

Abstract
Aberrant activation of nuclear factor κB (NF-κB)/RELA is often found in lung adenocarcinoma (LUAD). In this study, we determined that microRNA-3613-5p (miR-3613-5p) plays a crucial role in RELA-mediated post-transcriptional regulation of LUAD cell proliferation. Expression of miR-3613-5p in clinical LUAD specimens is associated with poor prognosis in LUAD. Upregulation of miR-3613-5p promotes LUAD cell proliferation in vitro and in vivo. Our results suggested a mechanism whereby miR-3613-5p expression is induced by RELA through its direct interaction with JUN, thereby stimulating the AKT/mitogen-activated protein kinase (MAPK) pathway by directly targeting NR5A2. In addition, we also found that phosphorylation of AKT1 and MAPK3/1 co-transactivates RELA, thus constituting a RELA/JUN/miR-3613-5p/NR5A2/AKT1/MAPK3/1 positive feedback loop, leading to persistent NF-κB activation. Our findings also revealed that miR-3613-5p plays an oncogenic role in LUAD by promoting cell proliferation and acting as a key regulator of the positive feedback loop underlying the link between the NF-κB/RELA and AKT/MAPK pathways.
Graphical Abstract
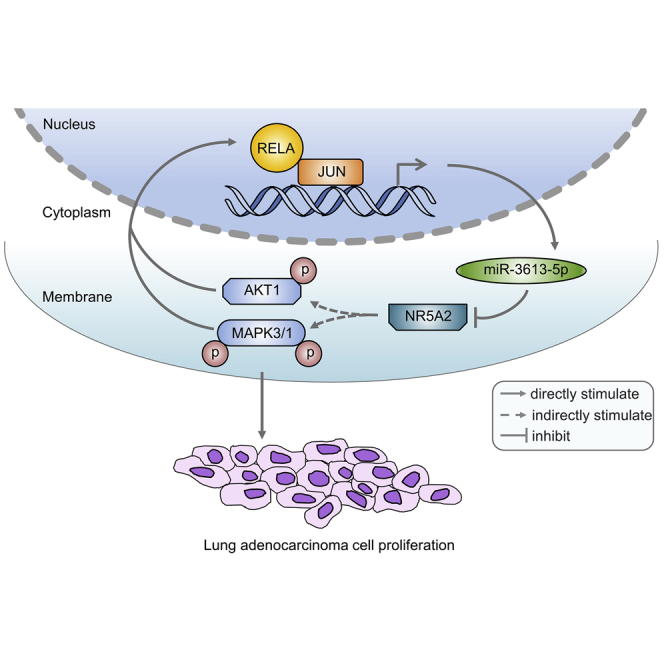
Aberrant activation of nuclear factor κB (NF-κB)/RELA is often found in lung adenocarcinoma (LUAD). However, RELA as a direct therapeutic target for lung cancer remains a challenge. This study investigated that microRNA-3613-5p (miR-3613-5p) plays a crucial role in RELA-mediated post-transcriptional regulation of LUAD cell proliferation.
Introduction
Lung cancers, most of which are classified as non-small-cell lung cancer (NSCLC), have been the leading cause of cancer incidence and deaths worldwide according to the latest statistics.1 Despite the outstanding advances in novel therapeutic approaches and drug development, the clinical prognosis for patients with lung cancer remains poor, and their 5-year overall survival rate is a disappointing 15%.2, 3, 4 Among NSCLCs, lung adenocarcinoma (LUAD) is one of the important histological subtypes with high incidence and mortality.5, 6, 7 In early-stage LUAD patients, tumor size and lymph node metastasis are considered to be major factors in the development of treatment strategies, as well as prognostic factors.8,9 However, the diagnosis and treatment of LUAD patients are frequently delayed due to lack of specific early symptoms. Accordingly, it is essential and urgent to further elucidate the molecular mechanisms involved in LUAD progression to identify more effective diagnostic biomarkers and precise therapeutic targets.
Nuclear factor κB (NF-κB) is a ubiquitously expressed transcription factor involved in the regulation of multiple biological progresses.10,11 Aberrant or uncontrolled activation of NF-κB is critical in most of lung cancer initiation and progression.12, 13, 14 NF-κB induces the expression of various proteins that are involved in inflammation and carcinogenesis, such as FAS, cyclins, interleukin (IL)-2, tumor necrosis factor (TNF), CXCL1, and matrix metallopeptidases (MMPs), which promote lung tumor cell survival and proliferation, inflammatory response, anti-apoptosis, angiogenesis, invasion, and metastasis.15, 16, 17 Activation of the canonical (or classical) NF-κB pathway leads to the heterodimerization of the RELA (also known as p65) subunit with the p50 subunit and its translocation to the nucleus, where it regulates the transcription of target genes.15 The persistent activation of NF-κB (especially RELA) in lung tumorigenesis has been reported.18,19 However, RELA as a direct therapeutic target for lung cancer remains a challenge, since it regulates a plethora of genes related to fundamental physiological processes. Consequently, it is necessary to elucidate the underlying mechanisms of RELA post-transcriptional regulation involved in lung cancer progression.
MicroRNAs (miRNAs) are endogenous small 19- to 25-nt-long non-coding RNAs, which post-transcriptionally regulate the expression of target mRNAs by imperfectly binding to their 3′ untranslated regions (UTRs), ultimately leading to translational repression or degradation. miRNAs play important regulatory roles in cellular processes, including cell growth, migration, and invasion,20, 21, 22 along with chemoresistance.23, 24, 25 Indeed, accumulating evidence demonstrates that miRNA dysregulation is implicated in many human malignancies, including LUAD.26,27
Mature miR-3613-3p and miR-3613-5p are encoded by a common primary gene. miR-3613-3p is also designated as miR-3613 in the miRBase database. In a small number of studies, miR-3613-3p was reported to function as an oncogene or tumor suppressor in various cancers. In particular, miR-3613-3p is reported to participate in neuroblastoma pathogenesis and differentiation of neuronal cells.28 Additionally, miR-3613-3p also acts as an oncogene by inhibiting the expression of RB1, thereby promoting retinoblastoma progression.29 Conversely, EGFR pathway genes are upregulated by suppression of miR-3613-3p expression during the epithelial-mesenchymal transition (EMT) process in LUAD.30 However, the role of miR-3613-5p and its molecular mechanisms underlying the progression of LUAD remain entirely unclear.
Therefore, this study is aimed to explore the relationship between the expression of RELA and miR-3613-5p, clinical correlation, and dysregulated mechanism in LUAD progression. Overall, our results provide a mechanism by which miR-3613-5p plays an oncogenic role in promoting LUAD cell proliferation.
Results
Overexpression of RELA Positively Modulates miR-3613-5p Transcription In Vitro
Analysis of cell proliferation by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and cell cycle progression by an ethynyl deoxyuridine (EdU) incorporation assay revealed that RELA overexpression in A549 and H1299 cells significantly promoted cell proliferation (Figure S1A) and G1 to S cell cycle phase transition (Figure S1B), respectively. The evaluation of the effect of RELA overexpression on the expression of miRNAs in human LUAD A549 cells by miRNA sequencing (miRNA-seq) analysis identified 12 markedly increased miRNAs, which included miR-3613-5p, miR-1277-5p, miR-5010-3p, and miR-34b-5p (Figure 1A). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis revealed that the target genes of differentially expressed miRNAs were related to the mitogen-activated protein kinase (MAPK) signaling pathway (Figure S1C). Consistent with the results of the miRNA sequencing analysis, miR-3613-5p was confirmed as a positive regulator of RELA by quantitative real-time PCR analysis in A549 and H1299 cells transfected with a negative control (NC) vector or the RELA plasmid (Figure 1B).
Figure 1.

Clinicopathological Features of miR-3613-5p Expression
(A) The portion of the differentially expressed miRNAs in RELA-overexpressing A549 cells and its fold change. Red frame, miR-3613-5p. (B) Quantitative real-time PCR analysis of miR-3613-5p expression in A549 and H1299 cells after treatment with negative control (NC) or RELA plasmid, normalized to U6. Mean ± SD. ∗∗p < 0.01, ∗∗∗p < 0.001, by Student’s t test. (C) miR-3613-5p expression was detected by quantitative real-time PCR in normal lung samples and LUAD samples, normalized to U6. Mean ± SD. ∗∗∗p < 0.001, by Student’s t test. (D) miR-3613-5p expression in LUAD TMA. (a) Weak expression of miR-3613-5p in normal lung samples; (b) strong expression of miR-3613-5p in normal lung samples; (c) weak expression of miR-3613-5p in LUAD samples; and (d) strong expression of miR-3613-5p in LUAD samples (original magnification, ×200). Scale bars, 20 μm. (E) Kaplan-Meier survival analysis of overall survival of 92 LUAD patients based on miR-3613-5p expression. The log-rank test was used to calculate p values.
Clinicopathological Features of miR-3613-5p Expression
High miR-3613-5p expression was detected in primary fresh LUAD samples by quantitative real-time PCR analysis (Figure 1C) as well as in the LUAD tissue microarray (TMA) by in situ hybridization assays (Figure 1D; Table 1). Clinical features associated with miR-3613-5p are summarized in Table 2. High levels of miR-3613-5p were positively correlated with the age of the patient (χ2 test, p = 0.017) and tumor size (T classification) (χ2 test, p = 0.003), but not other clinical factors. Additionally, Kaplan-Meier survival analysis demonstrated that patients with high expression of miR-3613-5p had poorer overall survival rates (Figure 1E).
Table 1.
The Expression of miR-3613-5p in LUAD Compared to Normal Lung Tissues
| Group | Cases (n) | miR-3613-5p Expression (%) |
p Value | |
|---|---|---|---|---|
| Low Expression | High Expression | |||
| Cancer | 92 | 31 (33.7) | 61 (66.3) | 0.005 |
| Normal | 88 | 48 (54.5) | 40 (45.5) | |
A χ2 test was applied to access the expression of miR-3613-5p in LUAD samples and normal lung samples. LUAD, lung adenocarcinoma.
Table 2.
Correlation between the Clinicopathologic Characteristics and Expression of miR-3613-5p in LUAD
| Characteristics | Cases (n) | miR-3613-5p Expression (%) |
p Value | |
|---|---|---|---|---|
| High Expression | Low Expression | |||
| Age | ||||
| <55 | 21 | 11 (52.4) | 10 (47.6) | 0.017 |
| ≥55 | 71 | 56 (78.9) | 15 (21.1) | |
| Type | ||||
| LUAD | 86 | 57 (66.3) | 29 (33.7) | 0.985 |
| Other | 6 | 4 (66.7) | 2 (33.3) | |
| Sex | ||||
| Male | 51 | 32 (62.7) | 19 (37.3) | 0.421 |
| Female | 41 | 29 (70.7) | 12 (29.3) | |
| Clinicopathologic Stage | ||||
| I | 1 | 0 | 1 (100) | 0.362 |
| II | 56 | 38 (67.9) | 18 (32.1) | |
| III | 35 | 23 (65.7) | 12 (34.3) | |
| TNM classification | ||||
| T | ||||
| T1 | 28 | 11 (39.3) | 17 (60.7) | 0.003 |
| T2 | 42 | 32 (76.2) | 10 (23.8) | |
| T3 | 14 | 12 (85.7) | 2 (14.3) | |
| T4 | 8 | 6 (75) | 2 (25) | |
| N | ||||
| N0 | 48 | 30 (62.5) | 18 (37.5) | 0.591 |
| N1 | 25 | 17 (68) | 8 (32) | |
| N2 | 16 | 11 (68.8) | 5 (31.2) | |
| N3 | 3 | 3 (100) | 0 | |
| M | ||||
| M0 | 90 | 59 (65.6) | 31 (34.4) | 0.308 |
| M1 | 2 | 2 (100) | 0 | |
| Clinical stage | ||||
| I | 39 | 23 (59) | 16 (41) | 0.443 |
| II | 24 | 16 (66.7) | 8 (33.3) | |
| III | 27 | 20 (74.1) | 7 (25.9) | |
| IV | 2 | 2 (100) | 0 | |
A χ2 test was applied to access the associations between miR-3613-5p expression and the clinicopathological parameters. LUAD, lung adenocarcinoma; TNM, tumor, node, and metastasis.
Upregulation of miR-3613-5p Promotes LUAD Cell Proliferation
The analysis of the biological effects of miR-3613-5p on LUAD cells (A549 and H1299) separately transfected with mimics, inhibitor, precursor (hsa-miR-3613-5p) lentivirus particles, and their negative controls by the MTT assay (Figure S2A), EdU incorporation assay (Figure S2B), and the colony formation assay (Figure S2C) revealed that miR-3613-5p overexpression markedly promotes cell growth and G1-to-S cell cycle transition in A549 and H1299 cells in vitro. Conversely, the miR-3613-5p inhibitor clearly decreased cell viability and delayed G1-to-S cell cycle transition in A549 and H1299 cells (Figures S2D and S2E).
In vivo tumorigenesis assay after subcutaneously injecting LV-miR-3613-5p-enhanced green fluorescent protein (eGFP) A549 and H1299 cells or control cells (Figures S2F and S2G) into nude mice showed clearly increased xenograft growth in the miR-3613-5p overexpression group relative to the control group (Figures 2A and 2B). Immunohistochemistry (IHC) analysis confirmed that these tumor tissues also expressed higher levels of proliferating cell nuclear antigen (PCNA) and Ki-67 compared with control tissues (Figure 2C), suggesting that miR-3613-5p significantly promotes LUAD cell growth in vivo.
Figure 2.

miR-3613-5p Promotes LUAD Cell Proliferation In Vitro and In Vivo by Activating the AKT/MAPK Pathway
(A) LV-miR-3613-5p markedly increased xenograft growth (n = 5/group). (B) The tumor volume was periodically measured for each mouse. Mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Student’s t test, one-way ANOVA, and Dunnett’s multiple comparison test. (C) Representative H&E staining as well as IHC staining of Ki-67 and PCNA of xenografts in groups indicated in (A) (original magnification, ×200). Scale bars, 20 μm. (D) Western blot assay was used to analyze the expression of AKT1, p-AKT1, MAPK3/1, p-MAPK3/1, CCND1, CDKN1B, and CDKN1A in the AKT/MAPK pathway after treatment with miR-3613-5p mimics in LUAD cells. GAPDH was used as a NC. (E) The expression of relevant proteins in the AKT/MAPK pathway was measured by western blot experiments after transfection with miR-3613-5p inhibitor, as shown. GAPDH served as a NC. (F) The downregulation of p-AKT1, p-MAPK3/1, and CCND1 and upregulation of CDKN1B and CDKN1A were detected by western blot assay after administration of miR-3613-5p inhibitor in RELA-overexpressing LUAD cells.
RELA Positively Regulates the AKT/MAPK Pathway via miR-3613-5p
Western blot analysis of the major mediators of AKT/MAPK signaling in miR-3613-5p-overexpressing A549 and H1299 cells revealed upregulated levels of phosphorylated (p-)AKT1, p-MAPK3/1, and CCND1 and downregulated levels of CDKN1B and CDKN1A (Figure 2D). In contrast, p-AKT1, p-MAPK3/1, and CCND1 expression was downregulated, and CDKN1B and CDKN1A expression was upregulated (Figure 2E), in LUAD cells treated with the miR-3613-5p inhibitor. These findings suggest the likely involvement of these proteins in the molecular mechanism by which miR-3613-5p positively regulates LUAD cell proliferation through activation of the AKT/MAPK signaling pathway.
In addition, we also found, in rescue experiments, that transient expression of the miR-3613-5p inhibitor in RELA proliferation-promoted A549 and H1299 cells remarkably restored the suppression of cell growth (Figure S2H) and G1-to-S cell cycle transition (Figure S2I). The western blot analysis results showed that the miR-3613-5p inhibitor prevented the RELA-mediated upregulation of p-AKT1, p- MAPK3/1, and CCND1, as well as downregulation of CDKN1B and CDKN1A (Figure 2F). These findings reveal that miR-3613-5p is essential to RELA-mediated promotion of LUAD cell proliferation and upregulates the AKT/MAPK signaling pathway.
miR-3613-5p Directly Targets NR5A2 to Promote Cell Proliferation by Activating the AKT/MAPK Pathway
NR5A2 was predicted to be a direct target gene of miR-3613-5p by the TargetScan and miRPathDB algorithms. The predicted binding site was among the miR-3613-5p seed sequence and NR5A2 3′ UTR region (Figure S3A). The UALCAN software (http://ualcan.path.uab.edu/index.html) analysis reported that NR5A2 is lowly expressed in primary LUAD tumor compared with normal lung tissue (Figure S3B). Furthermore, LUAD patients with low expression of NR5A2 had poorer overall survival rates (Figure S3C) by analysis of the Kaplan-Meier plotter datasets (http://kmplot.com/analysis/). These findings indicated that low expression of NR5A2 is correlated with LUAD progression. In addition, overexpression of miR-3613-5p downregulated NR5A2 protein levels in A549 and H1299 cells (Figure 3A), whereas the opposite result was obtained by suppressing miR-3613-5p expression with an inhibitor. Also, IHC analysis revealed that the expression of NR5A2 was significantly downregulated in xenografts generated from A549 and H1299 cells overexpressing miR-3613-5p (Figure 3B). The results of the luciferase reporter assay (Figure 3C) revealed that the luciferase activity of wild-type NR5A2 3′ UTR was downregulated by miR-3613-5p mimics compared to the negative group (∗∗∗p < 0.001), but it was upregulated by the miR-3613-5p inhibitor (∗∗∗p < 0.001). Taken together, these findings indicate that miR-3613-5p promotes LUAD progression through direct suppression of NR5A2 expression.
Figure 3.

miR-3613-5p Directly Targets NR5A2 to Activate the AKT/MAPK Pathway
(A) NR5A2 protein levels in miR-3613-5p-overexpressing or miR-3613-5p-suppressing LUAD cells by western blot analysis; β-tubulin served as a NC. (B) IHC staining of NR5A2 in xenografts derived from miR-3613-5p or NC-transfected LUAD cells. Scale bars, 20 μm. (C) miR-3613-5p directly targets NR5A2 as confirmed by a luciferase reporter assay. Mean ± SD. ∗∗∗p < 0.001, one-way ANOVA and Dunnett’s multiple comparison test. (D) Western blot analyzed the expression of AKT1, p-AKT1, MAPK3/1, p-MAPK3/1, CCND1, CDKN1B, and CDKN1A in LUAD cells following NR5A2 siRNA transfection. GAPDH was used as a NC. (E and F) MTT assay (E) and EdU incorporation assay (F) in NR5A2-overexpressing LUAD cells treated with miR-3613-5p mimics. Mean ± SD. ∗∗∗p < 0.001, by Student’s t test, one-way ANOVA. (G) Downregulation of NR5A2 expression in NR5A2-overexpressing LUAD cells was detected by western blot after transfection with miR-3613-5p mimics. GAPDH served as a NC.
Transient expression of NR5A2 small interfering RNA (siRNA) in A549 and H1299 cells resulted in increased protein levels of p-AKT1, p-MAPK3/1, and CCND1 and decreased levels of NR5A2, CDKN1B, and CDKN1A by western blot analysis (Figure 3D). Additionally, the analysis of cell proliferation and cell cycle progression by the MTT assay (Figure 3E) and EdU incorporation assay (Figure 3F) demonstrated that the transient expression of miR-3613-5p in LUAD cells overexpressing NR5A2 enhanced cell proliferation. Also, the level of NR5A2 was reduced in the same rescue experiment (Figure 3G). These results suggest that NR5A2 overexpression can reverse the promotion of LUAD cell proliferation by miR-3613-5p.
JUN Activates miR-3613-5p Transcription by Binding to Its Promoter Region
The regulatory mechanisms of miR-3613-5p were investigated at the transcriptional level by various approaches. The ALGGEN (http://alggen.lsi.upc.es) and JASPAR (http://jaspar.genereg.net) bioinformatics software predicted that a 2-kb region upstream of the transcription initiation site of miR-3613-5p contains two putative JUN-binding sites (Figure 4A), referred to as P1 (−752 to −764) and P2 (−815 to −821). The analysis of the role of JUN in the regulation of miR-3613-5p expression by quantitative real-time PCR showed that miR-3613-5p expression was significantly increased in JUN-overexpressing A549 and H1299 cells (Figure S4A), indicating that JUN is an upstream regulator of miR-3613-5p expression.
Figure 4.

RELA Directly Interacts with and Upregulates JUN expression, Which Activates miR-3613-5p Transcription by Binding to Its Promoter Region
(A) Schematic diagram of the promoter regions of miR-3613-5p with two putative JUN-binding sites (P1 and P2). (B) PCR gel exhibiting amplification of JUN-binding sites P1 and P2 after ChIP using antibody against JUN. IgG antibody served as a NC. (C) EMSA of JUN binding to the miR-3613-5p promoter in LUAD cells. The biotin-labeled wild-type probe was incubated without (lanes 1 and 6) or with (lanes 4 and 9) cell nuclear extract in the absence or presence of unlabeled probe (lanes 2, 3, 7, and 8). Unlabeled JUN wild-type probe (lanes 2 and 7) and JUN mutant probe (lanes 3 and 8) were used to compete with JUN binding, each at 100-fold excess. A supershift assay (lanes 5 and 10) was performed using an anti-JUN antibody. (D) Western blot analysis showing interaction between RELA and JUN in endogenous (A549 and H1299 cells) and exogenous (HEK293T cells) coIP assays. IgG antibody was used as a NC. (E) Nucleus colocalization of RELA protein and JUN protein in LUAD cells by immunofluorescence analysis (original magnification, ×400). RELA protein labeled with DyLight 594 (red fluorescence). JUN protein labeled with DyLight 488 (green fluorescence). Nuclear protein labeled with DAPI (blue fluorescence). (F) Upregulation of JUN expression in RELA-overexpressing LUAD cells was detected by western blot; β-tubulin served as a NC. (G) The expression of JUN, NR5A2, AKT1, p-AKT1, MAPK3/1, p-MAPK3/1, CCND1, CDKN1B, and CDKN1A in LUAD cells after RELA plasmid and JUN siRNA co-treatment by western blot. GAPDH was used as a NC.
Additionally, chromatin immunoprecipitation (ChIP) analysis confirmed that JUN can bind to the promoter region of miR-3613-5p in LUAD cells. In particular, compared with the negative control (immunoglobulin G [IgG]), positive results (Figure 4B) indicated the presence of JUN in products from the ChIP assays, detected with the JUN antibody by DNA electrophoresis assays. The analysis of the functionality of the JUN-binding sites P1 and P2 by an electrophoretic mobility shift assay (EMSA), as shown in Figure 4C, detected shifted bands when the LUAD cell nuclear extracts were incubated with the biotin-labeled probe (lanes 4 and 9), but not with the unlabeled JUN wild-type competitor (lanes 2 and 7). Shifted bands did not disappear in the presence of mutated P1 or P2 competitors (lane 3 and 8), whereas supershifted bands appeared in the presence of the JUN antibody (lanes 5 and 10). Collectively, these results revealed that JUN binds directly to the promoter sequence of miR-3613-5p and regulates its expression.
RELA Directly Interacts with JUN and Stimulates Its Expression
The evaluation of the interaction between RELA and JUN through the analysis of the STRING (https://string-db.org/) and BioGRID (https://thebiogrid.org/) databases predicted that RELA interacts with JUN in the same interaction network (Figures S4B and S4C). We also confirmed that RELA and JUN interact in A549, H1299, and HEK293T cells by exogenous and endogenous coimmunoprecipitation (coIP) assays (Figure 4D). Immunofluorescence analysis demonstrated that RELA and JUN colocalize in the nucleus (Figure 4E). Taken together, these data indicated that RELA directly interacts with JUN in LUAD cells.
In further research, western blot analysis found increased JUN expression in RELA-overexpressing A549 and H1299 cells (Figure 4F), indicating that RELA upregulates JUN expression. Rescue experiments results (Figure 4G) confirmed the downregulation of JUN, p-AKT1, p-MAPK3/1, and CCND1 and upregulation of NR5A2, CDKN1B, and CDKN1A in LUAD cells after co-transfection with RELA plasmid and siRNA JUN. These findings suggested that RELA promotes LUAD cell proliferation by stimulating JUN expression.
RELA Is Co-transactivated by Phosphorylation of AKT1 and MAPK3/1, Constituting a Positive Feedback Loop
The results (Figure 5A) of the quantitative real-time PCR analysis of RELA-overexpressing A549 and H1299 cells, treated with the specific AKT1 inhibitor MK-2206 2HCl and MAPK3/1 inhibitor SCH772984, demonstrated that both inhibitors significantly decreased miR-3613-5p expression, especially in co-treated cells, compared with similar cells without inhibitor treatment. Furthermore, western blot analysis also showed that the miR-3613-5p inhibitor and combination of MK-2206 2HCl and SCH772984 notably reversed the expression of p-AKT1 or p-MAPK3/1, RELA, and JUN (Figure 5B).
Figure 5.

RELA Is Co-transactivated by Phosphorylation of AKT1 and MAPK3/1
(A) Quantitative real-time PCR analysis showed that miR-3613-5p upregulation was abrogated by treatment with MK-2206 2HCl, SCH772984, or MK-2206 2HCl and SCH772984 in RELA-overexpressing LUAD cells, normalized to U6. Mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, by Student’s t test. (B) Western blot analysis demonstrates the downregulation of p-AKT1, p-MAPK3/1, RELA, and JUN in RELA-overexpressing LUAD cells after MK-2206 2HCl, SCH772984, a combination of MK-2206 2HCl and SCH772984, or miR-3613-5p inhibitor treatment. GAPDH served as a NC.
Discussion
This study aimed to investigate the function of miR-3613-5p in LUAD carcinogenesis and identify the signaling pathway mediating the cell proliferation-promoting effect of miR-3613-5p on LUAD cells. Our results demonstrated that miR-3613-5p promotes LUAD cells proliferation via a RELA/JUN/NR5A2/AKT/MAPK positive feedback loop. In addition, blockade of this feedback loop by miR-3613-5p inhibitor reverses the proliferation of LUAD cells.
The 5-year survival rate of NSCLC (mostly LUAD) patients who undergo surgical resection after being diagnosed with early stage can surpass 53%.31 Thus, patients’ overall survival could be improved by detection and therapy for LUAD as early as possible. In the early stage of lung carcinogenesis, uncontrolled cell proliferation is a major feature. Aberrant activation of NF-κB/RELA and dysregulation of miRNAs are correlated with cell proliferation in NSCLC. Subsequent studies reported that miRNAs influence cell proliferation in NSCLC by degrading RELA or inhibiting its expression. For instance, miR-506 inhibits RELA expression to selectively eliminate lung cancer cells by generating active oxygen and p53 activation.32 In KRAS-mutated NSCLC, miR-124 disrupts autophagy and reduces cell viability by suppressing RELA expression.33 Although many studies have focused on the mechanism by which miRNAs regulate RELA, very little is known about the mechanism by which RELA modulates miRNAs at the post-transcriptional level.
In this study, miR-3613-5p, which was screened by miRNA sequencing and verified by quantitative real-time PCR analysis of RELA-overexpressing LUAD cells, was upregulated by RELA in vitro. High miR-3613-5p expression was detected in primary fresh LUAD specimens by quantitative real-time PCR analysis. In LUAD TMAs, in situ hybridization analysis showed that miR-3613-5p levels were significantly increased in LUAD tissue samples compared with adjacent normal lung tissue samples. We also found that high expression of miR-3613-5p was positively correlated with the age of the patient and tumor size in accordance with the analysis of the LUAD TMA data. In addition, patients with a high level of miR-3613-5p had poorer overall survival compared to patients with a low level. These results indicate that miR-3613-5p acts as an oncogene in LUAD and is associated with the progression of LUAD.
In this study, miR-3613-5p was first clearly shown to promote LUAD cell proliferation in vitro and xenograft tumor growth in vivo. The crosstalk of NF-κB/RELA with other transcription factors and signaling proteins creates a complex functional network between the tumor and its microenvironment.34,35 Cell proliferation is driven by interdependent complicated signal transduction networks. The MAPK/extracellular signal-regulated kinase (ERK) and phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathways are two of the major cell proliferation pathways.36,37 The analysis of the molecular mechanism in this study showed that miR-3613-5p promotes LUAD cell proliferation through stimulation of the AKT/MAPK pathway and cell cycle G1-to-S transition. The KEGG pathway enrichment analysis predicted that the target genes of differentially expressed miRNAs were associated with MAPK signaling pathways, which is also verified by the experimental results of this study.
We found that NR5A2, a downstream target gene of miR-3613-5p, is essential for mediating oncogenic signaling in LUAD cells in vitro and LUAD carcinogenesis in vivo. To the best of our knowledge, this study is the first to demonstrate the involvement of NR5A2 downregulation in in vitro and in vivo models of LUAD cell proliferation. NR5A2 is an orphan nuclear receptor member of the NR5A (Ftz-F1) subfamily and is expressed in endoderm-derived tissues, including the lung.38 It plays important roles in various biological processes, such as development, differentiation, cholesterol metabolism, and steroidogenesis.39, 40, 41, 42 Accumulating evidence has demonstrated that NR5A2 is implicated in the pathogenesis of various cancers, especially pancreatic cancer, breast cancer, gastric cancer, and intestinal cancer.43, 44, 45, 46 To date, very few studies have been reported on the association between NR5A2 and LUAD. The Cancer Genome Atlas (TCGA) (LUAD) database and IHC analysis showed that NR5A2 is downregulated in LUAD and xenografts tumor tissues, and its low level is associated with LUAD progression. Accordingly, we speculated that NR5A2 acts as a tumor suppressor gene in LUAD. Our study demonstrated that NR5A2 expression is directly suppressed by miR-3613-5p, which leads to the promotion of the proliferation of LUAD cells through activation of the AKT/MAPK signaling pathway. Future research on NR5A2-mediated activation of the the AKT/MAPK signaling pathway ought to fully clarify the mechanism underlying the NR5A2 function in LUAD tumorigenesis.
The expression of miRNAs is modulated via the binding of transcription factors to sites in sequences within the upstream promoter region of miRNA genes.47 According to the prediction from bioinformatics datasets and quantitative real-time PCR analysis, JUN might be related with transcriptional upregulation of miR-3613-5p. As a crucial transcription factor, JUN (an AP-1 family member) plays a role in cell proliferation, metastasis, and drug resistance in a variety of cancers by affecting the expression of miRNAs.48,49 Additional experimental results proved that JUN can directly bind to the promoter region of miR-3613-5p and stimulate its expression. RELA can crosstalk with various transcription factors. Based on database predictions and some reports,50,51 we hypothesized that RELA might interact with JUN in LUAD cells. Further experiments revealed that RELA directly interacts with JUN and promotes JUN-mediated activation of the AKT/MAPK signaling pathway. Several studies have reported that the transactivation of NF-κB is involved in the AKT or MAPK signaling pathway. Moreover, phosphorylation of AKT1 has been shown to specifically activate NF-κB through degradation of the NF-κB inhibitor IκB.52 In melanoma cells, the NF-κB-inducing kinase induces NF-κB activation by increasing MAPK3/1 phosphorylation and IκB kinase activity.53 In this study, experimental results verified that the phosphorylation of AKT1 and MAPK3/1 also enhances the co-transactivation of RELA, constituting a RELA/JUN/NR5A2/AKT/MAPK positive feedback loop. Also, the AKT1 inhibitor or MAPK3/1 inhibitor impeded the activation of miR-3613-5p and the positive feedback loop. In contrast, the miR-3613-5p inhibitor or a combination of AKT1 and MAPK3/1 inhibitors is more effective in dual pathway blockade. Moreover, the level of suppression of the miR-3613-5p inhibitor is close to that of the combination of AKT1 and MAPK3/1 inhibitors.
Materials and Methods
Cell Culture
The human LUAD cell lines A549 and H1299 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and authenticated by short tandem repeat (STR) profiling. The HEK293T cell line was acquired from the Integrated Hospital of Traditional Chinese Medicine (TCM-Integrated Hospital) of Southern Medical University (Guangzhou, P.R. China). These cell lines were analyzed for mycoplasma and were verified to be mycoplasma-free. A549 and H1299 cells were cultured in RPMI 1640 medium (Biological Industries, Bet Haemek, Israel) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO, USA). HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Biological Industries) supplemented with 10% FBS. All cells were incubated in a humidified incubator at 37°C with 5% CO2. The AKT1 inhibitor MK-2206 2HCl and the MAPK3/1 inhibitor SCH772984 were purchased from Selleck Chemicals (Houston, TX, USA). For inhibitor treatment, 5 mmol/L MK-2206 2HCl and/or 5 mmol/L SCH772984, or dimethyl sulfoxide (DMSO; Sigma-Aldrich) alone for control, were freshly added to the cell culture every 24 h.
Clinical Sample Collection
A total of 26 fresh tissue samples of primary LUAD specimens and the paired fresh normal tissue samples were collected from the TCM-Integrated Hospital of Southern Medical University. Then, all of these samples were immediately frozen and stored in liquid nitrogen for subsequent RNA extraction. Informed consent was obtained from all patients. Approval for the collection and use of human tissue for research purposes was obtained from the Medical Ethics Committee of the TCM-Integrated Hospital of Southern Medical University. A TMA that included 92 paraffin-embedded primary LUAD specimens and 88 adjacent normal lung specimens was purchased from Outdo Biotech (Shanghai, P.R. China). Clinical and statistical data were obtained from the patients’ medical records.
Cell Transfection
RELA and JUN plasmids were purchased from Vigene Biosciences (Shandong, P.R. China). miR-3613-5p mimics and its inhibitor or siRNA for NR5A2 and JUN were designed and synthesized by RiboBio (Guangzhou, P.R. China) (Table S1). Lentiviral particles containing the hsa-miR-3613-5p precursor and its flanking control sequence, as well as the NR5A2 plasmid, were constructed by GeneChem (Shanghai, P.R. China). For plasmid and oligonucleotide transfection, A549 and H1299 cells were plated into 6- or 96-well plates (Corning, Corning, NY, USA) and cultured for 24 h until they reached a confluence of 30%–50% and then transfected with Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) in Opti-MEM (Gibco/Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturers’ instructions. Cells were collected after 48–72 h for further experiments. LUAD cells were infected with lentiviral vector with an eGFP tag, and expression of miR-3613-5p was confirmed by quantitative real-time PCR.
RNA Preparation and Quantitative Real-Time PCR
Total RNA from the LUAD tissue samples and cell lines was extracted using RNAiso Plus reagent (Takara Bio, Shiga, Japan). The extracted RNA was reverse transcribed into cDNA. The mRNA and miRNA expression levels were measured using the ChamQ Universal SYBR qPCR master mix kit (Vazyme Biotech, Nanjing, P.R. China) with specific primers (Table S2) according to the manufacturer’s protocols. ARF5 and U6 were selected as internal controls for genes and miRNAs, respectively. Independent experiments were performed in triplicate.
High-Throughput miRNA Sequencing and Data Analysis
miRNA sequencing was carried out by Genesky Bio-Tech (Shanghai, P.R. China). Total RNA was isolated and purified from RELA overexpressing or control A549 cells. At least 2 μg of qualified RNA samples with a high integrity number (RIN ≥ 7.0) was used for library construction using the TruSeq small RNA sample library preparation kit (Illumina, San Diego, CA, USA) and sequenced on an Illumina HiSeq 2500 sequencing system (Illumina). The MiRDeep2 software was used to analyze miRNAs and their expression data. Differential gene expression analysis was estimated with the DESeq2 Bioconductor software package. p < 0.05 and log2 fold change (FC) ≠ 0 were defined as the differential expression of miRNA between the two groups, in which log2FC > 0 was labeled up and log2FC < 0 was labeled down.
Cell Proliferation Assay
Cellular proliferation was measured using the MTT assay. For transient transfection, the LUAD cells transfected with RELA plasmid, NR5A2 plasmid, miR-3613-5p mimics, and inhibitor were separately cultured in 96-well plates at a density of 3,000 cells/well for 1–4 days. A 20-μL aliquot of MTT solution (Sigma-Aldrich) was added to each well and incubated at 37°C for 4 h. Crystals of MTT-formazan, formed in viable cells, were dissolved in 150 μL of DMSO. Absorbance (optical density [OD]) was measured at 490 nm using a microplate reader (BioTek Instruments, Winooski, VT, USA). All assays included three biological replicates.
EdU Incorporation Assay
EdU incorporation was detected using the Cell-Light EdU Apollo 567 in vitro imaging kit (RioBio, Guangzhou, P.R. China) according to the manufacturer’s instructions. LUAD cells were incubated with EdU for 2 h. Subsequently, cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and stained with 1× Apollo fluorescent cocktail. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Beyotime, Shanghai, P.R. China). Images were captured and the number of stained cells was counted in five random fields under a fluorescence microscope (Olympus, Tokyo, Japan). Each experiment was performed in triplicates.
Colony Formation Assay
LUAD cells were seeded into six-well plates at a density of 100 cells/well and cultured with 5% CO2 at 37°C for 2 weeks. Cells were fixed with 4% paraformaldehyde and stained with crystal violet solution. The colonies containing more than 50 cells were counted and imaged under an inverted optical microscope (Olympus). All experiments included three biological replicates.
In Situ Hybridization
The LUAD TMAs were deparaffinized in xylene, rehydrated with gradient ethanol, and then treated with 0.1% hydrochloric acid alcohol for 15 min. The TMA was incubated with diluted pepsin in 3% fresh citrate buffer at 37°C for 20 min and then washed with phosphate-buffered saline (PBS). In addition, hybridization with a digoxigenin (DIG)-labeled locked nucleic acid (LNA) miRNA probe complementary to hsa-miR-3613-5p (Biosense Bioscience, Guangzhou, P.R. China) was performed for 22 h at 37°C after pre-hybridization for 2 h at 37°C in pre-hybridization solution. The LUAD TMAs were sequentially washed with 2× SSC for 5 min, 0.5× SSC for 15 min, and 0.2× SSC for 15 min at 37°C. After incubation with and removal of the blocking solution, the TMA was incubated with anti-DIG- biotinylated mouse antibody at 37°C for 1 h. Then, the TMA was sequentially incubated with a streptavidin-biotin complex (SABC) for 20 min and biotin-labeled peroxidase for 20 min at 37°C. Finally, hematoxylin was used for counterstaining of the nucleus after staining with diaminobenzidine (DAB) reagent.
Tumorigenesis Experiments in Nude Mice
The animal studies were performed in strict accordance with protocols approved by the Institutional Animal Care and Use Committee of Southern Medical University (Guangzhou, P.R. China). Twenty female 4-week-old BALB/c athymic nude mice purchased from GemPharmatech (Nanjing, P.R. China) were housed in a specific pathogen-free (SPF) animal facility. To establish a xenograft model, 3.0 × 106 LUAD cells stably expressing miR-3613-5p and control cells, which were suspended in 100 μL of PBS, were subcutaneously injected into the symmetric hindlimb of the nude mice (n = 5 mice per group). The tumors of the nude mice were measured every 5 days and tumor volumes were calculated by using the formula V (mm3) = 0.5 × (length × width2). After 3 weeks, all nude mice were sacrificed and tumor tissues were collected for further analysis.
IHC Analysis
The antigen retrieval from paraffin-embedded sections prepared from tumorigenesis experiments was performed by deparaffinization, rehydration, and boiling in a microwave oven with citrated or EDTA buffer. Hematoxylin and eosin (H&E) staining was performed using a standard protocol. IHC staining was performed using the SP-9000 SPlink detection kit (ZSGB-Bio Technologies, Beijing, P.R. China) following the manufacturer’s protocols. The primary antibodies used in the IHC analysis were against mouse Ki-67, PCNA, and NR5A2.
Western Blot Analysis
Western blot analysis was performed according to a standard procedure. Briefly, total protein was extracted from the cultured cells, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were washed and blocked before incubation with primary antibodies, which included anti-AKT1, anti-p-AKT1, anti-MAPK3/1, anti-p-MAPK3/1, anti-CCND1, anti-CDKN1A, anti-CDKN1B, anti-RELA, anti-JUN, and anti-NR52A. Anti-glyderaldehyde-3-phosphate dehydrogenase (GAPDH) and β-tubulin were used as negative controls. Then, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies. The signal of the immobilized target proteins on the membranes was developed by enhanced chemiluminescence (ECL) using ECL reagent (Merck Millipore, Darmstadt, Germany). The images were captured with a chemiluminescence imager (Sage Creation Science, Beijing, P.R. China). The antibodies are described in Table S3.
Luciferase Reporter Assay
NR5A2 was predicted as a potential target gene of miR-3613-5p by TargetScan (http://www.targetscan.org/vert_72/) and miRPathDB (https://mpd.bioinf.uni-sb.de/overview.html) software. The sequence of the 3′ UTR of NR5A2 wild-type or mutant binding sites for miR-3613-5p were synthesized and cloned into the psiCHECK-2 vector plasmid (Promega, Madison, WI, USA). The psiCHECK-2 vector plasmid, the wild-type or mutant plasmids, and miR-3613-5p mimics or inhibitor were co-transfected into HEK293T cells using Hieff trans liposomal transfection reagent (Yeasen Biotech, Shanghai, P.R. China). The psiCHECK-2 vector and mimics or psiCHECK-2 vector and inhibitor were used as negative controls. After 48 h in culture, the firefly and Renilla luciferase activities were analyzed using the Dual-Luciferase reporter assay system (Promega) in accordance with the manufacturer’s protocols. Each assay was performed in triplicate.
ChIP Assay
The promoter region sequence of miR-3613-5p was searched in the Ensembl database (http://www.ensembl.org/index.html). The PROMO (http://alggen.lsi.upc.es/) and JASPAR (http://jaspar.genereg.net/) bioinformatics tools predicted three putative JUN binding sites on the miR-3613-5p promoter region. The ChIP assay was performed using the Pierce agarose ChIP kit (Thermo Fisher Scientific), according to the manufacturer’s protocols, using anti-JUN or IgG antibody. The IgG antibody was used as a negative control. The JUN-bound chromatin was specifically amplified by PCR and analyzed by electrophoresis on agarose gels. The PCR-specific primers for each of the three predicted sites are listed in Table S2.
EMSA
The EMSA kit (Biosense Bioscience) was used to detect the binding activity between JUN and the promoter region of miR-3613-5p following the manufacturer’s instructions. The miR-3613-5p specific probes for EMSA were designed and synthetized by Biosense Bioscience. The binding assays were performed using a reaction mixture system containing 1.5 μL of binding reaction reagent, 1 μL of poly(dI:dC), 5 μL of nuclear extracts, 5 μL of biotin-labeled probes, and 100-fold specific oligonucleotide competitor (unlabeled wild-type or mutant JUN probes) for 20 min at room temperature. LUAD cells extracts without nucleoprotein were used as a negative control. Anti-JUN antibody was used as supershift band control. Before development and fixing in X-rays, the target bands were detected with the ECL Plus reagent. The sequences of the probes used are listed in Table S2.
CoIP
STRING (https://string-db.org/) and BioGRID (https://thebiogrid.org/) database analysis predicted the interaction between RELA and JUN. CoIP was performed using protein A/G magnetic beads for IP (Bimake, Houston, TX, USA) in accordance with the manufacturer’s protocols. Briefly, total proteins were extracted from LUAD cells and HEK293T cells overexpressing RELA and incubated with anti-JUN or IgG antibodies overnight at 4°C. Beads were mixed with the complexes and rotated for 30 min at room temperature. After washing and resuspending in radioimmunoprecipitation assay (RIPA) buffer and SDS loading buffer, beads were boiled for 10 min at 100°C. CoIP samples were analyzed by western blotting. IgG antibody was used as a negative control.
Immunofluorescence
LUAD cells were seeded into a 20-mm glass-bottom dish and incubated overnight. Subsequently, LUAD cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100, prior to blocking with normal goat serum. Before culturing with fluorescence-tagged secondary antibodies, LUAD cells were incubated with anti-RELA and anti-JUN primary antibodies. Cell nuclei were visualized using DAPI. Ultimately, images were acquired with an LSM800 confocal microscope (Carl Zeiss, Jena, Germany).
Statistical Analysis
All statistical analyses were performed using the SPSS 20.0 software (IBM, Armonk, NY, USA) and GraphPad Prism 6 (GraphPad, La Jolla, CA, USA). The data in this study are expressed as the mean ± SD. A Student’s t test, χ2 test, or Fisher exact probability was used to analyze the statistical differences. Spearman correlation analysis was used for correlation analysis. Kaplan-Meier survival analysis was compared by log-rank analysis. p values <0.05 were considered to indicate a statistically significant difference.
Author Contributions
T.H., H.S., and S.W. conducted experiments and analyzed data. T.H. designed some experiments and wrote the manuscript. L.Z. assisted with the experiments and analyzed data. Y.W., X.D., D.W., and J.L. assisted with the experiments. Z.H. designed and supervised the experiments. S.Z., W.H., and H.Y. were responsible for the conception and design, data analysis, study supervision, writing of the manuscript, and final approval of the manuscript. All authors approved the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by grants from the National Key R&D Program of China (2017YFC1103400) and the National Natural Science Foundation of China (61427807 and 81272334).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.09.024.
Contributor Information
Shizhen Zhong, Email: zhszh@fimmu.com.
Wenhua Huang, Email: hwh@fimmu.com.
Huiling Yang, Email: 112699355@qq.com.
Supplemental Information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Yuan M., Huang L.L., Chen J.H., Wu J., Xu Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019;4:61. doi: 10.1038/s41392-019-0099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstraw P., Ball D., Jett J.R., Le Chevalier T., Lim E., Nicholson A.G., Shepherd F.A. Non-small-cell lung cancer. Lancet. 2011;378:1727–1740. doi: 10.1016/S0140-6736(10)62101-0. [DOI] [PubMed] [Google Scholar]
- 4.Chen Z., Fillmore C.M., Hammerman P.S., Kim C.F., Wong K.K. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat. Rev. Cancer. 2014;14:535–546. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beasley M.B., Brambilla E., Travis W.D. The 2004 World Health Organization classification of lung tumors. Semin. Roentgenol. 2005;40:90–97. doi: 10.1053/j.ro.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Warth A., Muley T., Meister M., Stenzinger A., Thomas M., Schirmacher P., Schnabel P.A., Budczies J., Hoffmann H., Weichert W. The novel histologic International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification system of lung adenocarcinoma is a stage-independent predictor of survival. J. Clin. Oncol. 2012;30:1438–1446. doi: 10.1200/JCO.2011.37.2185. [DOI] [PubMed] [Google Scholar]
- 7.Collisson E.A., Campbell J.D., Brooks A.N., Berger A.H., Lee W., Chmielecki J., Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito M., Miyata Y., Kushitani K., Yoshiya T., Mimae T., Ibuki Y., Misumi K., Takeshima Y., Okada M. Prediction for prognosis of resected pT1a-1bN0M0 adenocarcinoma based on tumor size and histological status: relationship of TNM and IASLC/ATS/ERS classifications. Lung Cancer. 2014;85:270–275. doi: 10.1016/j.lungcan.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 9.Sakao Y., Kuroda H., Mun M., Uehara H., Motoi N., Ishikawa Y., Nakagawa K., Okumura S. Prognostic significance of tumor size of small lung adenocarcinomas evaluated with mediastinal window settings on computed tomography. PLoS ONE. 2014;9:e110305. doi: 10.1371/journal.pone.0110305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaturvedi M.M., Sung B., Yadav V.R., Kannappan R., Aggarwal B.B. NF-κB addiction and its role in cancer: “one size does not fit all”. Oncogene. 2011;30:1615–1630. doi: 10.1038/onc.2010.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y., Wang L., Lv Y., Jiang C., Wu G., Dull R.O., Minshall R.D., Malik A.B., Hu G. The GTPase Rab1 is required for NLRP3 inflammasome activation and inflammatory lung injury. J. Immunol. 2019;202:194–206. doi: 10.4049/jimmunol.1800777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang X., Liu D., Shishodia S., Ozburn N., Behrens C., Lee J.J., Hong W.K., Aggarwal B.B., Wistuba I.I. Nuclear factor-κB (NF-κB) is frequently expressed in lung cancer and preneoplastic lesions. Cancer. 2006;107:2637–2646. doi: 10.1002/cncr.22315. [DOI] [PubMed] [Google Scholar]
- 13.Blakely C.M., Pazarentzos E., Olivas V., Asthana S., Yan J.J., Tan I., Hrustanovic G., Chan E., Lin L., Neel D.S. NF-κB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 2015;11:98–110. doi: 10.1016/j.celrep.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rotow J., Bivona T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer. 2017;17:637–658. doi: 10.1038/nrc.2017.84. [DOI] [PubMed] [Google Scholar]
- 15.Karin M., Greten F.R. NF-κB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 16.Taniguchi K., Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol. 2018;18:309–324. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 17.Liu T., Zhang L., Joo D., Sun S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017;2:17023. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meylan E., Dooley A.L., Feldser D.M., Shen L., Turk E., Ouyang C., Jacks T. Requirement for NF-κB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bassères D.S., Ebbs A., Levantini E., Baldwin A.S. Requirement of the NF-κB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70:3537–3546. doi: 10.1158/0008-5472.CAN-09-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winter J., Jung S., Keller S., Gregory R.I., Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 21.Li Y., Liu X., Lin X., Zhao M., Xiao Y., Liu C., Liang Z., Lin Z., Yi R., Tang Z. Chemical compound cinobufotalin potently induces FOXO1-stimulated cisplatin sensitivity by antagonizing its binding partner MYH9. Signal Transduct. Target. Ther. 2019;4:48. doi: 10.1038/s41392-019-0084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang M., Yu F., Ding H., Wang Y., Li P., Wang K. Emerging function and clinical values of exosomal microRNAs in cancer. Mol. Ther. Nucleic Acids. 2019;16:791–804. doi: 10.1016/j.omtn.2019.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhen Y., Fang W., Zhao M., Luo R., Liu Y., Fu Q., Chen Y., Cheng C., Zhang Y., Liu Z. miR-374a-CCND1-pPI3K/AKT-c-JUN feedback loop modulated by PDCD4 suppresses cell growth, metastasis, and sensitizes nasopharyngeal carcinoma to cisplatin. Oncogene. 2017;36:275–285. doi: 10.1038/onc.2016.201. [DOI] [PubMed] [Google Scholar]
- 24.Deng X., Liu Z., Liu X., Fu Q., Deng T., Lu J., Liu Y., Liang Z., Jiang Q., Cheng C., Fang W. miR-296-3p negatively regulated by nicotine stimulates cytoplasmic translocation of c-Myc via MK2 to suppress chemotherapy resistance. Mol. Ther. 2018;26:1066–1081. doi: 10.1016/j.ymthe.2018.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang Z., Liu Z., Cheng C., Wang H., Deng X., Liu J., Liu C., Li Y., Fang W. VPS33B interacts with NESG1 to modulate EGFR/PI3K/AKT/c-Myc/P53/miR-133a-3p signaling and induce 5-fluorouracil sensitivity in nasopharyngeal carcinoma. Cell Death Dis. 2019;10:305. doi: 10.1038/s41419-019-1457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joshi P., Middleton J., Jeon Y.J., Garofalo M. MicroRNAs in lung cancer. World J. Methodol. 2014;4:59–72. doi: 10.5662/wjm.v4.i2.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inamura K., Ishikawa Y. MicroRNA in lung cancer: novel biomarkers and potential tools for treatment. J. Clin. Med. 2016;5:36. doi: 10.3390/jcm5030036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boratyn E., Nowak I., Horwacik I., Durbas M., Mistarz A., Kukla M., Kaczówka P., Łastowska M., Jura J., Rokita H. Monocyte chemoattractant protein-induced protein 1 overexpression modulates transcriptome, including microRNA, in human neuroblastoma cells. J. Cell. Biochem. 2016;117:694–707. doi: 10.1002/jcb.25354. [DOI] [PubMed] [Google Scholar]
- 29.Castro-Magdonel B.E., Orjuela M., Camacho J., García-Chéquer A.J., Cabrera-Muñoz L., Sadowinski-Pine S., Durán-Figueroa N., Orozco-Romero M.J., Velázquez-Wong A.C., Hernández-Ángeles A. miRNome landscape analysis reveals a 30 miRNA core in retinoblastoma. BMC Cancer. 2017;17:458. doi: 10.1186/s12885-017-3421-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song J., Wang W., Wang Y., Qin Y., Wang Y., Zhou J., Wang X., Zhang Y., Wang Q. Epithelial-mesenchymal transition markers screened in a cell-based model and validated in lung adenocarcinoma. BMC Cancer. 2019;19:680. doi: 10.1186/s12885-019-5885-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldstraw P., Chansky K., Crowley J., Rami-Porta R., Asamura H., Eberhardt W.E.E., Nicholson A.G., Groome P., Mitchell A., Bolejack V., International Association for the Study of Lung Cancer Staging and Prognostic Factors Committee, Advisory Boards, and Participating Institutions. International Association for the Study of Lung Cancer Staging and Prognostic Factors Committee Advisory Boards and Participating Institutions The IASLC Lung Cancer Staging Project: proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J. Thorac. Oncol. 2016;11:39–51. doi: 10.1016/j.jtho.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Yin M., Ren X., Zhang X., Luo Y., Wang G., Huang K., Feng S., Bao X., Huang K., He X. Selective killing of lung cancer cells by miRNA-506 molecule through inhibiting NF-κB p65 to evoke reactive oxygen species generation and p53 activation. Oncogene. 2015;34:691–703. doi: 10.1038/onc.2013.597. [DOI] [PubMed] [Google Scholar]
- 33.Mehta A.K., Hua K., Whipple W., Nguyen M.T., Liu C.T., Haybaeck J., Weidhaas J., Settleman J., Singh A. Regulation of autophagy, NF-κB signaling, and cell viability by miR-124 in KRAS mutant mesenchymal-like NSCLC cells. Sci. Signal. 2017;10:10. doi: 10.1126/scisignal.aam6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grivennikov S.I., Karin M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–19. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu W., Wang S., Sun Q., Yang Z., Liu M., Tang H. DCLK1 promotes epithelial-mesenchymal transition via the PI3K/Akt/NF-κB pathway in colorectal cancer. Int. J. Cancer. 2018;142:2068–2079. doi: 10.1002/ijc.31232. [DOI] [PubMed] [Google Scholar]
- 36.Wang X., Cao X., Dong D., Shen X., Cheng J., Jiang R., Yang Z., Peng S., Huang Y., Lan X. Circular RNA TTN acts as a miR-432 sponge to facilitate proliferation and differentiation of myoblasts via the IGF2/PI3K/AKT signaling pathway. Mol. Ther. Nucleic Acids. 2019;18:966–980. doi: 10.1016/j.omtn.2019.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L., Guo P., He Y., Chen Z., Chen L., Luo Y., Qi L., Liu Y., Wu Q., Cui Y. HCC-derived exosomes elicit HCC progression and recurrence by epithelial-mesenchymal transition through MAPK/ERK signalling pathway. Cell Death Dis. 2018;9:513. doi: 10.1038/s41419-018-0534-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishimura M., Naito S., Yokoi T. Tissue-specific mRNA expression profiles of human nuclear receptor subfamilies. Drug Metab. Pharmacokinet. 2004;19:135–149. doi: 10.2133/dmpk.19.135. [DOI] [PubMed] [Google Scholar]
- 39.Zhang C., Large M.J., Duggavathi R., DeMayo F.J., Lydon J.P., Schoonjans K., Kovanci E., Murphy B.D. Liver receptor homolog-1 is essential for pregnancy. Nat. Med. 2013;19:1061–1066. doi: 10.1038/nm.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stein S., Schoonjans K. Molecular basis for the regulation of the nuclear receptor LRH-1. Curr. Opin. Cell Biol. 2015;33:26–34. doi: 10.1016/j.ceb.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 41.Holmstrom S.R., Deering T., Swift G.H., Poelwijk F.J., Mangelsdorf D.J., Kliewer S.A., MacDonald R.J. LRH-1 and PTF1-L coregulate an exocrine pancreas-specific transcriptional network for digestive function. Genes Dev. 2011;25:1674–1679. doi: 10.1101/gad.16860911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hale M.A., Swift G.H., Hoang C.Q., Deering T.G., Masui T., Lee Y.K., Xue J., MacDonald R.J. The nuclear hormone receptor family member NR5A2 controls aspects of multipotent progenitor cell formation and acinar differentiation during pancreatic organogenesis. Development. 2014;141:3123–3133. doi: 10.1242/dev.109405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schoonjans K., Dubuquoy L., Mebis J., Fayard E., Wendling O., Haby C., Geboes K., Auwerx J. Liver receptor homolog 1 contributes to intestinal tumor formation through effects on cell cycle and inflammation. Proc. Natl. Acad. Sci. USA. 2005;102:2058–2062. doi: 10.1073/pnas.0409756102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benod C., Vinogradova M.V., Jouravel N., Kim G.E., Fletterick R.J., Sablin E.P. Nuclear receptor liver receptor homologue 1 (LRH-1) regulates pancreatic cancer cell growth and proliferation. Proc. Natl. Acad. Sci. USA. 2011;108:16927–16931. doi: 10.1073/pnas.1112047108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiruchelvam P.T.R., Lai C.F., Hua H., Thomas R.S., Hurtado A., Hudson W., Bayly A.R., Kyle F.J., Periyasamy M., Photiou A. The liver receptor homolog-1 regulates estrogen receptor expression in breast cancer cells. Breast Cancer Res. Treat. 2011;127:385–396. doi: 10.1007/s10549-010-0994-9. [DOI] [PubMed] [Google Scholar]
- 46.Wang S.L., Zheng D.Z., Lan F.H., Deng X.J., Zeng J., Li C.J., Wang R., Zhu Z.Y. Increased expression of hLRH-1 in human gastric cancer and its implication in tumorigenesis. Mol. Cell. Biochem. 2008;308:93–100. doi: 10.1007/s11010-007-9616-1. [DOI] [PubMed] [Google Scholar]
- 47.Fu Q., Song X., Liu Z., Deng X., Luo R., Ge C., Li R., Li Z., Zhao M., Chen Y. miRomics and proteomics reveal a miR-296-3p/PRKCA/FAK/Ras/c-Myc feedback loop modulated by HDGF/DDX5/β-catenin complex in lung adenocarcinoma. Clin. Cancer Res. 2017;23:6336–6350. doi: 10.1158/1078-0432.CCR-16-2813. [DOI] [PubMed] [Google Scholar]
- 48.Zhao M., Xu P., Liu Z., Zhen Y., Chen Y., Liu Y., Fu Q., Deng X., Liang Z., Li Y. Dual roles of miR-374a by modulated c-Jun respectively targets CCND1-inducing PI3K/AKT signal and PTEN-suppressing Wnt/β-catenin signaling in non-small-cell lung cancer. Cell Death Dis. 2018;9:78. doi: 10.1038/s41419-017-0103-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Echevarría-Vargas I.M., Valiyeva F., Vivas-Mejía P.E. Upregulation of miR-21 in cisplatin resistant ovarian cancer via JNK-1/c-Jun pathway. PLoS ONE. 2014;9:e97094. doi: 10.1371/journal.pone.0097094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shyu Y.J., Suarez C.D., Hu C.D. Visualization of AP-1 NF-κB ternary complexes in living cells by using a BiFC-based FRET. Proc. Natl. Acad. Sci. USA. 2008;105:151–156. doi: 10.1073/pnas.0705181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rolland T., Taşan M., Charloteaux B., Pevzner S.J., Zhong Q., Sahni N., Yi S., Lemmens I., Fontanillo C., Mosca R. A proteome-scale map of the human interactome network. Cell. 2014;159:1212–1226. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kane L.P., Shapiro V.S., Stokoe D., Weiss A. Induction of NF-κB by the Akt/PKB kinase. Curr. Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 53.Dhawan P., Richmond A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-κB activity in melanoma cells. J. Biol. Chem. 2002;277:7920–7928. doi: 10.1074/jbc.M112210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.