

Abstract
In an attempt to search for selective inhibitors against the SARS-CoV-2 which caused devastating of lives and livelihoods across the globe, 415 natural metabolites isolated from several plants, fungi and bacteria, belonging to different classes, were investigated. The drug metabolism and safety profiles were computed in silico and the results showed seven compounds namely fusaric acid, jasmonic acid, jasmonic acid methyl ester, putaminoxin, putaminoxin B and D, and stagonolide K were predicted to having considerable absorption, metabolism, distribution and excretion parameters (ADME) and safety indices. Molecular docking against the receptor binding domain (RBD) of spike glycoprotein (S1) and the main protease (Mpro) exposed the compounds having better binding affinity to main protease as compared to the S1 receptor binding domain. The docking results were compared to an antiviral drug penciclovir reportedly of clinical significance in treating the SARS-CoV-2 infected patients. The results demonstrated the test compounds jasmonic acid, putaminoxins B and D bound to the HIS-CYS catalytic dyad as well as to other residues within the MPro active site with much greater affinity than penciclovir. The findings of the study suggest that these compounds could be explored as potential SARS-CoV-2 inhibitors, and could further be combined with the experimental investigations to develop effective therapeutics to deal with the present pandemic.
Keywords: Secondary metabolites, Putaminoxins, SARS-CoV-2, Main protease, Spike glycoprotein
1. Introduction
The devastating pandemic emerged due to the highly infectious SARS-CoV-2 virus continues to wreak havoc on the already fragile global health care systems and the human health. Hitting most vulnerable the hardest, the virus has infected millions of human populations across the globe with a progressive rise in the projected death toll (WHO, https://www.who.int/emergencies/diseases/novel-coronavirus-2019). Despite being declared by WHO as global health emergency, an effective and specific therapeutic intervention to prevent or arrest this deadly viral infection is largely challenging. On the other hand, the viral proteins that are responsible for the human cell infection and replication of this virus which could be treated as drug targets have been unveiled by a number of experimental studies (Lan et al., 2020; Hall and Ji 2020). Among them S1 glycoprotein and Mpro are two such protein targets being investigated extensively for their alleged role in the interaction with human ACE2 (angiotensin converting enzyme 2) receptor which is reported to be fundamental in the entry of SARS-CoV-2 into human cell. Indeed, a structure motif known as receptor binding motif or RBM present on the surface of RBD on S1 glycoprotein is eventually held responsible for the S1-ACE2 interaction and cell entry (Lan et al., 2020). In addition, transcription and translation of the viral genomic RNA is necessary for its survival and production of new generation viruses inside the cell, and is regulated by the main protease. Therefore, inhibiting these target proteins could discover novel inhibitors that could further lead to development of effective therapeutics against SARS-CoV-2.
Plant and microbial metabolites have been serving as an excellent and prolific source for a variety of medical agents since generations (Newman and Cragg 2016, 2020). Many such metabolites have been developed as latent therapeutic applications for human ailments including infectious diseases (Sohail et al., 2011; Shabat et al., 2019; Pham et al., 2019). Furthermore, development of bioinformatics tools and services makes it easier and advantageous to screen a large number of compounds for their pharmacokinetics features and to predict their possible affinity towards several drug targets. Herein, a total of 415 compounds of plant and microbial origin were screened for their drug metabolism and pharmacokinetics, and the selected compounds were used for computational docking against two of the best possible SARS-CoV-2 target proteins: the S1 RBD and Mpro. The present investigation focuses on searching potent and specific inhibitors of the SARS-CoV-2 with a reference to its entry and replication inside the host.
2. Materials and methods
2.1. Plant and microbial metabolites
A library of 415 bioactive secondary metabolites, belonging to several classes of natural compounds, isolated from different plant and microbial organic extracts at the Organic Chemistry Laboratory, University of Naples Federico II, Italy were optimized and treated as ligands for use in docking against the SARS-CoV-2 target proteins.
2.2. SARS-CoV-2 target proteins
In this study, two major SARS-CoV-2 proteins: the RBD of S1 glycoprotein and the Mpro involved respectively in the host cell infection and viral replication were used as receptors. The 3D structure of RBD having 193 amino acids was prepared from the crystal structure of RBD-ACE2 complex (PDB ID: 6M0J). The receptor structure comprises of a core region which is stabilized by three pairs of cysteine residues and five beta plated sheets organized in anti-parallel way, and a functional motif region known as RBM consisting 68 residues and supported by 2 α helices, 2 β sheets and connecting loops (Lan et al., 2020). Similarly, Mpro3D structure bearing 306 amino acids was prepared from the crystal structure Mpro– Feline complex (PDB ID: 6LU7). The overall structure was organized into three domains; domain I, II and III. Domain I and II have anti-parallel β barrel structure whereas Domain III is a large anti-parallel globular structure containing five α-helices. Domains II and III are connected by a 15 amino acids long loop (Khan et al., 2020).
2.3. Drug metabolism and pharmacokinetics
The 2D structures of the chemical compounds were optimized and screened for the drug metabolism and pharmacokinetics features using the ADME. These include aqueous solubility (in water at 25 °C), human intestinal absorption, ability to transport across blood brain barrier (BBB), plasma protein binding affinity (PPB), cytochrome P450 family 2 subfamily D member 6 (CYP2D6) binding and Hepatotoxicity (Zhou et al., 2016). The selected molecules were also computed for the drug safety using different datasets such as Ames mutagenicity (v3.1), rodent carcinogenicity (based on US National Toxicity Program (NTP v3.2)) and Development Toxicity Potential (DTP, v3.1) properties using Toxicity Prediction using Komputer Assisted Technology (TOPKAT) program (Zhou et al., 2018). The candidates cleared the screening process were considered as safe and used for further analyses. All operations were carried out in Discovery Studio (DS) Client v20.1.0.19,295.
2.4. Molecular docking
A virtual screening of the selected compounds including a reference antiviral drug penciclovir (PubChem CID: 135398748) (Razonable 2011) was carried out using the DS Libdock, a rigid based program that calculates hotspots for the receptor with placing a grid into the binding site, as well as using polar and apolar probes (Singh et al., 2016; Kang et al., 2018). The libdock score determines the binding affinity of the ligands towards a receptor, and is a cumulative count of van der Waals forces, H-bonds, pi interactions and other parameters. The RBM on the S1 RBD and the Mpro active site were considered as binding targets for the compounds which were prepared and optimized using the DS ‘prepare ligand’ protocol. The docking experiment with libdock was performed setting the ‘Docking Preferences’ user Specified and ‘Max Hits to Save’ to 10. The docked complex with higher libdock score assumed to have higher binding affinity (Kang et al., 2018) and based on the score, docking conformations of selected compounds were optimized using the CDOCKER program which is an implementation of a CHARMm based docking tool (Gagnon et al., 2014). Docking optimization was carried out with a root mean square threshold (RMSD) 0.5 Å and pose cluster radius 0.5 to ensure the docked poses are diverse. The pose with highest negative interaction energy was selected as best conformation and the results were compared to that of penciclovir.
3. Results
3.1. Drug metabolism and pharmacokinetics
In this study, 415 natural compounds isolated from different sources such as plants, bacteria and fungi, and belonging to different classes of secondary metabolites were screened for their drug metabolism and pharmacokinetics features. Among the screened metabolites, 332 compounds predicted to be aqueous soluble and 193 were predicted to be able to transport across the BBB. Similarly, 175 compounds were found to be non-inhibitors of CYT P450 2D6, an important enzyme involved in the metabolism of drugs. In addition, of the total compounds tested, 363 were shown to have considerable absorption level for the human intestine and 223 were having no hepatotoxic potential. Furthermore, 187 compounds were found to be highly bound with the plasma protein. Above all, the ADME properties of 31 compounds were predicted to be satisfactory (Supplementary Table 1). These selected compounds were further evaluated for their structure based toxicity profiles using six different reference datasets. Any compound that is computed to be toxic with reference to any one of the database is omitted and as such the results showed 7 compounds namely fusaric acid (Bani et al., 2014), putaminoxin (Evidente et al., 1995) putaminoxins B and D (Evidente et al., 1997, 1998), jasmonic acid methyl ester, jasmonic acid (Andolfi et al., 2014) and stagonolide K (Danilova et al., 2019) to be of no-toxic and having characteristic ADME profile (Table 1, Table 2 ).
Table 1.
Drug metabolism and pharmacokinetics of selected compounds and the reference drug penciclovir used in this study.
| Compound | Isolation source | Solubility levela | BBB Levelb |
CYP2D6c | Hepato-toxicityd | Absorption levele | PPB levelf |
|---|---|---|---|---|---|---|---|
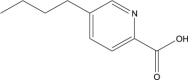 Fusaric acid |
Fusarium oxysporum f. sp. pisi (fungus) | 3 | 2 | 0 | 0 | 0 | 1 |
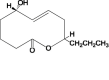 Putaminoxin Putaminoxin |
Phoma putaminum (fungus) | 3 | 2 | 0 | 0 | 0 | 1 |
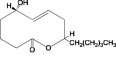 Putaminoxin B |
Phoma putaminum | 3 | 1 | 0 | 0 | 0 | 1 |
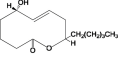 Putaminoxin D |
Phoma putaminum | 3 | 1 | 0 | 0 | 0 | 1 |
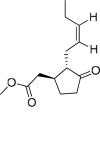 Jasmonic cid methyl ester |
Lasiodiplodia mediterranea (fungus) | 3 | 2 | 0 | 0 | 0 | 1 |
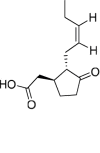 Jasmonic acid |
Lasiodiplodia mediterranea | 4 | 3 | 0 | 0 | 0 | 1 |
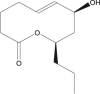 Stagonolide K |
Stagonospora cirsii (fungus) | 3 | 2 | 0 | 0 | 0 | 1 |
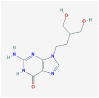 Penciclovir |
Reference antiviral drug |
4 | 4 | 0 | 1 | 1 | 0 |
*aAqueous solubility level: 0 (extremely low), 1 (very low), 2 (low), 3 (good), 4 (optimal); bBlood Brain Barrier level: 0 (very high), 1 (high), 2 (medium), 3 (low), 4 (undefined); cCytochrome P450 2D6 level: 0 (non-inhibitor), 1(inhibitor); dHepatotoxicity: 0 (non-toxic), 1 (toxic); eHuman intestinal absorption level: 0 (good absorption), 1 (moderate absorption), 2 (low absorption), 3 (very low absorption); fPlasma Protein Binding: 0 (binding <90%), 1 (binding>90%).
Table 2.
Computed probabilities of mutagenicity and carcinogenicity of selected compounds used in this study.
| Name of the compound | Probabilities of mutagenicity and carcinogenicity |
|||||
|---|---|---|---|---|---|---|
| Ames mutagenicity |
NTP Carcinogenicity |
Developmental Toxicity Potential | ||||
| Male rat | Female rat | Male mouse | Female mouse | |||
| Fusaric acid | 0 | 0 | 0.037 | 0 | 0 | 0.437 |
| Putaminoxin | 0.535 | 0 | 0 | 0 | 0.180 | 0 |
| Putaminoxin B | 0.537 | 0.005 | 0 | 0 | 0.157 | 0 |
| Putaminoxin D | 0.537 | 0.005 | 0 | 0 | 0.157 | 0 |
| Jasmonic acid methyl ester | 0 | 0 | 0 | 0 | 0 | 0 |
| Stagonolide K | 0.601 | 0 | 0 | 0 | 0.211 | 0 |
| Jasmonic acid | 0.001 | 0 | 0 | 0 | 0 | 0 |
| Penciclovir | 0.000 | 0.016 | 0 | 1 | 1 | 0 |
*values from ≤0.30 are considered low probabilities, and are likely to produce a negative response in an experimental assay; values ≥ 0.70 are considered high, and are likely to produce a positive response in an experimental assay; whereas 0.30 ≤ values ≥ 0.70 are considered indeterminate.
3.2. Molecular docking
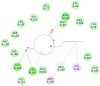
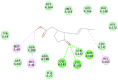
The selected seven compounds were screened for possible binding affinity against two of the best characterized drug targets of SARS-CoV-2: S1 RBD and Mpro. Libdock was used for virtual screening of the test compounds where the docking score was used for determining the binding affinity. The outcomes were compared to that of an experimental antiviral drug penciclovir. The results showed penciclovir having higher affinity towards the RBM with a libdock score 96.8718 followed by the test compounds jasmonic acid (81.4371), putaminoxin D (libdock score 79.4063) and jasmonic acid methyl ester (78.2874). The virtual screening of the test compounds against the Mpro active site displayed their considerable binding affinity as compared to the penciclovir (libdock score 87.9996). The libdock score was highest in putaminoxin D (105.099) followed by putaminoxin B (94.4192) and jasmonic acid methyl ester (92.3017) (Table 3 ). Therefore, the structures for which the libdock score was significant were further optimized using CDOCKER protocol. Briefly, the test compounds putaminoxins B and D, jasmonic acid and jasmonic acid methyl ester were docked against the Mpro active site and the interaction energies were computed. The results demonstrated putaminoxin B and D formed 7 favorable non covalent contacts with 6 MPro active site residues. Similarly, jasmonic acid and jasmonic acid methyl ester formed 6 favorable contacts with 6 of the active site amino acids. The interaction energy for putaminoxin D was computed to be minimum (-36.1847 kcal/mol) followed by jasmonic acid (-36.1283 kcal/mol), putaminoxin B (-35.4201 kcal/mol) and jasmonic acid methyl ester (-33.3747 kcal/mol). The reference drug penciclovir bound to the enzyme active site by 8 favorable and 1 unfavorable non covalent interactions, and the CDOCKER interaction energy for penciclovir was found to be -30.9096 kcal/mol. The test ligands were found to be interacted with HIS41 and CYS145 in common besides the other active site residues (MET49, LEU141, SER 144, MET 165 and GLU 166). Penciclovir interacted with PHE140, ASN142, SER144, CYS145, HIS163, HIS164 and GLU166. The details of interaction, types and active site residues involved are presented in Table 4 .
Table 3.
Receptor ligand interactions between Mpro and selected compounds.
| Ligands | Docking of the ligand on the Mpro active site | Mpro-ligand interaction | Libdock Score | CDOCKER Interaction energy (kcal/mol) |
|---|---|---|---|---|
| Putaminoxin D | 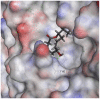 |
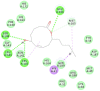 |
105.099 | −36.1847 |
| Jasmonic acid | 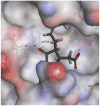 |
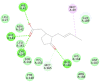 |
88.0807 | −36.1283 |
| Putaminoxin B | 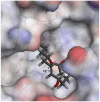 |
 |
94.4192 | −35.4201 |
| Jasmonic acid methyl ester | 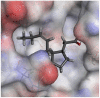 |
 |
92.3017 | −33.3747 |
| Penciclovir | 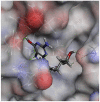 |
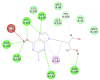 |
87.9996 | −30.9096 |
Table 4.
Details of the ligand interactions with SARS-CoV-2 MPr.o.
| Name of the compound | MPro amino acids | Interaction types | Bond distance (Å) |
|---|---|---|---|
| Putaminoxin D | HIS 41 | Pi-sigma | 2.83 |
| LEU141 | Conventional Hydrogen | 2.20 | |
| SER 144 | Conventional Hydrogen | 2.41 | |
| CYS 145 | Conventional Hydrogen, Alkyl | 2.43, 4.47 | |
| MET 165 | Alkyl, Carbon Hydrogen | 5.26, 2.49 | |
| GLU 166 | Conventional Hydrogen | 2.42 | |
| Jasmonic acid | HIS41 | Conventional Hydrogen | 2.29 |
| MET49 | Alkyl | 4.11 | |
| GLY143 | Conventional Hydrogen | 2.40 | |
| CYS145 | Conventional Hydrogen | 2.31 | |
| MET165 | Carbon–Hydrogen Bond | 2.34 | |
| GLU166 | Conventional Hydrogen | 2.50 | |
| Putaminoxin B | HIS 41 | Pi-alkyl | 3.85 |
| ASN 142 | Conventional Hydrogen | 2.78 | |
| CYS 145 | Conventional Hydrogen, Alkyl | 2.48, 5.16 | |
| HIS 164 | Conventional Hydrogen | 2.63 | |
| MET 165 | Alkyl | 5.37 | |
| GLN 189 | Carbon Hydrogen | 2.52 | |
| Jasmonic acid methyl ester | HIS41 | Pi-Alkyl | 4.43 |
| MET49 | Alkyl | 3.72 | |
| GLY143 | Conventional Hydrogen | 2.13 | |
| SER144 | Conventional Hydrogen | 2.89 | |
| CYS145 | Conventional Hydrogen | 2.45 | |
| HIS163 | Pi-Alkyl | 4.52 | |
| Penciclovir | PHE140 | Conventional Hydrogen | 2.43 |
| ASN142 | Conventional Hydrogen | 2.25 | |
| SER144 | Conventional Hydrogen | 2.44 | |
| CYS145 | Pi-Alkyl, Alkyl | 5.03, 4.64 | |
| HIS163 | Conventional Hydrogen | 2.77, 2.50 | |
| HIS164 | Carbon–Hydrogen Bond | 2.75 | |
| GLU166 | Conventional Hydrogen | 2.94 | |
| HIS172 | Unfavorable |
4. Discussion
Natural products of plant and microbial origin have been proven as potential inhibitors of many infectious agents including viruses since decades (Mukhtar et al., 2008; Ben-Shabat et al., 2020; Roy 2017; Linnakoski et al., 2018). In the ongoing pandemic situation of SARS-CoV-2 infection where no specific treatments are available, these natural metabolites are being extremely exploited in silico for their inhibiting ability for major proteins (Das et al., 2020; Gurung et al., 2020). A library of 415 natural compounds, belonging to several classes of natural compounds, isolated from different plant, fungal and bacterial extracts at the Department of Chemical Sciences, University of Naples Federico II, Italy were used in this investigation for their drug metabolism and pharmacokinetics properties screening, and selected candidates were examined for possible inhibition of SARS–CoV-2-S1 RBD and Mpro. The analyses of ADME parameters revealed 31 compounds predicted to have good quality features including their aqueous solubility, human intestinal absorption, plasma protein binding ability and others. Moreover, 7 of them were predicted to be safe and have negligible or no toxicity. Interestingly, the selected compounds are of fungal origin and most of them are phytotoxic in nature. Putaminoxins (including B and D) were isolated from the fungus Phoma putaminum and were reported to be phytotoxic to Erigeron annuus, a common weed found in fields and pastures (Evidente et al., 1995, 1997, 1998). Similarly, stagonolide K is another phytotoxic metabolite isolated from fungus Stagonospora cirsii. This is also established that phytochemicals and microbial metabolites reportedly exhibiting manifold biological activities including anticancer, antimicrobial etc. (Nisar et al., 2008; Khurm et al., 2016). Therefore, inclusion of these metabolites as proposed drugs is not surprising. Furthermore, jasmonic acid and jasmonic acid methyl ester were isolated from a fungus Lasiodiplodia mediterranea and were reported to be plant stress hormones. In addition, jasmonic acid methyl ester has been proven as an anticancer metabolite and also as a proteinase inhibitor in plant leaves (Farmer et al., 1990; Fingrut and Flescher 2002). Fusaric acid is produced by many Fusarium species and is known for its various bioactivities including antioxidant, antifungal and more particularly as a quorum sensing inhibitors to many gram negative bacteria (Tung et al., 2017). Moreover, the prediction ADME parameters thought to play an important task in the drug designing process and account for the failure of most drugs in the clinical phases (Zhong 2017). For any proposed drug to reach a tissue, it must be absorbed by the blood stream or more often by the mucosal surfaces such as the digestive tract before being taken up by the target cells. Few important factors like poor solubility, intestinal transit time and inability to penetrate the intestinal wall could lower the extent to which a drug is absorbed after oral absorption (Singh 2006). Further, the drug is subjected to several distribution processes followed by its metabolism where the parent drug is broken down into many metabolites which is usually takes place in liver by the redox enzymes especially CYT 450. Pharmacologically inert metabolites need to be removed from the body to reduce any side effects which is done through excretion process via kidney (Balani et al., 2005). The test compounds used in this investigation are predicted to be having good solubility and intestinal absorption that could make them easier to be taken up by the blood stream or the intestinal wall for further their action. The compounds are also predicted to show CYT450 2D6 binding ability suggesting ease of their metabolism. Furthermore, interaction of these molecules with the plasma proteins may lead to elimination of a fraction of drug in a protein bound state and the unbound portion could exert therapeutic action (Osakwe 2016). In addition, safety concerns linking to mutagenicity and carcinogenicity are believed to be significant aspects of drug research. And also with the discovery of new tools, techniques and generation of new data helps the scientific community to understand much deeper about the pharmacological profiles of a larger number of compounds. This in turn leads to removal of molecules with poor metabolism and pharmacokinetics from the drug pipeline, and saves research cost as well as time (Benfenati, 2016).
Based on drug metabolism, pharmacokinetics and safety profiles, the selected 7 compounds (fusaric acid, putaminoxin, putaminoxins B and D, jasmonic acid, jasmonic acid methyl ester and stagonolide K) were subjected to virtual screening using libdock program to predict the possible binding affinity against two SARS-CoV-2 S1 RBD (RBM in particular) and Mpro. The libdock score was used as an estimation to select the best binding pose and the scores were compared to a reference antiviral drug ‘penciclovir’ which is being used for the treatment of Herpes Simplex Virus (I and II) and Varicella Zoster Virus (VZV) (Razonable 2011). In addition, the same has been recently proven to be clinically significant in treating SARS-CoV-2 infected patients (Zhang et al., 2020). The findings revealed our test compounds have less affinity towards the receptor binding motif (RBM) as the libdock scores were found to be less significant while comparing with the reference penciclovir. However, binding affinity in some of the test compounds (putaminoxins B and D, jasmonic acid, jasmonic acid methyl ester) against the active site of the Mpro enzyme observed to be significant as compared to the penciclovir. Libdock has been widely used as a tool for virtual screening of small molecules against protein or enzyme targets (Rao et al., 2007; Singh et al., 2016; Alam and Khan 2018). Libdock score which is a cumulative count of all non covalent interactions including the van der Waals has been adopted in many instances for determining the binding affinity (Zhou et al., 2016; Kang et al., 2018). Taking the binding affinity against the SARS-CoV-2 main protease into account, putaminoxins B and D, jasmonic acid, jasmonic acid methyl ester were docked against the active site using CDOCKER protocol where the interaction energy of the complex was used as an estimate to select the best binding complex. In many instances, CDOCKER has been used for docking of small molecules against the protein or enzyme targets and therein the docked complexes were ranked relating to their interaction energies (Lee et al., 2015; Zhang et al., 2019). The interaction energies of the ligand bound MPro complexes were computed to be minimum than that of the pencivlovir-MPro docked complex suggesting the binding involving test ligands were strong and substantial. The structures also made contacts with the active site CYS145-HIS41 catalytic dyad located in a cleft between Domains I and II which represents the substrate binding site reportedly vital to the proteolytic activity of the enzyme (Khan et al., 2020; Suarez and Diaz 2020). We hypothesize that interacting with CYS145–HIS41 dyad as well as blocking it could prevent the substrate access and interfere with enzyme's proteolytic action.
Among all, putaminoxin D and jasmonic acid seemed to be largely favorable in inhibiting the SARS-CoV-2 main protease both in terms of lower interaction energy and blocking the catalytic dyad. The non covalent forces including conventional hydrogen bonds, Pi-Sigma interactions (Pi-anion, Pi-cation, Pi-alkyl) and salt bridges could further maximize the binding affinity of the ligands towards the target in a physiological environment (Rahman et al., 2016; Ajoundi et al., 2020). Therefore, in keeping view of the quality pharmacokinetics and safety profiles and binding affinity in comparison to the experimental inhibitor penciclovir, the structures could be explored as potential SARS-CoV-2 inhibitors. This in silico study may swiftly be combined with in vitro experiments to unveil the efficacy of these metabolites against this deadly virus. It is further suggested that clinical significance of our findings could be established by evaluating these compounds for the SARS-CoV-2 infection in transgenics, xenograft models and animal studies.
5. Conclusions
The present study encourages the utilization of phytochemicals and microbial secondary metabolites for effective inhibition of SARS-CoV-2 S1 the main protease which are reported to be key players of viral cell proliferation. The findings further inferred that the natural compounds putaminoxin B and D, jasmonic acid and jasmonic methyl ester which were computed to have good pharmacokinetics, safety profiles and effective binding against the target protein could be explored as prospective inhibitor of SARS-CoV-2 subject to more in vitro and in vivo investigations.
CRediT authorship contribution statement
Srichandan Padhi: Investigation, Formal analysis, Methodology, Writing - original draft. Marco Masi: Resources, Investigation. Rounak Chourasia: Data curation. Yallappa Rajashekar: Resources, Validation. Amit Kumar Rai: Writing - review & editing, Software, Investigation, Supervision. Antonio Evidente: Conceptualization, Resources, Investigation, Supervision.
Declaration of competing interest
The authors declare no conflict of interests.
Acknowledgements
The authors are thankful to Director, Institute of Bioresources and Sustainable Development, India for providing necessary facilities to carry out the work. The institutional manuscript number for this work is IBSD/2020/01/034.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejphar.2020.173648.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- Alam S., Khan F. Virtual screening, docking, ADMET and system pharmacology studies on Garcinia caged xanthone derivatives for anticancer activity. Sci. Rep. 2018;8:5524. doi: 10.1038/s41598-018-23768-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljoundi A., Bjij I., El Rashedy A., Soliman M.E.S. Covalent versus non-covalent enzyme inhibition: which route should we take? a justification of the good and bad from molecular modelling perspective. Protein J. 2020;39:97–105. doi: 10.1007/s10930-020-09884-2. [DOI] [PubMed] [Google Scholar]
- Andolfi A., Maddau L., Cimmino A., Linaldeddu B.T., Basso S., Deidda A., Serra S., Evidente A. Lasiojasmonates A–C, three jasmonic acid esters produced by Lasiodiplodia sp., a grapevine pathogen. Phytochemistry (Oxf.) 2014;103:145–153. doi: 10.1016/j.phytochem.2014.03.016. [DOI] [PubMed] [Google Scholar]
- Balani S.K., Devishree V.S., Miwa G.T., Gan L.S., Wu J.T., Lee F.W. Strategy of utilizing in vitro and in vivo ADME tools for lead optimization and drug candidate selection. Curr. Top. Med. Chem. 2005;5(11):1033–1041. doi: 10.2174/156802605774297038. [DOI] [PubMed] [Google Scholar]
- Bani M., Rispail N., Evidente A., Rubiales D., Cimmino A. Identification of the main toxins isolated from Fusarium oxysporum f. sp pisi Race 2 and their relation with isolates’ pathogenicity. J. Agric. Food Chem. 2014;62(12):2574–2580. doi: 10.1021/jf405530g. [DOI] [PubMed] [Google Scholar]
- Ben-Shabat S., Yarmolinsky L., Porat D., Dahan A. Antiviral effect of phytochemicals from medicinal plants: applications and drug delivery strategies. Drug. Deliv. Transl. Res. 2020;10(2):354–367. doi: 10.1007/s13346-019-00691-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati E. Springer; 2016. In Silico Methods for Predicting Drug Toxicity. [Google Scholar]
- Dalinova A., Dubovik V., Chisty L., Kochura D., Ivanov A., Smirnov S. Stagonolides J and K and stagochromene A, two new natural substituted nonenolides and a new disubstituted chromene-4,5-dione isolated from Stagonospora cirsii S-47 proposed for the biocontrol of Sonchus arvensis. Agric. Food Chem. 2019;67(47):13040–13050. doi: 10.1021/acs.jafc.9b04573. [DOI] [PubMed] [Google Scholar]
- Das S., Sarmah S., Lyndem S., Roy A.S. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1763201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evidente A., Lanzetta R., Capasso R., Andolfi A., Bottalico A., Vurro M., Zonno M.C. Putaminoxin, a phytotoxic nonenolide from Phoma putaminum. Phytochemistry. 1995;40(6):1637–1641. [Google Scholar]
- Evidente A., Lanzetta R., Capasso R., Andolfi A., Vurro M., Zonno M.C. Putaminoxin B and C from Phoma putaminum. Phytochemistry. 1997;44:1041–1045. [Google Scholar]
- Evidente A., Capasso R., Andolfi A., Vurro M., Zonno M.C. Putaminoxin D and E from Phoma putaminum. Phytochemistry. 1998;48:941–945. [Google Scholar]
- Farmer E.E., Ryan C.A. Interplant communication: airborne methyl jasmonate induces synthesis of proteinase inhibitors in plant leaves. Proc. Nat. Aca. Sci. USA. 1990;87:7713–7716. doi: 10.1073/pnas.87.19.7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingrut O., Flescher E. Plant stress hormones suppress the proliferation and induce apoptosis in human cancer cells. Leukemia. 2002;16:608–616. doi: 10.1038/sj.leu.2402419. [DOI] [PubMed] [Google Scholar]
- Gagnon J.K., Law S.M., Brooks C.L. 3rd. Flexible CDOCKER: development and application of a pseudo-explicit structure-based docking method within CHARMM. J. Comput. Chem. 2014;106(2):646A. doi: 10.1002/jcc.24259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D.C., Ji H.F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease, Travel. Med. Infect. Dis. 2020;35 doi: 10.1016/j.tmaid.2020.101646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D., Pang X., Lian W., Xu L., Wang J., Jia H., Zhang B., Liu A.L., Du G.H. Discovery of VEGFR2 inhibitors by integrating natıve Bayesian classification, molecular docking and drug screening approaches. RSC Adv. 2018;8:5286–5297. doi: 10.1039/c7ra12259d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S., Siddique R., Shereen M.A., Ali A., Liu J., Bai Q., Bashir N., Xue M. Emergence of a novel coronavirus, severe acute respiratory syndrome coronavirus 2: biology and therapeutic options. J. Clin. Microbiol. 2020;58(8) doi: 10.1128/JCM.00187-20. e01297-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurm M., Chaudhry B.A., Uzair M., Janbaz K.H. Antimicrobial, cytotoxic, phytotoxic and antioxidant potential of Heliotropium strigosum Willd. Medicines. 2016;3(3):20. doi: 10.3390/medicines3030020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y., Kim S., Kim J.Y., Arooj M., Kim S., Hwang S., Kim B.W., Park K.H., Lee K.W. Interaction energy and negative CDOCKER energy of compound 12 obtained from molecular docking simulation. PloS One. 2015 doi: 10.1371/journal.pone.0085827.t002. Dataset. [DOI] [Google Scholar]
- Linnakoski R., Reshamwala D., Veteli P., Cortina-Escribano M., Vanhanen H., Marjomäki V. Antiviral agents from fungi: diversity, mechanisms and potential applications. Front. Microbiol. 2018;9:23–25. doi: 10.3389/fmicb.2018.02325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtar M., Arshad M., Ahmad M., Pomerantz R.J., Wigdahl B., Parveen Z. Antiviral potentials of medicinal plants. Virus Res. 2008;131(2):111–120. doi: 10.1016/j.virusres.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman D.J., Cragg G.M. Natural products as sources of new drugs from 1981 to 2014. Nat. Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Newman D.J., Cragg G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat. Prod. 2020;83:770–803. doi: 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Nisar, M., Tariq, S.A., Marwat, I.K., Shah, M.R., Khan, I.A., Antibacterial, antifungal, insecticidal, cytotoxicity and phytotoxicity studies on Indigofera gerardiana. J. Enzym. Inhib. Med. Chem. 24, 224-229. [DOI] [PubMed]
- Osakwe O. Preclinical in vitro studies: development and applicability. In: Osakwe O., Rizvi S., editors. Social Aspects of Drug Discovery, Development and Commercialization. Elsevier; 2016. pp. 129–148. [DOI] [Google Scholar]
- Pham J.V., Yilma A., Feliz A., Majid M.T., Maffetone N., Walker J.R., Kim E., Cho H.J., Reynolds J.M., Song M.C. A review of the microbial production of bioactive natural products and biologics. Front. Microbiol. 2019;10:1404. doi: 10.3389/fmicb.2019.01404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A., Hoque M.M., Khan M.A., Sarwar M.G., Halim M.A. Non-covalent interactions involving halogenated derivatives of capecitabine and thymidylate synthase: a computational approach. SpringerPlus. 2016;5:146. doi: 10.1186/s40064-016-1844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.N., Head M.S., Kulkarni A., LaLonde J.M. Validation studies of the site-directed docking program LibDock. J. Chem. Inf. Model. 2007;4:2159–2171. doi: 10.1021/ci6004299. [DOI] [PubMed] [Google Scholar]
- Razonable R.R. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo Clin. Proc. 2011;86(10):1009–1026. doi: 10.4065/mcp.2011.0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B.G. Potential of small-molecule fungal metabolites in antiviral chemotherapy. Antivir. Chem. Chemother. 2017;25(2):20–52. doi: 10.1177/2040206617705500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.S. Preclinical pharmacokinetics: an approach towards safer and efficacious drugs. Curr. Drug Metabol. 2006;7(2):165–182. doi: 10.2174/138920006775541552. [DOI] [PubMed] [Google Scholar]
- Singh J., Kumar M., Mansuri R., Sahoo G.C., Deep A. Inhibitor designing, virtual screening, and docking studies for methyltransferase: a potential target against dengue virus. J. Pharm. BioAllied Sci. 2016;8(3):188–194. doi: 10.4103/0975-7406.171682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail M.N., Rasul F., Karim A., Kanwal U., Attital I. Plant as a Source of natural antiviral agents. Asian J. Anim. Vet. Adv. 2011;6(12):1125–1152. [Google Scholar]
- Suárez D., Díaz N. SARS-CoV-2 main protease: a molecular dynamics study. J. Chem. Inf. Model. 2020 doi: 10.1021/acs.jcim.0c00575. [DOI] [PubMed] [Google Scholar]
- Truong T.T., Jakobsen K.H., Trong T.D., Fuglsang A.T., Givskov M., Christensen S.B., Nielsen J. Fusaric acid and analogues as Gram-negative bacterial quorum sensing inhibitors. Eur. J. Pharm. Chem. 2017;126:1011–1020. doi: 10.1016/j.ejmech.2016.11.044. [DOI] [PubMed] [Google Scholar]
- Zhang W.F., Stephen P., The J.F., Wang R., Lin S.X. Novel Coronavirus polymerase and nucleotidyl-transferase structures: potential to target new outbreaks. J. Phys. Chem. Lett. 2020;11:4430–4435. doi: 10.1021/acs.jpclett.0c00571. [DOI] [PubMed] [Google Scholar]
- Zhong H.A. ADMET properties: overview and current topics. In: Grover A., editor. Drug Design: Principles and Applications. Springer; Singapore: 2017. [Google Scholar]
- Zhou X., Yu S., Su J., Sun L. Computational study on new natural compound inhibitors of pyruvate dehydrogenase kinases. Int. J. Mol. Sci. 2018;17(3):340. doi: 10.3390/ijms17030340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.