

Summary
During brain development, neural stem cells (NSCs) initially produce neurons and change their fate to generate glias. While the regulation of neurogenesis is well characterized, specific markers for glial precursor cells (GPCs) and the master regulators for gliogenesis remain unidentified. Accumulating evidence suggests that RNA-binding proteins (RBPs) have significant roles in neuronal development and function, as they comprehensively regulate the expression of target genes in a cell-type-specific manner. We systematically investigated the expression profiles of 1,436 murine RBPs in the developing mouse brain and identified quaking (Qk) as a marker of the putative GPC population. Functional analysis of the NSC-specific Qk-null mutant mouse revealed the key role of Qk in astrocyte and oligodendrocyte generation and differentiation from NSCs. Mechanistically, Qk upregulates gliogenic genes via quaking response elements in their 3′ untranslated regions. These results provide crucial directions for identifying GPCs and deciphering the regulatory mechanisms of gliogenesis from NSCs.
Keywords: RNA-binding protein, quaking/Qk, neural stem cells/NSCs, glial precursor cells/GPCs, gliogenesis, mRNA stabilization, 3′ untranslated region, regulon, endocytosis, RNA-seq
Graphical Abstract
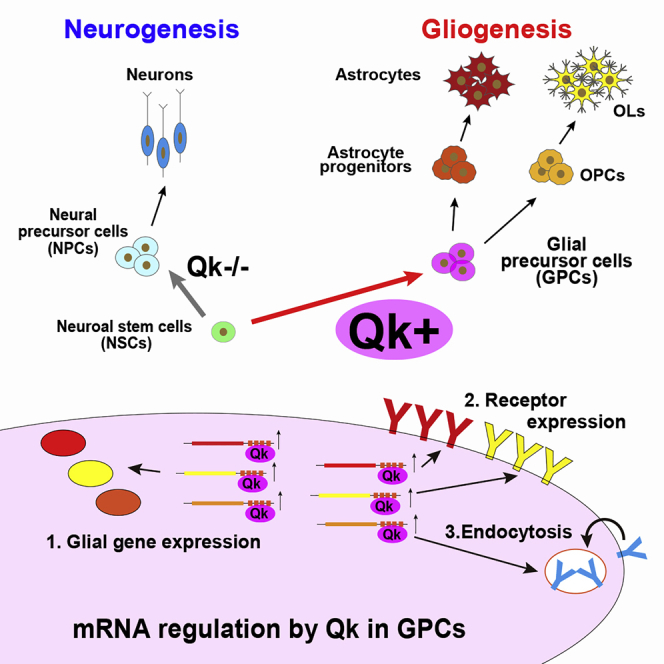
Highlights
-
•
Differential expression analysis identified Qk as a glial precursor cell marker
-
•
Loss of Qk ablated both astrocyte and OL production from neural stem cells
-
•
Qk−/− NSCs failed to become glia and aberrantly expressed neural genes
-
•
Qk comprehensively upregulates essential genes for gliogenesis as regulons via QREs
This study sought to determine whether there is a common glial precursor cell (GPC), what the GPC markers are, and what molecular mechanisms govern gliogenesis. Takeuchi and colleagues identified the RNA-binding protein Qk as a GPC marker. Functional analysis revealed that Qk comprehensively upregulates essential genes for gliogenesis and that loss of Qk disrupts the generation of glial cells from NSCs.
Introduction
In early embryonic stages, multipotent neural stem cells (NSCs) generate only neurons, while in later stages they halt neurogenesis and shift to gliogenesis, namely the production of astrocytes and oligodendrocytes (OLs). Fate change and transition timing of pluripotent NSCs to neurogenesis are regulated by the master bHLH (basic-helix-loop-helix) transcription factors of Hes1/Hes5 (Hairy and enhancer of split 1/5), Asc1, and Neurog2 (Neurogenin 2) (Imayoshi and Kageyama, 2014). In addition, epigenetic regulators, such as the polycomb group (PcG) complex (Corley and Kroll, 2015) or Hmga1/2 (high-mobility-group AT-hook 1/2) (Ozturk et al., 2014), and Notch signaling are essential for neurogenesis. In contrast, the regulation of gliogenesis is unresolved. PcG complex, Hmga2, and fibroblast growth factor (FGF) signaling are required for fate transition of NSCs from neuro- to gliogenesis (Bronstein et al., 2017; Dinh Duong et al., 2019; Hirabayashi et al., 2009). However, studies to identify parallel mechanisms for gliogenesis, particularly gliogenic gene regulation, have yet to be successful (Imayoshi and Kageyama, 2014), suggesting an elusive molecular mechanism for gliogenesis other than transcription factor regulation. Furthermore, no marker for a common glial precursor cell (GPC; or a bipotent OL-astrocyte glial-restricted precursor cell) of the two glial cell types has been identified (Rowitch and Kriegstein, 2010), leaving unanswered questions regarding the regulation of gliogenesis.
Accumulating evidence suggests that RNA-binding proteins (RBPs) play significant roles in brain development. RBPs directly bind to target mRNAs via consensus sequences and comprehensively regulate the expression of target genes in functional clusters called “regulons” (Keene, 2007; Takeuchi et al., 2018). RBPs generate spatially and temporally regulated gene expression patterns in a cell-type-specific manner and regulate fate specification and differentiation of cells in neuronal tissues (Gerstberger et al., 2014; Kapeli and Yeo, 2012; Lennox et al., 2018). In this study, we focused on RBPs and identified Qk as a novel GPC marker and a key regulator of gliogenesis. Qk has long been known to regulate OL differentiation, based on research conducted on viable quaking-hypomorphic mutant mice (qkv) (Chenard and Richard, 2008). To our knowledge, this is the first report to indicate that Qk is required for comprehensive gliogenic gene expression, therefore making it essential for inducing GPCs from NSCs. The findings indicate the broader impact of Qk on gliogenesis in its entirety and have redefined the overall picture of glial cell development from NSCs.
Results
Identification of RBPs Highly Expressed in NSCs yet Shut Off after Neural Differentiation
Given that a certain population of RBPs show tissue-, cell-type-, and spatiotemporal-specific expression patterns, we aimed to identify a novel RBP marker of GPCs that could potentially be the essential regulator of GPC genesis from NSCs. Master transcription factors that induce neuronal differentiation are expressed both in NSCs and in differentiating neurons, but not in glial cell lineages (Imayoshi and Kageyama, 2014). Hence, we aimed to identify RBPs that are highly expressed in NSCs with no expression in neuronal cell lineages. For this purpose, we utilized laser capture microdissection to separate the developing cerebral cortex into NSC- and neuron-specific areas: the ventricular zone (VZ) and the cortical plate (CP) that arises from the VZ, respectively. Next, RNA was extracted from these regions and a transcriptome analysis was carried out using the GeneChip Mouse Exon 1.0 ST Array (Figure 1A). Compared with RNA sequencing (RNA-seq), array-based transcriptome assays require a reduced amount of RNA with linear amplification, making this technique suitable for the limited sample volumes recovered from specific regions of embryonic brains or laser-captured specimens. In our transcriptome study, Pax6 (paired box 6), Neurog1/2, Hes5, and Gli3 showed NSC-enriched expression (fold change [FC] in neuron/NSC <0.1, p < 0.01), and Reln (reelin), CamK (Ca2+/calmodulin-dependent protein kinase) family members, glutamate receptors, ion channels, and cadherins demonstrated neuron-enriched expression (FC in neuron/NSC >5.0, p < 0.01), indicating the successful separation of NSC- and neuron-specific genes. Using these transcriptome data, we identified NSC-enriched RBPs with expression significantly higher than in neurons (FC in neuron/NSC <0.33, p < 0.01) (Figure 1B). By referencing a database containing the tissue-specific expression profiles of 1,542 human RBPs (Gerstberger et al., 2014), we further characterized the selective expression of the NSC-enriched RBPs in neural tissues. Among all identified NSC-enriched RBPs, Qk was the most abundantly expressed in NSCs and was selectively expressed in fetal and adult brains (Figure 1B, fetal and adult brain indices), suggesting its specific role in neural development or in brain-specific cells. Next, we histologically examined QK expression in the E15.5 cerebral cortex. QK expression was detected in the VZ, where it was positively stained with bromodeoxyuridine (BrdU). QK expression was negative above the VZ, where Tuj1-positive neurons that had differentiated from NSCs were accumulated (Figure 1C). These results indicate that the laser capture microdissection and transcriptome study successfully identified a candidate RBP with potential to be a marker or regulator of GPCs.
Figure 1.

Identification of RBPs Highly Expressed in NSCs Yet Shut Off After Neural Differentiation
(A) Scheme for extracting RNA from NSC and neuron layers in cerebral cortices using laser capture microdissection and transcriptome analysis with ExonArray. Scale bar, 100 μm. Abbreviations: MZ, marginal zone; CP, nascent cortical plate; SP, subplate; IZ/SVZ, intermediate zone/subventricular zone; VZ, ventricular zone.
(B) Identification of NSC-specific RBPs. Mean expression levels in NSC and neuronal layers, fold change (FC) of expression levels between NSC and neuronal layers (neuron/NSC), and p values were calculated (n = 3 each for NSC and neuronal layers). Embryonic and adult brain indices were calculated based on RBP expression levels in human fetal and adult brains and compared against those of other organs (expression level in embryonic or adult brain/mean expression level in other organs) using a human RBP database that covers 1,542 RBPs (Gerstberger et al., 2014). Putative target RNAs are shown.
(C) Co-immunostaining of QK (red), BrdU (green), and Tuj1 (turquoise) in coronal sections of the cranial regions of E15.5 embryos (top). Scale bar, 250 μm. Boxed areas are shown at higher magnification in the bottom row. Scale bar, 50 μm. Abbreviations are the same as in (A).
Loss of Qk Causes Severe Brain Atrophy with Hypomyelination in Qk-Null Brains
We next investigated the function of Qk in gliogenesis. During embryogenesis, QK is selectively expressed in the VZ; in neuronal cells, its expression is lost (Figure 1C). In histological examination at P0, QK was expressed throughout the VZ (or ependymal layer). QK was also densely expressed in the intermediate zone/subventricular zone (IZ/SVZ), especially in regions close to the VZ, and sparsely in the CP, with a gradient of high apical density to low basal density (Figure 2A). This expression pattern suggests that QK is expressed in the early GPC population, which is differentiated from NSCs and migrates from the stem cell niche into the IZ/SVZ and later to the cortical layers that harbor more differentiated cells. In addition, QK expression was detected in endothelial and smooth muscle cells of cortical vessels, and in the pia mater in the developing cerebral cortex (Figure 2A), as previously reported (Li et al., 2003).
Figure 2.

Loss of Qk Causes Severe Brain Atrophy with Hypomyelination in Qk-Null Brains
(A) Immunostaining of QK (red) in coronal sections of P0 brain (leftmost image). Scale bar, 500 μm. The boxed area is shown at higher magnification with DAPI counterstaining in the two right images. Arrowheads indicate QK expression in endothelial and smooth muscle cells. Scale bar, 100 μm. Abbreviations are the same as in Figure 1A.
(B) Nissl staining of the coronal sections of head regions from E18.5 control (Cont) and Qk f/f;Nestin-Cre (Qk-KO) embryos. Scale bar, 500 μm. Boxed areas in the two left images are shown at higher magnification in the two right images. Scale bar, 100 μm. Abbreviations are the same as in Figure 1A.
(C) Co-immunostaining of MBP (green) and QK (red) in coronal sections of P14 wild-type cortex (three left images). Scale bar, 250 μm. The boxed area in the left MBP/QK image is shown at higher magnification in the rightmost image. Scale bar, 50 μm.
(D) Immunostaining of MBP in the coronal sections of the P14 brain from Cont and Qk-KO mice (two left images). Scale bar, 500 μm. Boxed areas indicated in Cont and Qk-KO cortices are shown at higher magnification in the image to their immediate right. Scale bar, 250 μm. Abbreviations: 3-V, third ventricle; LV, lateral ventricle.
To elucidate the function of Qk in glial cell regulation, we generated conditional knockout (KO) mice for Qk (Qk flox) (Figure S1) and crossed them with Nestin-Cre mice to delete the floxed Qk gene in both NSCs and their offspring cells. Qk-KO embryos were obtained at E18.5, 1 day before birth, with the expected Mendelian ratio, exhibiting no detectable abnormalities in brain morphology. In histological analysis, no gross abnormalities were detected in structure or cellular distribution; the thicknesses of the VZ and the CP in the developing cortex of the Qk-KO brain were comparable to those of the control (Cont) brain (Figure 2B), indicating that the loss of Qk in NSCs does not cause severe defects in brain formation during the embryonic period. As Qk is known to regulate OL production, we further examined the role of Qk in generating OLs from NSCs. Expression of the mature OL marker MBP (myelin basic protein) became evident in the wild-type cerebral cortex around P14, particularly in the white matter of the developing cortex and the corpus callosum (Figure 2C, MBP). QK was expressed throughout the cortex at this stage. Co-localization of QK with MBP was detected, indicating that the OLs that form the myelin sheath express QK (Figure 2C, MBP/QK). In P14 QK-KO brains, MBP expression was significantly decreased throughout the brain, including in the corpus callosum, indicating severe hypomyelination of OLs (Figure 2D, Qk-KO). These brains exhibit loose cerebral parenchyma with greatly dilated lateral and third ventricles (Figure 2D, Qk-KO), indicating that the loss of Qk caused hypomyelination with severe brain atrophy. In the quaking viable (qkv) mouse, a spontaneous autosomal recessive Qk mutant, hypomorphic Qk expression reduced the number of mature OLs in the CNS (Hardy, 1998). These, and our results, indicate that Qk is essential for the generation and differentiation of OLs from NSCs.
QK Is Expressed in Putative GPCs
We further investigated QK-expressing cells via marker analysis, by examining the expression of OLIG2, a bHLH transcription factor that regulates the specification and differentiation of NSCs to OL precursor cells (OPCs) (Meijer et al., 2012) and astrocyte progenitors (Marshall et al., 2005). In P0 Cont brains, QK was densely expressed in the VZ (or ependymal layer) (Figure 3A) as previously observed (Figure 2A). In the area above the white line (Figure 3A), scattered QK-expressing cells were well overlapped by OLIG2-positive cells, indicating that QK is expressed in OPCs and astrocyte progenitors. We further examined the expression of the OPC marker platelet-derived growth factor receptor alpha (PDGF-Rα), which is essential for the proliferation, differentiation, and survival of OPCs (Funa and Sasahara, 2014). PDGF-Rα-positive OPCs were detected in the same area as QK and OLIG2 double-positive cells (Figure 3A). Dual immunostaining for QK and PDGF-Rα revealed that PDGF-Rα-expressing cells were largely positive for QK (Figure 3B), indicating that OPCs express QK. Accumulation of QK-positive cells, negative for OLIG2 and PDGF-Rα, was detected between the VZ and the OLIG2-positive cell layers (Figure 3A). As no markers for the GPC population have been identified, it could not be definitively concluded that these cells were, in fact, GPCs. However, considering that QK expression is lost in neuronal cell lineages after they differentiate from NSCs, these data strongly suggest that NSC-derived GPCs express QK and further differentiate into OPCs and, presumably, astrocyte progenitors. Next, we examined the expression of known glial progenitor markers in these putative GPC populations using anti-CNP (2ʹ,3ʹ-cyclic nucleotide 3ʹ-phosphodiesterase) and anti-NG2 antibodies. CNP is expressed during OL development (Groteklaes et al., 2020). In P0, CNP expression was not observed in the developing cortex (Figure S2A), and was weakly detected in the corpus callosum of P7 cortex (Figure S2B), consistent with a previous report. NG2 proteoglycan (the product of the CSPG4 [chondroitin sulfate proteoglycan 4] gene) is the OL lineage marker (Nishiyama et al., 2009), and recent studies suggest multipotency of NG2 cells (Tsoa et al., 2014). In P0 brains, a small number of NG2-positive cells showing highly branched morphology were scattered, while the vascular cells in the brain parenchyma were highly positive (Figure S2C), as was previously reported. Whereas QK expression overlapped with these NG2 cells, the vast majority of putative GPCs did not express NG2, which is consistent with the observation indicating that NG2 is expressed later than PDGF-Rα (Nishiyama et al., 2009). Overall, these results indicate that QK is a good marker for the earlier GPC population that still does not express CNP and NG2.
Figure 3.

QK Is Expressed in Putative GPCs
(A) Co-immunostaining of QK (red) and OLIG2 (green) in coronal sections of P0 cortices from control (Cont) (upper leftmost) and Qk-KO (lower leftmost) mice. Scale bar, 250 μm. The boxed areas in QK/OLIG2 images are shown at higher magnification in the three images to their right. PDGF-Rα labeling was performed using adjacent sections. The white line in Cont demarks the area where QK-positive/OLIG2-negative cells accumulated (lower left) and the area where QK-positive/OLIG2-positive cells scattered (upper right). The white line in Qk-KO corresponds to the area in Cont. Scale bar, 100 μm.
(B) Co-immunostaining of QK (red) and PDGF-Ra (green) in coronal section of P0 Cont cortex. Arrows indicate QK/PDGF-Ra double-positive cells. Scale bar, 50 μm.
(C) The number of OLIG2+ and PDGF-Rα+ cells in P0 cortices is shown (n = 3 in each genotype). Data are presented as mean ± SD. ∗p < 0.05 and ∗∗p < 0.01 (Student's t test).
(D) Representative results of WB for QK, OLIG2, PDGF-Rα, and α-TUBULIN in P0 cortices from Cont and Qk-KO mice. Signal intensities of OLIG2 and PDGF-Rα in WB were quantified by densitometry, normalized by α-tubulin, and indicated using a bar graph (n = 3 in each genotype). Data are presented as mean ± SD. ∗∗p < 0.01 (Student's t test).
We further analyzed the progenitors of astrocytes and OPCs in P0 Qk-KO brains. The number of QK-expressing cells was greatly reduced and the remaining Qk expression was primarily detected in the endothelial and smooth muscle cells of cortical vessels (Figure 2A). As the efficiency of Cre-induced inversion and inactivation of Lox66/Lox71 via the FlipRosaβgeo trapping vector (Schnutgen et al., 2005) is known to be lower than Cre-induced excision of LoxP floxed alleles (Oberdoerffer et al., 2003), we were able to detect the remaining QK-expressing cells in which the Qk gene was not inactivated in Nestin-Cre-expressing NSCs. The distribution of OLIG2-positive cells in Qk-KO cortices was slightly, yet significantly, increased compared with Cont (Figures 3A and 3C). Western blotting (WB) of Cont and Qk-KO cortices confirmed disruption of QK expression in Qk-KO cortices. Meanwhile, total OLIG2 protein expression in Qk-KO cortices was slightly, yet significantly, increased compared with Cont (Figure 3D), suggesting a compensatory mechanism triggered by decreased number of OLs. In contrast, expression of PDGF-Rα was significantly downregulated in Qk-KO cortices compared with Cont (Figures 3A and 3C), and total PDGF-Rα protein expression in Qk-KO cortices was significantly downregulated compared with Cont (Figure 3D). Although we detected CNP expression in OLs within the corpus callosum in P7 Cont, it became much weaker in P7 QK-KO (Figure S2B). In addition, NG2 cells were nearly absent from P0 QK-KO cortices (Figure S2D). These data indicate that loss of Qk failed to induce PDGF-Rα-positive OPCs from NSCs, subsequently decreasing the number of OLs and causing severe hypomyelination (Figure 2D). As expression of OLIG2 precedes PDGF-Rα in OPCs (Stolt et al., 2002), it is postulated that OLIG2-positive glial progenitors were generated from NSCs in Qk-KO brains; however, these cells had already lost their ability to differentiate into PDGF-Rα-positive OPCs.
As the expression profiling of QK+ cells suggested QK as a good marker of the GPC population, we broadened our analysis and evaluated QK-expressing cells (irrespective of the type) using single-cell transcriptomic data reported in the context of developing mouse neocortex (Loo et al., 2019). In P0, QK-expressing cell populations include immature astrocytes (including GPCs and astrocyte progenitors), OLs (OPCs), endothelial cells, and choroid plexus cells (Figures S3A and S3B), in line with our immunohistochemistry studies. Of note, QK expression was also detected in the microglia and interneurons; thus we further analyzed the distribution of these cells. QK expression was detected in IBAI (induction of brown adipocytes 1)-positive cells in the pia mater and in the septal area, but not in the IZ/SVZ (Figure S3C, Cont), consisten with the recent findings that brain microglia originates from embryonic mesenchymal cells in the pia or from fetal macrophages in the yolk sac (for more details, please see the review by Mosser et al., 2017) and these microglia express QK. Interneurons are known to be generated in the VZ of the medial ganglionic eminence and to migrate tangentially into the CP. In this study, a weak QK expression was detected in the subpallium, where GAD65/GAD67 (glutamic acid decarboxylase-65 and -67)-positive cells were accumulating, suggesting that QK is expressed by interneurons (Figure S3D, subpallium in Cont). However, we did not observe QK expression in migrating interneurons in the CP (Figure S3D, CP in Cont). Overall these data suggest that newly generated interneurons express QK but then lose its expression during differentiation, similar to that observed for cortical neurons (Figure 1C). Altogether, these results attest that QK is a novel marker of the early GPC population; a substantial number of cells in the IZ/SVZ still negative for OLIG2, PDGF-Rα, CNP, and NG2 can be identified via QK (Figures 3A, 3B, S2, and S3E).
QK Is Expressed in Astrocyte Progenitor Cells, and Loss of Qk Impairs Astrocyte Generation from NSCs
We next investigated whether QK is expressed in astrocyte progenitor cells and is involved in astrocyte genesis. We utilized acyl-CoA synthetase bubblegum family member 1 (ACSBG1) as an astrocyte marker, as it is specifically expressed in astrocytes and their progenitors (Cahoy et al., 2008). In expression analyses of wild-type cerebral cortices, ACSBG1 expression became evident near P3 and was detected throughout the cortex, particularly in cortical gray matter (Figure 4A). Scattered QK-positive cells in the CP were more prevalent at P3 than P0 and were uniformly detected throughout the cortex (Figure 4A), suggesting the migration and proliferation of QK-positive cells from the VZ to the developing CP. Moreover, ACSBG1-expressing cells were largely positive for QK (Figure 4A), indicating that astrocyte progenitor cells express QK.
Figure 4.

QK Is Expressed in Astrocyte Progenitor Cells, and Loss of Qk Impairs Astrocyte Generation from NSCs
(A) Co-immunostaining of ACSBG1 (green) and QK (red) in the coronal sections of P3 wild-type cortex (left two). The boxed area in the ACSBG1 image is shown at higher magnification in the rightmost image. Arrows indicate cells that are double positive for red (QK) and green (ACSBG1). Scale bar, 50 μm.
(B) Immunostaining of ACSBG1 in coronal sections of P3 cortices from control (Cont) and Qk-KO mice (left two). Scale bar, 250 μm. Boxed areas in Cont and Qk-KO mouse cortices are shown at higher magnification in the next two images. Scale bar, 50 μm. The number of ACSBG1+ cells in P0 cortices is shown using a bar graph in the rightmost image (n = 3 in each genotype). Data are presented as mean ± SD. ∗∗∗p < 0.001 (Student's t test).
(C) Representative results of WB for QK, ACSBG1, and α-TUBULIN in P3 cortices from control and Qk-KO mice. Signal intensities of ACSBG1 were quantified by densitometry (n = 3 in each genotype), normalized by α-TUBULIN, and indicated using a bar graph. Data are presented as mean ± SD. ∗∗p < 0.01 (Student's t test).
(D) Co-immunostaining of GFAP (green) and QK (red) in coronal sections of the P7 wild-type cortex. Scale bar, 250 μm. The boxed area in the GFAP image is shown at higher magnification in the rightmost image. Arrows indicate cells that are double positive for red (QK) and green (GFAP). Scale bar, 50 μm.
(E) Immunostaining of GFAP in coronal sections of P7 brains from Cont and Qk-KO mice (left). Scale bar, 500 μm. Asterisk indicates dilated lateral and third ventricles detected in Qk-KO brains. The boxed areas in the two left images are shown at higher magnification in the two right images. Scale bar, 50 μm. The number of GFAP+ cells in P7 cortices is shown using a bar graph at the rightmost bottom (n = 2 in each genotype). Data are presented as mean ± SD. ∗p < 0.05 (Student's t test).
(F) Representative results of WB for QK, GFAP, and α-TUBULIN in P7 cortices from Cont and Qk-KO mice are shown. Signal intensities of GFAP were quantified by densitometry (n = 3 in each genotype), normalized by α-tubulin, and indicated using a bar graph. Data are presented as mean ± SD. ∗∗∗p < 0.001 (Student's t test).
Next, we investigated the generation of astrocytes in Qk-KO brains. In P3 Qk-KO brains, ACSBG1 expression was downregulated throughout the cortex, which was confirmed by the significantly reduced number of ACSBG1-positive cells (Figure 4B). Disruption of QK expression and significant downregulation of ACSBG1 protein expression in Qk-KO cortices was confirmed by WB (Figure 4C). Hence, the genesis of astrocyte progenitor cells from GPCs was disrupted in Qk-KO mouse brains and was analogous to a defect in OL differentiation. To further confirm this hypothesis, we investigated the production of astrocytes by glial fibrillary acidic protein (GFAP) immunostaining. The expression of GFAP in astrocytes became evident in the cerebral cortex around P7, and its expression in radial glial cells was particularly apparent in their radial fibers (Figure 4D). At P7, QK-expressing cells were scattered widely throughout the entire cortex. GFAP-expressing astrocytes were largely positive for QK, indicating that astrocytes express QK.
Next, we examined the phenotype of astrocytes in Qk-KO brains. In P7 Qk-KO brains, GFAP immunostaining was severely decreased throughout the cortex, which was confirmed by the significantly decreased number of GFAP-positive cells (Figure 4E). We confirmed the disruption of QK expression and significant downregulation of GFAP protein expression in Qk-KO cortices by WB (Figure 4F). In Qk-KO brains, atrophy of brain parenchyma alongside the expansion of the lateral and third ventricles was detected (Figure 4E). Among several possible explanations for this phenomenon, this may be explained by a loss of astrocytes. Our Qk-floxed mice could not completely inactivate the Qk genes in VZ by P0, thus we estimated the KO efficacy by counting the remaining QK+/OLIG2+ cells that escaped from Cre-mediated inactivation and comparing with the numbers in Cont. The KO efficacy at P7 cortices was 84.8%; this frequency was larger than that characterizing the decrease of GFAP-positive cells or GFAP protein levels, indicating that the loss of Qk did not completely abolish astrocytes at this time point, but significantly disrupted the generation of astrocytes from NSCs. On the other hand, the OL production examined via MBP expression was more severely downregulated, as shown in Figure 2D, thus indicating that Qk is indispensable for the generation and maturation of OLs, as previously reported (please see review by Chenard and Richard, 2008). Qk is known to regulate OL production, as observed in hypomorphic qkv mice. However, these studies using NSC-specific null mutant mice for Qk revealed the extended role of Qk in regulating gliogenesis.
Qk Is Required for Fate Specification of NSCs to GPCs
As the staining of markers in cells cannot reliably identify whether expression has been completely lost or is downregulated, we established an in vitro culture system of NSCs and conducted transcriptome analysis to capture cell fate change. NSCs were isolated from cerebral cortices of Qk-KO and Cont mice, and glial differentiation was induced by mimicking in vivo gliogenesis (Figure 5A). Prior to induction, we examined the proliferative capability of Qk−/− NSCs in the presence of basic FGF (bFGF) and no significant difference was observed in proliferation between Qk-KO and Cont, consistent with the finding that VZ thickness and neuronal cell production from NSCs were intact in Qk-KO brains (Figure 2B). Next, differentiation of NSCs to glial cells was induced using media containing 10% fetal bovine serum but lacking bFGF. Cont NSCs expressed the stem cell marker SOX2 on day 1; however, they began to express ACSBG1 on day 2 and GFAP on day 4, indicating successful astrocyte differentiation from NSCs (Figure 5B). Qk-KO NSCs also expressed SOX2 on day 1; however, expression of ACSBG1 and GFAP was much lower than in Cont NSCs (Figure 5B), consistent with our earlier findings in Qk-KO brains. Quantitative analysis indicated fewer GFAP-positive cells in Qk-KO NSCs than in Cont NSCs, although total cell numbers were comparable (Figure 5C), supporting our hypothesis that loss of Qk inhibited NSC differentiation into GFAP-positive astrocytes without causing cell loss.
Figure 5.

Qk Is Required for the Fate Specification of NSCs to GPCs
(A) Scheme for isolating NSCs from embryonic mouse cortices and inducing differentiation into glial cells in vitro.
(B) Expression of SOX2 (green), ACSBG1 (red), and GFAP (red) during glial differentiation of control (Cont) and Qk-KO NSCs. Scale bar, 100 μm.
(C) GFAP-positive and total cell numbers counted during glial differentiation of Cont and Qk-KO NSCs (counted on days 1, 2, 3, and 4 after induced differentiation using four samples from Cont and Qk-KO NSCs). Data are represented as the mean ± SEM (n = 4 in each genotype, ∗∗∗p < 0.001 using Student's t test).
(D) GSEA plots of Cont and Qk-KO NSCs at day 1 after glial differentiation. Differentially expressed genes were analyzed using glia-, OPC-, and neuron-enriched gene sets. NES, normalized enrichment score.
Hypothesizing that Qk is required for fate specification of NSCs to GPCs, we carried out a transcriptome analysis at an early time point, 24 h after inducing glial differentiation (day 1). To comprehensively detect transcriptomic changes in differentiating Qk−/− NSCs, we performed gene set enrichment analysis (GSEA), which uses statistical approaches to identify significantly enriched or depleted groups of genes. We referenced the cell-type-specific enriched gene lists for astrocytes, OLs, OPCs, glial cells (astrocytes + OPCs + OLs), and neurons (Zhang et al., 2014). GSEA showed that glia- and OPC-enriched genes were induced in Cont NSCs, indicating that Cont NSCs differentiate into GPCs. In contrast, loss of Qk significantly downregulated glia- and OPC-enriched genes compared with Cont NSCs (Figure 5D), indicating that marker expression was downregulated rather than completely abolished. Surprisingly, we found that neuron-enriched genes, including several types of neuron-specific genes, were aberrantly upregulated in Qk−/− NSCs (Figure 5D), suggesting that loss of Qk disrupted NSC differentiation to GPCs and instead induced these cells to become neural precursor cells. Together, GSEA results clearly indicate that Qk is required for fate specification of NSCs to GPCs.
Qk Upregulates Astrocyte/OL Genes, Cell-Surface Signaling Receptors, and Endocytosis Pathway Genes as Gliogenic Regulons
Next, we attempted to elucidate the molecular function of Qk in mRNA regulation of gliogenesis. To specify the target mRNAs of Qk, we identified genes with significantly altered expression in Qk−/− NSCs using the thresholds of FC > 1.5 (log2 FC > 0.58) and FC < 0.67 (log2 FC < −0.58) with adjusted p < 0.01 in DESeq2; a total of 383 genes were downregulated and 570 genes were upregulated in Qk−/− NSCs compared with Cont. Qk contains RNA-binding motifs known as STAR domains. Several studies using SELEX and CLIP-seq have identified the unique UACUAAH (H = not G) motif as the RNA recognition sequence of Qk and designated it the quaking response element (QRE) (Fagg et al., 2017; Galarneau and Richard, 2005; Hafner et al., 2010; Hayakawa-Yano et al., 2017). Previous studies have indicated that Qk binds directly to the QRE(s) in the 3′ untranslated region (3′ UTR) and stabilizes target mRNAs (Larocque et al., 2005; Li et al., 2000; Zearfoss et al., 2011; Zhang et al., 2003; Zhao et al., 2006). Based on these findings, we examined whether Qk globally regulates expression of genes possessing a QRE(s) in their 3′ UTR. We analyzed the relationship between FC of all expressed genes under Qk-KO conditions and the presence of QREs in 3′ UTRs. Compared with genes that lack QRE, the cumulative curves of genes possessing QREs were shifted to the left according to the number of QREs they possessed, indicating that the loss of Qk decreased expression of target mRNAs (Figure 6A).
Figure 6.

Qk Upregulates Astrocyte/OL Genes, Cell-Surface Signaling Receptors, and Endocytosis Pathway Genes as Gliogenic Regulons
(A) The cumulative distribution showing the relationship between changes in mRNA expression of all expressed genes and the presence of QRE in 3′ UTRs in control (Cont) and Qk-KO NSCs at day 1 after glial differentiation.
(B) Top: Scheme of 3′ UTR reporter vectors expressing eGFP cDNA with 3′ UTR of Rab5a (Rab5a wt 3′ UTR), Snx5 (Snx5 wt 3′ UTR), Cbl (Cbl wt 3′ UTR), Cav1 (Cav1 wt 3′ UTR), or eGFP lacking a 3′ UTR (eGFP). Bottom: Reporter vectors were co-transfected into HeLa cells with a Qk-6 expression vector and the expressed mRNA from each vector was examined by qPCR. Relative mRNA levels to eGFP lacking a 3′ UTR (eGFP) are shown. Data are presented as mean ± SD (experiments were performed with biological triplicate (n = 3), ∗∗∗p < 0.01 using Student's t test).
(C) Pathway analysis of regulatory Qk target genes. The table lists the pathways for which the p value is <0.001 and FDR (false discovery rate) is <0.1. Genes highlighted with yellow are categorized as cell-surface receptors or ligands.
We next examined whether QREs in the 3′ UTR of transcripts are responsible for Qk-dependent mRNA expression. Endocytosis has been identified as a regulatory target of Qk (Shingu et al., 2017), and two endocytosis pathway genes, Rab5a and Snx5 (Sorting nexin 5), were identified as targets of Qk containing QREs in their 3′ UTR and were downregulated in Qk-KO NSCs (log2 FC = −1.12 for Rab5a and −1.49 for Snx5). Two non-target control endocytosis pathway genes Cbl and Cav1 (Caveolin 1) do not possess QREs in their 3′ UTR, yet were also downregulated in Qk-KO NSCs (log2 FC = −0.63 for Cbl and −1.44 for Cav1). We generated reporter vectors that express eGFP (enhanced green fluorescent protein) mRNA with the 3′ UTR of these genes (Figure 6B). Each reporter construct was co-transfected with the cytoplasmic isoform of Qk-6 (Qk full). To examine whether the presence of Qk is sufficient to stabilize target mRNAs in a non-cell-type-specific manner, HeLa cells were used due to their low, or undetectable, levels of endogenous Qk mRNA and protein compared with brain samples (data not shown). The levels of eGFP mRNA of Rab5a and Snx5 reporters were significantly higher than Cont eGFP lacking a 3′ UTR (1.80- and 1.88-fold for Rab5a and Snx5 wt 3′ UTRs, respectively; Figure 6B), while levels of Cbl and Cav1 reporters lacking QREs were comparable to Cont eGFP (1.01- and 1.08-fold for Cbl and Cav1 wt 3′ UTRs, respectively; Figure 6B), suggesting that Qk stabilized Rab5a and Snx5 reporter mRNAs via QREs. We further examined the ability of QREs to stabilize mRNAs by introducing a mutation in which the putative QRE was changed from ACTAAH to TCGAGH. The mRNA levels of QRE-mutated Rab5a and Snx5 reporters (Rab5a mut 3′ UTR and Snx5 mut 3′ UTR) decreased to a level comparable to Cont eGFP, Cbl, and Cav1 reporters lacking QRE, indicating that the mutation rendered them insensitive to Qk (1.18- and 1.26-fold for Rab5a and Snx5 mut 3′ UTR, respectively; Figure 6B). These results indicate that a QRE(s) in the 3′ UTR is essential for Qk-dependent upregulation of target mRNAs.
To further identify the target genes of Qk that regulate gliogenesis, the significantly downregulated genes in the Qk-KO NSC-induced glial differentiation were separated according to the presence or absence of a QRE(s) in their 3′ UTR. A total of 279 genes were identified as putative Qk targets. These were further classified according to astrocyte-, OL-, OPC-, glia-, or neuron-enriched genes using gene lists for GSEA. The putative Qk target genes contained numerous glia-, astrocyte-, OL-, and OPC-enriched genes. These include essential molecules for the proliferation or development of astrocytes or OLs, such as S100β (Zimmer et al., 1995) and PDGF-Rα (Funa and Sasahara, 2014) (Table S2), indicating that several gliogenic genes possess QREs and that Qk comprehensively upregulates them during gliogenesis. To further identify the Qk-targeted regulons of gliogenesis, we carried out a gene ontology pathway analysis of the 279 putative target genes of Qk. We identified two biological processes with significant enrichment scores (p < 1 × 10−3 and q < 0.1): “positive regulation of peptidyl-tyrosine phosphorylation” and “positive regulation of cell migration” (Figure 6C). Although there was no clear evidence that these processes are essential for gliogenesis, genes in these categories are highly enriched for cell-surface signaling receptors or ligands (9/11 and 11/14, respectively) (Figure 6C) such as PDGF-Rα or FGF receptor 3, which was recently shown to be essential for the neurogenic-to-gliogenic fate transition of NSCs (Dinh Duong et al., 2019). These data indicate that Qk is necessary for the upregulation of several cell-surface signaling molecules that mediate glial differentiation. The gene set involved in the “endocytosis pathway” was identified in the Kyoto Encyclopedia of Genes and Genomes (KEGG) with a significant enrichment score (p < 1 × 10−3 and q < 0.1) (Figure 6C), and this pathway is essential for glial differentiation (please see Discussion). Taking our results together, we have indicated that Qk is required for comprehensive upregulation of (1) astrocyte/OL-specific genes, (2) cell-surface signaling receptors, and (3) endocytosis pathway genes as gliogenic regulons (Figure 7). As previously mentioned, Qk is known to regulate OL production, as observed in hypomorphic qkv mice. However, these studies, using NSC-specific null mutant mice for Qk, revealed the extended role of Qk in regulating gliogenesis.
Figure 7.

Scheme for the Identified Qk Function in Regulation of Glial Differentiation from NSCs
Discussion
Growing evidence indicates the significance of RBPs in neuronal development and brain functions. NOVA1/2 (Ule et al., 2005), PTBP1 (polypyrimidine tract binding protein 1) (Boutz et al., 2007; Licatalosi et al., 2012; Zhang et al., 2016), SRRM4 (serine/arginine repetitive matrix 4) (Calarco et al., 2009), RBFOX1/2 (RNA-binding protein fox-1 homolog 1/2) (Gehman et al., 2011, 2012), TDP-43 (TAR DNA binding protein-43) (Polymenidou et al., 2011), Fus (fused in sarcoma) (Lagier-Tourenne et al., 2012), and SFPQ/PSF (splicing factor proline/glutamine rich or PTB-associated splicing factor) (Takeuchi et al., 2018) have been shown to be essential for neurogenesis or neural functions. In contrast, PTBP2 (Boutz et al., 2007; Licatalosi et al., 2012; Zhang et al., 2016), ELAVl2 (ELAV-like RNA-binding protein 2) (Akamatsu et al., 1999), and musashi (Sakakibara et al., 2002) have been demonstrated to be required for the proliferation and stemness of NSCs. These studies show that specific subsets of RBPs are important in neurons and NSCs encompassing neurogenesis, yet RBPs with specific roles in gliogenesis were not identified. The systematic exploration of all RBPs herein revealed Qk function in astrocytes as well as in the fate decision of NSCs to glial cells (Figure 7), while Qk is widely accepted as the specific regulator for OL differentiation.
Genetic studies have identified mutations in RBP genes as causes of neurodegenerative or psychiatric diseases, indicating that RBPs play important roles in brain function (Kapeli and Yeo, 2012). Alterations in isoform expression of the human homolog of Qk, QKI, were demonstrated in approximately 30% (6/20) of glioblastomas (Li et al., 2002), and QKI was identified through a genotype-phenotype study as a tumor suppressor that affects the stemness of glioma stem cells (Hu et al., 2013; Shingu et al., 2017). Here, we reveal that the loss of Qk selectively disturbed the glial differentiation of NSCs. These findings provide insight into why Qk mutations contribute to poorly differentiated malignant glial cell tumors, as well as into the molecular pathogenesis of glioblastomas and their treatment. Interestingly, QKI mutation or dysregulation of QKI expression is frequently observed in schizophrenia patients, and decreased transcript levels of QKI-7 and QKI-7b splice variants (or their aberrant splicing) were reported (please see review by Chenard and Richard, 2008). These results suggest an association between the dysregulation of gliogenesis and the pathological mechanism of schizophrenia. In addition, a number of studies have investigated the nature of unique postnatal stem cells of NG2 cells, which give rise to both glial cells and interneurons (Tsoa et al., 2014). We observed Qk expression in NG2 cells and their ablation in P0 Qk-KO cortices (Figure S2D), suggesting an essential function of Qk in gliogenesis and also in neurogenesis. We also found that QK is expressed in microglial cells; of note, its expression is not inactivated in Nestin-Cre mice (Figure S3C, Qk-KO), since microglial cells are not generated from NSCs in brain but originate from non-neuronal fetal tissues. As our Qk-KO mice experienced premature death, similar to that observed in qke5 mice (Noveroske et al., 2005), and microglial cells still expressed Qk in this context, further studies using time- and site-specific gene-targeting techniques would be expected to decipher the function of Qk in adult brains and to elucidate its roles in NG2 cells, microglial cells, and the disease-causing mechanisms in adult brains.
The mechanism responsible for directly inducing glial cells from NSCs requires further investigation. Qk regulates the differentiation of NSCs to GPCs by comprehensively regulating gliogenic regulons that include glia-, astrocyte-, OPC-, and OL-specific genes, cell-surface signaling receptors, and endocytosis pathway genes (Figure 7). Endocytosis pathways facilitate signaling receptor expression and replacement (Haglund and Dikic, 2012). Downregulation of Qk causes persistent expression of the receptors responsible for maintaining pluripotency and prevents differentiation of NSCs in Qk/Pten/Trp53 triple-KO mouse brains (Shingu et al., 2017). Mechanistically, Qk stabilizes the target mRNAs of the genes listed above. Thus, transcriptional regulation and Qk may synergistically regulate gliogenic gene expression. In addition, we noted that Qk−/− NSCs failed to differentiate into GPCs and aberrantly expressed neural genes; however, these Qk−/− NSCs were unable to become mature neurons (data not shown), which is consistent with the observation that cortical formation was intact at E18.5. This result indicates that the mechanism of fate switching between neurogenesis and gliogenesis could not be explained simply by the presence or absence of Qk in precursor cells. The identification of Qk as the RBP that drives GPC development allows Qk to be used as a marker for the isolation of GPCs and is an ideal tool for the characterization of GPCs and further investigations into the regulatory mechanisms of gliogenesis. This would lead to an enriched understanding of the global structure of the regulation of glial differentiation from NSCs during brain development, as well as the disease-causing mechanism and pathological consequence of schizophrenia.
Experimental Procedures
Mice
Conditional KO mice for Qk were generated (Figures S1A and S1B). Detailed information is given in the Supplemental Experimental Procedures. All animal care and experiments were conducted in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals, and all experimental protocols were approved by the Institutional Animal Care and Use Committee of the Kyoto University Graduate School of Medicine and the RIKEN Kobe Branch.
Primary Neural Stem/Precursor Cell (NS/PC) Culture and Differentiation into Astrocytes
NS/PCs were isolated from E14 mouse embryo (male and female) cortices of Qk-KO and Cont brains. To induce glial differentiation, cells were seeded on dishes in N2 medium lacking bFGF but containing 10% fetal bovine serum.
Exon Array Analysis of NSC- and Neuron-Specific Layers and Bioinformatics Analysis
Cranial regions were isolated from E15.5 C57BL/6 mouse embryos. The lower and upper areas of the entire cortex (harboring NSC- and neuron-specific regions, respectively) from rostral to caudal were isolated using a laser capture microdissection system (LMD6; Leica) (Figure 1A). Exon array experiments were performed using the GeneChip Mouse Exon 1.0 ST Exon Array (Affymetrix, Santa Clara, CA, USA) as previously described (Ishigaki et al., 2012). Bioinformatics analysis of exon array data was performed as previously described (Yamashita et al., 2012).
Comparative Analysis of RNA-Seq Data from NSCs
RNA-seq was performed as previously described (Takeuchi et al., 2018). For differential gene expression analysis, we used DEseq2 (Love et al., 2014). Genes that met the following criteria were considered as differentially expressed: log2 FC > 0.58 (FC > 1.5) or log2 FC < −0.58 (FC < 0.67) and an adjusted p < 0.01 for primary cells; raw tag count ≥31 for at least one condition (Cont or Qk-KO); and TPM ≥2 for at least one condition. Raw data from mRNA-seq were deposited in the Gene Expression Omnibus (GEO) under accession no. GSE117018.
Data and Code Availability
All data are available from the GEO under accession no. GSE117018. In-house scripts were used to calculate RPKM and TPM. All data and scripts not included here are available from the corresponding author upon reasonable request.
Author Contributions
A.T. conceived and designed the study, generated Qk-KO mice, performed the experiments, and wrote the manuscript. Y.T. contributed to the initial phenotypic analysis. K. Iida performed bioinformatic analysis of high-throughput sequencing data. M. Hosokawa assisted with wet experiments. K. Irie and K.N. provided technical supervision and assisted with in vitro experiments using NSCs. M.I. and K.O. performed the exon array experiment. J.B.B. assisted in writing the manuscript. M. Hagiwara assisted in project and manuscript preparation.
Acknowledgments
We thank the Kyoto University Medical Research Support Center for performing the high-throughput sequencing analysis and its Institute of Laboratory Animals for providing animal care; A. Hagiwara, A. Hirose, and K. Makigaya for technical assistance; Dr. T. Awaya for critical reading of the manuscript and helpful comments; and Editage (www.editage.jp) for English language editing of this article. This work was supported in part by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT, JSPS KAKENHI 19500269, 25500288, 21249013, 15H05721, 19K06907) (to M. Hagiwara and A.T.); Innovative Cell Biology by Innovating Technology (Cell Innovation) (to M. Hagiwara, K.O., and A.T.); a Core Research for Evolutional Science and Technology grant from the Japan Science and Technology Agency (to M. Hagiwara); a grant from the Japan Agency for Medical Research and Development (to M. Hagiwara); the Asian CORE Program of JSPS (to M. Hagiwara); iCeMS Cross-Disciplinary Research Promotion Project of Kyoto University (to A.T.); and the Fujiwara Memorial Foundation (to A.T.).
Published: September 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.08.010.
Supplemental Information
References
- Akamatsu W., Okano H.J., Osumi N., Inoue T., Nakamura S., Sakakibara S., Miura M., Matsuo N., Darnell R.B., Okano H. Mammalian ELAV-like neuronal RNA-binding proteins HuB and HuC promote neuronal development in both the central and the peripheral nervous systems. Proc. Natl. Acad. Sci. U S A. 1999;96:9885–9890. doi: 10.1073/pnas.96.17.9885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz P.L., Stoilov P., Li Q., Lin C.H., Chawla G., Ostrow K., Shiue L., Ares M., Jr., Black D.L. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–1652. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronstein R., Kyle J., Abraham A.B., Tsirka S.E. Neurogenic to gliogenic fate transition perturbed by loss of HMGB2. Front. Mol. Neurosci. 2017;10:153. doi: 10.3389/fnmol.2017.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy J.D., Emery B., Kaushal A., Foo L.C., Zamanian J.L., Christopherson K.S., Xing Y., Lubischer J.L., Krieg P.A., Krupenko S.A. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calarco J.A., Superina S., O'Hanlon D., Gabut M., Raj B., Pan Q., Skalska U., Clarke L., Gelinas D., van der Kooy D. Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell. 2009;138:898–910. doi: 10.1016/j.cell.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Chenard C.A., Richard S. New implications for the QUAKING RNA binding protein in human disease. J. Neurosci. Res. 2008;86:233–242. doi: 10.1002/jnr.21485. [DOI] [PubMed] [Google Scholar]
- Corley M., Kroll K.L. The roles and regulation of Polycomb complexes in neural development. Cell Tissue Res. 2015;359:65–85. doi: 10.1007/s00441-014-2011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh Duong T.A., Hoshiba Y., Saito K., Kawasaki K., Ichikawa Y., Matsumoto N., Shinmyo Y., Kawasaki H. FGF signaling directs the cell fate switch from neurons to astrocytes in the developing mouse cerebral cortex. J. Neurosci. 2019;39:6081–6094. doi: 10.1523/JNEUROSCI.2195-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagg W.S., Liu N., Fair J.H., Shiue L., Katzman S., Donohue J.P., Ares M., Jr. Autogenous cross-regulation of Quaking mRNA processing and translation balances Quaking functions in splicing and translation. Genes Dev. 2017;31:1894–1909. doi: 10.1101/gad.302059.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funa K., Sasahara M. The roles of PDGF in development and during neurogenesis in the normal and diseased nervous system. J. Neuroimmune Pharmacol. 2014;9:168–181. doi: 10.1007/s11481-013-9479-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau A., Richard S. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat. Struct. Mol. Biol. 2005;12:691–698. doi: 10.1038/nsmb963. [DOI] [PubMed] [Google Scholar]
- Gehman L.T., Meera P., Stoilov P., Shiue L., O'Brien J.E., Meisler M.H., Ares M., Jr., Otis T.S., Black D.L. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev. 2012;26:445–460. doi: 10.1101/gad.182477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman L.T., Stoilov P., Maguire J., Damianov A., Lin C.H., Shiue L., Ares M., Jr., Mody I., Black D.L. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat. Genet. 2011;43:706–711. doi: 10.1038/ng.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S., Hafner M., Tuschl T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groteklaes A., Bonisch C., Eiberger B., Christ A., Schilling K. Developmental maturation of the cerebellar white matter-an instructive environment for cerebellar inhibitory interneurons. Cerebellum. 2020;19:286–308. doi: 10.1007/s12311-020-01111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jr., Jungkamp A.C., Munschauer M. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund K., Dikic I. The role of ubiquitylation in receptor endocytosis and endosomal sorting. J. Cell Sci. 2012;125:265–275. doi: 10.1242/jcs.091280. [DOI] [PubMed] [Google Scholar]
- Hardy R.J. Molecular defects in the dysmyelinating mutant quaking. J. Neurosci. Res. 1998;51:417–422. doi: 10.1002/(SICI)1097-4547(19980215)51:4<417::AID-JNR1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Hayakawa-Yano Y., Suyama S., Nogami M., Yugami M., Koya I., Furukawa T., Zhou L., Abe M., Sakimura K., Takebayashi H. An RNA-binding protein, Qki5, regulates embryonic neural stem cells through pre-mRNA processing in cell adhesion signaling. Genes Dev. 2017;31:1910–1925. doi: 10.1101/gad.300822.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y., Suzki N., Tsuboi M., Endo T.A., Toyoda T., Shinga J., Koseki H., Vidal M., Gotoh Y. Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron. 2009;63:600–613. doi: 10.1016/j.neuron.2009.08.021. [DOI] [PubMed] [Google Scholar]
- Hu J., Ho A.L., Yuan L., Hu B., Hua S., Hwang S.S., Zhang J., Hu T., Zheng H., Gan B. From the Cover: neutralization of terminal differentiation in gliomagenesis. Proc. Natl. Acad. Sci. U S A. 2013;110:14520–14527. doi: 10.1073/pnas.1308610110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I., Kageyama R. bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron. 2014;82:9–23. doi: 10.1016/j.neuron.2014.03.018. [DOI] [PubMed] [Google Scholar]
- Ishigaki S., Masuda A., Fujioka Y., Iguchi Y., Katsuno M., Shibata A., Urano F., Sobue G., Ohno K. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci. Rep. 2012;2:529. doi: 10.1038/srep00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeli K., Yeo G.W. Genome-wide approaches to dissect the roles of RNA binding proteins in translational control: implications for neurological diseases. Front. Neurosci. 2012;6:144. doi: 10.3389/fnins.2012.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene J.D. RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 2007;8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C., Polymenidou M., Hutt K.R., Vu A.Q., Baughn M., Huelga S.C., Clutario K.M., Ling S.C., Liang T.Y., Mazur C. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012;15:1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocque D., Galarneau A., Liu H.N., Scott M., Almazan G., Richard S. Protection of p27(Kip1) mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nat. Neurosci. 2005;8:27–33. doi: 10.1038/nn1359. [DOI] [PubMed] [Google Scholar]
- Lennox A.L., Mao H., Silver D.L. RNA on the brain: emerging layers of post-transcriptional regulation in cerebral cortex development. Wiley Interdiscip. Rev. Dev. Biol. 2018;7 doi: 10.1002/wdev.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Takakura N., Oike Y., Imanaka T., Araki K., Suda T., Kaname T., Kondo T., Abe K., Yamamura K. Defective smooth muscle development in qkI-deficient mice. Dev. Growth Differ. 2003;45:449–462. doi: 10.1111/j.1440-169x.2003.00712.x. [DOI] [PubMed] [Google Scholar]
- Li Z., Zhang Y., Li D., Feng Y. Destabilization and mislocalization of myelin basic protein mRNAs in quaking dysmyelination lacking the QKI RNA-binding proteins. J. Neurosci. 2000;20:4944–4953. doi: 10.1523/JNEUROSCI.20-13-04944.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.Z., Kondo T., Murata T., Ebersole T.A., Nishi T., Tada K., Ushio Y., Yamamura K., Abe K. Expression of Hqk encoding a KH RNA binding protein is altered in human glioma. Jpn. J. Cancer Res. 2002;93:167–177. doi: 10.1111/j.1349-7006.2002.tb01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi D.D., Yano M., Fak J.J., Mele A., Grabinski S.E., Zhang C., Darnell R.B. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev. 2012;26:1626–1642. doi: 10.1101/gad.191338.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo L., Simon J.M., Xing L., McCoy E.S., Niehaus J.K., Guo J., Anton E.S., Zylka M.J. Single-cell transcriptomic analysis of mouse neocortical development. Nat. Commun. 2019;10:134. doi: 10.1038/s41467-018-08079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall C.A., Novitch B.G., Goldman J.E. Olig2 directs astrocyte and oligodendrocyte formation in postnatal subventricular zone cells. J. Neurosci. 2005;25:7289–7298. doi: 10.1523/JNEUROSCI.1924-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer D.H., Kane M.F., Mehta S., Liu H., Harrington E., Taylor C.M., Stiles C.D., Rowitch D.H. Separated at birth? The functional and molecular divergence of OLIG1 and OLIG2. Nat. Rev. Neurosci. 2012;13:819–831. doi: 10.1038/nrn3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser C.A., Baptista S., Arnoux I., Audinat E. Microglia in CNS development: shaping the brain for the future. Prog. Neurobiol. 2017;149-150:1–20. doi: 10.1016/j.pneurobio.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Nishiyama A., Komitova M., Suzuki R., Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- Noveroske J.K., Hardy R., Dapper J.D., Vogel H., Justice M.J. A new ENU-induced allele of mouse quaking causes severe CNS dysmyelination. Mamm. Genome. 2005;16:672–682. doi: 10.1007/s00335-005-0035-x. [DOI] [PubMed] [Google Scholar]
- Oberdoerffer P., Otipoby K.L., Maruyama M., Rajewsky K. Unidirectional Cre-mediated genetic inversion in mice using the mutant loxP pair lox66/lox71. Nucleic Acids Res. 2003;31:e140. doi: 10.1093/nar/gng140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozturk N., Singh I., Mehta A., Braun T., Barreto G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front. Cell Dev. Biol. 2014;2:5. doi: 10.3389/fcell.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M., Lagier-Tourenne C., Hutt K.R., Huelga S.C., Moran J., Liang T.Y., Ling S.C., Sun E., Wancewicz E., Mazur C. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch D.H., Kriegstein A.R. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468:214–222. doi: 10.1038/nature09611. [DOI] [PubMed] [Google Scholar]
- Sakakibara S., Nakamura Y., Yoshida T., Shibata S., Koike M., Takano H., Ueda S., Uchiyama Y., Noda T., Okano H. RNA-binding protein Musashi family: roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc. Natl. Acad. Sci. U S A. 2002;99:15194–15199. doi: 10.1073/pnas.232087499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnutgen F., De-Zolt S., Van Sloun P., Hollatz M., Floss T., Hansen J., Altschmied J., Seisenberger C., Ghyselinck N.B., Ruiz P. Genomewide production of multipurpose alleles for the functional analysis of the mouse genome. Proc. Natl. Acad. Sci. U S A. 2005;102:7221–7226. doi: 10.1073/pnas.0502273102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingu T., Ho A.L., Yuan L., Zhou X., Dai C., Zheng S., Wang Q., Zhong Y., Chang Q., Horner J.W. Qki deficiency maintains stemness of glioma stem cells in suboptimal environment by downregulating endolysosomal degradation. Nat. Genet. 2017;49:75–86. doi: 10.1038/ng.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt C.C., Rehberg S., Ader M., Lommes P., Riethmacher D., Schachner M., Bartsch U., Wegner M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 2002;16:165–170. doi: 10.1101/gad.215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A., Iida K., Tsubota T., Hosokawa M., Denawa M., Brown J.B., Ninomiya K., Ito M., Kimura H., Abe T. Loss of sfpq causes long-gene transcriptopathy in the brain. Cell Rep. 2018;23:1326–1341. doi: 10.1016/j.celrep.2018.03.141. [DOI] [PubMed] [Google Scholar]
- Tsoa R.W., Coskun V., Ho C.K., de Vellis J., Sun Y.E. Spatiotemporally different origins of NG2 progenitors produce cortical interneurons versus glia in the mammalian forebrain. Proc. Natl. Acad. Sci. U S A. 2014;111:7444–7449. doi: 10.1073/pnas.1400422111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J., Ule A., Spencer J., Williams A., Hu J.S., Cline M., Wang H., Clark T., Fraser C., Ruggiu M. Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 2005;37:844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- Yamashita Y., Matsuura T., Shinmi J., Amakusa Y., Masuda A., Ito M., Kinoshita M., Furuya H., Abe K., Ibi T. Four parameters increase the sensitivity and specificity of the exon array analysis and disclose 25 novel aberrantly spliced exons in myotonic dystrophy. J. Hum. Genet. 2012;57:368–374. doi: 10.1038/jhg.2012.37. [DOI] [PubMed] [Google Scholar]
- Zearfoss N.R., Clingman C.C., Farley B.M., McCoig L.M., Ryder S.P. Quaking regulates Hnrnpa1 expression through its 3' UTR in oligodendrocyte precursor cells. PLoS Genet. 2011;7:e1001269. doi: 10.1371/journal.pgen.1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Chen M.H., Wu X., Kodani A., Fan J., Doan R., Ozawa M., Ma J., Yoshida N., Reiter J.F. Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell. 2016;166:1147–1162.e15. doi: 10.1016/j.cell.2016.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Chen K., Sloan S.A., Bennett M.L., Scholze A.R., O'Keeffe S., Phatnani H.P., Guarnieri P., Caneda C., Ruderisch N. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Lu Z., Ku L., Chen Y., Wang H., Feng Y. Tyrosine phosphorylation of QKI mediates developmental signals to regulate mRNA metabolism. EMBO J. 2003;22:1801–1810. doi: 10.1093/emboj/cdg171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Ku L., Chen Y., Xia M., LoPresti P., Feng Y. QKI binds MAP1B mRNA and enhances MAP1B expression during oligodendrocyte development. Mol. Biol. Cell. 2006;17:4179–4186. doi: 10.1091/mbc.E06-04-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer D.B., Cornwall E.H., Landar A., Song W. The S100 protein family: history, function, and expression. Brain Res. Bull. 1995;37:417–429. doi: 10.1016/0361-9230(95)00040-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the GEO under accession no. GSE117018. In-house scripts were used to calculate RPKM and TPM. All data and scripts not included here are available from the corresponding author upon reasonable request.