

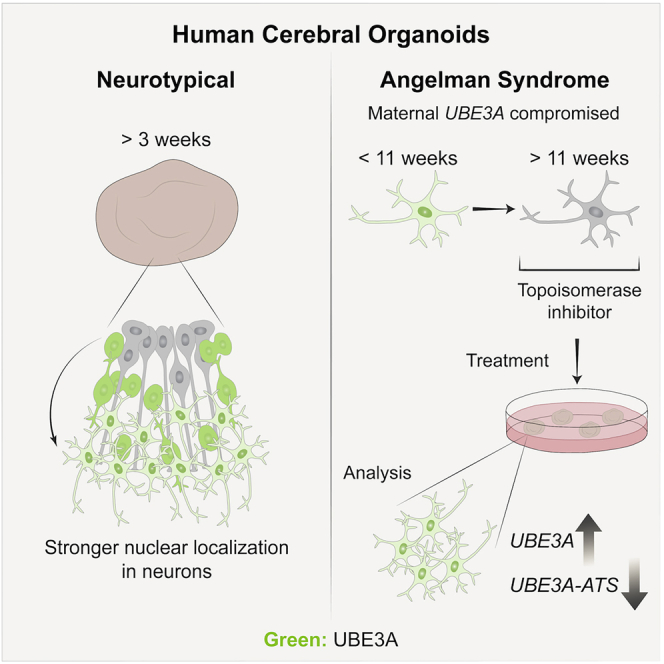
Summary
Angelman syndrome is a complex neurodevelopmental disorder characterized by delayed development, intellectual disability, speech impairment, and ataxia. It results from the loss of UBE3A protein, an E3 ubiquitin ligase, in neurons of the brain. Despite the dynamic spatiotemporal expression of UBE3A observed in rodents and the potential clinical importance of when and where it is expressed, its expression pattern in humans remains unknown. This reflects a common challenge of studying human neurodevelopment: prenatal periods are hard to access experimentally. In this work, human cerebral organoids reveal a change from weak to strong UBE3A in neuronal nuclei within 3 weeks of culture. Angelman syndrome human induced pluripotent stem cell-derived organoids also exhibit early silencing of paternal UBE3A, with topoisomerase inhibitors partially rescuing UBE3A levels and calcium transient phenotypes. This work establishes human cerebral organoids as an important model for studying UBE3A and motivates their broader use in understanding complex neurodevelopmental disorders.
Keywords: cerebral organoid, neurodevelopment, imprinting, epigenetic, UBE3A, Angelman syndrome, neurogenesis, pluripotent stem cell, autism
Graphical Abstract
Highlights
-
•
UBE3A signals in neuronal nuclei in hCOs correlate to early stages of development
-
•
UBE3A exhibits a change from weakly to strongly nuclear in cortical layers
-
•
UBE3A is imprinted and aberrantly localized in Angelman syndrome hCOs
-
•
Topoisomerase inhibitors partially rescue UBE3A and neuronal function in AS hCOs
In this article Keung and colleagues use human cerebral organoids to show that important spatiotemporal dynamics of the imprinted gene UBE3A occur early in human neurodevelopment. Organoids exhibit stronger UBE3A in the neuronal nuclei within 3 weeks and imprinting of paternal UBE3A within 6 weeks of culture, with topoisomerase inhibitors partially rescuing UBE3A and neuronal function in Angelman syndrome organoids.
Introduction
Angelman syndrome (AS) is characterized by delayed development, severe speech impairment, ataxia, and intellectual disability (Lopez et al., 2019). It results from mutations, deletions, or imprinting defects that negatively affect the levels or activities of UBE3A (Kishino et al., 1997), an E3 ubiquitin ligase (LaSalle et al., 2015). In neurotypical development, UBE3A is initially expressed biallelically and then becomes paternally silenced in neurons of the brain, which leaves the maternal allele the only source of UBE3A and the reason its specific maternal loss or mutation results in AS.
Rodent studies have revealed additional key molecular features of UBE3A important in disease etiology. These features share a common characteristic in that they occur at relatively early periods in neurodevelopment. For example, epigenetic silencing of paternal Ube3a and loss of UBE3A in AS mouse models was observed perinatally (Judson et al., 2014). Furthermore, early ablation or rescue of UBE3A in AS mouse models induced or rescued behavioral phenotypes, respectively (Silva-Santos et al., 2015; Sonzogni et al., 2019).
In addition to its imprinted expression, one of the salient molecular features of UBE3A is its nuclear localization in neurons, which also occurs perinatally and in the first couple postnatal weeks of murine neurodevelopment (Burette et al., 2017; Judson et al., 2014). This localization may be regulated by shifts in the expression levels of UBE3A isoforms (Sirois et al., 2020). It was recently shown that mice lacking a nuclear UBE3A isoform exhibited electrophysiological and behavioral deficits similar to those in other AS model mice (Avagliano Trezza et al., 2019). Apart from its ubiquitin ligase activity, UBE3A also has a putative role in transcriptional regulation, implying that these two independent functions could be influenced by its localization and contribute to disease phenotypes (LaSalle et al., 2015).
This work motivates three important and interrelated questions. In which cell types are these molecular features occurring, when are they occurring, and how do these features map to human neurodevelopment, if they do at all? Studying early pre- and perinatal periods, even in animal models, is challenging given the restricted availability and experimental tractability of human fetal tissue. Furthermore, there are significant differences in mouse and human imprinting centers (Johnstone et al., 2006) and UBE3A isoforms (LaSalle et al., 2015). To address these challenges, human stem cell-derived neurons (Fink et al., 2017; Hsiao et al., 2019) and cerebral organoids (hCOs) (Sun et al., 2019) are promising experimental models for AS research. hCOs in particular provide access to early prenatal periods of human neurodevelopment in an experimentally tractable and abundant form, as they have been shown to accurately model the cell types and transcriptomes of early human neurodevelopment (Camp et al., 2015; Kanton et al., 2019; Quadrato et al., 2017).
In this work, hCOs reveal the complex spatiotemporal dynamics of UBE3A in a range of neurodevelopmental cell types, identify key prenatal developmental windows for the subcellular localization and imprinted expression of UBE3A, and capture transcriptional and functional responses to candidate small-molecule therapeutics.
Results
hCOs Reveal an Early Change from Weak to Strong Nuclear UBE3A in Neuronal Nuclei
In this work, “whole-brain” hCOs (Lancaster et al., 2013) were first used to efficiently map when and in which cell types UBE3A was expressed. Neurotypical hCOs derived from H9 human embryonic stem cells (hESCs) were fixed over a broad time range (1–12 weeks to approximate the first trimester) and stained for nuclei, UBE3A, and a panel of cell-type-specific markers (Figures 1, 2, S1, and S3). Interestingly, UBE3A was prominently nuclear in a substantial number of neurons after only 3 weeks in culture, and this localization increased over time (Figures 1A, 1B, and S1A). This matched the transition observed in P0-P7 mice on an absolute timescale (Judson et al., 2014). Neuronal differentiation tracked the change in UBE3A localization: TUJ1+ neuronal areas showed much stronger nuclear UBE3A staining compared with SOX2+ stem cells, and roughly half of PAX6+ progenitors exhibited strong nuclear UBE3A (Figures 1A, 1C, 1D, and S1A–S1C), with similar results observed in two additional pluripotent cell lines (Figure 1E). Supporting these immunostaining patterns, the ratio of cytoplasmic to nuclear UBE3A measured through subcellular fractionation and western blot decreased over the course of 2, 6, and 9 weeks in H9 hCOs (Figure 1F). While subcellular fractionation results cannot be used to definitively conclude an increase in nuclear UBE3A in neurons given the heterogeneous cell type composition of hCOs, immunostaining results from 3D sectioned (Figures 1A and 1D) and 2D dissociated hCOs (Figures 1G and S2D) also showed no evidence of increased nuclear UBE3A in progenitors.
Figure 1.

hCOs Reveal an Early Change from Weak to Strong Nuclear UBE3A in Neurons
(A) Immunostaining of neurotypical hCO neurodevelopment. Strong nuclear UBE3A in neurons (arrows) and weak nuclear UBE3A in SOX2+ cells (arrowheads) are seen. Cytoplasmic UBE3A decreases over time (double arrows). Dotted white lines delineate boundaries between TUJ1+ and SOX2+ cells.
(B) Percentage of strong nuclear UBE3A increases during hCO development. Immunostaining quantification. ∗p < 0.05 between all groups, one-way ANOVA with Tukey-Kramer post hoc analysis, n = 3 independent experiments with two organoids in each. Error bars, 95% confidence intervals.
(C) Strong (arrows) and weak (double arrows) nuclear UBE3A in PAX6+ cells. Strong nuclear UBE3A in PAX6−/weak cells (arrowheads) is seen.
(D) UBE3A localization by cell type identified by immunostaining. Error bars, 95% confidence intervals. n = 3 independent experiments with two organoids in each.
(E) H1- and hiPSC-derived hCOs. Strong nuclear UBE3A in neurons (arrows) is seen.
(F) Immunoblot analysis of UBEA, GAPDH, and H3 using nuclear (NE) and cytoplasmic (CE) extracts isolated from H9 hCOs. n = 2 independent experiments with 15–25 organoids in each. Error bars, 95% confidence intervals.
(G) 2D immunostaining of dissociated H9 hCOs. Strong nuclear UBE3A in neurons (arrows) and weaker diffuse staining in progenitors (double arrows) are seen.
(See also Figures S1 and S2)
Figure 2.

Strong UBE3A Signal in Neuronal Nuclei in hCOs Correlates to Early Stages of Prenatal Neurodevelopment
(A) Schematics illustrating the simplified cellular and laminar organization of the developing human fetal cortex and that of a typical hCO.
(B) Summary of dynamic UBE3A localization in neurotypical hCOs.
(C) Dotted white lines delineate boundaries between TBR1+ and EOMES+ regions. Strong nuclear UBE3A in TBR1+ cells (arrows), weak nuclear UBE3A in EOMES+ cells (arrowheads), and weak UBE3A in cytoplasm of TBR1+ cells (double arrows) are seen.
(D) Strong nuclear UBE3A colocalizes with CTIP2+/TBR1+ cells (arrows).
(E) Strong nuclear UBE3A in TBR1+/SATB2− (arrowheads) and TBR1+/SATB2+ (double arrows) cells and weaker UBE3A in TBR1−/SATB2+ cells (arrows).
(F) UBE3A localization by cortical cell type identified by immunostaining. Error bars, 95% confidence intervals. n = 3 independent experiments with two organoids in each.
(G) Strong nuclear UBE3A in TBR1+/Calretinin+ (arrows), TBR1+/Calretinin weak (double arrows), and TBR+/Calretinin− (arrowheads) neurons.
(See also Figure S3.)
Strong UBE3A Signal in Neuronal Nuclei Correlates to Early Stages of Prenatal Neurodevelopment
In addition to absolute timescales, the presence of specific cell types in hCOs can be correlated to distinct stages of fetal neurodevelopment. The fetal cortex comprises cell layers representing different stages of differentiation, including a sequential transition from neural precursors (SOX2), to radial glia and intermediate progenitors (EOMES), to postmitotic neurons (TBR1, CTIP2, SATB2) (Figure 2A) (Englund et al., 2005). Interestingly, a striking boundary formed between layers of TBR1+ and EOMES+ cells, with strong nuclear UBE3A only in TBR1+ cells (Figures 2B and 2C). CTIP2+ (precursors of early-born deep-layer neurons) and SATB2+ (precursors of late-born superficial-layer neurons) cells also expressed strong nuclear UBE3A (Figures 2B, 2D, 2E, and S3A–S3C). Furthermore, mature SATB2+/TBR1− cells (late-born superficial-layer neurons) exhibited a relative loss in nuclear UBE3A compared with their more immature SATB2+/TBR1+ counterparts (Figures 2B, 2E, 2F, and S3B), consistent with observations in P0-P7 mice (Judson et al., 2014). Interestingly, strong coexpression of TBR1 with SATB2 and CTIP2 correlates with periods before human postconception week 20 (PCW20), with separation of these markers occurring closer to PCW30 in human fetal tissue (Saito et al., 2011); this suggests these tissue-like structures in hCOs may reflect PCW20–30 and that UBE3A is already stronger in the neuronal nuclei at this stage of neurodevelopment.
It was previously reported that Calretinin in the early fetal brain is specifically coexpressed with TBR1 only in the first excitatory projection neurons of the cortex during human PCW7–7.5 and diminishes shortly after PCW8 (Gonzalez-Gomez and Meyer, 2014). Both Calretinin+/TBR1+ and Calretinin−/TBR1+ neurons appeared in hCOs, and in both cell types in hCOs UBE3A was localized primarily to the nucleus, but the expression of UBE3A was higher in Calretinin+ neurons (Figures 2G and S3D). Collectively these results indicate that the nuclear localization of UBE3A in hCOs correlates with at least the mid-to-late first trimester of human gestation.
UBE3A Is Imprinted and Aberrantly Localized in Angelman Syndrome hCOs
In addition to its subcellular localization, the dosage of UBE3A, controlled by the epigenetic silencing of its paternal allele in neurons, is a primary driver of AS. However, it is not known when paternal UBE3A silencing occurs during human neurodevelopment. UBE3A-ATS is a long non-coding RNA whose paternal expression is known to increase during development and to silence paternal UBE3A (Hsiao et al., 2019; Stanurova et al., 2016) (Figure 3A). To track the timing of imprinting in hCOs, UBE3A and UBE3A-ATS transcripts were measured by RT-qPCR (Figures 3B and 3C). hCOs were generated from H9 cells as well as AS human induced pluripotent stem cells (hiPSCs) that harbor a large maternal UBE3A deletion, previously generated and characterized by Chamberlain and colleagues (Chamberlain et al., 2010). Direct comparisons between these cell lines remained phenomenological in this study as they are not isogenic; however, AS hCOs provided a method to unambiguously determine when paternal UBE3A is silenced, as maternal UBE3A is absent. Both hCOs exhibited a monotonic increase in UBE3A-ATS transcripts starting at 3 weeks in culture (Figure 3B). UBE3A transcripts decreased in AS hCOs, but only after 6 weeks (Figure 3C). This ~3 week delay is similar to previous observations in hiPSC-derived neurons (Hsiao et al., 2019; Stanurova et al., 2016).
Figure 3.

UBE3A Is Imprinted and Aberrantly Localized in Angelman Syndrome hCOs
(A) The UBE3A locus.
(B and C) qRT-PCR measurements of mRNA levels of UBE3A-ATS (B) and UBE3A (C) in neurotypical and AS hCOs, normalized to HPRT, ratioed to 1 week AS hCOs. Error bars are 95% confidence intervals. (B) p < 0.05 t test against null-slope hypothesis. n = 3 independent experiments with three to five organoids in each. (C) ∗p < 0.05, full tick marks compared with half tick marks by one-way ANOVA with Tukey-Kramer post hoc. n = 3 independent experiments with three to five organoids in each.
(D, E, and G) UBE3A expression and localization in AS hCOs. (D) Salient nuclear UBE3A in SOX2+ progenitors of 4–12 week AS hCOs (arrows). Salient nuclear UBE3A in 4–7 week TUJ1+/SOX2−neurons (arrowheads) is lost in 12 week AS hCOs (double arrows).
(E) Strong nuclear UBE3A in 7 week EOMES+ cells (arrows) is weakened at 10 weeks in AS hCOs.
(F) Summary of dynamic UBE3A localization in AS hCOs.
(G) UBE3A is absent in 17 week MAP2+/SOX2− neurons (arrows). SOX2+ progenitors still express some paternal UBE3A (arrowheads).
(H) Percentage of nuclear UBE3A in 7–12 week AS hCOs. ∗p < 0.05, full tick marks compared with half tick marks by one-way ANOVA with Tukey-Kramer post hoc analysis, n = 3 independent experiments with two organoids in each. Error bars are 95% confidence intervals.
(See also Figure S4.)
The subcellular localization of paternal UBE3A in AS hCOs was also tracked over time (Figures 3D–3H and S4A–S4E). Interestingly, unlike in neurotypical hCOs, a salient nuclear UBE3A localization pattern was observed in SOX2+ and EOMES+ progenitors during early hCO development (4–7 weeks) (Figures 3D–3F and S4A). In older AS hCOs (10–12 weeks), UBE3A expression became substantially more diffuse in EOMES+ cells (Figures 3E, 3F, 3H, and S4B). Similarly, in neurons of 3–7 week hCOs, UBE3A was prominently nuclear, but upon extended culture (10–17 weeks) UBE3A intensity weakened, indicating that the paternal allele was silenced during this time interval (Figures 3D, 3F–3H, S4A, S4C, and S4D). Interestingly, immature SOX2+/TUJ1+ neurons did exhibit nuclear UBE3A in 10–12 week AS hCOs (Figure S4E), consistent with previous reports of paternal UBE3A expression in immature neurons (Judson et al., 2014).
Topoisomerase Inhibitors Partially Rescue UBE3A Levels and Neuronal Function in AS hCOs
Since AS hCOs successfully silence paternal UBE3A, they represent a potentially useful system to study therapeutic strategies. Prior work found that topoisomerase inhibitors (topotecan and indotecan) could suppress UBE3A-ATS and reactivate paternal UBE3A in mice and in human cell cultures (Fink et al., 2017; Huang et al., 2012; Lee et al., 2018) to compensate for the absent maternal copy. To assess their activity in hCOs, 1 μM topotecan or indotecan was added to hCOs at different ages and dosing regimens. These drugs were added to 11 week hCOs, as significant silencing of UBE3A was observed at that time point (Figures 3C, 3D, and 3H). UBE3A-ATS and UBE3A transcripts were measured 3 days after treatment. Both topotecan and indotecan were able to knock down UBE3A-ATS 7- and 4-fold and increased UBE3A 1.8- and 1.75-fold, respectively (Figure 4A). Importantly, nuclear UBE3A in individual neurons identified with a CamKIIa-GFP reporter increased with treatment as well (Figures 4B and 4C). Interestingly, in addition to neurons, SOX2+ neural precursor cells also showed increased UBE3A levels (Figure S4G), suggesting the effect of topoisomerase inhibition may affect other cell types, and that there may be further room for UBE3A levels to increase even when already actively transcribed at basal levels. Indeed, UBE3A levels also increased in neurotypical hCOs treated with topotecan (Figure S4G).
Figure 4.

Topoisomerase Inhibitors Partially Rescue UBE3A Levels and Neuronal Function in AS hCOs
(A, D, F, and G) qRT-PCR measurements of mRNA levels of UBE3A (red) and UBE3A-ATS (gray) after vehicle (DMSO), 1 μM topotecan, or 1 μM indotecan treatment. Signals normalized to TATA-box binding protein (TBP) and ratioed to vehicle-treated AS hCOs.
(A) mRNA 3 days after a single drug treatment in 11 week hCOs.
(B) 11 week AS hCOs with CamKIIa-GFP neurons. Insets zoom in on arrowheads.
(C) Quantification of (B).
(D) mRNA 3 days after a single drug treatment in 4 week hCOs.
(E) mRNA after 1–9 days of continuous 1 μM indotecan treatment in 11 week AS hCOs, ratioed to untreated day 0 AS hCOs. No significant change in vehicle-treated samples (Figure S4C).
(F) mRNA 10 days after a single drug treatment in 11 week hCOs.
(G) mRNA 17 days after a single drug treatment in 11 week hCOs. Statistics: ∗p < 0.05, n.s., not significant, full tick marks compared with half tick marks by one-way ANOVA with Tukey-Kramer post hoc. For (A, D, E, F, G) n = 3 independent experiments with three to five organoids in each; error bars, 95% confidence intervals. For (C) A.U., arbitrary fluorescence units; n = 3 independent experiments with two organoids in each.
(H–J) (H) Quantification of the calcium transient frequencies. (I) Quantification of the amplitudes of calcium transients. (J) Representative sets of calcium transient traces extracted from individual neurons of H9 DMSO, AS DMSO, and AS indotecan-treated hCOs.
Statistics for (H and I): ∗p < 0.05, two-tailed unpaired Student's t test. n = 22, 41, and 34 neurons from H9 DMSO, AS DMSO, and AS indotecan-treated hCOs, respectively. Note: statistical comparison between H9 and AS hCOs shown only for completeness, as these are non-isogenic cell lines.
(See also Figure S4.)
A crucial set of questions in the treatment of neurodevelopmental disorders is at what time point, how frequently, and for how long should potential therapeutics be delivered; furthermore, how persistent are therapeutic effects? To address these questions, topoisomerase inhibitors were delivered to AS hCOs at 4, 11, or 15 weeks followed by qRT-PCR 3 days after treatment. Both inhibitors knocked down UBE3A-ATS and increased UBE3A at 11 and 15 weeks (Figures 4A and S4H). However, at 4 weeks, only UBE3A-ATS decreased (Figure 4D), likely attributable to the fact that UBE3A transcripts were still high at that early time point (Figure 3B).
Next, 1 μM indotecan was added to AS hCOs every day for 9 days and analyzed at days 1, 3, 6, and 9. This experiment asked if the rescue of UBE3A could be enhanced by persistent and longer-term indotecan delivery. UBE3A transcripts increased up to day 6 but decreased on day 9. UBE3A-ATS transcripts remained low throughout this analysis with no significant differences between time points (Figures 4E and S4I).
The decrease in UBE3A transcripts after 9 days of repeated treatments may have been due to the toxicity of topoisomerase inhibitors (Lee et al., 2018). It would therefore be advantageous if fewer doses could still elicit a persistent response. To test this, 11 week AS hCOs were exposed to a single treatment of topotecan or indotecan, changing to fresh medium without inhibitor after 3 days, and measuring transcript levels 10 and 17 days later. While topotecan was unable to elevate UBE3A levels, indotecan persistently rescued UBE3A (Figures 4F and 4G). The increased “memory” of indotecan response could be due to the compound's increased chemical stability or an as yet unknown epigenetic mechanism.
In addition to paternal UBE3A activation, indotecan was also able to partially rescue calcium transient phenotypes in AS hCOs. AS hCOs exhibited shorter interevent intervals and higher calcium transient frequencies compared with neurotypical hCOs, in agreement with recent work (Sun et al., 2019). Fourteen days post treatment by indotecan, transient amplitudes were rescued to levels nearing that of neurons from neurotypical hCOs (Figures 4H–4J).
Discussion
The excitement surrounding hCOs derives from their potential to fill important gaps, in this case in prenatal human development, that are difficult to access by other experimental systems. One of the most important aspects of UBE3A biology is the fact that the salient changes both in subcellular localization and in UBE3A-ATS/UBE3A expression occurred in what is the hCO equivalent of the first human trimester. The potential implications of these early dynamics are profound. Although hCOs cannot capture behavioral phenotypes, recent work through conditional UBE3A knockout or reinstatement shows that at least a subset of behaviors in mice are affected by perinatal UBE3A levels and cannot be rescued later in neurodevelopment (Rotaru et al., 2018; Silva-Santos et al., 2015; Sonzogni et al., 2019). Collectively, both hCO and mouse studies support a scenario in which early, even prenatal, treatment in humans may be necessary to have maximal therapeutic effects, although significant benefits may still be achieved through interventions later in life.
Another major advantage of using hCOs is their ability to generate a diverse range of human cell types from very early points in neurodevelopment that may be important in disease etiology. In our experiments we observed aberrant nuclear UBE3A in neural precursor cells of early AS hCOs (Figures 3D and 3E, 3H, S4A, S4B). Intriguingly there is some evidence of impacts on neurogenesis implicated in autism spectrum disorder, which shares some comorbidities with AS. Furthermore, prior work has reported partial paternal imprinting in progenitor cell types (Herzing et al., 2002). However, while nuclear expression of UBE3A is a hallmark of neuronal differentiation, and it is critical for proper function, the precise mechanistic role of nuclear UBE3A is not well understood even in neurons. Thus, although the clinical significance of this aberrant localization is unclear, it may hint at a potential role for neurogenesis in AS etiology. Additional work in this area is needed to identify not only the subcellular localization of UBE3A in distinct cell types, but also the absolute and graded levels of cytoplasmic and nuclear UBE3A and the levels of each UBE3A isoform in different cell types with improved temporal resolution. Overall, this work motivates the broader use of hCOs in future work to unlock important and highly relevant prenatal time periods in investigating imprinted genes, complex epigenetic phenomena, and their related neurodevelopmental disorders.
Experimental Procedures
Cell Culture and Cerebral Organoid Generation
Feeder-independent cell lines were H9 and H1 hESCs (WA09 and WA01, WiCell) and hiPSCs (cat. no. SC102-A1, Systems Biosciences). AS hiPSCs were developed in the Chamberlain and Lalande groups and obtained from Kerafast (Chamberlain et al., 2010). UBE3A double-knockout H9 cells (H9UBE3A m-/p-) with a 66 kb deletion (chr15: 25,338,949–25,405,676) were provided by Dr. Stormy Chamberlain (UCONN) (Sirois et al., 2020). Cells were maintained in tissue culture dishes (Fisher Scientific Corning Costar) coated with 0.5 mg/cm2 vitronectin (VTN-N; Thermo Fisher Scientific) in E8 medium (Thermo Fisher Scientific) and passaged using standard protocols. The imprinting status of H9, H1, and AG1-0 cell lines was confirmed previously (Chamberlain et al., 2010; Stanurova et al., 2016). hCOs were generated and maintained using the same protocol as described at 37°C with 5% CO2 (Lancaster et al., 2013).
Immunofluorescence, Immunoblot, and qRT-PCR Analyses
Standard methods were used. Detailed protocols are provided in the Supplemental Information.
Topotecan and Indotecan Treatment
Topotecan (Sigma Aldrich) and indotecan (NCI) were directly added to AS hCOs at 1 μM final concentration in culture medium. hCOs were cultured for 72 h without a fresh medium change. For long-term effects of single-drug exposure experiments, the first drug-free medium change was performed 3 days after the single-drug administration. Samples were collected 7 and 14 days after the fresh medium change (total of 10 and 17 days from initial drug exposure). When testing the effects of drug exposure time, hCO culture medium was replaced daily with fresh medium containing drugs. For live imaging experiments, hCO cells were treated with 1 μM indotecan or vehicle and cultured for 72 h without a fresh medium change. Live Ca2+ imaging was carried out 2 weeks after the 72 h treatment.
Live Ca2+ Imaging
Live imaging was performed using a Nikon AR confocal laser-scanning microscope (Nikon) equipped with temperature and CO2 control. For calcium imaging, Fluo-4 direct (Life Technologies) was prepared according to the manufacturer's protocol. hCOs were dissociated using Accutase (STEMCELL Technologies) and plated on reduced growth factor Matrigel (Corning) for 2–3 weeks before experiments were conducted. hCO cells were incubated with Fluo-4 60 min prior to start of imaging. Frames were taken every 2 s for 150 frames. Data analysis of calcium imaging was performed using FIJI. Regions of interest were manually selected, and mean fluorescence was calculated for each time frame. Change in fluorescence was calculated as follows: ΔF/F = (F − F0)/F0, in which F0 was the mean fluorescence value recorded at t = 0.
Data Availability and Code Availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
No custom codes or mathematical algorithms were used in this work.
Author Contributions
D.S. and A.J.K. conceived the study. D.S. planned and performed the wet lab experiments with guidance from A.J.K. and experimental support from A.V. and Z.D. D.S. and A.J.K. wrote the paper.
Acknowledgments
This work was supported by a Simons Foundation SFARI Explorers grant (495112), a Faculty Research and Professional Development Program and Research Innovation Seed Fund Grant from NCSU, the NSF Emerging Frontiers in Research and Innovation program (NSF-1830910), an NIH Avenir Award (DP1-DA044359), and a fellowship from the American Association of University Women (to D.S.). The Developmental Therapeutics Program branch, National Cancer Institute, provided indotecan (LMP400). We thank Drs. Stormy Chamberlain (University of Connecticut Genetics and Genome Sciences, Farmington, CT) and Benjamin Philpot (University of North Carolina School of Medicine, Chapel Hill, NC) for graciously gifting the UBE3A double-knockout H9 hESC line and wild-type and AS mouse brain sections, respectively. We thank Drs. Mark Zylka and G. Aneeshkumar Arimbasseri for their helpful discussions.
Published: September 10, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.08.006.
Supplemental Information
References
- Avagliano Trezza R., Sonzogni M., Bossuyt S.N.V., Zampeta F.I., Punt A.M., van den Berg M., Rotaru D.C., Koene L.M.C., Munshi S.T., Stedehouder J. Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat. Neurosci. 2019;22:1235–1247. doi: 10.1038/s41593-019-0425-0. [DOI] [PubMed] [Google Scholar]
- Burette A.C., Judson M.C., Burette S., Phend K.D., Philpot B.D., Weinberg R.J. Subcellular organization of UBE3A in neurons. J. Comp. Neurol. 2017;525:233–251. doi: 10.1002/cne.24063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp J.G., Badsha F., Florio M., Kanton S., Gerber T., Wilsch-Bräuninger M., Lewitus E., Sykes A., Hevers W., Lancaster M. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. U S A. 2015;112:15672–15677. doi: 10.1073/pnas.1520760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain S.J., Chen P.-F., Ng K.Y., Bourgois-Rocha F., Lemtiri-Chlieh F., Levine E.S., Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. U S A. 2010;107:17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C., Fink A., Lau C., Pham D., Daza R., Bulfone A., Kowalczyk T., Hevner R. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J. Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink J.J., Robinson T.M., Germain N.D., Sirois C.L., Bolduc K.A., Ward A.J., Rigo F., Chamberlain S.J., Levine E.S. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun. 2017;8:15038. doi: 10.1038/ncomms15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Gomez M., Meyer G. Dynamic expression of calretinin in embryonic and early fetal human cortex. Front. Neuroanat. 2014;8:41. doi: 10.3389/fnana.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzing L.B.K., Cook E.H., Ledbetter D.H. Allele-specific expression analysis by RNA-FISH demonstrates preferential maternal expression of UBE3A and imprint maintenance within 15q11-q13 duplications. Hum. Mol. Genet. 2002;11:1707–1718. doi: 10.1093/hmg/11.15.1707. [DOI] [PubMed] [Google Scholar]
- Hsiao J.S., Germain N.D., Wilderman A., Stoddard C., Wojenski L.A., Villafano G.J., Core L., Cotney J., Chamberlain S.J. A bipartite boundary element restricts UBE3A imprinting to mature neurons. Proc. Natl. Acad. Sci. U S A. 2019;116:2181–2186. doi: 10.1073/pnas.1815279116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.S., Allen J.A., Mabb A.M., King I.F., Miriyala J., Taylor-Blake B., Sciaky N., Dutton J.W., Lee H.M., Chen X. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2012;481:185–191. doi: 10.1038/nature10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone K.A., DuBose A.J., Futtner C.R., Elmore M.D., Brannan C.I., Resnick J.L. A human imprinting centre demonstrates conserved acquisition but diverged maintenance of imprinting in a mouse model for Angelman syndrome imprinting defects. Hum. Mol. Genet. 2006;15:393–404. doi: 10.1093/hmg/ddi456. [DOI] [PubMed] [Google Scholar]
- Judson M.C., Sosa-Pagan J.O., Del Cid W.A., Han J.E., Philpot B.D. Allelic specificity of Ube3a expression in the mouse brain during postnatal Development. J. Comp. Neurol. 2014;522:1874–1896. doi: 10.1002/cne.23507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanton S., Boyle M.J., He Z., Santel M., Weigert A., Sanchís-Calleja F., Guijarro P., Sidow L., Fleck J.S., Han D. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature. 2019;574:418–422. doi: 10.1038/s41586-019-1654-9. [DOI] [PubMed] [Google Scholar]
- Kishino T., Lalande M., Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- Lancaster M.A., Renner M., Martin C.-A., Wenzel D., Bicknell L.S., Hurles M.E., Homfray T., Penninger J.M., Jackson A.P., Knoblich J.A. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaSalle J.M., Reiter L.T., Chamberlain S.J. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics. 2015;7:1213–1228. doi: 10.2217/epi.15.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.M., Clark E.P., Kuijer M.B., Cushman M., Pommier Y., Philpot B.D. Characterization and structure-activity relationships of indenoisoquinoline-derived topoisomerase i inhibitors in unsilencing the dormant Ube3a gene associated with Angelman syndrome. Mol. Autism. 2018;9:45. doi: 10.1186/s13229-018-0228-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez S.J., Segal D.J., LaSalle J.M. UBE3A: an E3 ubiquitin ligase with genome-wide impact in neurodevelopmental disease. Front. Mol. Neurosci. 2019;11:476. doi: 10.3389/fnmol.2018.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrato G., Nguyen T., Macosko E.Z., Sherwood J.L., Min Yang S., Berger D.R., Maria N., Scholvin J., Goldman M., Kinney J.P. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545:48–53. doi: 10.1038/nature22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotaru D.C., van Woerden G.M., Wallaard I., Elgersma Y. Adult Ube3a gene reinstatement restores the electrophysiological deficits of prefrontal cortex layer 5 neurons in a mouse model of angelman syndrome. J. Neurosci. 2018;38:8011–8030. doi: 10.1523/JNEUROSCI.0083-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T., Hanai S., Takashima S., Nakagawa E., Okazaki S., Inoue T., Miyata R., Hoshino K., Akashi T., Sasaki M. Neocortical layer formation of human developing brains and lissencephalies: consideration of layer-specific marker expression. Cereb. Cortex. 2011;21:588–596. doi: 10.1093/cercor/bhq125. [DOI] [PubMed] [Google Scholar]
- Silva-Santos S., van Woerden G.M., Bruinsma C.F., Mientjes E., Jolfaei M.A., Distel B., Kushner S.A., Elgersma Y. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Invest. 2015;125:2069–2076. doi: 10.1172/JCI80554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirois C.L., Bloom J.E., Fink J.J., Gorka D., Keller S., Germain N.D., Levine E.S., Chamberlain S.J. Abundance and localization of human UBE3A protein isoforms. Hum. Mol. Genet. 2020 doi: 10.1093/hmg/ddaa191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonzogni M., Hakonen J., Bernabé Kleijn M., Silva-Santos S., Judson M.C., Philpot B.D., van Woerden G.M., Elgersma Y. Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mol. Autism. 2019;10:23. doi: 10.1186/s13229-019-0277-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanurova J., Neureiter A., Hiber M., De Oliveira Kessler H., Stolp K., Goetzke R., Klein D., Bankfalvi A., Klump H., Steenpass L. Angelman syndrome-derived neurons display late onset of paternal UBE3A silencing. Sci. Rep. 2016;6:30792. doi: 10.1038/srep30792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A.X., Yuan Q., Fukuda M., Yu W., Yan H., Lim G.G.Y., Nai M.H., D’Agostino G.A., Tran H.D., Itahana Y. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science. 2019;366:1486–1492. doi: 10.1126/science.aav5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
No custom codes or mathematical algorithms were used in this work.