

Abstract
Purpose
To report population pharmacokinetic (PK) analysis of the phase 1 study (FPA144-001, NCT02318329) and to select a clinical dose and schedule that will achieve an empirical target trough concentration (Ctrough) for an anti-fibroblast growth factor receptor 2b antibody, bemarituzumab.
Methods
Nonlinear mixed-effect modeling was used to analyse PK data. In vitro binding affinity and receptor occupancy of bemarituzumab were determined. Simulation was conducted to estimate dose and schedule to achieve an empirical target Ctrough in a phase 2 trial (FIGHT, NCT03694522) for patients receiving first-line treatment combined with modified 5-fluourouracil, oxaliplatin and leucovorin (mFOLFOX6) for gastric and gastroesophageal junction adenocarcinoma.
Results
Bemarituzumab PK is best described by a two-compartment model with parallel linear and nonlinear (Michaelis–Menten) elimination from the central compartment. Albumin, gender, and body weight were identified as the covariates on the linear clearance and/or volume of distribution in the central compartment, and no dose adjustment was warranted. An empirical target of bemarituzumab Ctrough of ≥ 60 µg/mL was projected to achieve > 95% receptor occupancy based on in vitro data. Fifteen mg/kg every 2 weeks, with a single dose of 7.5 mg/kg on Cycle 1 Day 8, was projected to achieve the target Ctrough on Day 15 in 98% of patients with 96% maintaining the target at steady state, which was confirmed in the FIGHT trial.
Conclusion
A projected dose and schedule to achieve the target Ctrough was validated in phase 1 of the FIGHT trial which supported selection of the phase 2 dose and schedule for bemarituzumab.
Electronic supplementary material
The online version of this article (10.1007/s00280-020-04139-4) contains supplementary material, which is available to authorized users.
Keywords: Bemarituzumab, Anti-fibroblast growth factor receptor 2b, Population PK analysis, Target-mediated clearance, Dose selection, Gastric and gastroesophageal junction adenocarcinoma
Introduction
Gastric and gastroesophageal junction adenocarcinoma (GEA) represents the fourth most common cancer worldwide and is a highly lethal disease, with 5-year overall survival (OS) rates below 33% in the United States (US) [1–3]. Chemotherapy as standard first-line treatment has demonstrated an improvement in OS compared to best supportive care and additional progress has been made with the use of targeted therapies, such as trastuzumab and ramucirumab. However, even with these therapies, prognosis is poor. Therefore, the identification of new therapeutics with acceptable toxicities is important for this patient population [4, 5].
The fibroblast growth factor/fibroblast growth factor receptor (FGF/FGFR) pathway can stimulate the transformation and proliferation of tumor cells and angiogenesis. FGF signaling is mediated by a family of transmembrane tyrosine kinase receptors encoded by four distinct genes producing FGF receptor subtypes termed FGFR1–4 [6]. Fibroblast growth factor receptor 2b (FGFR2b), one of 2 FGFR2 splicing variants, is expressed in tissues of epithelial origin (e.g., stomach, skin) with FGF7, FGF10, and FGF22 as three of its major ligands [7]. Alterations in signaling in the FGF/FGFR2 pathway (e.g., overexpression of FGFR2 protein or amplification of FGFR2 gene) have been associated with GEA, breast, and other cancers and with a decreased prognosis [6, 8–11], suggesting that inhibition of FGFR2 may be a rational target for cancer therapy [12, 13].
Bemarituzumab (FPA144) is a first-in-class humanized Immunoglobulin G1 (IgG1) monoclonal antibody specific to the human FGFR2b receptor that blocks FGF binding to the receptor. It is also glycoengineered for increased affinity for human Fc gamma receptor IIIA (FcγRIIIa) expressed on natural killer cells, resulting in enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) against FGFR2b-overexpressing tumors. These two mechanisms of action of bemarituzumab may together lead to improved OS in patients with GEA whose tumors overexpress FGFR2b.
Bemarituzumab has been evaluated in patients receiving treatment for late-line solid tumors, including those with GEA with or without overexpression of FGFR2b in the FPA144-001 trial (NCT02318329). Promising single agent activity with acceptable tolerability was demonstrated in patients whose GEA overexpressed FGFR2b [14]. GEA tends to be highly heterogeneous within the same tumor, and when present, FGFR2b may not be uniformly distributed throughout the tumor specimen [15, 16]. Combining bemarituzumab with chemotherapeutic agents that will target FGFR2 negative tumor cells is likely to improve the clinical benefit over bemarituzumab alone. Currently, bemarituzumab is being evaluated in combination with modified 5-fluourouracil, oxaliplatin, and leucovorin (mFOLFOX6) in the phase 2 placebo-controlled trial, A Study of Bemarituzumab Combined with Modified FOLFOX6 in Gastric/Gastroesophageal Junction Cancer (FIGHT, NCT03694522).
Dose selection is frequently a challenge for novel biological oncology drugs because the maximum tolerated dose approach may identify a dose that is significantly higher than the dose required to achieve maximum efficacy [17]. There are additional challenges for an orphan drug like bemarituzumab because of the lower prevalence of tumors expressing the target FGFR2b, which may limit the feasibility of an extensive clinical dose-ranging study. Therefore, an efficient approach was needed to advance bemarituzumab dose selection. To achieve this goal, multiple factors had to be considered, including population pharmacokinetic analysis (PopPK), which is the focus of this manuscript. On the basis of the mechanism of action for bemarituzumab (including blocking of FGFR2b binding to its ligands), it is projected that keeping trough concentration (Ctrough) above a threshold to saturate the target between-dose interval might provide maximum anti-tumor activity. Therefore, an empirical target Ctrough estimated from in vitro data was included as an exposure target to support dose selection. This approach was used in multiple targeted antibody therapies including bevacizumab and trastuzumab as well as immunotherapies, such as ipilimumab [18–20]. In addition, a concentration associated with a high percentage of saturation of target-mediated clearance for bemarituzumab can be used as the target Ctrough.
Here, we describe results from the PopPK analysis of bemarituzumab using serum concentration data from a first-in-human phase 1 study (FPA144-001) to support dose selection for the phase 2 trial. The key objectives of the analysis were to assess pharmacokinetic (PK) and covariates of bemarituzumab in humans using PopPK analysis, to describe the in vitro data used to derive an empirical target Ctrough, to report the simulation results which identified the dose and regimen necessary to achieve an empirical target Ctrough in the majority of patients, and to provide clinical data to support the projection from the simulation.
Materials and methods
Antibody reagents
Bemarituzumab was produced in a Chinese hamster ovary cell line that lacks the FUT8 gene (α1, 6-fucosyltransferase) at Five Prime Therapeutics, Inc. (South San Francisco, CA) for nonclinical studies, at AbbVie Bioresearch Center (Worcester, MA) for the FPA144-001 trial, and at Patheon (St. Louis, MO) for the FIGHT trial.
Bemarituzumab serum concentration in humans
Human serum concentration data were collected from 75 patients enrolled in the FPA144-001 clinical trial; 22 patients with a variety of solid tumors and 53 patients with GEA. Among the 75 patients, 27 were enrolled in the dose escalation portion of the trial and received a 30-min intravenous (IV) infusion of bemarituzumab once every 2 weeks (Q2W) in a range of doses [0.3 (n = 3), 1 (n = 4), 3 (n = 4), 6 (n = 4), 10 (n = 9), and 15 (n = 3) mg/kg; Part 1]. An additional 48 patients received 15 mg/kg Q2W in a 30-min IV infusion (Part 2). Each cycle had two doses with a Q2W dosing regimen. Three to five serum concentration samples were collected within 8 h on the day of dosing followed by collections on Days 2, 4, and 8 post-first dose of Cycle 1. Additional collections were done both before and at the end of infusion for the second dose of Cycle 1 and the first dose of Cycles 2 through 5, every other cycle thereafter, and at the End of Treatment Follow-up Visit. Part 2 had the same serum concentration sample collection time points without collection of samples on Days 2 and 4 post-first dose.
FIGHT is a phase 2 trial preceded by dose-finding in phase 1. Ctrough data collected during Cycle 1 through Cycle 5 from ten patients (n = 3 for 6 mg/kg and n = 7 for 15 mg/kg Q2W with a single dose of 7.5 mg/kg on Cycle 1 Day 8) in phase 1 of the FIGHT trial made up the observed serum concentration data displayed in Fig. 4.
Fig. 4.
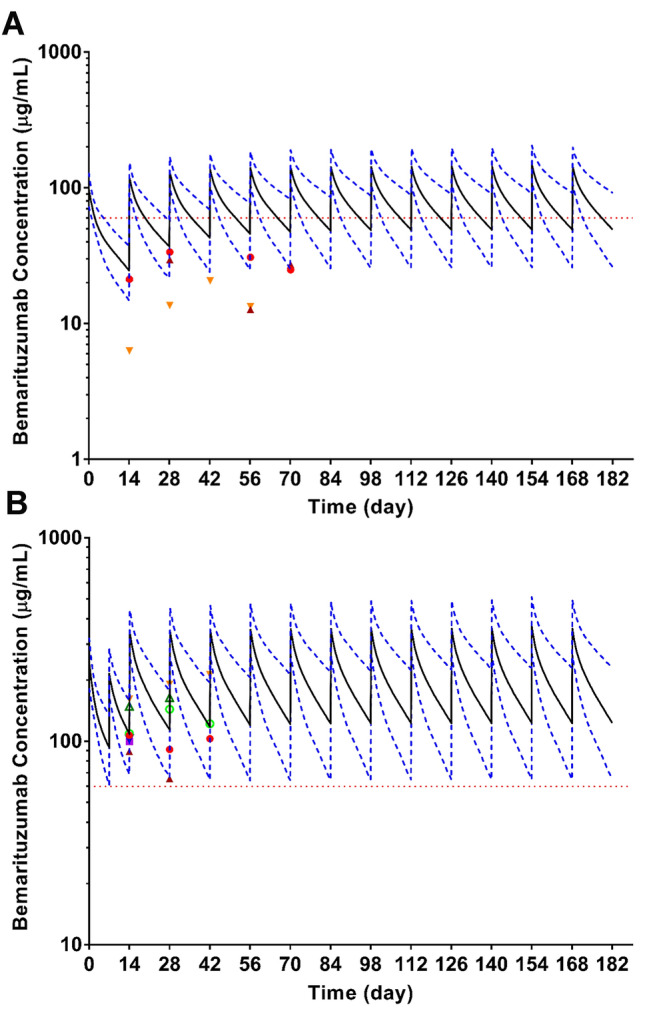
Simulation results using PK parameters obtained from PopPK analysis versus observed Ctrough from phase 1 of the FIGHT trial. Simulated serum concentration versus time profiles of bemarituzumab after continuous Q2W dosing of bemarituzumab at 6 mg/kg (a) or 15 mg/kg Q2W with a single dose of 7.5 mg/kg on Cycle 1 Day 8 (b) for 6 months based on the final PopPK model. For each dose regimen, 1000 simulated model parameters constructed the distribution of model predictions for a typical population (61 kg male patients with albumin of 3.7 g/dL). In each figure, solid black line represents the median predicted value and dashed blue lines represent the 5th and 95th percentile of the predicted concentrations and dashed red line is for Ctrough of 60 µg/mL. Each symbol represents the individual bemarituzumab Ctrough (µg/mL) in the presence of mFOLFOX6 at 6 mg/kg (a, n = 3) and 15 mg/kg Q2W with a single dose of 7.5 mg/kg on Cycle 1 Day 8 (b, n = 7), respectively
Anti-drug antibody in humans
Anti-drug antibody (ADA) samples were collected from all patients in study FPA144-001 prior to dosing on Day 1 of Cycles 1 to 5, every other cycle after Cycle 5, and at the End of Treatment Follow-up Visit.
Determination of serum concentration of bemarituzumab in humans
The bemarituzumab serum concentration in humans was quantitatively measured at ICON Laboratory Services, Inc. (Whitesboro, NY) with a validated ELISA in which bemarituzumab was captured by plate-bound FGFR2b-Fc and detected by an anti-idiotypic antibody coupled to HRP. The LLOQ of the assay was 0.125 µg/mL. The accuracy expressed as percent relative error ranged from 0.250% to 4.80%. The intra- and inter-assay ranges were − 0.240% to − 4.80% and 5.74% to 7.45%, respectively.
Measurement of anti-drug antibody in patients
A validated bridging electrochemiluminescence assay that utilizes Meso Scale Discovery (MSD) technology was used to detect ADA in patient serum samples. The samples were acidified, then neutralized in a reaction mixture containing ruthenylated bemarituzumab and biotinylated bemarituzumab. After a 75-min incubation, the samples were transferred onto a streptavidin coated MSD assay plate. The plate was washed and a tripropylamine (TPA)-containing MSD read buffer added. In the presence of TPA, ruthenium produces a chemiluminescent signal when voltage is applied. Only samples that contained antibody bound to both biotinylated bemarituzumab and ruthenylated bemarituzumab produced the chemiluminescent signal, which was proportional to the amount of anti-bemarituzumab antibody present. The assay sensitivity was 32.7 ng/mL relative to the rabbit anti-FPA144 positive control. Drug tolerance was 2 µg/mL in the presence of 800 ng/mL of rabbit anti-bemarituzumab antibody.
Human population pharmacokinetic analysis
A PopPK model was developed to describe serum bemarituzumab concentration–time profiles. Various PK structural models were tested, including a one-compartment, two-compartment, two-compartment with time-varying clearance, and two-compartment with linear and nonlinear elimination components. The PK of bemarituzumab in the tested clinical dose range was best described by a two-compartment model with parallel linear and nonlinear (Michaelis–Menten) elimination pathways from the central compartment using the differential equations below [21, 22]:
| 1 |
| 2 |
where Ac and Ap are the amount of drug in central and peripheral compartments, respectively. Vmax represents the maximum drug elimination by nonlinear clearance, and Km, Michaelis–Menten constant, indicates the drug concentration at 50% Vmax. CL and Q represent linear clearance and distribution clearance, respectively, while Vc and Vp represent volume of distribution in central and peripheral compartments, respectively.
After an optimal structural model was identified (final base model), the effect of covariates (Table 1) including baseline demographic information, renal function, hepatic function, disease status, baseline tumor measurements, Eastern Cooperative Oncology Group (ECOG) status, and FGFR2b expression in patients with GEA were evaluated for their impacts on the CL and Vc. FGFR2b high was defined as ≥ 10% of tumor cells with 3 + membranous staining using a centrally performed validated laboratory-developed prototype immunohistochemistry assay (LabCorp, Burlington, NC). Correlations between the PK parameters and the covariates were explored graphically, followed by linear regression (continuous covariates) and analysis of variance (ANOVA) testing (categorical covariates) using R software. These analyses were conducted on individual random effects (ETAs) for CL and Vc. Only covariates that showed a significant (p < 0.01) effect on the random effect, and that could be meaningfully explained from both a clinical and scientific perspective, were examined further using NONMEM. Covariates were selected based on their potential clinical relevance. The possible physiological basis for commonly evaluated covariates such as body size, age, gender, race, ADA, serum albumin, creatinine clearance, etc., were summarized by Thomas and Balthasar [23]. Selection of the final covariate model (final PopPK model) was determined for its significance on the basis of likelihood ratio test at the p < 0.01 for forward inclusion and p < 0.001 for backward deletion.
Table 1.
Covariate values of bemarituzumab population pharmacokinetic dataset
| Variable | Study FPA144-001 (n = 75) |
|---|---|
| Age (year), median (range) | 58 (25, 86) |
| Weight (kg), median (range) | 61.4 (35.5, 148) |
| Gender, n (%) | |
| Female | 42 (56%) |
| Male | 33 (44%) |
| Race, n (%) | |
| White | 29 (38.7%) |
| American Indian or Alaska Native | 1 (1.33%) |
| Asian | 44 (58.7%) |
| Black or African American | 1 (1.33%) |
| Albumin (g/dL), median (range) | 3.7 (1.9, 4.6) |
| Creatinine Clearance (mL/min), median (range) | 75.3 (26.4, 200) |
| Total Bilirubin (mg/dL), median (range) | 0.4 (0.1, 2.0) |
| ALT (U/L), median (range) | 19 (6, 74) |
| AST (U/L), median (range) | 24 (7, 106) |
| Baseline Tumor Size (mm), median (range) | 50 (10, 244) |
| Tumor type, n (%) | |
| Gastric cancer | 53 (70.7%) |
| Other solid tumors | 22 (29.3%) |
| ECOG status, n (%) | |
| 0 | 23 (30.7%) |
| 1 | 52 (69.3%) |
| FGFR2b Expression in patients with gastric and gastroesophageal junction adenocarcinoma, n (%) | |
| FGFR2b high | 26 (49%) |
| FGFR2b other | 27 (51%) |
The final PopPK model was evaluated with multiple internal model validations, including goodness-of-fit diagnostics, prediction-corrected visual predictive check (pcVPC) plots, numerical predictive check, bootstrap, and shrinkage assessments. The pcVPC was created to assess the predictive ability of the model. A total of 1000 replicates of the trials were simulated using the individual dosing history and covariates, the typical parameter estimates, and random sampled interindividual variability and residual errors. The 2.5th, 50th, and 97.5th percentiles of the observed data were overlaid on the 90% confidence interval (CI) of the 2.5th, 50th, and 97.5th simulated percentiles, and a visual inspection was performed. The sensitivity analysis was performed for the final PopPK model to examine the contribution of significant baseline covariates to the overall variability of the steady-state exposures including area under concentration–time curve at steady state (AUCss), maximum serum concentration at steady state (Cmax ss), and trough concentration at steady state (Ctrough ss) after Q2W dosing of 15 mg/kg bemarituzumab.
The PopPK analysis was performed using the nonlinear mixed effects modeling approach with the first-order conditional estimation with interaction method. Model parameter estimation and evaluation were implemented with NONMEM 7 (v. 7.3.0; ICON Development Solutions, Ellicott City, MD) with an Intel Fortran Compiler (v. 10.1.021; Intel, Santa Clara, CA), Perl-speaks-NONMEM (PsN, v. 3.2.12; Uppsala University, Uppsala, Sweden), and R 3.3.1.
Binding affinity using surface plasmon resonance
The binding affinity of bemarituzumab for human FGFR2b ECD-IgG1 Fc fusion protein (FGFR2b-Fc) was measured using surface plasmon resonance (SPR, Biacore T100, GE Healthcare Life Science, Marlborough, MA). Bemarituzumab was immobilized on a dextran chip using an amine coupling kit and 100 mM ethylenediamine in 100 mM Sodium Borate, pH 8.0, was used as the blocking reagent. Six different concentrations (0 nM to 500 nM) of FGFR2b-Fc proteins were diluted in HEPES buffered saline with 0.05% surfactant P20 running buffer and flowed over the immobilized antibody.
To determine binding affinity for FcγRIIIa (V158) by SPR, bemarituzumab was captured on the chip via protein A. Protein A was covalently attached to a dextran chip using the same protocol as above. Five concentrations (0 nM to 1000 nM) of FcγRIIIa (V158) were diluted in running buffer and flowed over the captured antibody.
The association constant, dissociation constant, and affinity for bemarituzumab binding to human FGFR2b and human FcγRIIIa (V158) were calculated using the Biacore T100 Evaluation Software 1:1 binding model.
Cell lines
OCUM-2M is a FGFR2 gene-amplified, FGFR2b protein overexpressing gastric cancer cell line obtained from Public University Corporation Osaka City University, Japan (Source: Dr Masakazu Yashiro). HSC-39 is a FGFR2-amplified gastric cell line and was kindly provided by Rebecca Fitzgerald (Hutchison/MRC Research Centre, Cambridge, UK). Gastric cell lines SNU-16 (with FGFR2 amplification) and NCI-N87 (without FGFR2 amplification or overexpression) were commercially acquired (ATCC, Manassas, VA). All four cell lines were mycoplasma negative tested by IDEXX Laboratories (Columbia, MO) using a real-time polymerase chain reaction.
Gastric cell lines-based binding
The cell surface expression of FGFR2b on OCUM-2 M, HSC-39, SNU-16, and NCI-N87 cells was calculated after incubation with 50 µg/mL of either bemarituzumab or human IgG1 (human anti-hen egg lysozyme, Five Prime Therapeutics, Inc). Unbound antibody was washed with 1 × phosphate-buffered saline with 2% fetal bovine serum (VWR Visalia, CA) and the cells were incubated with mouse anti-human IgG1-PE (Clone HP6001, lot L3117-M839, SouthernBiotech, Birmingham, AL). Cells were washed and data acquired on the BD LSRII Flow Cytometer (BD, San Jose, CA). Quantum Simply Cellular anti-mouse IgG Bang Beads (815B lot 12,380, Bangs Laboratories, Fishers, IN) were stained in parallel according to the manufacturer’s instructions and acquired on the BD LSRII Flow Cytometer. The antibody binding capacity (ABC) was calculated using the Bang Laboratories QuickCal Template.
Separately, 2 × 105 OCUM-2 M, HSC-39, SNU-16, or NCI-N87 cells were incubated with varying concentrations of bemarituzumab or human IgG1, washed, stained with a mouse anti-human IgG1 PE secondary antibody (clone HP6001; SouthernBiothech, Birmingham, AL), washed, and acquired on the BD LSRII Flow Cytometer. A 4-parameter logistical regression curve fit for acquired mean fluorescence intensity versus bemarituzumab concentration data in semi-log plot was applied in GraphPad PRISM (La Jolla, CA) to estimate receptor occupancy.
Human pharmacokinetic simulation
Bemarituzumab exposures were simulated using the final PopPK model with different dose regimens. For each dose regimen, 1000 simulated model parameters constructed the distribution of model predictions for a typical population (61 kg male patients with albumin of 3.7 g/dL). The area under the concentration–time curve (AUC), Cmax, and Ctrough after single and multiple treatment for each of the dose regimens were computed. The simulation was done for the dose cohorts tested in the safety lead-in phase 1 portion of the FIGHT trial: 6 mg/kg Q2W and 15 mg/kg Q2W with a single dose of 7.5 mg/kg on Cycle 1 Day 8.
Results
Bemarituzumab serum concentration in humans
Bemarituzumab serum concentration versus time data (group mean ± SD) from Cycle 1 dose 1 in the FPA144-001 trial displayed a typical antibody serum concentration profile with a short distribution phase followed by a long elimination phase [14]. Bemarituzumab demonstrated dose dependent clearance in patients with solid tumors including GEA, with faster clearance at lower doses, suggesting target-mediated drug disposition. Clearance appeared nonlinear from 0.3 mg/kg to 1 mg/kg and approximately linear from 1 mg/kg to 15 mg/kg.
The anti-drug antibody impact on bemarituzumab pharmacokinetics
A total of 75 patients from the FPA144-001 trial were tested for ADA. The patients were exposed to bemarituzumab for up to 966 days (median = 55 days). No patient sample was confirmed to be ADA positive after administration of bemarituzumab. A predose sample before bemarituzumab administration from one patient was ADA positive; however, all tested ADA samples post-bemarituzumab administration for this patient were negative. The predose ADA positive signal did not have a visible impact on the PK profile of this patient in comparison with the PK profiles from the other patients treated at the same dose level.
Population pharmacokinetic analysis
The development dataset for the final model included 814 bemarituzumab serum concentration data from 75 participants in the phase 1 study FPA144-001. A two-compartment model with parallel linear and nonlinear (Michaelis–Menten) elimination from the central compartment best described the bemarituzumab serum concentration data. No time-varying CL was identified after bemarituzumab administration. Covariate analysis was conducted to understand the impact of the covariate values (Table 1) on the CL and Vc of bemarituzumab. The forward addition and backward deletion based on the final base model identified body weight, albumin, and gender as statistically significant covariates for bemarituzumab disposition parameters of CL and Vc, and this model is referred to as the final PopPK model:
| 3 |
| 4 |
where CLi is the individual linear clearance; Vci is the individual volume of distribution in the central compartment, ηCL and ηVc are interindividual variability of CL and Vc, respectively.
The final PopPK model estimated a typical CL, saturation of target-mediated drug disposition, of 0.331 L/day, Vc of 3.70 L, Q of 0.788 L/day, Vp of 2.05 L, Vmax of 1.70 µg/day, and Km of 4.58 µg/mL. The estimated linear clearance half-life was 12.8 days. The interindividual variability on CL, Vc, and Vp were 27.1%, 17.3%, and 60.0%, respectively.
Bootstrapping of 1000 datasets resulted in median parameter estimates and 95% CI similar to the estimates from the original dataset (Table 2), indicating that the final PopPK model provided good precision for parameter estimation. Goodness-of-fit plots showed agreement between predicted and observed concentrations of bemarituzumab with no apparent bias in residual plots over time or across population-predicted concentrations (Supplementary Fig. 1). pcVPC plots (Fig. 1) demonstrated that the final PopPK model could reasonably describe the central tendency and variability of the bemarituzumab pharmacokinetic data.
Table 2.
Summary of population pharmacokinetic parameters
| Parameter description | Base model estimates (%RSE) | Final model estimates (%RSE) | Bootstrap estimate median (2.5–97.5%tiles) |
|---|---|---|---|
| Vmax (μg/day) | 2.75 (21.6%) | 1.70 (14.3%) | 1.82 (0.0901; 7.55) |
| KM (μg/mL) | 4.64 (42.5%) | 4.58 (15.1%) | 5.24 (0.533; 24.7) |
| Linear clearance, CL (L/day) | 0.331 (4.73%) | 0.331 (3.55%) | 0.327 (0.275; 0.364) |
| Influence of body weight on CL | – | 0.601 (19.5%) | 0.641 (0.355; 0.991) |
| Influence of albumin on CL | – | − 0.776 (19.9%) | − 0.777 (− 1.31; − 0.262) |
| Volume of central compartment, Vc (L) | 3.43 (2.76%) | 3.70 (2.98%) | 3.70 (3.51; 3.92) |
| Influence of body weight on Vc | – | 0.303 (24.8%) | 0.306 (0.109; 0.471) |
| Influence of sex on Vc | – | − 0.191 (23.4%) | − 0.194 (− 0.284; − 0.114) |
| Distribution clearance, Q (L/day) | 0.772 (10.6%) | 0.788 (8.99%) | 0.791 (0.599; 1.11) |
| Volume of peripheral compartment, Vp (L) | 2.07 (8.25%) | 2.05 (7.73%) | 2.08 (1.76; 2.42) |
| Interindividual variability of Vmax | 113 (32.6%) | 128 (23.3%) | 122 (65.9; 437) |
| Interindividual variability of CL | 36.0 (20.1%) | 27.1 (19.7%) | 25.5 (18.4; 31.7) |
| Interindividual variability of Vc | 22.2 (18.2%) | 17.3 (19.1%) | 16.8 (13.7; 19.9) |
| Interindividual variability of Vp | 59.3 (25.1%) | 60.0 (24.1%) | 56.0 (38.3; 74.9) |
| Covariance between CL and Vc | 0.0319 (36.2%) | 0.0141 (46.1%) | 0.0137 (0.00111; 0.0254) |
| Residual variability (%CV) | 14.4 (5.61%) | 14.5 (5.89%) | 14.4 (13.0; 15.7) |
Fig. 1.

Prediction-corrected VPC of bemarituzumab serum concentration–time profile across dose groups. Black open circles are observed serum concentrations, solid red line represents the median observed value, and dashed red lines represent 2.5th and 97.5th percentile of the observed values, respectively. Pink shaded areas represent the spread of the median predicted values (5th to 95th percentile), and blue shaded areas represent the spread (5th and 95th percentile) of the 2.5th and 97.5th predicted percentile concentrations
Body weight was identified as a covariate for both CL and Vc, while albumin was a covariate for CL and gender was a covariate for Vc. Patients with higher body weight had statistically larger Vc and faster CL. Lower albumin had statistically faster CL and male patients had a higher Vc. Due to the limited number of patients (n = 75), the covariate relationship should be confirmed in future studies. The sensitivity analysis suggested that albumin is the most important factor influencing the Ctrough ss of bemarituzumab. The population-predicted 5th and 95th percentile of total Ctrough ss for the actual patients’ Q2W dosing of the 15 mg/kg were 65 and 196 µg/mL, respectively, corresponding to − 47.5% and 58.3% variation around the Ctrough ss predicted for a typical patient (123.8 µg/mL). Of note, the extreme albumin (5th and 95th percentiles) corresponded to as high as − 31.3% and 24.4% variation for Ctrough ss (Fig. 2). Body weight had a mild impact based on the sensitivity analysis (− 9.2% and 8.7% variation on Ctrough ss; Fig. 2). Gender was also an influential factor for the Ctrough ss with a − 6.2% variation for female compared to male patients (Fig. 2). Albumin was the most influential factor for AUCss with − 21.7% and 16.4% variation while body weight had the greatest impact on Cmax ss with − 20% to 25.7% variation (data not shown).
Fig. 2.
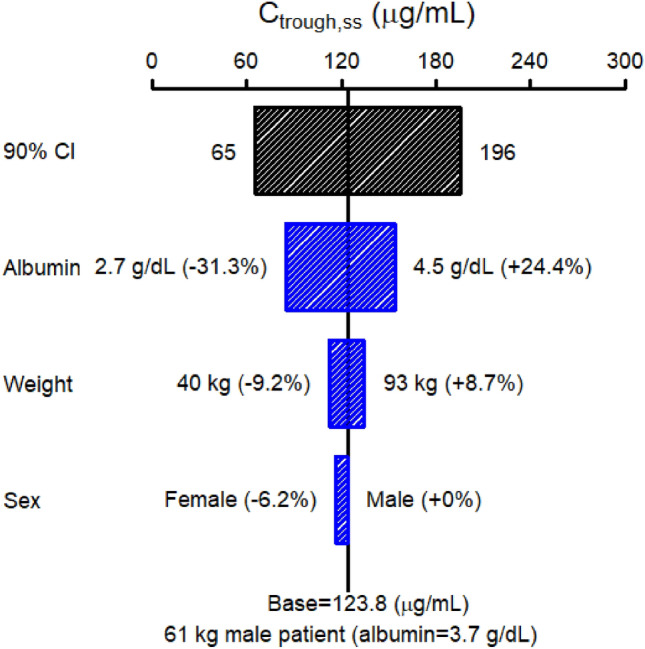
Sensitivity plot comparing the effect of covariates on Ctrough ss of bemarituzumab. Base, as represented by the black vertical line and value, refers to the predicted typical Ctrough ss of bemarituzumab in a 61 kg male patient with an albumin of 3.7 g/dL kg after continuous Q2W dosing of 15 mg/kg bemarituzumab for 6 months. The black horizontal bar with values at each end shows the 5th to 95th percentile Ctrough ss range across the entire population. Each blue bar represents the influence of a single covariate on the Ctrough ss. The label at the left end of the bar represents the covariate being evaluated. The upper and lower values for each covariate capture 90% of the plausible range in the population. The length of each bar describes the potential impact of that particular covariate on bemarituzumab Ctrough ss with the percentage value in the parentheses at each end representing the percent change of Ctrough ss from the base. The most influential covariate is at the top of the tornado plot
An empirical target trough concentration based on in vitro data
The affinity of bemarituzumab for FGFR2b and FcγRIIIa (V158) was measured by SPR. The binding affinity of bemarituzumab for FGFR2b was 0.58 nM and for FcγRIIIa (V158) was 9.2 nM. Therefore, 95% and 99% receptor occupancy by bemarituzumab for FGFR2b is estimated to be achieved at concentrations ≥ 1.59 µg/mL and ≥ 8.26 µg/mL, respectively, and 95% and 99% receptor occupancy for FcγRIIIa (V158) is estimated to be achieved at concentrations of 25.1 µg/mL and 131 µg/mL, respectively, using the following equation:
| 1 |
where C is the concentration of the molecule and KD is the affinity of the molecule for its ligand/binding partner.
Among 3 FGFR2-amplified cell lines tested, FGFR2 expression level on the cell surface demonstrated the following order: OCUM-2 M, SNU-16, and HSC-39. The mean EC50 is similar among these three cell lines with values at 1.37 ± 0.0525 µg/mL, 1.81 ± 0.0289 µg/mL, and 2.53 ± 0.170 µg/mL for OCUM-2 M, HSC-39, and SNU-16, respectively (Fig. 3). The EC95 is 26.1 ± 1.00 µg/mL, 34.4 ± 0.548 µg/mL, and 48.1 ± 3.24 µg/mL, respectively.
Fig. 3.

In vitro cell-based binding for bemarituzumab using gastric cell lines. Mean fluorescence intensity (ABC) versus bemarituzumab concentration profile from receptor occupancy study in vitro are presented for FGFR2b-amplified cell lines OCUM-2 M, HSC-39, and SNU-16 in Figs. 3A, 3B, and 3C, respectively. NCI-N87 is presented in 3D as a negative control. Symbols represent observed data (n = 3 per time point). L.D. limit of detection, which is at 62 for ABC
Therefore, an empirical targeted Ctrough of 60 µg/mL was selected, which would achieve > 95% receptor occupancy based on binding affinity and in vitro cell-based binding data from multiple FGFR2-amplified cell lines.
Simulation to support dose and regimen selection for bemarituzumab in combination with modified FOLFOX6 in the FIGHT trial
Using the final PopPK model, simulations were performed to determine whether alternative dosing schedules would allow patients to more rapidly reach target Ctrough of ≥ 60 µg/mL for a typical population (61 kg male patients with albumin of 3.7 g/dL). Accordingly, the dose cohorts tested in the safety lead-in phase 1 portion of the FIGHT trial were 6 mg/kg Q2W and 15 mg/kg Q2W with a single dose of 7.5 mg/kg on Cycle 1 Day 8. If the last dose cohort was not tolerable, then the Cycle 1 Day 8 dose was to be omitted and 15 mg/kg Q2W would be tested as a de-escalation dose cohort. The serum concentration versus time profiles for the two dose cohorts in the phase 1 portion of the FIGHT trial were projected (Fig. 4). The addition of a single dose of 7.5 mg/kg on Cycle 1 Day 8 for 15 mg/kg Q2W was predicated to allow a majority of patients (98%) to achieve target Ctrough by Day 15, compared to 96% achieving target Ctrough by week 10 with good tolerance, thereby shortening the time to reach the target Ctrough.
PK profile of bemarituzumab is not affected by addition of mFOLFOX6
Bemarituzumab in combination with mFOLFOX6 had an acceptable toxicity profile in previously treated patients with GEA in the phase 1 safety lead-in portion of the FIGHT trial at its highest dose tested-15 mg/kg Q2W with a single dose of 7.5 mg/kg on Cycle 1 Day 8 [24]. Bemarituzumab concentration was not affected by the presence of mFOLFOX6. In addition, Ctrough concentration from all seven patients who received 15 mg/kg with a single dose of 7.5 mg/kg on Cycle 1 Day 8 achieved target Ctrough of 60 µg/mL (Fig. 4b).
Discussion
Bemarituzumab, an FGFR2b inhibitor, demonstrated promising monotherapy activity in late-line GEA [14]. A phase 2 trial (FIGHT) was designed to evaluate bemarituzumab combined with mFOLFOX6 chemotherapy in patients with previously untreated GEA whose tumors overexpress FGFR2b or have FGFR2 amplification, however, efficient dose selection was required. A PK simulation in combination with an empirical target Ctrough obtained from in vitro data was used to support the selection of the maximum test dose at 15 mg/kg Q2W with a single dose of 7.5 mg/kg on Cycle 1 Day 8 with mFOLFOX6 in the phase 1 safety lead-in portion of the FIGHT trial. This dose and schedule were selected for the phase 2 evaluation following the observed PK and safety data from phase 1 portion of FIGHT trial [24].
This is the first report of bemarituzumab PopPK analysis and covariate assessment of the phase 1 data. Bemarituzumab PK was best described by a two-compartment model with parallel linear and nonlinear (Michaelis–Menten) elimination from the central compartment. Target-mediated clearance observed with bemarituzumab has been reported for multiple other antibodies, such as cetuximab, panitumumab, and anti-NRP1 [25–27].
Covariates impacting CL and Vc are variable for targeted antibodies. For example, body weight and gender were identified as the most significant covariates for CL and Vc of bevacizumab while baseline serum albumin and lean body weight were identified as significant covariates for pertuzumab CL [28, 29]. None of these covariates for bevacizumab or pertuzumab are significant enough to require a dose adjustment in the clinic for a specific patient population. The covariate analyses for bemarituzumab similarly did not identify any covariates for exposure which would require a dose adjustment. Among 13 covariates evaluated (Table 1), only 3 had a significant impact on either CL or Vc. Body weight impacted both CL and Vc, albumin impacted CL, and gender impacted Vc. Sensitivity analyses indicated that the effects of these covariates on steady-state exposures including Ctrough ss, AUCss, and Cmax ss were small compared to the overall between-subject variability in the population. Therefore, dose adjustment based on any of these parameters is not warranted. Due to the limited number of patients (n = 75) evaluated in the PopPK analysis, the covariate relationship will be further evaluated at the end of the phase 2 FIGHT trial. FGF2b expression was tested predose to select patients with GEA in the trial. FGFR2b status at baseline (high vs others) in patients with GEA was not a covariate for PK (Supplementary Fig. 2). In addition, there were no time-dependent changes in PK. Therefore, the PK profile should not be affected by changes in FGFR2b expression during treatment. Because no patients developed ADA after administration of bemarituzumab, there is no data to perform analysis of potential ADA impact on PK. When the impact of body weight is considered, gender does not play a significant role to impact PK parameters in general. However, the variability of CL and V1 for bemarituzumab cannot be fully explained by either body weight or gender and, therefore, both are significant according to the method used for the PopPK analysis and statistical criteria described in the Materials and Methods section. This result suggests that gender had impact beyond body weight although the underlying reason is not understood. This observation was previously reported for other antibodies [28, 30, 31].
The empirical target Ctrough of 60 µg/mL for bemarituzumab was to achieve > 95% receptor occupancy based on in vitro data, including receptor occupancy using multiple FGFR2-amplified gastric cancer cell lines. Although there is no robust clinical data to support the selection of the empirical target Ctrough for bemarituzumab, clinical data indicated that at 60 µg/mL, > 90% of nonlinear clearance was projected to be saturated based on Km obtained from the PopPK analysis. When Km data from the clinic is available, it can be used directly to estimate target Ctrough. In addition, all 6 patients with GEA and confirmed partial responses in the phase 1 monotherapy trial (5 with high FGFR2b overexpression), regardless of dose level, achieved the desired target Ctrough ss of ≥ 60 µg/mL although no dose-efficacy relationship was identified because of limited data [14 and Five Prime Therapeutics data on file]. It is understood that more clinical data is needed to support the empirical target Ctrough and it is expected that the exposure–response relationship analysis post FIGHT trial should provide that data. For blocking antibodies, the Ctrough has generally been used to select the target dose to maximize receptor occupancy throughout the treatment cycles [18–20]. Using trastuzumab as an example, two mechanisms of action are described: inhibition of the proliferation of human tumor cells that overexpress Her2 and ADCC. The minimum desired Ctrough for trastuzumab in the clinic was > 10 µg/mL based on nonclinical data, which theoretically achieved ≥ 99% receptor saturation in blood based on its affinity for Her2 (0.1 nM) and < 50% receptor saturation based on its affinity for FcγRIIIa (V158) (252 nM) [19, 32, 33], respectively. In the clinic, the observed mean Ctrough ss is 79 µg/mL, which is projected to achieve 99% receptor saturation of Her2, but not of FcγRIIIa [34]. Similar to trastuzumab, bemarituzumab also has two mechanisms of action: blocking FGFR2b binding to its FGF ligands and enhanced ADCC. Therefore, Ctrough was used as an exposure target to saturate FGFR2b receptors during the dose interval.
The two dose cohorts in the phase 1 part of the FIGHT trial were 6 mg/kg Q2W and 15 mg/kg Q2W with a single dose of 7.5 mg/kg on Cycle 1 Day 8. As advanced stage GEA is an aggressive disease with standard chemotherapy providing a median of only 6 months of disease control with a worse prognosis expected in patients whose tumors overexpress FGFR2b, a method to shorten the time to achieve target Ctrough levels was deemed clinically important. In the single agent phase 1 FPA144-001 trial, 15 mg/kg was the highest dose tested. Simulations using PopPK parameters were performed to estimate the doses required to achieve target Ctrough. To maximize efficacy, simulation of the dosing regimen of 15 mg/kg q2w with a single dose of 7.5 mg/kg on Cycle 1 Day 8 showed that 98% of patients could be expected to achieve the target Ctrough level of 60 μg/mL on Day 15 and 96% of patients could be expected to maintain the target Ctrough ss. To minimize a potential safety risk which could be associated with an increase in Cmax, the addition of a single 7.5 mg/kg dose on Day 8 was instituted to maintain the projected Cmax for this regimen within the level previously shown to be safe during the single agent study FPA144-001. It was anticipated that the addition of a single dose of 7.5 mg/kg on Cycle 1 Day 8 would shorten the time to achieve the target Ctrough which was thought to be clinically important in the early control of an aggressive tumor such as GEA. Indeed, target Ctrough was achieved by all patients in the phase 1 portion of the FIGHT trial on Day 15 using this dose and regimen. This PopPK analysis and clinical findings supported the selection of dose and schedule for initiation of the phase 2 randomized, double-blind portion of the global FIGHT trial which is currently ongoing worldwide [24].
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank all the investigators, the patients, and their families. The authors also acknowledge the entire bemarituzumab team at Five Prime Therapeutics, Inc. The authors appreciate Drs. Lee Clark, Marina Neyssa, Siddhartha Mitra, and Wei Deng for their inputs. The authors thank Dr. Masakazu Yashiro from Public University Corporation Osaka City University for providing the OCUM-2M cell line.
Funding
This work was funded by Five Prime Therapeutics, Inc.
Compliance with ethical standards
Conflict of interest
H. Xiang, A. Ahene, L. Dreiling, and H. Collins are employed with Five Prime Therapeutics, Inc. and M. Macal and AW Hsu are former Five Prime employees. They have ownership interest in the Five Prime stock. L. Liu and Y. Gao declare no potential conflicts of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sitarz R, Skierucha M, Mielko J, Offerhaus GJA, Maciejewski R, Polkowski WP. Gastric cancer: epidemiology, prevention, classification, and treatment. Cancer Manag Res. 2018;10:239–248. doi: 10.2147/CMAR.S149619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Stat Facts: Esophageal Cancer. National Cancer Institute (2016) https://seer.cancer.gov/statfacts/html/esoph.html.
- 3.Cancer Stat Facts: Stomach cancer. National Cancer Institute (2016) https://seer.cancer.gov/statfacts/html/stomach.html4.
- 4.Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE. Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate Date. J Clin Oncol. 2006;24(18):2903–2909. doi: 10.1200/JCO.2005.05.0245. [DOI] [PubMed] [Google Scholar]
- 5.Digklia A, Wagner AD. Advanced gastric cancer: current treatment landscape and future perspectives. World J Gastroenterol. 2016;22(8):2403–2414. doi: 10.3748/wjg.v22.i8.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 7.Orr-Urtreger A, Bedford MT, Burakova T, Arman E, Zimmer Y, Yayon A, Givol D, Lonai P. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2) Dev Biol. 1993;158(2):475–486. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- 8.Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, Lonigro RJ, Vats P, Wang R, Lin SF, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3(6):636–647. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su X, Zhan P, Gavine PR, Morgan S, Womack C, Ni X, Shen D, Bang YJ, Im SA, Kim WH, et al. FGFR2 amplification has prognostic significance in gastric cancer: results from a large international multicentre study. Br J Cancer. 2014;110(4):967–975. doi: 10.1038/bjc.2013.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seo S, Park SJ, Ryu MH, Park SR, Ryoo BY, Park YS, Na YS, Lee CW, Lee JK, Kang YK. Prognostic impact of fibroblast growth factor receptor 2 gene amplification in patients receiving fluoropyrimidine and platinum chemotherapy for metastatic and locally advanced unresectable gastric cancers. Oncotarget. 2017;8(20):33844–33854. doi: 10.18632/oncotarget.12953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn S, Lee J, Hong M, Kim ST, Park SH, Choi MG, Lee JH, Sohn TS, Bae JM, Kim S, et al. FGFR2 in gastric cancer: protein overexpression predicts gene amplification and high H-Index predicts poor survival. Mod Pathol. 2016;29(9):1095–1103. doi: 10.1038/modpathol.2016.96. [DOI] [PubMed] [Google Scholar]
- 12.Matsumoto K, Arao T, Hamaguchi T, Shimada Y, Kato K, Oda I, Taniguchi H, Koizumi F, Yanagihara K, Sasaki H, et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br J Cancer. 2012;106(4):727–732. doi: 10.1038/bjc.2011.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung EJ, Jung EJ, Min SY, Kim MA, Kim WH. Fibroblast growth factor receptor 2 gene amplification status and its clinicopathologic significance in gastric carcinoma. Hum Pathol. 2012;43(10):1559–1566. doi: 10.1016/j.humpath.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Catenacci DVT, Rasco D, Lee J, Rha SY, Lee KW, Bang YJ, Bendell J, Enzinger P, Marina N, Xiang H, et al. Phase I escalation and expansion study of bemarituzumab (FPA144) in patients with advanced solid tumors and FGFR2b-selected gastroesophageal adenocarcinoma. J Clin Oncol. 2020 doi: 10.1200/JCO.19.01834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alsina M, Gullo I, Carneiro F. Intratumoral heterogeneity in gastric cancer: a new challenge to face. Ann Oncol. 2017;28(5):912–913. doi: 10.1093/annonc/mdx134. [DOI] [PubMed] [Google Scholar]
- 16.Deshpande AM, Palencia S, Bellovin DI, Gemo AT, Giese T, Stohr B, Pierce KL, Los G. Expression of FGFR2b in gastric cancer as measured by immunohistochemistry with a highly specific monoclonal antibody. Cancer Res. 2014;74(19 Suppl):2845. doi: 10.1158/1538-7445.AM2014-2845. [DOI] [Google Scholar]
- 17.Ji Y, Jin JY, Hyman DM, Kim G, Suri A. Challenges and opportunities in dose finding in oncology and immuno-oncology. Clin Trans Sci. 2018;11(4):345–351. doi: 10.1111/cts.12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergsland E, Dickler MN. Maximizing the potential of bevacizumab in cancer treatment. Oncologist. 2004;9(Suppl 1):36–42. doi: 10.1634/theoncologist.9-suppl_1-36. [DOI] [PubMed] [Google Scholar]
- 19.Leyland-Jones B, Arnold A, Gelmon K, Verma S, Ayoub J-P, Seidman A, Dias R, Howell J, Rakhit A. Pharmacologic insights into the future of trastuzumab. Ann Oncol. 2001;12(Suppl. 1):S43–S47. doi: 10.1093/annonc/12.suppl_1.s43. [DOI] [PubMed] [Google Scholar]
- 20.Feng Y, Masson E, Dai D, Parker SM, Berman D, Roy A. Model-based clinical pharmacology profiling of ipilimumab in patients with advanced melanoma. Br J Clin Pharmcol. 2014;78(1):106–117. doi: 10.1111/bcp.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mager DE. Target-mediated drug disposition and dynamics. Biochem Pharmacol. 2006;72(1):1–10. doi: 10.1016/j.bcp.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 22.Gibiansky L, Gibiansky E, Kakkar T, Ma P. Approximations of the target-mediated drug disposition model and identifiability of model parameters. J Pharmacokinet Pharmacodyn. 2008;35(5):573–591. doi: 10.1007/s10928-008-9102-8. [DOI] [PubMed] [Google Scholar]
- 23.Thomas VA, Balthasar JP. Understanding inter-individual variability in monoclonal antibody disposition. Antibodies. 2019;8(4):56. doi: 10.3390/antib8040056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tejani MA, Cheung E, Eisenberg PD, Scott AJ, Tesfaye AA, Dreiling L, Eng C, Marina N, Mitra S, Xiang H, et al. Phase 1 results from the phase 1/3 FIGHT study evaluating bemarituzumab and mFOLFOX6 in advanced gastric/GEJ cancer (GC) J Clin Oncol. 2019;37:91–91. doi: 10.1200/JCO.2019.37.4_suppl.91. [DOI] [Google Scholar]
- 25.Dirks NL, Nolting A, Kovar A, Meibohm B. Population pharmacokinetics of cetuximab in patients with squamous cell carcinoma of the head and neck. J Clin Pharmacol. 2008;48(3):267–278. doi: 10.1177/0091270007313393. [DOI] [PubMed] [Google Scholar]
- 26.Ma P, Yang BB, Wang YM, Peterson M, Narayanan A, Sutjandra L, Rodriguez R, Chow A. Population pharmacokinetic analysis of panitumumab in patients with advanced solid tumors. J Clin Pharmacol. 2009;49(10):1142–1156. doi: 10.1177/0091270009344989. [DOI] [PubMed] [Google Scholar]
- 27.Xin Y, Bai S, Damico-Beyer LA, Jin D, Liang WC, Wu Y, Theil FP, Joshi A, Lu Y, Lowe J, et al. Anti-neuropilin-1 (MNRP1685A): unexpected pharmacokinetic differences across species, from preclinical models to humans. Pharm Res. 2012;29(9):2512–2521. doi: 10.1007/s11095-012-0781-x. [DOI] [PubMed] [Google Scholar]
- 28.Lu JF, Bruno R, Eppler S, Novotny W, Lum B, Gradreault J. Clinical pharmacokinetics of bevacizumab in patients with solid tumors. Cancer Chemother Pharmacol. 2008;62(5):779–786. doi: 10.1007/s00280-007-0664-8. [DOI] [PubMed] [Google Scholar]
- 29.Garg A, Quartino A, Li J, Jin J, Wada DR, Li H, Cortés J, McNally V, Ross G, Visich J, et al. Population pharmacokinetic and covariate analysis of pertuzumab, a HER2-targeted monoclonal antibody, and evaluation of a fixed, non-weight-based dose in patients with a variety of solid tumors. Cancer Chemother Pharmacol. 2014;74(4):819–829. doi: 10.1007/s00280-014-2560-3. [DOI] [PubMed] [Google Scholar]
- 30.Baverel PG, Dubois VFS, Jin CY, Zheng Y, Song X, Jin X, Mukhopadhyay P, Gupta A, Dennis PA, Ben Y, et al. Population pharmacokinetics of durvalumab in cancer patients and association with longitudinal biomarkers of disease status. Clin Pharmacol Therap. 2018;103(4):631–642. doi: 10.1002/cpt.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang E, Kang D, Bae KS, Marshall MA, Pavlov D, Parivar K. Population pharmacokinetic and pharmacodynamic analysis of tremelimumab in patients with metastatic melanoma. J Clin Pharmacol. 2014;54(10):1108–1116. doi: 10.1002/jcph.309. [DOI] [PubMed] [Google Scholar]
- 32.Baselga J. Phase I and II clinical trials of trastuzumab. Ann Oncol. 2001;12:S49–S55. doi: 10.1093/annonc/12.suppl_1.s49. [DOI] [PubMed] [Google Scholar]
- 33.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. PNAS. 2006;103(11):4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Genentech Inc . HERCEPTIN® (trastuzumab) [Package insert] South San Francisco: Genentech Inc; 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.