CREKA-functionalized RBC membrane-derived vesicles were mixed with the DTC NPs and then extruded to yield the T-RBC-DTC NPs [37]. Similarly, the Cy 5.5-tagged DTC NPs, RBC-DTC NPs, and T-RBC-DTC NPs were prepared with the aforementioned processes by using Cy 5.5-tagged DTC.
2.5.3. Characterization
The hydrodynamic size and zeta potential of the nanoparticles were measured by dynamic light scattering (DLS, ZetaSizer Nano ZS90, Malvern Instruments, USA) at a 90° detection angle at a concentration of 0.1 mg/mL. The morphology of different nanoparticles was visualized by transmission electronic microscopy (TEM, FEI Tecnai G2 F30 TWIN 300 KV, E.A. Fischione Instruments, Inc. USA) after negative staining with a phosphotungstic acid solution (1%, w/w). The particle size of the T-RBC-DTC NPs in PBS containing 10% FBS was monitored by DLS over one week to evaluate the stability of the nanoparticles. Dissociation of the nanoparticles triggered by H2O2 was performed by incubation with 100 μM H2O2 at 37 °C. Particle size was monitored by DLS at 1 h after incubation.
In vitro release kinetics of tirofiban from the T-RBC-DTC NPs were studied by a dialysis method. 1.5 mL of T-RBC-DTC NPs suspension (1 mg/mL of tirofiban) was sealed in a dialysis bag with a molecular weight cutoff of 3.5 kDa and immersed in 50 mL of 1% Tween 80 containing PBS (0.1 M, pH 7.4) with or without 100 μM H2O2 under gentle shaking at 37 °C. At certain intervals (0.5, 1, 2, 4, 6, 8, 10, 12, 24, and 48 h), 0.2 mL of the solution outside the bag was collected to determine the released tirofiban amount by reversed-phase high-performance liquid chromatography (HPLC) with a UV detector measuring absorbance at 227 nm. An equal volume of pre-warmed fresh medium was added back to the release medium.
The ability of the T-RBC-DTC NPs to scavenge H2O2 was evaluated by the Amplex Red assay (Invitrogen, #A22188) [38]. Various amounts of the T-RBC-DTC NPs were added to a H2O2 solution with an initial H2O2 concentration of 100 μM. The resulting solution was incubated at 37 °C under gentle shaking. At 1 h post-treatment, the solution was centrifuged to precipitate the nanoparticles. The residual level of H2O2 was measured by the Amplex Red assay according to the manufacturer’s protocol.
2.6. Cytotoxicity assay
The cytotoxicity of various formulations on RAW 264.7 cells and HUVECs was tested by an MTT assay. Cells were cultured in a 96-well plate (1 × 104 cells/well) and incubated till around 80% confluence. The cells were then treated with various concentrations of nanoparticles and incubated with or without H2O2 (100 μM) for 24 h. Then, the cell media was discarded, and the cells were washed with PBS for 3 times. Subsequently, 200 μL of MTT solution (0.5 mg/mL in PBS) was added to each well and incubated at 37 °C for another 4 h. The resulting purple precipitates were dissolved in 150 μL of DMSO. Cell viability was measured by monitoring the difference between the absorbance at 560 nm and 650 nm using a GloMax-Multi Microplate Multimode Reader (Promega, WI, USA). Cell viability was calculated as a percentage relative to untreated cells.
2.7. Intracellular ROS detection
The intracellular ROS was monitored using the 2,7-dichlorofluorescein diacetate (DCFH-DA) assay kit. RAW 264.7 cells and HUVECs were seeded in 24-well culture plates and incubated for 24 h. Then, cells were treated with various formulations in the presence of H2O2 (100 μM). After incubation at 37 °C for 24 h, cells were washed and treated with 10 μM DCFH-DA for 30 min in the dark at 37 °C. Then, cells were harvested and washed to measure the intracellular ROS concentration by detecting the dihydrodichlorofluorescein (DCF) fluorescence through flow cytometry (Attune NxT flow cytometer system, ThermoFisher, USA) and analyzed with FlowJo 7.6. The untreated cells were used as a negative control. 1 × 104 events in total were counted for each sample. For fluorescence microscopy imaging, cells were washed with PBS, fixed with 4% (w/v) paraformaldehyde, and stained with DAPI. The cells were imaged under a fluorescence microscope.
2.8. Animal studies
2.8.1. A FeCl3-induced carotid thrombosis mouse model
All animal studies conform to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) and protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Wisconsin. ICR mice (female, 22–24 g, Charles River) were maintained in an SPF level animal room at 22 °C under a 12 h light-dark cycle and food and water ad libitum. A FeCl3-induced carotid artery thrombosis model was established as previously described [39]. Briefly, after the mouse was anesthetized with isoflurane (3% for inducing and 1% for maintaining anesthesia), the hair of the whole neck was shaved off. Thereafter, a longitudinal incision of the neck skin was made, and the carotid artery was exposed. A 1 mm × 2 mm piece of filter paper soaked with 10% FeCl3 was used to wrap around the carotid artery for 3 min to generate vascular injury. The body temperature of the mice was maintained at around 37 °C using a heating pad during the surgery.
2.8.2. Thrombus imaging
The Cy 5.5-labeled nanoparticles were tail-vein injected immediately after vascular injury. At scheduled time points, the Cy 5.5 fluorescence images at the carotid artery were captured via an in vivo imaging system (IVIS) equipped with an excitation bandpass filter at 676 nm and emission at 705 nm. After euthanasia of the mice, injured and non-injured carotid arteries and major organs including heart, liver, spleen, lung, and kidney were collected.
2.8.3. Histological examination
To assess the in vivo therapeutic potential of the T-RBC-DTC NPs, mice were divided into six groups including saline, tirofiban, DTC NPs, RBC-DTC NPs, T-RBC-DTC NPs, and the sham group. The dose of tirofiban in all tirofiban-containing groups was constant at 80 μg/kg. After the FeCl3-induced thrombosis mouse model was established, the different formulations were immediately injected via the tail vein. In the sham group, the carotid arteries were exposed and incubated with saline-soaked filter paper instead of FeCl3-soaked one. After 24 h of therapy, carotid arteries excised from mice were embedded in optimal cutting temperature (OCT) compound blocks. Then, the arterial tissue blocks were sectioned at a microtome setting of 7 μm. To observe the antithrombotic effect, the sections were stained with hematoxylin and eosin (H&E) and examined under an optical microscope. The thrombosis degree of the carotid artery was analyzed using Image J software and calculated by the blood clot area/total vascular area. For dihydroethidium (DHE) staining, tissue sections were incubated with 5 μM DHE at 37 °C for 30 min in the dark. Then, DAPI was applied to the sections. The sectioned carotid artery tissues were observed using a fluorescence microscope.
2.8.4. Measurement of the levels of TNF-α and sCD40L
Blood was collected into a tube freshly pretreated with 1% heparin and centrifuged at 3000 rpm for 10 min at 4 °C to obtain plasma. The levels of TNF-α and sCD40L were measured with a mouse TNF-α ELISA kit (R&D Systems, MTA00B) and a mouse sCD40L ELISA kit (Invitrogen, BMS6010), respectively, according to manufacturer’s instructions.
2.9. Biocompatibility
Mice were administrated either with saline or T-RBC-DTC NPs (containing tirofiban at 80 μg/kg) every day for a week. After euthanasia, organs including heart, liver, spleen, lung, and kidney were excised for histological examination. Organs were fixed with paraformaldehyde for 24 h and embedded in OCT and the blocks were sectioned for H&E staining. The image of tissues was acquired using an optical microscope.
2.10. Statistical analysis
Data are presented as mean ± standard deviation (SD). Statistical analysis was conducted using a One-way ANOVA or t-test by GraphPad Prism software version 7.0. Significance was reported as * when p < 0.05, ** when p < 0.01, and *** when p < 0.001.
3. Results and Discussion
3.1. Biomimetic thrombus-targeting T-RBC-DTC NPs were successfully synthesized and well characterized
To develop thrombus-targeting and H2O2-scavenging antithrombotic nanoparticles, a H2O2-responsive prodrug polymer was synthesized by conjugating tirofiban to dextran through a H2O2-cleavable phenylboronic ester linkage (Fig. S1). We chose dextran as a substrate to synthesize the prodrug polymer because it is an FDA-approved volume expander with good biodegradability and biocompatibility. Tirofiban was conjugated to phenylboronic acid via an amine alkylation reaction. After deprotection of pinacol ester, the prodrug was then conjugated to dextran through a coupling reaction between the phenylboronic acid functional group on the prodrug and the diol group on dextran. The structure of all intermediate and final products (Fig. S2–4) were confirmed by 1H NMR. The tirofiban conjugation ratio quantified by NMR was 60% (i.e., 60 tirofiban molecules were conjugated per 100 dextran saccharide units).
For rapid and specific thrombus-targeting, CREKA was chosen as a targeting ligand because of its high affinity for fibrins which are abundantly present at thrombus sites. DSPE-PEG-CREKA was synthesized via a Michael addition reaction between the thiol group of CREKA and the maleimide group of DSPE-PEG-Mal. It is used to facilitate the integration of the CREKA peptide into the RBC membrane-derived vesicles. The successful synthesis of DSPE-PEG-CREKA was confirmed by 1H NMR (Fig. S5–6). Chemical shifts of the methoxy group (−OCH2) from PEG were observed at 3.6 ppm, while those from the C-H lipid chain were found at 0.8, 1.2 and 1.5 ppm. After CREKA was conjugated to DSPE-PEG-Mal, the original chemical shifts of maleimide groups (at 6.92 ppm) disappeared, suggesting the successful conjugation of CREKA.
The T-RBC-DTC NPs were prepared using a three-step process based on a previously published method [37]. In the first step, the RBC membrane was extracted by the combination of hypotonic lysis and mechanical disruption to yield RBC membrane-derived vesicles. To achieve the targeting capability, DSPE-PEG-CREKA was integrated into the RBC membrane-derived vesicles by co-incubation. Meanwhile, due to its amphiphilic nature, DTC can self-assemble into a nanoparticle core with an anionic surface via hydrophobic interactions between the phenylboronic acid-tirofiban segments of the polymers. Thereafter, the CREKA-decorated RBC membrane-derived vesicles were subsequently coated onto the surface of DTC NPs through an extrusion process. The hydrophilic glycans and the negatively charged sialic acid residues on the surface of the RBC membranes contribute to the unilamellar coating of the RBC membranes onto the nanoparticles surface in a right-side-out orientation [40]. DLS measurements revealed that with RBC membrane coating, the average hydrodynamic diameter of the RBC-DTC NPs increased from 160.6 ± 3.9 to 176.5 ± 2.6 nm and the surface zeta potential decreased from −13.5 ± 1.0 to −28.5 ± 1.2 mV (Fig. 2A). An increase around 16 nm in diameter is consistent with the addition of a lipid bilayer membrane onto the exterior of a polymeric nanoparticle [41]. Additionally, the surface zeta potential of the RBC-DTC NPs was more negative than that of the cores but comparable to the RBC membrane-derived vesicles (−31.1 ± 0.8 mV). This phenomenon is commonly observed after membrane coating to nanoparticles and indicates shielding of the less negatively charged DTC NPs with the more negatively charged outer cell membrane surface [41]. As shown in the TEM images, the RBC-DTC NPs and T-RBC-DTC NPs exhibited a characteristic core-shell structure, which further demonstrated a successful and complete cell membrane coating on the DTC NPs (Fig. 2B). In addition, the drug loading content of the T-RBC-DTC NPs was 13% by weight. The sizes of the T-RBC-DTC NPs in PBS containing 10% FBS remained relatively stable for one week at both room temperature (i.e., 20 °C) and 37 °C, indicating good stability.
Fig. 2.
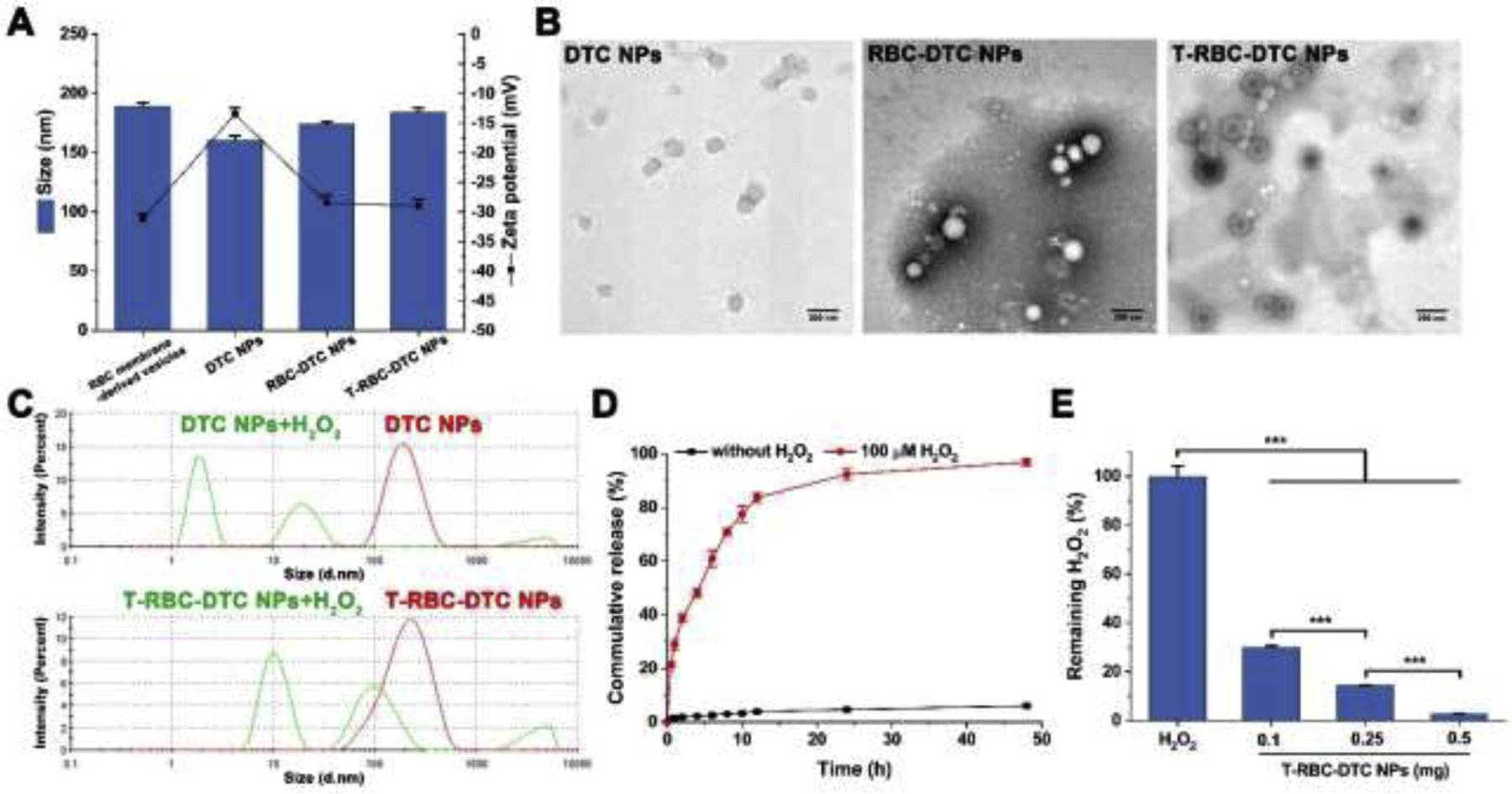
Characterizations of the nanoparticles. (A) Particle size and zeta potential of various nanoparticles. Data is shown as mean ± SD (n=3). (B) TEM images of the DTC NPs, RBC-DTC NPs, and T-RBC-DTC NPs. Scale bar: 200 nm. (C) Size distribution of the DTC NPs and T-RBC-DTC NPs in response to 100 μM H2O2. (D) Kinetics of tirofiban release from the T-RBC-DTC NPs in PBS containing 1% Tween 80 with or without 100 μM H2O2. Data is shown as mean ± SD (n=3). (E) H2O2-scavenging capability of the T-RBC-DTC NPs. Data is shown as mean ± SD (n=3). ***p < 0.001.
3.2. The T-RBC-DTC NPs effectively released drugs in response to an elevated H2O2 level, and efficiently scavenged H2O2 to protect cells in vitro
In order to evaluate the H2O2 sensitivity of the T-RBC-DTC NPs, the morphology change of the nanoparticles was monitored in the presence of H2O2 (100 μM) at 37 °C by DLS. As shown in Fig. 2C, the sizes of the nanoparticles changed dramatically in response to the presence of H2O2. The addition of H2O2 resulted in rapid dissociation of the DTC NPs because the phenylboronic ester functional groups in the polymer were oxidized by H2O2 and were quickly cleaved, leading to a decrease of hydrophobic interaction between polymers. Moreover, the RBC membrane coating may be ruptured by H2O2-induced hydrophobic to hydrophilic conversion of the polymer. As presented in Fig. S1, oxidation of H2O2 can lead to DTC degradation, which generate three types of water soluble small molecules, i.e., tirofiban, 4-(hydroxymethyl)phenol, and boronic acid. These compounds together with the recovered hydrophilic dextran dramatically raised the osmotic pressure within the RBC vesicles, thereby possibly leading to the rupture of the RBC membrane coating. The in vitro drug release profile of the T-RBC-DTC NPs was evaluated in the 1% Tween 80 containing PBS (0.1 M, pH 7.4) with or without H2O2 at 37 °C (Fig. 2D). There was only about 14% drug release within 48 h in the absence of H2O2. However, almost all the drug (95%) loaded in the T-RBC-DTC NPs was released with a rapid initial burst release in the presence of H2O2 (100 μM). The H2O2-dependent drug release behavior further confirmed the cleavage of the phenylboronic ester linker between the drug and the polymer. The H2O2 scavenging capability of the T-RBC-DTC NPs was also investigated. A H2O2 solution was treated with various amounts of the T-RBC-DTC NPs and the concentration of remaining H2O2 was measured using the Amplex Red assay. As shown in Fig. 2E, the T-RBC-DTC NPs were able to efficiently scavenge H2O2 in a concentration-dependent manner. The H2O2 scavenging capability was attributed to the phenylboronic ester linkages of the prodrug polymer that undergo H2O2-triggered oxidation to consume H2O2.
To investigate the cytotoxicity of the nanoparticles, the MTT assay was performed using RAW 264.7 cells and HUVECs. DTC NPs showed negligible toxicity at concentrations less than 150 μg/mL (Fig. 3A and Fig. 4A). No obvious cytotoxicity was observed in the tirofiban, RBC-DTC NPs, and T-RBC-DTC NPs treatment groups, demonstrating their excellent biocompatibility at the tested concentrations. We also assessed the protective effects of the nanoparticles from the H2O2-induced oxidative stress. Stimulation with H2O2 (100 μM) induced a remarkable reduction in cell viability for untreated cells. Tirofiban also exhibited no effect on the viability of H2O2-stimulated cells. However, the DTC NPs effectively protected cells from H2O2-induced cell death in a DTC concentration-dependent manner because of the H2O2-scavenging ability of the phenylboronic ester functional groups. In the presence of DTC NPs (150 μg/mL DTC), the viability of RAW 264.7 cells increased from 35% to 69%. The viability of HUVECs increased from 31% to 61% after the treatment of DTC NPs (150 μg/mL DTC). The RBC-DTC NPs and T-RBC-DTC NPs showed similar protective effects with the DTC NPs when the DTC concentration was 150 μg/mL. However, an equivalent amount of free tirofiban did not show any protective effect on cells subjected to H2O2 treatment.
Fig. 3.
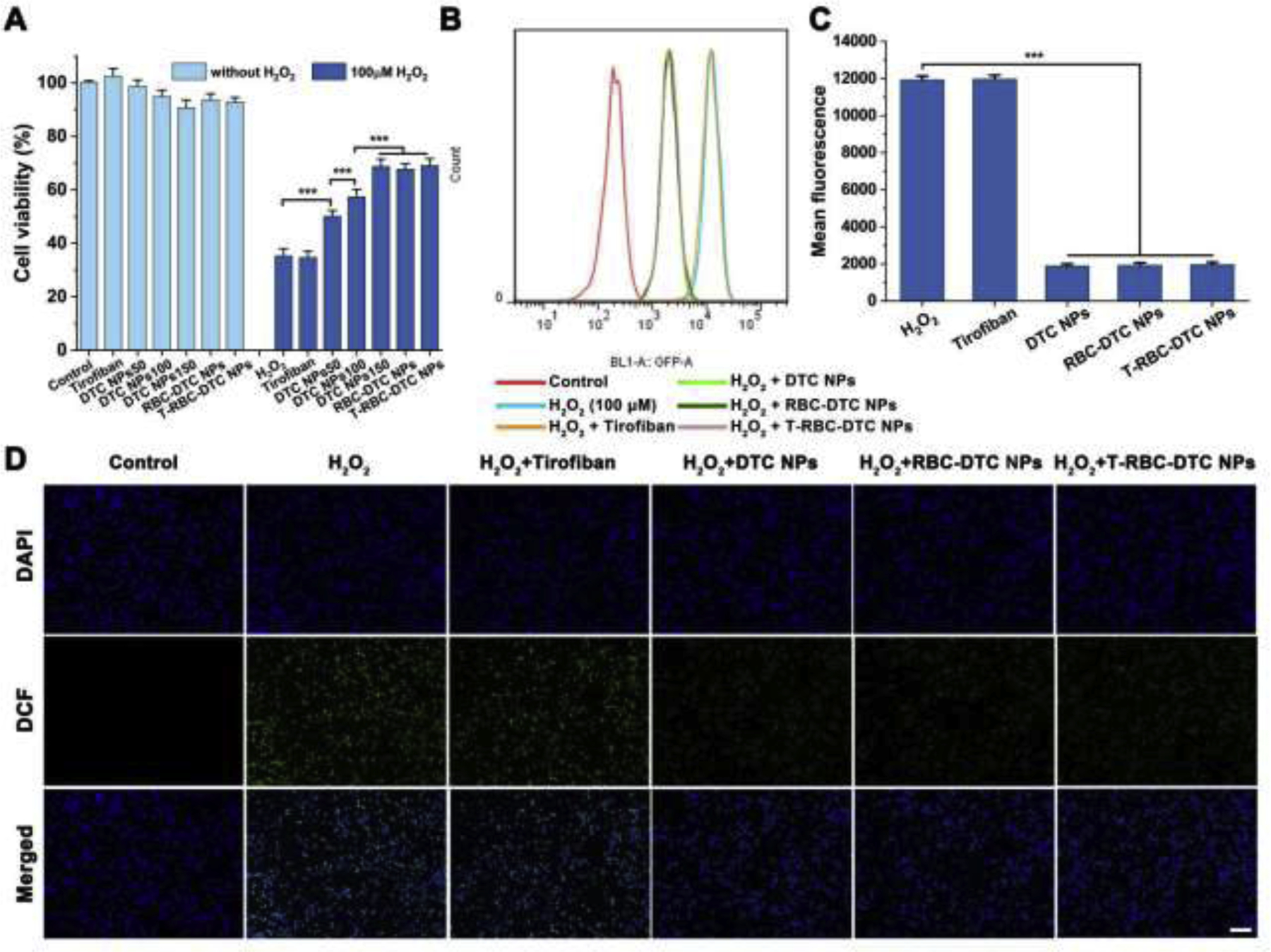
Anti-oxidative effects of the T-RBC-DTC NPs in H2O2-stimulated RAW 264.7 cells. (A) Cytotoxicity and protective effects of different treatments on H2O2-induced RAW 264.7 cells. 50,100, and 150 μg/mL is the concentration of dextran-drug conjugate in the DTC NPs. In the RBC-DTC NPs and T-RBC-DTC NPs groups, the cells were treated with the nanoparticles with the dextran-tirofiban conjugate (DTC) concentration of 150 μg/mL. In the tirofiban group, the cells were treated with 78 μg/mL of tirofiban, an equivalent amount of drug in 150 μg/mL of DTC. Data is shown as mean ± SD (n=6). (B) Flow cytometry histogram of intracellular ROS level in H2O2-stimulated RAW 264.7 cells subjected to different treatments. (C) Quantitative analysis of the intracellular ROS level in RAW 264.7 cells. Data is shown as mean ± SD (n=3). (D) Fluorescence microscopy images of intracellular ROS in RAW 264.7 cells subjected to different treatments. Blue channel: 4’,6-diamidino-2-phenylindole (DAPI) stained nucleus. Green channel: dihydrodichlorofluorescein (DCF) fluorescence illustrated intracellular ROS. Scale bar: 100 μm. ***p < 0.001.
Fig. 4.
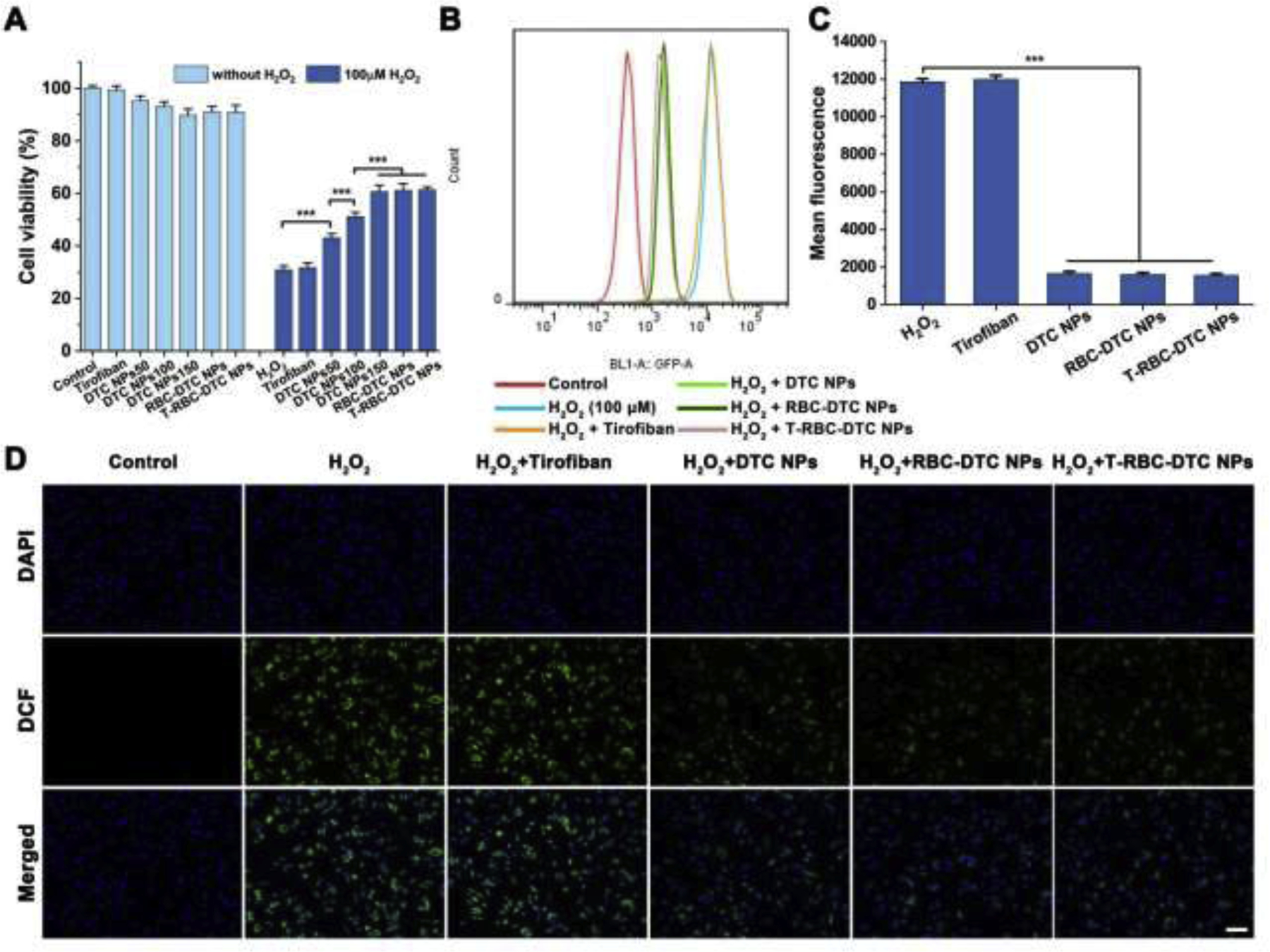
Anti-oxidative effects of the T-RBC-DTC NPs in H2O2-stimulated vascular endothelial cells. (A) Cytotoxicity and protective effects of different formulations on H2O2-induced vascular endothelial cells. Data is shown as mean ± SD (n=6). (B) Flow cytometry analysis of intracellular ROS in H2O2-stimulated HUVECs subjected to different treatments. (C) Quantitative analysis of the intracellular ROS level in HUVECs. Data is shown as mean ± SD (n=3). (D) Detection of intracellular ROS using an ROS indicator in HUVECs subjected to different treatments via fluorescence imaging. Blue channel: 4’,6-diamidino-2-phenylindole (DAPI) stained nucleus. Green channel: dihydrodichlorofluorescein (DCF) fluorescence illustrated intracellular ROS. Scale bar: 100 μm. ***p < 0.001.
We next investigated the effects of the nanoparticles on the intracellular ROS level in H2O2-stimulated RAW 264.7 cells and HUVECs using DCFH-DA as an ROS probe, which can be oxidized by ROS and subsequently become fluorescent DCF. The intracellular fluorescence intensity of DCF was measured by flow cytometry. As demonstrated in Fig. 3B and Fig. 4B, H2O2-stimulated cells showed a dramatically higher level of intracellular ROS compared to normal cells. However, the DTC NPs, RBC-DTC NPs, and T-RBC-DTC NPs significantly reduced the intracellular ROS levels (Fig. 3C and Fig. 4C). In contrast, an equivalent amount of free tirofiban showed no anti-oxidative ability. The ability of the nanoparticles in reducing intracellular ROS was further substantiated by fluorescence microscopy (Fig. 3D and Fig. 4D). ROS production is related to both cellular and tissue injuries induced by oxidative stress. These results suggested that judiciously designed nanoparticles can be applied to relieve the pathological conditions by consuming excessive ROS, which was in agreement with recent reports that certain nanoparticles can function as scavengers of ROS [42].
3.3. The T-RBC-DTC NPs effectively homed to the injured carotid artery
The fibrin-targeting CREKA peptide is decorated on the surface of the T-RBC-DTC NPs to enhance the accumulation of the nanoparticles at the fibrin-abundant thrombus site. To evaluate the targeting efficiency, a mouse carotid arterial thrombosis model was used in this study. One of the carotid arteries was isolated and incubated with filter paper soaked with FeCl3 to induce the formation of thrombus. The Cy 5.5-tagged RBC-DTC NPs or T-RBC-DTC NPs prepared by the Cy 5.5-tagged DTC was intravenously injected. In vivo fluorescence imaging of the carotid arteries were carried out at various time points post-injection. A weak fluorescence signal was observed in the non-injured carotid artery, likely due to the circulating nanoparticles. In contrast, the FeCl3-treated artery exhibited a very strong fluorescence signal only 5 min after T-RBC-DTC NPs administration (Fig. 5A). The fluorescence intensity gradually increased within the first 30 min post-injection because more circulating T-RBC-DTC NPs bound to fibrin and accumulated at the thrombus site. After that, the fluorescence signal slightly reduced as shown in Fig. 5B and Fig. 5C. The fluorescence intensity in the injured carotid artery in the T-RBC-DTC NPs treatment group was 5 times higher than that in the RBC-DTC NPs treatment group at 30 min, demonstrating the fibrin-targeting ability of the T-RBC-DTC NPs through surface modification with the CREKA peptide (Fig. 5B).
Fig. 5.
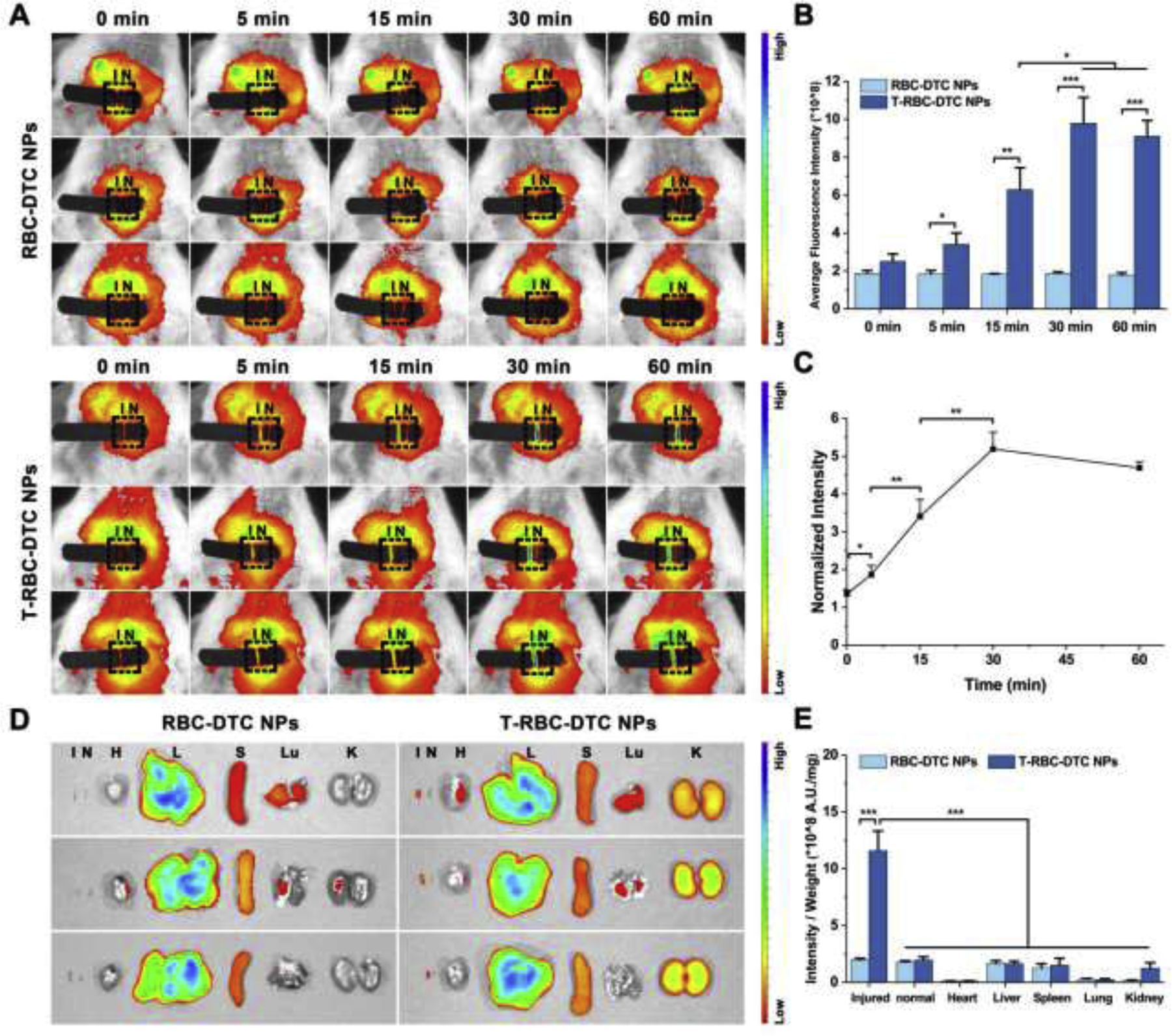
Targeting ability of the T-RBC-DTC NPs to the thrombosed vessels. (A) In vivo fluorescence images of the carotid arteries treated with the Cy 5.5-labeled RBC-DTC NPs and T-RBC-DTC NPs, respectively. I and N represent the injured and non-injured artery (outlined by the black rectangle), respectively. (B) Quantification of fluorescence intensity of carotid arteries in (A). Data is shown as mean ± SD (n=3). (C) The ratio of normalized fluorescence intensity between the FeCl3-treated artery and non-injured artery as a function of time in the T-RBC-DTC NPs group. Data is shown as mean ± SD (n=3). (D) Ex vivo fluorescence image of the Cy 5.5-labeled RBC-DTC NPs and T-RBC-DTC NPs in the major organs, and injured and non-injured carotid artery. H, L, S, Lu, K, I and N represent heart, liver, spleen, lung, kidney, injured artery, and non-injured artery, respectively. (E) Quantitative analysis of the mean fluorescence intensity per unit mass in each organ or tissue shown in the ex vivo images. Data is shown as mean ± SD (n=3). *p < 0.05, **p < 0.01, ***p < 0.001.
Although the surfaces of the T-RBC-DTC NPs were functionalized with the fibrin-targeting CREKA peptide, these nanoparticles also accumulated in liver, spleen, and kidney, since these organs are responsible for the elimination of xenobiotic substances (Fig. 5D). In fact, this is very common for nanoparticle-based targeted delivery [43–45]. However, as shown in Fig. 5E, the FeCl3-injured artery showed a drastically higher fluorescence intensity per unit mass in comparison with all major organs as well as the non-injured artery, demonstrating the superior targeting capability of the T-RBC-DTC NPs to the thrombus site.
3.4. The T-RBC-DTC NPs effectively inhibited thrombosis in vivo with good biocompatibility
We then examined the antithrombotic activity of the prodrug nanoparticles via histological examination. The DTC NPs, RBC-DTC NPs, and T-RBC-DTC NPs were intravenously injected immediately after vascular injury. In parallel, additional controls including saline and free drug were injected. The sham group is used to study whether the surgery could induce vascular injury. As shown in Fig. 6A and Fig. 6B, no thrombus was found in the sham-operated mice, suggesting that the surgery did not induce mechanical vascular damage, while a large thrombus was formed in FeCl3-treated mice. Compared to the DTC NPs-treated group (80.3% thrombosis degree), the RBC-DTC NPs showed better antithrombotic efficiency (64.4% thrombosis degree). More remarkably, treatment with the T-RBC-DTC NPs effectively inhibited the thrombus development (14.9% thrombosis degree) compared to the equivalent amount of tirofiban (88.1% thrombosis degree), despite its potent antiplatelet activity. This result indicated that the delivery of tirofiban in a targeted manner using the T-RBC-DTC NPs can enhance the antithrombotic effect.
Fig. 6.
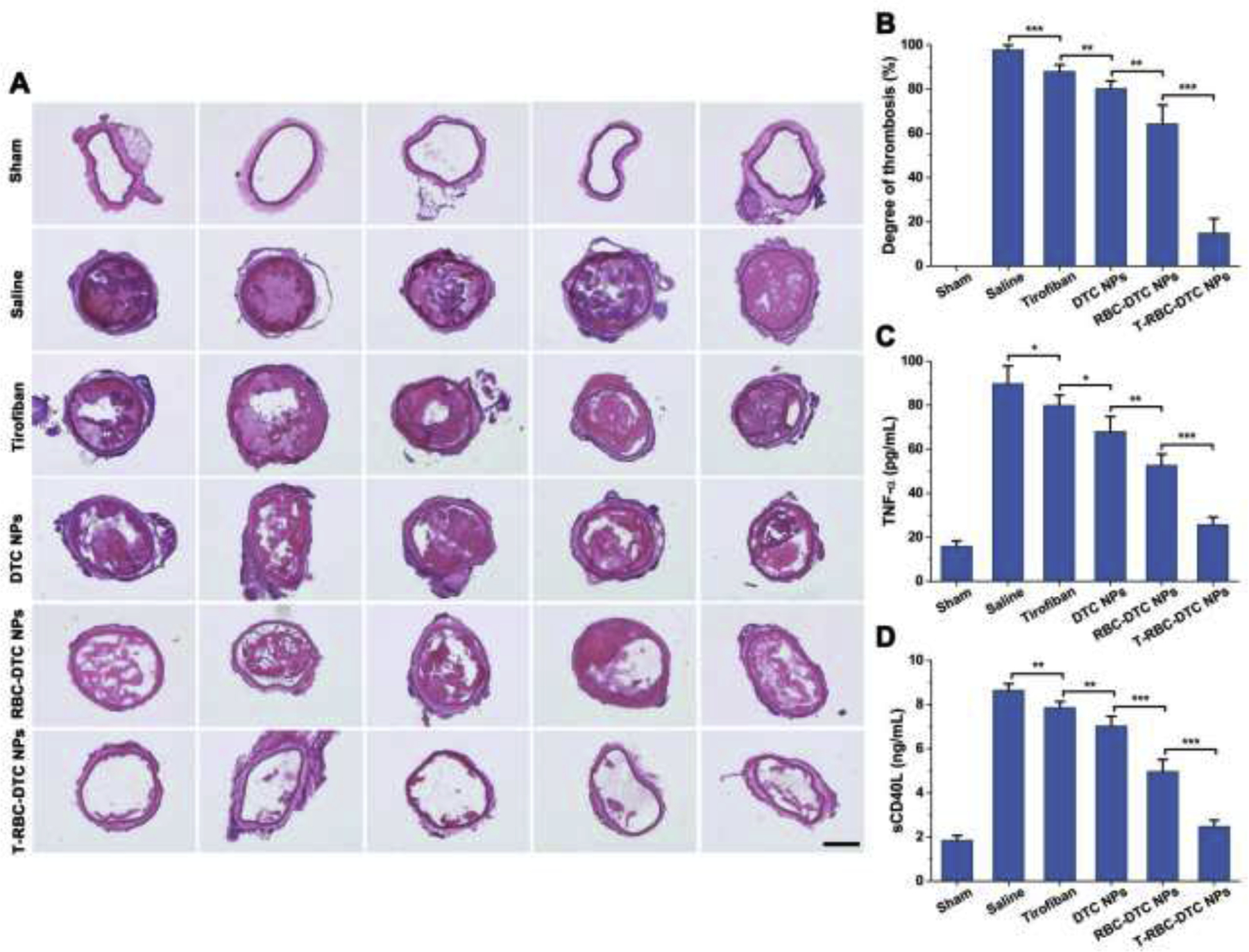
Therapeutic potential of the T-RBC-DTC NPs in the FeCl3-induced carotid artery thrombosis mouse model. (A) H&E staining of the carotid arteries from the mice subjected to various treatments. Scale bar: 200 μm. (B) Quantitative analysis of the thrombosis degree. Data is shown as mean ± SD (n=5). (C) The levels of TNF-α after various treatments as detected by ELISA. Data is shown as mean ± SD (n=5). (D) The levels of sCD40L after various treatments. Data is shown as mean ± SD (n=5). *p < 0.05, **p < 0.01, ***p < 0.001.
The DHE staining was also performed to investigate the impact of the prodrug nanoparticles on the level of ROS in thrombosed vessels. DHE, a fluorescent probe, can be oxidized by ROS to form ethidium oxide with red fluorescence. A qualitative analysis of the red fluorescence signal was conducted by fluorescence microscopy. As revealed in Fig. 7, the thrombus and endothelium of the FeCl3-treated carotid artery showed a high intensity of DHE fluorescence, indicating that a large number of platelets were recruited to the thrombosed blood vessel and were activated to generate massive ROS. The T-RBC-DTC NPs significantly suppressed ROS generation compared to the equivalent amount of tirofiban. The superior anti-oxidative activity of the T-RBC-DTC NPs than the free drug (i.e., tirofiban) can be attributed to the multifunctionalities offered by the nanoplatform including thrombus targeting, H2O2 scavenging, and H2O2-triggered release of the therapeutic drug at the thrombus site.
Fig. 7.

Dihydroethidium (DHE) staining of the sectioned carotid arteries from the mice subjected to various treatments. Scale bar: 200 μm.
We next measured the level of TNF-α to evaluate the anti-inflammatory effect of the prodrug nanoparticles (Fig. 6C). Treating the carotid artery with FeCl3 significantly increased the level of TNF-α (89.9 ± 8.0 pg/mL). As expected, T-RBC-DTC NPs exhibited much stronger inhibitory effects on the level of TNF-α (25.7 ± 3.5 pg/mL) than free tirofiban (80.0 ± 4.7 pg/mL), DTC NPs (68.1 ± 6.8 pg/mL), and RBC-DTC-NPs (52.7 ± 5.1 pg/mL), suggesting that T-RBC-DTC NPs possess highly potent anti-inflammatory activity. Moreover, sCD40L is produced primarily by activated platelets and interacts with CD40 on endothelial cells or immune cells to mediate the thrombotic and inflammatory processes [46]. We therefore investigated the inhibitory effects of the nanoparticles on the expression of sCD40L (Fig. 6D). FeCl3 treatment induced a significant upregulation of sCD40L (8.7 ± 0.3 ng/mL). T-RBC-DTC NPs suppressed the expression of sCD40L drastically (2.5 ± 0.3 ng/mL) and was much more effective than free tirofiban (7.9 ± 0.3 ng/mL), DTC NPs (7.0 ± 0.4 ng/mL), and RBC-DTC-NPs (4.9 ± 0.5 ng/mL). These observations implied that T-RBC-DTC NPs can simultaneously provide anti-inflammatory, antiplatelet, and anti-oxidation effects via thrombus targeted delivery of tirofiban and an anti-oxidation effect via the ROS-scavenging nanocarriers.
To further evaluate the in vivo biocompatibility of the T-RBC-DTC NPs, the T-RBC-DTC NPs were intravenously administered to mice every day for one week. H&E staining of the tissue samples including heart, liver, spleen, lung, and kidney after the treatment with saline and T-RBC-DTC NPs were conducted. There was no evidence of pathological differences or inflammatory cell infiltration in the tissue sections, demonstrating the good in vivo biocompatibility of the T-RBC-DTC NPs (Fig. 8).
Fig. 8.
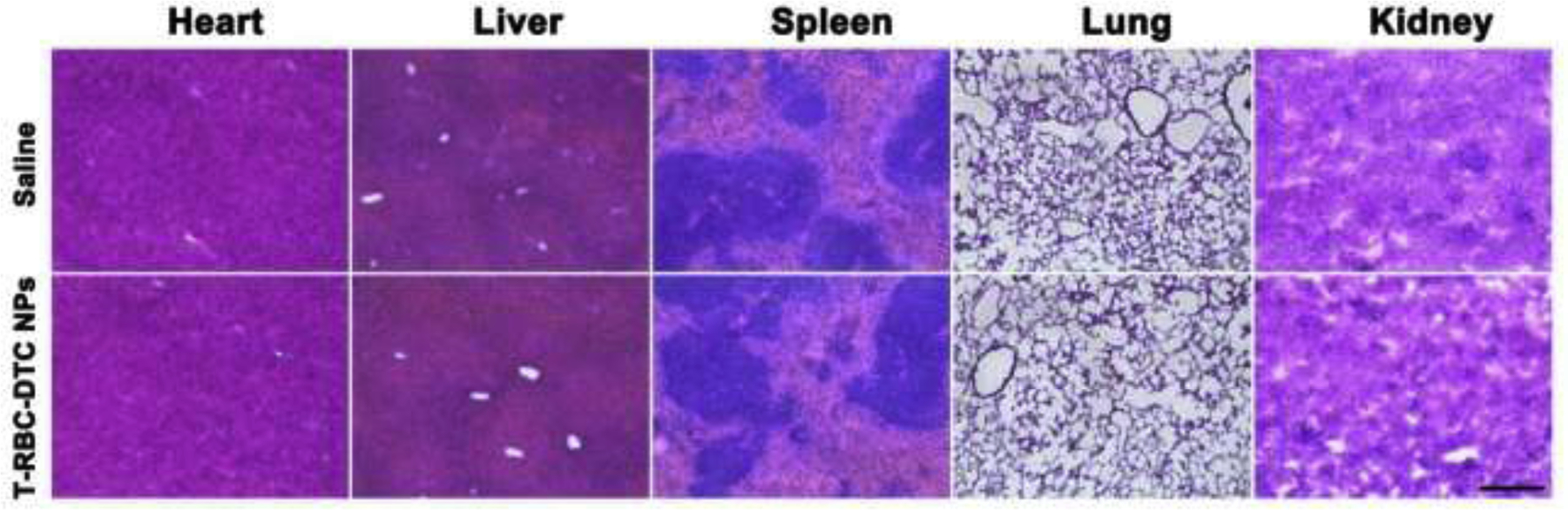
Representative histological images of different organs from mice with daily injection of saline or the T-RBC-DTC NPs for a week. Scale bar: 200 μm.
Regarding the clinical translation of the T-RBC-DTC NPs, many opportunities and challenges lie ahead. The T-RBC-DTC NPs hold a great potential for translation due to the thrombus-targeted and controlled drug delivery capability, enhanced antithrombotic efficacy, reduction in the drug dosage, and good biocompatibility. The excellent accessibility (the most abundant cell in the human body) of RBC membrane can further facilitate the translation of T-RBC-DTC NPs. Furthermore, surface functionalization including targeting ligand conjugation can be conveniently achieved by inserting modified lipids to the RBC membranes, thereby offering versatility for various potential clinical applications. The T-RBC-DTC NPs can also enable personalized therapy for enhanced therapeutic outcome with little risk of immunogenicity by using RBC membranes from individual patients. The fabrication process of the RBC membrane-coated nanoparticles includes RBC membrane extraction, preparation of the nanocores, and membrane coating procedures. This is an environmentally friendly fabrication process as it does not involve any organic solvents. However, each fabrication step requires a proper protocol to ensure scalability and reproducibility of the final product. More efforts are thus needed to alleviate the gap between preclinical research and clinical translation of the RBC membrane-coated nanotherapeutics.
In addition, for thrombus targeting, platelet membrane can also be used as the nanocarrier surface coating due to its thrombus-homing property and the role of platelet in the thrombotic processes. However, in the present study, tirofiban was selected as the model drug for antithrombotic therapy, which is an antagonist of the platelet glycoprotein IIb/IIIa receptor. Tirofiban is likely to compromise the receptor on the platelet membrane and thus impact the targeting capability. Instead, we used RBC membrane coating and incorporated fibrin-targeting peptides to achieve thrombus targeting. The T-RBC-DTC NPs can accumulate at thrombus sites, where abundant fibrin is present, and it may be suitable for treating various thrombus-associated diseases, including arterial thrombosis, pulmonary embolism, ischemic stroke, acute myocardial infarction, and deep vein thrombosis. Furthermore, since our conjugation chemistry to achieve H2O2-responsive drug release is versatile for drugs that bear an amino group, we envision our nanoplatform can be applied for a broad range of applications by varying the types of drug, cell membrane coating and targeting ligand.
4. Conclusion
A biomimetic core-shell drug delivery system was designed for thrombus-targeting delivery of antithrombotic agent. The nanocarrier consisted of a H2O2-activatable dextran-drug conjugate nanocore and a RBC membrane coating integrated with the fibrin-targeting CREKA peptides. Upon intravenous administration, the T-RBC-DTC NPs were able to rapidly accumulate at the fibrin-rich blood clots and were also able to effectively suppress the thrombus development in the injured vessels through the combination effect of anti-oxidation enabled by the nanocarriers and the antithrombotic effect from tirofiban. Based on these unique features, including thrombus-targeting capability, H2O2 scavenging, and ROS-triggered therapeutic action at the thrombus site, the rationally engineered T-RBC-DTC NPs can be potentially used to treat various life-threatening thrombosis diseases.
Supplementary Material
Highlights.
H2O2-responsive thrombus-targeting red blood cell membrane-cloaked dextran-tirofiban conjugate nanoparticles (T-RBC-DTC NPs) were developed for antithrombotic therapy.
The T-RBC-DTC NPs conjugated with fibrin-targeting peptide, CREKA, efficiently accumulated at the injured carotid artery.
The T-RBC-DTC NPs effectively scavenged H2O2 and protected cells from H2O2-induced cytotoxicity.
The T-RBC-DTC NPs significantly suppressed thrombus formation, and the levels of H2O2 and inflammatory cytokine in a ferric chloride-induced carotid arterial thrombosis mouse model.
Acknowledgements
We would like to acknowledge the financial support from the NIH (R01 HL129785 and R01 HL143469).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no conflicts of interest.
References
- [1].Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, Circulation, 135 (2017) e146–e603. 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, Biller J, Brown M, Demaerschalk BM, Hoh B, Jauch EC, Kidwell CS, Leslie-Mazwi TM, Ovbiagele B, Scott PA, Sheth KN, Southerland AM, Summers DV, Tirschwell DL, Stroke, 50 (2019) e344–e418. 10.1161/STR.0000000000000211. [DOI] [PubMed] [Google Scholar]
- [3].Kearon C, Akl EA, Ornelas J, Blaivas A, Jimenez D, Bounameaux H, Huisman M, King CS, Morris TA, Sood N, Stevens SM, Vintch JRE, Wells P, Woller SC, Moores CL, Chest, 149 (2016) 315–352. [DOI] [PubMed] [Google Scholar]
- [4].Leopold JA, Loscalzo J, Free Radical Bio. Med, 47 (2009) 1673–1706. 10.1016/j.freeradbiomed.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dayal S, Wilson KM, Motto DG, Miller FJ, Chauhan AK, Lentz SR, Circulation, 127 (2013) 1308–1316. 10.1161/CIRCULATIONAHA.112.000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Krötz F, Sohn H, Pohl U, Arteriosclerosis, Thrombosis, and Vascular Biology, 24 (2004) 1988–1996. 10.1161/01.ATV.0000145574.90840.7d. [DOI] [PubMed] [Google Scholar]
- [7].Freedman JE, Arteriosclerosis, Thrombosis, and Vascular Biology, 28 (2008) s11–s16. 10.1161/ATVBAHA.107.159178. [DOI] [PubMed] [Google Scholar]
- [8].Cai H, Cardiovasc. Res, 68 (2005) 26–36. 10.1016/j.cardiores.2005.06.021. [DOI] [PubMed] [Google Scholar]
- [9].Vara D, Pula G, Curr. Mol. Med, 14 (2014) 1103–1125. 10.2174/1566524014666140603114010. [DOI] [PubMed] [Google Scholar]
- [10].Kim KS, Song CG, Kang PM, Antioxid. Redox Sign, 30 (2019) 733–746. 10.1089/ars.2017.7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gunawan ST, Kempe K, Bonnard T, Cui J, Alt K, Law LS, Wang X, Westein E, Such GK, Peter K, Hagemeyer CE, Caruso F, Adv. Mater, 27 (2015) 5153–5157. 10.1002/adma.201502243. [DOI] [PubMed] [Google Scholar]
- [12].Cui C, Yang Z, Hu X, Wu J, Shou K, Ma H, Jian C, Zhao Y, Qi B, Hu X, Yu A, Fan Q, ACS Nano, 11 (2017) 3298–3310. 10.1021/acsnano.7b00594. [DOI] [PubMed] [Google Scholar]
- [13].Li B, Chen R, Zhang Y, Zhao L, Liang H, Yan Y, Tan H, Nan D, Jin H, Huang Y, ACS Applied Bio Materials, 2 (2018) 437–446. 10.1021/acsabm.8b00644. [DOI] [PubMed] [Google Scholar]
- [14].Zhang N, Li C, Zhou D, Ding C, Jin Y, Tian Q, Meng X, Pu K, Zhu Y, Acta Biomater, 70 (2018) 227–236. 10.1016/j.actbio.2018.01.038. [DOI] [PubMed] [Google Scholar]
- [15].Huang Y, Yu L, Ren J, Gu B, Longstaff C, Hughes AD, Thom SA, Xu XY, Chen R, Control J. Release, 300 (2019) 1–12. 10.1016/j.jconrel.2019.02.033. [DOI] [PubMed] [Google Scholar]
- [16].Xu J, Zhou J, Zhong Y, Zhang Y, Liu J, Chen Y, Deng L, Sheng D, Wang Z, Ran H, Guo D, ACS Appl. Mater. Inter, 9 (2017) 42525–42535. 10.1021/acsami.7b12689. [DOI] [PubMed] [Google Scholar]
- [17].Juenet M, Aid-Launais R, Li B, Berger A, Aerts J, Ollivier V, Nicoletti A, Letourneur D, Chauvierre C, Biomaterials, 156 (2018) 204–216. 10.1016/j.biomaterials.2017.11.047. [DOI] [PubMed] [Google Scholar]
- [18].Li B, Aid-Launais R, Labour M, Zenych A, Juenet M, Choqueux C, Ollivier V, Couture O, Letourneur D, Chauvierre C, Biomaterials, 194 (2019) 139–150. 10.1016/j.biomaterials.2018.12.023. [DOI] [PubMed] [Google Scholar]
- [19].Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R, Kuriakose G, Perretti M, Farokzhad O, Tabas I, Sci Translational Medicine, 7 (2015) 220r–275r. 10.1126/scitranslmed.aaa1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kim J, Ryu JH, Schellingerhout D, Sun I, Lee S, Jeon S, Kim J, Kwon IC, Nahrendorf M, Ahn C, Kim K, Kim D, Theranostics, 5 (2015) 1098–1114. 10.7150/thno.11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhong Y, Zhang Y, Xu J, Zhou J, Liu J, Ye M, Zhang L, Qiao B, Wang Z, Ran H, Guo D, ACS Nano, 13 (2019) 3387–3403. 10.1021/acsnano.8b09277. [DOI] [PubMed] [Google Scholar]
- [22].Lee J, Jeong L, Jung E, Ko C, Seon S, Noh J, Lee D, Control J. Release, 304 (2019) 164–172. 10.1016/j.jconrel.2019.05.012. [DOI] [PubMed] [Google Scholar]
- [23].Palekar RU, Jallouk AP, Myerson JW, Pan H, Wickline SA, Arteriosclerosis, Thrombosis, and Vascular Biology, 36 (2016) 446–455. 10.1161/ATVBAHA.115.306697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Furie B, Furie BC, New Engl. J. Med, 359 (2008) 938–949. [DOI] [PubMed] [Google Scholar]
- [25].Ciesienski KL, Caravan P, Current Cardiovascular Imaging Reports, 4 (2011) 77–84. 10.1007/s12410-010-9061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Song Y, Huang Z, Xu J, Ren D, Wang Y, Zheng X, Shen Y, Wang L, Gao H, Hou J, Pang Z, Qian J, Ge J, Biomaterials, 35 (2014) 2961–2970. 10.1016/j.biomaterials.2013.12.038. [DOI] [PubMed] [Google Scholar]
- [27].Kang C, Gwon S, Song C, Kang PM, Park S, Jeon J, Hwang DW, Lee D, ACS Nano, 11 (2017) 6194–6203. 10.1021/acsnano.7b02308. [DOI] [PubMed] [Google Scholar]
- [28].Simberg D, Duza T, Park JH, Essler M, Pilch J, Zhang L, Derfus AM, Yang M, Hoffman RM, Bhatia S, Sailor MJ, Ruoslahti E, Proc Natl Acad Sci U S A, 104 (2007) 932–936. 10.1073/pnas.0610298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xu J, Wang X, Yin H, Cao X, Hu Q, Lv W, Xu Q, Gu Z, Xin H, ACS Nano, 13 (2019) 8577–8588. 10.1021/acsnano.9b01798. [DOI] [PubMed] [Google Scholar]
- [30].Wang X, Wei C, Liu M, Yang T, Zhou W, Liu Y, Hong K, Wang S, Xin H, Ding X, Adv. Funct. Mater, 27 (2017) 1701824 10.1002/adfm.201701824. [DOI] [Google Scholar]
- [31].Thomas MD, Wood C, Lovett M, Dembo L, O’Driscoll G, The Journal of Heart and Lung Transplantation, 27 (2008) 925–927. 10.1016/j.healun.2008.05.015. [DOI] [PubMed] [Google Scholar]
- [32].McClellan KJ, Goa KL, Drugs, 56 (1998) 1067–1080. [DOI] [PubMed] [Google Scholar]
- [33].Hu CJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L, Proceedings of the National Academy of Sciences, 108 (2011) 10980–10985. 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Luk BT, Jack Hu C, Fang RH, Dehaini D, Carpenter C, Gao W, Zhang L, Nanoscale, 6 (2014) 2730–2737. 10.1039/C3NR06371B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lv W, Xu J, Wang X, Li X, Xu Q, Xin H, ACS Nano, 12 (2018) 5417–5426. 10.1021/acsnano.8b00477. [DOI] [PubMed] [Google Scholar]
- [36].Yan J, Yu J, Wang C, Gu Z, Small Methods, 1 (2017) 1700270 10.1002/smtd.201700270. [DOI] [Google Scholar]
- [37].Chai Z, Hu X, Wei X, Zhan C, Lu L, Jiang K, Su B, Ruan H, Ran D, Fang RH, Zhang L, Lu W, Control J. Release, 264 (2017) 102–111. 10.1016/j.jconrel.2017.08.027. [DOI] [PubMed] [Google Scholar]
- [38].Ko E, Jeong D, Kim J, Park S, Khang G, Lee D, Biomaterials, 35 (2014) 3895–3902. 10.1016/j.biomaterials.2014.01.048. [DOI] [PubMed] [Google Scholar]
- [39].Pan Y, Ren X, Wang S, Li X, Luo X, Yin Z, Biomacromolecules, 18 (2017) 865–876. 10.1021/acs.biomac.6b01756. [DOI] [PubMed] [Google Scholar]
- [40].Hu CJ, Fang RH, Luk BT, Chen KNH, Carpenter C, Gao W, Zhang K, Zhang L, Nanoscale, 5 (2013) 2664–2668. 10.1039/c3nr00015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Su J, Sun H, Meng Q, Yin Q, Zhang P, Zhang Z, Yu H, Li Y, Adv. Funct. Mater, 26 (2016) 7495–7506. 10.1002/adfm.201603381. [DOI] [Google Scholar]
- [42].Yang B, Chen Y, Shi J, Chem. Rev, 119 (2019) 4881–4985. 10.1021/acs.chemrev.8b00626. [DOI] [PubMed] [Google Scholar]
- [43].Wang J, Hu S, Mao W, Xiang J, Zhou Z, Liu X, Tang J, Shen Y, Adv. Funct. Mater, 29 (2019) 1807446 10.1002/adfm.201807446. [DOI] [Google Scholar]
- [44].Peng J, Yang Q, Xiao Y, Shi K, Liu Q, Hao Y, Yang F, Han R, Qian Z, Adv. Funct. Mater, 29 (2019) 1900004 10.1002/adfm.201900004. [DOI] [Google Scholar]
- [45].Yang X, Hu C, Tong F, Liu R, Zhou Y, Qin L, Ouyang L, Gao H, Adv. Funct. Mater, 29 (2019) 1901896 10.1002/adfm.201901896. [DOI] [Google Scholar]
- [46].Lievens D, Zernecke A, Seijkens T, Soehnlein O, Beckers L, Munnix ICA, Wijnands E, Goossens P, van Kruchten R, Thevissen L, Boon L, Flavell RA, Noelle RJ, Gerdes N, Biessen EA, Daemen MJAP, Heemskerk JM, Weber C, Lutgens E, Blood, 116 (2010) 4317–4327. 10.1182/blood-2010-01. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.