

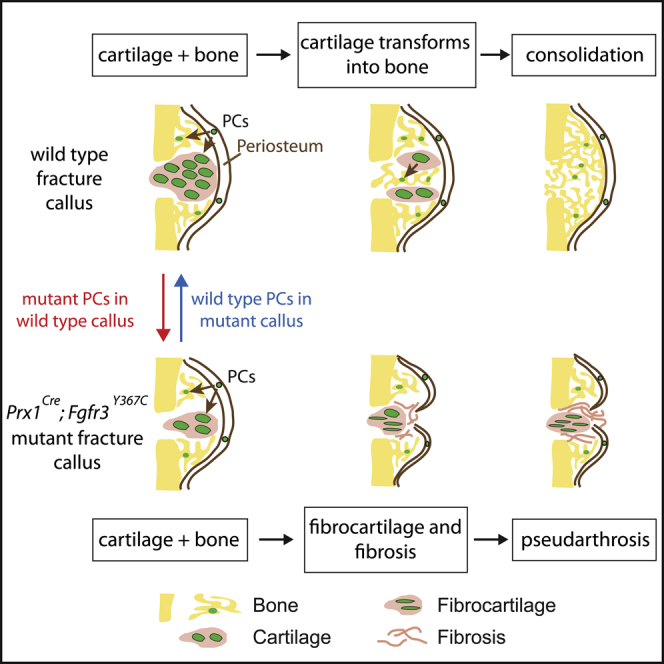
Summary
Most organs and tissues in the body, including bone, can repair after an injury due to the activation of endogenous adult stem/progenitor cells to replace the damaged tissue. Inherent dysfunctions of the endogenous stem/progenitor cells in skeletal repair disorders are still poorly understood. Here, we report that Fgfr3Y637C/+ over-activating mutation in Prx1-derived skeletal stem/progenitor cells leads to failure of fracture consolidation. We show that periosteal cells (PCs) carrying the Fgfr3Y637C/+ mutation can engage in osteogenic and chondrogenic lineages, but following transplantation do not undergo terminal chondrocyte hypertrophy and transformation into bone causing pseudarthrosis. Instead, Prx1Cre;Fgfr3Y637C/+ PCs give rise to fibrocartilage and fibrosis. Conversely, wild-type PCs transplanted at the fracture site of Prx1Cre;Fgfr3Y637C/+ mice allow hypertrophic cartilage transition to bone and permit fracture consolidation. The results thus highlight cartilage-to-bone transformation as a necessary step for bone repair and FGFR3 signaling within PCs as a key regulator of this transformation.
Keywords: bone repair, periosteum, pseudarthrosis, FGFR3, skeletal stem/progenitor cell, endochondral ossification
Graphical Abstract
Highlights
-
•
Fgfr3Y367C activating mutation in skeletal stem/progenitor cells prevents bone healing
-
•
Intrinsic deficiencies in transplanted Prx1Cre;Fgfr3Y637C/+ PCs cause pseudarthrosis
-
•
Prx1Cre;Fgfr3Y637C/+ PCs cannot support cartilage-to-bone transformation
-
•
Wild-type PCs can rescue the Prx1Cre;Fgfr3Y637C/+ pseudarthrosis phenotype
Julien et al. report a severe bone repair phenotype associated with pseudarthrosis and functional impairment of periosteal cells (PCs) in Prx1Cre;Fgfr3Y637C/+ mice. PCs carrying the Fgfr3Y367C activating mutation cannot undergo cartilage-to-bone transformation thus uncovering an essential role of this transformation step in bone healing.
Introduction
Bone fracture repair is an efficient regenerative process, which depends on a controlled inflammatory response, followed by the recruitment of skeletal stem/progenitor cells, deposition of cartilage and bone matrix and skeletal tissue remodeling. Skeletal stem/progenitor cells are recruited from multiple sources in the bone fracture environment, including the bone marrow, the periosteum, and the surrounding soft tissues but cannot be strictly distinguished using genetic lineage tracing (Abou-Khalil et al., 2015; Chan et al., 2015; Duchamp de Lageneste et al., 2018; Debnath et al., 2018; Matsushita et al., 2020; Ortinau et al., 2019; Van Gastel et al., 2012; Worthley et al., 2015; Zhang et al., 2005; Zhou et al., 2014a). However, several strategies have revealed that the periosteum comprises populations of skeletal stem/progenitor cells largely involved in callus formation through intramembranous and endochondral ossification (Debnath et al., 2018; Duchamp de Lageneste et al., 2018; Ortinau et al., 2019). These studies emphasized the periosteum as a key contributor to bone repair and likely affected in skeletal repair dysfunctions.
When bone repair is compromised by extensive injuries or pathological conditions, failure of the bone regenerative process can lead to delayed-union or non-union but the underlying cause of this failed healing is often undetermined. An adverse inflammatory environment or impaired revascularization of the fracture site may contribute to the bone repair deficit, but the role of the main players, the skeletal stem/progenitor cells, is less understood. Whether cells cannot be properly activated in response to injury or do not support the subsequent stages of skeletal tissue deposition and remodeling is difficult to assess in vivo due to the diversity of skeletal stem/progenitor cell populations that may participate in repair.
Here, we explored the role of periosteal cells (PCs) in bone repair and uncovered PC dysfunctions in Prx1Cre;Fgfr3Y367C/+ mice that exhibit a severe bone repair phenotype of pseudarthrosis. Fibroblast growth factor receptor 3 (FGFR3) is a key regulator of endochondral ossification during bone development. Gain-of-function point mutations in FGFR3 gene causing upregulation of FGFR3 signaling are associated with the human genetic disease achondroplasia or dwarfism, affecting the cartilage growth plate in developing long bones with abnormal chondrocyte proliferation and differentiation, loss of columnar organization, and overall reduction in long bone growth (Bonaventure et al., 1996; Mugniery et al., 2012; Pannier et al., 2009; Rousseau et al., 1994). We report that mice carrying the Fgfr3Y637C/+ activating mutation in the Prx1-derived skeletal stem/progenitor cells exhibit short limbs during post-natal growth and pseudarthrosis following a tibial fracture. The pseudarthrosis phenotype is marked by the accumulation of fibrotic tissue and un-resorbed cartilage in the center of the fracture callus. We show that impaired bone healing in Prx1Cre;Fgfr3Y367C/+ mice is correlated with intrinsic deficiencies in PCs affecting the endochondral ossification process, and more specifically cartilage-to-bone transformation that is taking place at a critical time when cartilage replacement by bone is crucial to consolidate the fracture gap. The requirement of this transformation step for bone repair is still debated and our results reveal that this transformation is required for effective healing (Hu et al., 2017; Yang et al., 2014; Zhou et al., 2014b).
Results
Impaired Bone Regeneration and Pseudarthrosis in Prx1Cre;Fgfr3Y367C/+ Mice
To investigate the role of FGFR3 in bone repair, we crossed Fgfr3Y637C/+ mice with the Prx1Cre mouse line to target the mesenchymal lineage in the limb (Logan et al., 2002; Pannier et al., 2009). This mesenchymal lineage gives rise to the skeletal stem/progenitor cells in the periosteum that contributes to fracture repair in the adult (Duchamp de Lageneste et al., 2018). While mice expressing the heterozygous Fgfr3Y637C/+ mutation ubiquitously recapitulate a severe form of dwarfism and die within 2 months after birth, the Prx1Cre;Fgfr3Y367C/+ mice also displayed shorter long bones in the limbs but survived until adulthood. Adult Prx1Cre;Fgfr3Y367C/+ mice exhibited a 65% reduction in tibia length (Figure 1A). Consistent with the known dwarfism phenotype, we observed a strong disorganization of the epiphyseal cartilage in 1- and 3-month-old Prx1Cre;Fgfr3Y367C/+ mice with an abnormal shape and size of hypertrophic chondrocytes compared with control mice (Figure 1B). Immunofluorescence staining on epiphyseal cartilage of 3-month-old Prx1Cre;Fgfr3Y367C/+ mice showed absence of Collagen X (COLX) and Osterix (OSX) expression in the area below the cartilage where trabeculae are missing compared with controls (Figure 1C). We also observed a significant reduction of vascularization and an absence of CD31/Endomucin (EMCN) double-positive vessels at the cartilage-to-bone transition zone in Prx1Cre;Fgfr3Y367C/+ mice, indicating an impairment of endochondral ossification (Figures 1C and 1D).
Figure 1.

Reduced Tibia Length and Abnormal Tibial Epiphyseal Cartilage Organization in Prx1Cre;Fgfr3Y367C/+ Mice
(A) Three-month-old Prx1Cre;Fgfr3Y367C/+ and Prx1Cre;Fgfr3+/+ mice (top), radiographs of hindlimbs (bottom), and quantification of tibia length (right) (n = 5 per group). Scale bar, 0.5 cm.
(B) Representative Safranin O (SO) staining of epiphyseal cartilage of uninjured tibia from 1- to 3 month-old Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ mice. Scale bar, 50 μm.
(C) Immunofluorescence of Collagen X (COLX), Osterix (OSX), and CD31/Endomucin (EMCN) at the transition zone between the epiphyseal hypertrophic cartilage (hc) and the metaphysis in uninjured tibia from 3 months old Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ mice (n = 3). Scale bar, 50 μm.
(D) Quantification of CD31 and EMCN immunofluorescence at the transition zone between the epiphyseal cartilage and the metaphysis from Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ mice (n = 6).
Values represent mean ± SD. ∗∗p < 0.01 using Mann-Whitney test.
We induced open non-stabilized tibial fractures in 3-month-old Prx1Cre;Fgfr3Y367C/+ mice and Prx1Cre;Fgfr3+/+ control mice. Histomorphometric analyses showed a severe impairment of bone healing as indicated by a marked reduction in callus and bone volumes in Prx1Cre;Fgfr3Y367C/+ mice throughout the stages of repair (Figure 2A). Cartilage volume was also reduced through to day 14 followed by cartilage accumulation from day 21 post-fracture. At 28 days post-fracture, micro-computed tomography (micro-CT) and histological analyses showed a complete absence of bone bridging of Prx1Cre;Fgfr3Y367C/+ calluses, whereas Prx1Cre;Fgfr3+/+ control mice displayed fully ossified calluses with reconstitution of the cortex to bridge the fracture gap. Although new bone formation was detected at the periphery of Prx1Cre;Fgfr3Y367C/+ calluses, there was a complete absence of bone deposition in the center of the callus (Figures 2B, 2C, and S1). Instead, we identified areas of unresorbed fibrocartilage with low Safranin O (SO) staining and abnormal COLX expression and areas of fibrous tissue deposition marked by the matrix protein Periostin in Prx1Cre;Fgfr3Y367C/+ mice that are characteristic of pseudarthrosis (Figures 2D and 2E). By day 56, micro-CT analyses showed partial bridging of the two ossified parts of the callus in Prx1Cre;Fgfr3Y367C/+ mice with a persistent decrease in bone volume (Figure S2).
Figure 2.

Altered Bone Regeneration and Pseudarthrosis in Prx1Cre;Fgfr3Y367C/+ Mice
(A) Histomorphometric analyses of callus, cartilage, and bone volumes at days 7, 10, 14, 21, and 28 post-fracture in Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ mice (n = 4 or 5 per group).
(B) Representative micro-CT images of Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ calluses at 28 days post-fracture. White arrow points to the absence of bone bridging in Prx1Cre;Fgfr3Y367C/+ callus (n = 4–5 per group). Scale bar, 1 mm.
(C) Longitudinal sections stained with Masson's trichrome (TC) of Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ calluses 28 days post-fracture. High magnification showing continuity of the newly formed cortex in Prx1Cre;Fgfr3+/+ callus (box 1, red arrows) and absence of new bone in the center of Prx1Cre;Fgfr3Y367C/+ callus (box 2, asterisk). Scale bars, 1 mm (left) and 300 μm (right).
(D) Longitudinal sections stained with SO of Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ calluses 28 days post-fracture (callus delimited with a black dotted line). Scale bar, 1 mm.
(E) High magnification of fibrocartilage (fc) stained with SO and COLX immunostaining showing few COLX+ cells adjacent to fibrocartilage and bone (b) in Prx1Cre;Fgfr3Y367C/+ callus (box 3, top). High magnification of fibrosis stained with Picrosirius (PS) and Periostin (POSTN) immunostaining in Prx1Cre;Fgfr3Y367C/+ callus (box 4, bottom). Scale bar, 100 μm. Values represent mean ± SD. ∗p < 0.05, ∗∗p < 0.01 using Mann-Whitney test.
Cartilage-to-Bone Transition Is Disrupted in Prx1Cre;Fgfr3Y367C/+ Fracture Callus
The pseudarthrosis phenotype in Prx1Cre;Fgfr3Y367C/+mice indicated a lack of endochondral ossification during bone repair. Indeed, when we evaluated bone repair strictly through intramembranous ossification using a unicortical defect repair model in Prx1Cre;Fgfr3Y367C/+ mice, we did not observe delayed healing compared with Prx1Cre;Fgfr3+/+ mice (Figure S3). Endochondral ossification depends on proper chondrocyte differentiation, followed by maturation to hypertrophic chondrocytes and finally cartilage-to-bone transition to allow the replacement of the cartilaginous matrix by bone. The volume and percentage of hypertrophic cartilage were markedly decreased in the callus of Prx1Cre;Fgfr3Y367C/+ mice compared with controls (Figure 3A). At day 7 post-fracture, we observed the presence of SOX9-expressing cells in the cartilage of control and mutant mice (Figure 3B). By day 14 post-fracture, although chondrocytes expressed markers of hypertrophy, such as vascular endothelial growth factor (VEGF) and COLX, we observed a reduced amount of COLX+ hypertrophic cartilage (Figure 3C). Hypertrophic chondrocytes also exhibited abnormal shape and reduced size in Prx1Cre;Fgfr3Y367C/+ mice that may also contribute to the reduced cartilage volume. These results suggest that mutant chondrocytes can initiate chondrogenic differentiation and maturation but failed to achieve hypertrophy. We then examined the crucial step of cartilage-to-bone transition in Prx1Cre;Fgfr3Y367C/+ mice. Further analyses of the transition zone at day 14 post-fracture showed no co-localization of COLX+ cells and OSX+ cells in the new bone area of the transition zone in Prx1Cre;Fgfr3Y367C/+ calluses. POSTN was undetected within cartilage but was highly expressed in the fibrotic area suggesting absence of terminal hypertrophy and fibrotic accumulation by day 14 in Prx1Cre;Fgfr3Y367C/+ calluses. This correlated with a poor vascularization as shown by the absence of CD31/EMCN double-positive vessels in Prx1Cre;Fgfr3Y367C/+ calluses compared with controls (Figure 3D). In previous studies, the presence of proliferating chondrocytes in the transition zone has been associated with the ability of terminal hypertrophic chondrocytes to transdifferentiate into osteoblasts (Hu et al., 2017; Yang et al., 2014; Zhou et al., 2014b). We observed a strong reduction in the number of KI67+ and SOX2+ chondrocytes in the transition zone of Prx1Cre;Fgfr3Y367C/+ calluses compared with controls (Figure 3E). These results indicate abnormal chondrocyte hypertrophy followed by lack of cartilage-to-bone transition that may lead to the pseudarthrosis phenotype in Prx1Cre;Fgfr3Y367C/+ mice.
Figure 3.

Impaired Cartilage Differentiation and Cartilage-to-Bone Transition in Prx1Cre;Fgfr3Y367C/+ Fracture Callus
(A) Histomorphometric quantification of hypertrophic cartilage volume and percentage of hypertrophic cartilage volume in total cartilage volume at days 7 and 14 post tibial fracture in Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ mice (n = 4 or 5 per group).
(B) Longitudinal sections of Prx1Cre;Fgfr3+/+ (control) and Prx1Cre;Fgfr3Y367C/+ (mutant) calluses (delimited with a black dotted line) at day 7 post-fracture stained with SO. High magnification of cartilage area (box 1, 2) and SOX9 immunostaining showing the presence of chondrogenic cells in the callus of control and mutant mice. Scale bars, 1 mm and 50 μm (boxes 1 and 2).
(C) Longitudinal sections of Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ calluses at day 14 post-fracture stained with SO. High magnification of hypertrophic cartilage (hc) area (box 3, 5) and COLX immunostaining on adjacent sections showing positive staining in control and mutant hc, and within new bone trabeculae (b) in control but not in mutant (asterisk). High magnification of hypertrophic chondrocytes (box 4, 6) and VEGF immunostaining showing abnormal cellular size and shape in mutant. Scale bars, 1 mm, 300 μm (boxes 3 and 5), and 25 μm (boxes 4 and 6).
(D) Immunostaining for COLX, OSX, Periostin (POSTN), and CD31/EMCN at the cartilage-to-bone transition zone in Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ calluses 14 days post-fracture (n = 5 per group). Scale bar, 50 μm.
(E) High magnification of cartilage area next to bone and immunostaining for KI67 and SOX2 (box 7 and 8). Quantification of KI67+ and SOX2+ hypertrophic chondrocytes in day 14 post-fracture calluses from Prx1Cre;Fgfr3+/+ and Prx1Cre;Fgfr3Y367C/+ mice (n = 5 per group) Scale bar, 25 μm. Values represent the mean ± SD. ∗∗p < 0.01 using Mann-Whitney test.
Prx1Cre;Fgfr3Y367C/+ PCs Fail to Undergo Cartilage-to-Bone Transformation
We sought to test the involvement of PCs in this defective endochondral ossification process during bone repair given their efficient contribution to this process (Duchamp de Lageneste et al., 2018; Colnot, 2009). We did not identify changes in the proportions of skeletal stem/progenitor cells, osteo-chondroprogenitors and stromal cells in the GFP+ PC populations from Prx1Cre;RosamTmG;Fgfr3Y367C/+ mutant mice compared with Prx1Cre;RosamTmG;Fgfr3+/+ control mice (Figures 4A and 4B) (Chan et al., 2015). Mutant PCs could engage into the osteogenic, adipogenic, and chondrogenic lineages in vitro as observed for control PCs (Figure 4C). When mutant PCs were transplanted at the fracture site of wild-type hosts, they integrated into the cartilage by differentiating into SOX9+ chondrocytes at day 10 (Figures 5A and 5B). However, unlike control PCs they failed to mature and undergo chondrocyte hypertrophy by day 14. Mutant PCs gave rise to cells producing fibrocartilage in the center of the fracture callus in place of hypertrophic cartilage (Figure 5C). By day 21, cartilage-to-bone transformation had occurred and osteocytes derived from control PCs were localized within new bone trabeculae (Figure 5D, top). Mutant PCs were not detected within hypertrophic cartilage or new bone by day 21 but localized within fibrous tissue and caused a pseudarthrosis-like phenotype (Figure 5D, bottom). Histomorphometric analysis confirmed that the transplantation of mutant PCs at the fracture site of wild-type mice impaired healing as shown by decreased callus and bone volumes by day 14 and increased fibrosis by days 14 and 21 compared with wild-type callus transplanted with control PCs (Figure 5E). Thus, transplanted PCs carrying an over-activating mutation in FGFR3 cannot form hypertrophic cartilage or bone via endochondral ossification during bone repair but cause pseudarthrosis through a cell-autonomous mechanism.
Figure 4.

In Vitro Characterization of Prx1Cre;Fgfr3Y367C/+ Periosteal Cells
(A) Experimental design of Prx1Cre;RosamTmG;Fgfr3+/+ (control) and Prx1Cre;RosamTmG;Fgfr3Y367C/+ (mutant) PC analyses via flow cytometry and in vitro differentiation.
(B) Representative fluorescence-activated cell sorting plots of GFP+ (Prx1-derived) PCs negative for hematopoietic and endothelial markers, and positive for ITGAV marker (left). Percentage of stem/progenitor (THY1−/6C3− cells), stromal (THY1−/6C3+ cells), and osteo/chondroprogenitor (THY1+/6C3− cells) sub-populations in GFP+ PCs as defined (Chan et al., 2015) (right) (n = 3 from 3 independent experiments per group). Values represent the mean ± SD.
(C) Osteogenic (alizarin red staining), adipogenic (oil red O staining), and chondrogenic (Alcian blue staining) in vitro differentiation of control and mutant PCs (n = 3 from 3 independent experiments per group). Values represent the mean ± SD.
Figure 5.

Transplantation of Prx1Cre;Fgfr3Y367C/+ Periosteal Cells Impairs Bone Healing
(A) Experimental design of Prx1Cre;RosamTmG;Fgfr3+/+ (control) and Prx1Cre;RosamTmG;Fgfr3Y367C/+ (mutant) PC isolation and transplantation at the fracture site of wild-type host.
(B) Lineage tracing of GFP+ control and mutant PCs at 10 days post-fracture. SO staining and SOX9 immunofluorescence on longitudinal sections of host calluses showing SOX9/GFP double-positive cells from control and mutant donor in the cartilage (box 1 and 2). Scale bars, 1 mm and 25 μm (high magnification).
(C and D) Lineage tracing of GFP+ control and mutant PCs at days 14 (C) and 21 (D) post-transplantation. SO staining and DAPI/GFP/Tomato fluorescence on longitudinal sections of host calluses (black dotted line). (C) Control PCs differentiate into hypertrophic chondrocytes (hc) (box 3, white arrowhead) but mutant PCs form elongated fibrocartilage cells by day 14 (fc, box 4). (D) Control PCs give rise to osteocytes within new bone trabeculae (b, box 5, white arrowhead) in the center of the callus, whereas mutant PCs form fibrotic (fib, box 6) cells by day 21 leading to pseudarthrosis (black arrow) (n = 5 per group). Scale bars: 1 mm, 100 μm (C, high magnification), and 25 μm (D, high magnification).
(E) Histomorphometric quantification of callus, cartilage, bone, and fibrosis volumes at days 14 and 21 post-fracture and PCs transplantation (n = 5 per group). Values represent mean ± SD. ∗p < 0.05, ∗∗p < 0.01 using Mann-Whitney test.
Exogenous PCs Can Rescue Pseudarthrosis in Prx1Cre;Fgfr3Y367C/+ Mice
We next attempted to rescue the pseudarthrosis phenotype in Prx1Cre;Fgfr3Y367C/+ mice with the tyrosine kinase inhibitor PD173074 that can arrest the G0/G1 phase of the FGFR3-expressing cells (Jonquoy et al., 2012; Martin et al., 2018). Although PD173074 treatment increased cartilage and hypertrophic cartilage volumes in the fracture callus of mutant mice, the treatment did not stimulate cartilage-to-bone transition and did not affect callus and bone volumes (Figure S4). Given the cell-autonomous defect of mutant PCs, we turned to a cell-based approach to rescue the pseudarthrosis phenotype (Figure 6A). PCs isolated from Prx1Cre;RosamTmG;Fgfr3+/+ mice integrated into the fracture callus of mutant hosts and were able to differentiate into hypertrophic chondrocytes by day 14 and subsequently gave rise to osteocytes within new bone trabeculae by day 21 (Figure 6B, top). Conversely, in mutant hosts treated with Tisseel matrix carrier alone, we observed the pseudarthrosis phenotype of mutant mice (Figure 6B, bottom). By day 28, the transplantation of Prx1Cre;RosamTmG;Fgfr3+/+ PCs in mutant hosts had a significant impact on bone healing as shown by the decrease in callus, unresorbed cartilage and persistent fibrosis volumes, and by the increase in bone volume (Figure 6C). The callus was fully ossified with complete resorption of cartilage and fibrous tissue in 7 out of 11 cases (Figure 6D). Transplanted PCs in mutant hosts were sufficient to support endochondral ossification by compensating for the failure of mutant hypertrophic cartilage to transform into bone, and preventing fibrocartilage and fibrosis accumulation. All together, these data establish that the pseudarthrosis phenotype in Prx1Cre;Fgfr3Y367C/+ mice is linked with an intrinsic deficiency of PCs to differentiate into late hypertrophic chondrocytes, leading to a defect in cartilage-to-bone transformation and absence of bone bridging, that can be prevented by exogenous PCs transplantation.
Figure 6.

Transplantation of PCs Corrects the Prx1Cre;Fgfr3Y367C/+ Pseudarthrosis Phenotype
(A) Experimental design of transplantation of Tisseel matrix or Prx1Cre;RosamTmG;Fgfr3+/+ PCs at the fracture site of Prx1Cre;Fgfr3Y367C/+ hosts.
(B) SO staining and DAPI/GFP/Tomato fluorescence on longitudinal sections of Prx1Cre;Fgfr3Y367C/+ calluses (delimited by a black dotted line) at days 14 and 21 post-fracture. GFP+ PCs form hypertrophic chondrocytes (box 1, white arrowheads) by day 14 and osteocytes (white arrow) within new bone (b, dotted line) by day 21 post-transplantation. By days 14 and 21, the center of mutant calluses transplanted with Tisseel matrix is composed of fibrocartilage (box 2, n = 4 or 5 per group). Scale bars, 1 mm, 100 μm (for d14, high magnification), and 25 μm (for d21, high magnification).
(C) Histomorphometric quantification of callus, cartilage, bone, and fibrosis volumes at day 28 post-fracture (n = 9 or 11 per group).
(D) Representative micro-CT images of Prx1Cre;Fgfr3Y367C/+ calluses by day 28 post-fracture showing bone bridging after PCs transplantation and absence of bone bridging (white arrow) after transplantation with Tisseel matrix. SO and PS staining on callus sections after transplantation of PCs showing complete ossification and absence of fibrocartilage and fibrous tissue accumulation (box 3, n = 7 cases out of 11). Representative callus sections confirm the presence of pseudarthrosis (black arrows) and at high magnification of fibrocartilage (fc) and fibrous tissue (fib) in mice transplanted with Tisseel matrix (box 4, n = 9 cases out of 9). Scale bars, 1 mm and 100 μm (high magnification). Values represent mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.005 using Mann-Whitney test.
Discussion
Here, we elucidate the underlying mechanisms of bone repair defect in the Prx1Cre;Fgfr3Y367C/+ mouse model that exhibit a pseudarthrosis phenotype. We identify inherent deficiencies of PCs carrying the Fgfr3Y367C heterozygous mutation and causing the pseudarthrosis phenotype after cell transplantation. These PCs deficiencies were not revealed using fluorescence-activated cell sorting analyses or in vitro differentiation assays that are classically used to identify functional impairment of skeletal stem/progenitor cells. Instead, we implanted mutant PCs into a bone fracture environment to study their behavior and to establish a correlation between the impaired regenerative potential of PCs in vivo and the bone repair deficit. Mutant PCs transplanted at the fracture site of wild-type hosts were detected in the cartilage within the fracture callus indicating that they did not exhibit deficiency in recruitment or migration. However, transplanted PCs could not differentiate into late hypertrophic chondrocytes and undergo cartilage-to-bone transformation, a process that cannot be evaluated in vitro. Mutant PCs were sufficient to impair bone healing in a wild-type host highlighting the cell-autonomous effect of exogenous PCs on bone repair and their involvement in the pseudarthrosis phenotype. The bone repair phenotype in Prx1Cre;Fgfr3Y367C/+ mice mimics the severe human condition of pseudarthrosis that can be associated with trauma or disease, suggesting that deficient PCs may be correlated with other pseudarthrosis phenotypes.
We also report the ability of control PCs to consolidate the bone fracture by producing hypertrophic cartilage that can transform into bone independent of the disease environment. This suggests that PCs could be beneficial in cell-based approaches to treat pseudarthrosis, given their positive effect on the very dynamic process of bone healing. Furthermore, in a cell-based therapy setting, it is generally accepted that long-term integration of transplanted cells is needed for efficient treatment (O'Keefe et al., 2020b; O'Keefe et al., 2020a). Our results illustrate that transient integration of PCs may be sufficient to correct a skeletal repair disorder.
The results show that cartilage-to-bone transformation mediated by PCs during bone repair is regulated by FGFR3 signaling. Fibroblast growth factors and their receptors are important regulators of bone development and are re-expressed in hypertrophic cartilage after bone fracture (Du et al., 2012; Nakajima et al., 2001; Schmid et al., 2009; Su et al., 2008). In the FGFR3 model carrying the Fgfr3G369C/+ gain-of-function mutation with milder achondroplasia phenotype, delayed healing of un-stabilized or semi-stabilized tibial fractures has been reported due to altered chondrogenesis and hypertrophic differentiation (Chen et al., 2017; Su et al., 2008). However, callus ossification was not inhibited in the Fgfr3G369C/+ model suggesting that cartilage-to-bone transformation was not affected. Thus, our results using the Fgfr3Y367C/+ model bring further insights on the essential role of FGFR3 during bone repair in regulating not only chondrocyte differentiation but also cartilage-to-bone transformation necessary for endochondral ossification. The functional requirement of this cartilage-to-bone transformation process for the success of bone repair is still underestimated. We provide evidence that when this process is disrupted bone healing fails. Although intramembranous ossification at the periphery of the callus is not delayed in the Prx1Cre;Fgfr3Y367C/+ model, healing is halted due to absence of cartilage-to-bone transformation in the center of the callus. In addition, abnormal chondrocyte hypertrophy and transition to bone is associated with fibrocartilage and fibrous tissue accumulation that also interferes with bone bridging, revealing a central role of FGFR3 signaling in controlling the balance between ossification and fibrosis. These findings have major impacts on the management of patients affected with achondroplasia that may require surgery for limb lengthening as managing the level of stabilization and endochondral ossification may be crucial to allow successful healing (Chilbule et al., 2016; Kitoh et al., 2014). Beyond this specific clinical setting, the results bring fundamental insights on the mechanisms of bone repair defects in pseudarthrosis and the role of PCs and FGFR3 in the essential step of cartilage-to-bone transformation during bone repair.
Experimental Procedures
Mice
C57BL/6ScNj, Prx1Cre, and Rosa-tdTomato-EGFP (RosamTmG) transgenic mice were obtained from Jackson Laboratory (Bar Harbor, ME). Fgfr3Y367C mice were generated in Dr. Legeai-Mallet’s laboratory (Pannier et al., 2009). In brief, the point mutation was inserted within exon 9 at position 367 of the Fgfr3 gene, directly followed by a floxed neo cassette. Under Cre recombination, the neo cassette is removed, allowing expression of the mutated allele of the Fgfr3 gene. Mice were bred and genotyped in our laboratory using primers purchased from Eurofins (Eurofins Scientific, Lux). Five- to 8-week-old mice were used for in vitro experiments and 3 month-old mice for in vivo experiments. Male and female mice were used and distributed homogenously within experimental groups. All procedures were approved by the Paris University and Creteil University Ethical Committees.
Primary Cultures of PCs
PCs were obtained as described previously (Duchamp de Lageneste et al., 2018). In brief, mouse hindlimbs (tibias and femurs) were harvested free of skin, and muscles were dissected using scissors and forceps, without damaging the periosteum. Epiphyses were cut and bone marrow was flushed using growth media (MEMα supplemented with 20% lot-selected non-heat-inactivated FBS, 1% penicillin-streptomycin, Life Technologies, Carlsbad, CA). The remaining flushed bones free of bone marrow were placed in culture and PCs migrated out of the explants after 3 days in growth medium. Explants were removed after 2 weeks and PCs were expanded after one passage in culture for in vivo and in vitro experiments.
In Vitro Differentiation
In vitro differentiation of PCs in the three mesenchymal lineages (osteoblasts, chondrocytes, and adipocytes) were performed as described previously (Duchamp de Lageneste et al., 2018). Osteogenic, adipogenic, and chondrogenic differentiation was performed for 4 weeks, 10 and 3 days, respectively. Cells were stained with alizarin red S, oil red O, or Alcian blue solutions to stain mineralization, lipid droplets, and glycosaminoglycans, respectively.
Flow Cytometry Analyses
PCs were isolated as described above and incubated with CD45-BV650 (563410, BD Biosciences), TER-119-BV650 (747739, BD Biosciences), TIE2-APC (124009, BioLegend), CD51-BV711 (740755, BD Biosciences), THY1-BV786 (564365, BD Biosciences), and 6C3-PeCy7 (BLE108313, BioLegend) in PBS-brilliant stain buffer (563794, BD Biosciences) for 15 min on ice protected from light. Cells were then washed by adding 1 mL of wash medium (MEMα supplemented with 2% lot-selected non-heat-inactivated FBS and 1% penicillin-streptomycin) and centrifuged for 10 min at 1,500 rpm. Supernatant was discarded and cell pellets were resuspended in 200 μL of wash medium and 7-AAD (559925, BD Biosciences) was added just before analysis as viability marker. Beads (01-2222-42, Thermo Fischer Scientific) were used for initial compensation set up and FMO (fluorescence minus one) controls were used to determine background level of each color. Analyses were performed on a BD LSRFortessa SORP (BD Biosciences) and results analyzed using FlowJo software, version 10.2.
Tibial Fracture, Unicortical Defect Injury, and Cell Transplantation
Open non-stabilized tibial fractures and unicortical defects were performed as previously described to study bone regeneration through endochondral and intramembranous ossification, respectively (Duchamp de Lageneste et al., 2018). For cell transplantation, 100,000 PCs were embedded in a fibrin gel (Baxter, France, Tisseel, composed of human fibrinogen 15 mg/mL and thrombin 9 mg/mL) and the cell pellet was transplanted at the time of fracture (Abou-Khalil et al., 2015; Duchamp de Lageneste et al., 2018).
PD173074 Treatment
Prx1Cre;Fgfr3Y367C/+ mice received intraperitoneal injections of PD173074 (Sigma, St. Louis, MO, ref. P2499) at a concentration of 10 mg/kg per injection. One injection per day was performed on the day of tibial fracture and every day until harvesting at days 7 and 14 post-fracture. Control mice were injected daily with 5% DMSO in PBS.
Histomorphometry and In Vivo Cell Tracing
Mice were euthanized, and tibias were harvested and fixed for 24 h in 4% paraformaldehyde followed by decalcification in 19% EDTA for 3 weeks. Tibias were embedded in paraffin or O.C.T. compound. Ten-micron-thick cryosections or paraffin sections were stained with Safranin O (SO), Masson's trichrome (TC), or Picrosirius (PS) for histomorphometric analyses to determine callus, cartilage, bone, and fibrosis volumes as described previously (Abou-Khalil et al., 2014, 2015). In brief, images of the stained sections were captured and analyzed using ZEN software to determine the surface of callus and cartilage on SO sections, bone on TC sections, and fibrosis on PS sections. Total volume of each component was determined using the dedicated formula (Abou-Khalil et al., 2014). Hypertrophic cartilage was quantified based on the cell morphology of hypertrophic chondrocytes (cells with a large cytoplasm in the SO+ area and specific organization) (Su et al., 2008). For cell transplantation experiments, Prx1-derived cells expressing GFP were detected by analysis of GFP signal on cryosections adjacent to SO and TC. Images were captured using a Zeiss Imager D1 AX10 light microscope and ZEN software (Carl Zeiss Microscopy, Gottinger, Germany).
Vascularization at the cartilage-to-bone transition zone was quantified at three different regions per section and on two sections per sample. The surface of AF488 and AF546 signal per μm2 was measured using ZEN software (Carl Zeiss Microscopy). Numbers of KI67+ and SOX2+ hypertrophic chondrocytes was determined by the average count of positive cells in three sections at the center of the callus per sample.
Immunofluorescence and Immunohistochemistry
For immunofluorescence, tibias and fracture calluses were processed as described previously (Kusumbe et al., 2015). Samples were fixed in ice-cold paraformaldehyde 4% for 4 h, decalcified in EDTA 19% at 4°C for 2–5 days and placed in sucrose 30% for 24 h before embedding in O.C.T. Thirty micron thick cryosections were collected for CD31/EMCN immunostaining and 10-μm-thick cryosections for the other markers. After blocking in 5% serum 0.25% Triton PBS for 1 h, sections were incubated with the primary antibody overnight at 4°C to label CD31 (AF3628, 1:100, BioTechne), EMCN (sc-65495, 1:100, Santa Cruz), COLX (ab58632, 1:200, Abcam), OSX (ab22552, 1:200, Abcam), SOX2 (ab97959, 1:400, Abcam), SOX9 (ab182530, 1:1,000, Abcam), KI67 (ab15580, 1:200, Abcam), and POSTN (AF2955,1:400, BioTechne). Sections were washed and incubated with secondary antibody Alexa Fluor 488 goat anti-rabbit (A11034, 1:1,000, Invitrogen), Alexa Fluor 488 donkey anti-goat (A11055, 1:1,000, Invitrogen), Alexa Fluor 546 goat anti-rabbit (A11056, 1:1,000, Invitrogen), Alexa Fluor 647 goat anti-rabbit (A-21245, 1:1,000, Invitrogen), and rhodamine donkey anti-rat (712-025-150, 1:400, Jackson ImmunoResearch). Sections were mounted with Fluoromount-G mounting medium with DAPI (00-4959-52, Life Technologies).
For immunohistochemistry staining, sections were rehydrated and incubated in 5% serum 0.25% Triton PBS for 1 h before incubation at 4°C overnight with primary antibody anti-VEGF (ab46154, 1:750, Abcam) and SOX9 (ab182530, 1:1,000, Abcam). Sections were incubated with biotin-coupled secondary antibody, followed by streptavidin-HRP (554066, 1:200, BD Biosciences) and staining was revealed using DAB-Plus Substrate Kit (002020, Invitrogen).
For COLX immunostaining a Dako EnVision Kit (N.2031501005; 1:50, BIOCYC) was used.
Micro-Computed Tomography Imaging
The injured tibia were dissected and micro-CT images were captured with a SkyScan 1172 micro-CT (Bruker, Hamburg, Germany) using the following settings: 80 kV, 100 μA, exposure time 100 ms, filter Al 0.5, and pixel size 19.98 μm. The scanned images were reconstructed as a stack of slices of each sample using Nrecon software (Bruker, Hamburg, Germany) and DataViewer software (Bruker, Hamburg, Germany). The callus bone volume was quantified by delimitating the callus area without the cortex using CTan software (Bruker, Hamburg, Germany).
Statistical Analyses
Statistical significance was determined with two-sided Mann-Whitney test and reported in GraphPad Prism v.6.0a. p values were determined as follows: ∗p ≤ 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005. All samples were included. All analyses were performed using a blind numbering system.
Author Contributions
C. Colnot conceived the project and wrote the paper. A.J., S.P., and O.D.d.L. performed the experiments, analyzed the data, and wrote the paper. C. Carvalho performed the experiments. M.B. assisted with micro-CT analyses. L.L.-M. provided the Fgfr3Y367C/+ mice, provided help to generate immunostaining data, and reviewed the manuscript.
Acknowledgments
We thank C. Benoit and T. Horville for technical assistance and advice, C. Cordier and J. Megret of the Imagine Institute Flow Cytometry Core, E. Panafieu and C. Dicu of the Imagine Institute Animal Facility. Micro-CT analyses were performed at the B3OA laboratory IMOSAR platform. This research was supported by Inserm ATIP-Avenir, France, Osteosynthesis and Trauma Care Foundation, Switzerland, Fondation de l'Avenir, France, Agence Nationale de la Recherche ANR-13-BSV1 and ANR-18-CE14-0033, France, NIAMS R01 AR072707, United States to C. Colnot. A.J., S.P., and O.D.d.L. were supported by PhD fellowships from Paris University.
Published: September 10, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.08.005.
Supplemental Information
References
- Abou-Khalil R., Yang F., Lieu S., Julien A., Perry J., Pereira C., Relaix F., Miclau T., Marcucio R., Colnot C. Role of muscle stem cells during skeletal regeneration. Stem Cells. 2015;33:1501–1511. doi: 10.1002/stem.1945. [DOI] [PubMed] [Google Scholar]
- Abou-Khalil R., Yang F., Mortreux M., Lieu S., Yu Y.Y., Wurmser M., Pereira C., Relaix F., Miclau T., Marcucio R.S., Colnot C. Delayed bone regeneration is linked to chronic inflammation in murine muscular dystrophy. J. Bone Miner Res. 2014;29:304–315. doi: 10.1002/jbmr.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaventure J., Rousseau F., Legeai-Mallet L., le Merrer M., Munnich A., Maroteaux P. Common mutations in the fibroblast growth factor receptor 3 (FGFR 3) gene account for achondroplasia, hypochondroplasia, and thanatophoric dwarfism. Am. J. Med. Genet. 1996;63:148–154. doi: 10.1002/(SICI)1096-8628(19960503)63:1<148::AID-AJMG26>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Chan C.K., Seo E.Y., Chen J.Y., Lo D., Mcardle A., Sinha R., Tevlin R., Seita J., Vincent-Tompkins J., Wearda T. Identification and specification of the mouse skeletal stem cell. Cell. 2015;160:285–298. doi: 10.1016/j.cell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Sun X., Yin L., Chen S., Zhu Y., Huang J., Jiang W., Chen B., Zhang R., Chen L. PTH 1-34 ameliorates the osteopenia and delayed healing of stabilized tibia fracture in mice with achondroplasia resulting from gain-of-function mutation of FGFR3. Int. J. Biol. Sci. 2017;13:1254–1265. doi: 10.7150/ijbs.21258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilbule S.K., Dutt V., Madhuri V. Limb lengthening in achondroplasia. Indian J. Orthop. 2016;50:397–405. doi: 10.4103/0019-5413.185604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C. Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J. Bone Miner Res. 2009;24:274–282. doi: 10.1359/jbmr.081003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath S., Yallowitz A.R., Mccormick J., Lalani S., Zhang T., Xu R., Li N., Liu Y., Yang Y.S., Eiseman M. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature. 2018;562:133–139. doi: 10.1038/s41586-018-0554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X., Xie Y., Xian C.J., Chen L. Role of FGFs/FGFRs in skeletal development and bone regeneration. J. Cell Physiol. 2012;227:3731–3743. doi: 10.1002/jcp.24083. [DOI] [PubMed] [Google Scholar]
- Duchamp de Lageneste O., Julien A., Abou-Khalil R., Frangi G., Carvalho C., Cagnard N., Cordier C., Conway S.J., Colnot C. Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat. Commun. 2018;9:773. doi: 10.1038/s41467-018-03124-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D.P., Ferro F., Yang F., Taylor A.J., Chang W., Miclau T., Marcucio R.S., Bahney C.S. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development. 2017;144:221–234. doi: 10.1242/dev.130807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonquoy A., Mugniery E., Benoist-Lasselin C., Kaci N., le Corre L., Barbault F., Girard A.L., le Merrer Y., Busca P., Schibler L. A novel tyrosine kinase inhibitor restores chondrocyte differentiation and promotes bone growth in a gain-of-function Fgfr3 mouse model. Hum. Mol. Genet. 2012;21:841–851. doi: 10.1093/hmg/ddr514. [DOI] [PubMed] [Google Scholar]
- Kitoh H., Mishima K., Matsushita M., Nishida Y., Ishiguro N. Early and late fracture following extensive limb lengthening in patients with achondroplasia and hypochondroplasia. Bone Joint J. 2014;96-B:1269–1273. doi: 10.1302/0301-620X.96B9.33840. [DOI] [PubMed] [Google Scholar]
- Kusumbe A.P., Ramasamy S.K., Starsichova A., Adams R.H. Sample preparation for high-resolution 3D confocal imaging of mouse skeletal tissue. Nat. Protoc. 2015;10:1904–1914. doi: 10.1038/nprot.2015.125. [DOI] [PubMed] [Google Scholar]
- Logan M., Martin J.F., Nagy A., Lobe C., Olson E.N., Tabin C.J. Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- Martin L., Kaci N., Estibals V., Goudin N., Garfa-Traore M., Benoist-Lasselin C., Dambroise E., Legeai-Mallet L. Constitutively-active FGFR3 disrupts primary cilium length and IFT20 trafficking in various chondrocyte models of achondroplasia. Hum. Mol. Genet. 2018;27:1–13. doi: 10.1093/hmg/ddx374. [DOI] [PubMed] [Google Scholar]
- Matsushita Y., Nagata M., Kozloff K.M., Welch J.D., Mizuhashi K., Tokavanich N., Hallett S.A., Link D.C., Nagasawa T., Ono W., Ono N. A Wnt-mediated transformation of the bone marrow stromal cell identity orchestrates skeletal regeneration. Nat. Commun. 2020;11:332. doi: 10.1038/s41467-019-14029-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugniery E., Dacquin R., Marty C., Benoist-Lasselin C., de Vernejoul M.C., Jurdic P., Munnich A., Geoffroy V., Legeai-Mallet L. An activating Fgfr3 mutation affects trabecular bone formation via a paracrine mechanism during growth. Hum. Mol. Genet. 2012;21:2503–2513. doi: 10.1093/hmg/dds065. [DOI] [PubMed] [Google Scholar]
- Nakajima A., Nakajima F., Shimizu S., Ogasawara A., Wanaka A., Moriya H., Einhorn T.A., Yamazaki M. Spatial and temporal gene expression for fibroblast growth factor type I receptor (FGFR1) during fracture healing in the rat. Bone. 2001;29:458–466. doi: 10.1016/s8756-3282(01)00604-4. [DOI] [PubMed] [Google Scholar]
- O'Keefe R.J., Tuan R.S., Lane N.E., Awad H.A., Barry F., Bunnell B.A., Colnot C., Drake M.T., Drissi H., Dyment N.A. American Society for Bone and Mineral Research—Orthopaedic Research Society joint task force report on cell-based therapies. J. Bone Miner Res. 2020;35:3–17. doi: 10.1002/jbmr.3839. [DOI] [PubMed] [Google Scholar]
- O'Keefe R.J., Tuan R.S., Lane N.E., Awad H.A., Barry F., Bunnell B.A., Colnot C., Drake M.T., Drissi H., Dyment N.A. American Society for Bone and Mineral Research—Orthopaedic Research Society joint task force report on cell-based therapies—secondary publication. J. Orthop. Res. 2020;38:485–502. doi: 10.1002/jor.24485. [DOI] [PubMed] [Google Scholar]
- Ortinau L.C., Wang H., Lei K., Deveza L., Jeong Y., Hara Y., Grafe I., Rosenfeld S.B., Lee D., Lee B. Identification of functionally distinct Mx1+alphaSMA+ periosteal skeletal stem cells. Cell Stem Cell. 2019;25:784–796 e5. doi: 10.1016/j.stem.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannier S., Couloigner V., Messaddeq N., Elmaleh-Berges M., Munnich A., Romand R., Legeai-Mallet L. Activating Fgfr3 Y367C mutation causes hearing loss and inner ear defect in a mouse model of chondrodysplasia. Biochim. Biophys. Acta. 2009;1792:140–147. doi: 10.1016/j.bbadis.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Rousseau F., Bonaventure J., Legeai-Mallet L., Pelet A., Rozet J.M., Maroteaux P., le Merrer M., Munnich A. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- Schmid G.J., Kobayashi C., Sandell L.J., Ornitz D.M. Fibroblast growth factor expression during skeletal fracture healing in mice. Dev. Dyn. 2009;238:766–774. doi: 10.1002/dvdy.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N., Yang J., Xie Y., Du X., Lu X., Yin Z., Yin L., Qi H., Zhao L., Feng J., Chen L. Gain-of-function mutation of FGFR3 results in impaired fracture healing due to inhibition of chondrocyte differentiation. Biochem. Biophys. Res. Commun. 2008;376:454–459. doi: 10.1016/j.bbrc.2008.08.165. [DOI] [PubMed] [Google Scholar]
- Van Gastel N., Torrekens S., Roberts S.J., Moermans K., Schrooten J., Carmeliet P., Luttun A., Luyten F.P., Carmeliet G. Engineering vascularized bone: osteogenic and proangiogenic potential of murine periosteal cells. Stem Cells. 2012;30:2460–2471. doi: 10.1002/stem.1210. [DOI] [PubMed] [Google Scholar]
- Worthley D.L., Churchill M., Compton J.T., Tailor Y., Rao M., Si Y., Levin D., Schwartz M.G., Uygur A., Hayakawa Y. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell. 2015;160:269–284. doi: 10.1016/j.cell.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Tsang K.Y., Tang H.C., Chan D., Cheah K.S. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. U S A. 2014;111:12097–12102. doi: 10.1073/pnas.1302703111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Xie C., Lin A.S., Ito H., Awad H., Lieberman J.R., Rubery P.T., Schwarz E.M., O'keefe R.J., Guldberg R.E. Periosteal progenitor cell fate in segmental cortical bone graft transplantations: implications for functional tissue engineering. J. Bone Miner Res. 2005;20:2124–2137. doi: 10.1359/JBMR.050806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.O., Yue R., Murphy M.M., Peyer J.G., Morrison S.J. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15:154–168. doi: 10.1016/j.stem.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., von der Mark K., Henry S., Norton W., Adams H., de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014;10:e1004820. doi: 10.1371/journal.pgen.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.