

Abstract
Necroptosis, a genetically programmed form of necrotic cell death, serves as an important pathway in human diseases. As a critical cell-killing mechanism, necroptosis is associated with cancer progression, metastasis, and immunosurveillance. Targeting necroptosis pathway by small molecule modulators is emerging as an effective approach in cancer therapy, which has the advantage to bypass the apoptosis-resistance and maintain antitumor immunity. Therefore, a better understanding of the mechanism of necroptosis and necroptosis modulators is necessary to develop novel strategies for cancer therapy. This review will summarize recent progress of the mechanisms and detecting methods of necroptosis. In particular, the relationship between necroptosis and cancer therapy and medicinal chemistry of necroptosis modulators will be focused on.
Keywords: Cell death, Necroptosis, Inducers, Inhibitors, Anticancer therapy
Graphical abstract
Targeting necroptosis pathway by small molecule modulators is emerging as an effective approach in cancer therapy

.
1. Introduction
A dynamic balance among cell death, proliferation and differentiation is of great significance in maintaining tissue homeostasis1,2. For a long time, apoptosis has been considered as the only pathway in the programmed cell death (PCD), which is characterized by condensed nuclei, shrinking but integral membrane, and rapid elimination by phagocytosis3,4. In contrast, necrosis refers to the process that cells swell, rupture and then release cellular contents and proinflammatory molecules in response to the overwhelming stress (e.g., heat)5, 6, 7. Necrosis is regarded as an uncontrollable process, and therefore it is highly challenging to identify small molecules that can interfere with this process5, 6, 7. In the late 1980s, cells under the treatment of tumor necrosis factor (TNF) were found to be characterized with either apoptotic or necrotic features in a cell type-dependent manner8. In 2004, Thompson et al.9 discovered that alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (1, Fig. 1) initiated cell necrosis depending on the expression of poly ADP-ribose polymerase (PARP), suggesting that necrosis could be regulated by small molecules. In 2005, the concept “necroptosis”, a combination of “necrosis” and “apoptosis”, was proposed by Yuan's group10, and Nec-1 (2, Fig. 1) was reported to be able to reverse cell death in this pathway. Since apoptosis-resistant cases were reported in clinical cancer therapy11, it was urgent and important to search for a new pathway to induce the death of cancer cells, while necroptosis is a promising alternative12,13. Therefore, intensive attention and efforts have been paid to investigate the mechanism of necroptosis and clarify its relationship and effectiveness to cancer therapy14, 15, 16. Recent studies indicated that necroptosis could be initiated by activation of specific receptors on cell membrane and regulated by cytokines and small molecules17, 18, 19, 20, 21, 22. While the relationship between necroptosis and cancer has not been clarified explicitly, both efforts are being focused on the factors relevant to the induction and inhibition of necroptosis23, 24, 25. Thus, a better understanding of the mechanism of necroptosis and necroptosis modulators is important to develop novel strategies for cancer therapy. From a medicinal chemist point of view, the present review will summarize recent progress of the mechanisms and detecting methods of necroptosis. In particular, the relationship between necroptosis and cancer therapy, and the discovery and biological activity of necroptosis modulators will be focused on.
Figure 1.
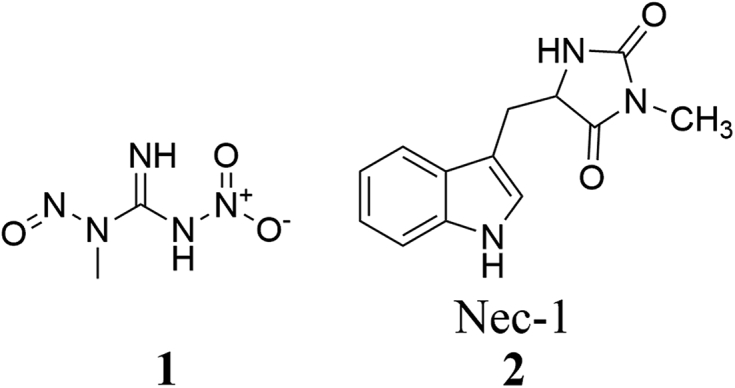
Chemical structures of necroptosis modulators 1 and 2.
2. Mechanism of necroptosis
2.1. Initiation of necroptosis
Necroptosis is initiated under several circumstances, including the combination of TNF cytokines and membrane receptors, virus infection and interferon stimulation or chemical compounds (Fig. 2)17−22,26. TNF cytokines include TNF-α, TNF-related apoptosis-inducing ligand, factor-associated suicide ligand (FASL), etc.27 When TNF-α binds to the membrane receptor, it can promote the formation of a plasma membrane-associated complex which is consisted of adaptor proteins tumor necrosis factor receptor 1 (TNFR1)-associated death domain protein (TRADD), receptor-interacting serine-threonine kinase 1 (RIPK1), TNFR-associated factor 2/5 (TRAF2/5), cellular inhibitor of apoptosis protein 1/2 (cIAP1/2) and the linear ubiquitin chain assembly complex (LUBAC complex)18. Subsequently, the complex results in RIPK1 polyubiquitination which is responsible for the activation of NF-κB pathway and mitogen-activated protein kinases (MAPKs)17, 18, 19. Inhibition of cIAPs or deubiquitination of RIPK1 by cylindromatosis (CYLD) can block the activation of NF-κB pathway, leading to the dissociation of RIPK1 and TRADD from the plasma membrane-associated complex28. Then RIPK1 and TRADD bind to FAS-associated death domain protein (FADD) and procaspase-8 to form a complex called cytosolic death-inducing signaling complex (DISC) which is able to activate caspase-8 and initiate apoptosis29, while caspase-8 activation can be inhibited by the cellular FLIP long isoform protein (cFLIPL). When receptor-interacting serine/threonine-protein kinase 3 (RIPK3) is overexpressed, it can be connected to RIPK1 through their own RIP homotypic interaction motif (RHIM) to form a complex called necrosome30. Then both or either of RIPK1 and RIPK3 are phosphorylated, resulting in recruitment and subsequent phosphorylation of mixed lineage kinase domain-like protein (MLKL), and activation of necroptosis31. It was reported that caspase-8 in the necrosome was able to cleave RIPK1–RIPK3 complex, and therefore, cFLIPL could promote necroptosis32.
Figure 2.

A brief pathway of necroptosis.
Other necroptotic stimulus, including FASL, lipopolysaccharide (LPS), double-stranded RNA (dsRNA), double-stranded DNA (dsDNA), interferon-γ (IFN-γ) and some chemical compounds, can be recognized by their corresponding receptors located on the cell membrane or sensors outside or inside the cell20,21,26,33. For example, the combination of FAS to FASL or combination of TRAIL to DR4/5 subsequently binds to FADD through intercellular DD domain, leading to the formation of DISC directly33. Then the downstream pathway is the same to that of TNF-α-induced necroptosis. In addition, LPS can activate Toll-like receptor 4 (TLR4)20, while viral dsRNA is able to activate TLR321. Their combination enables recruitment of Toll/IL-1 receptor domain-containing adaptor to elicit inflammatory cytokines and type 1 IFN responses. Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF⋅TICAM-1) is the sole adaptor protein for TLR322, while either TRIF or myeloid differentiation primary response gene (Myd88) can mediate TLR420. TRIF contains the RHIM motif that links RIPK1 and RIPK3, and then induces necroptosis34. Apart from these two pathways, DNA virus can trigger the DNA-dependent activator of interferon-regulatory factors (DAI) pathway35. DAI is a cytosolic DNA sensor that activates IRF3 and NF-κB pathways to elicit cytokines and type1 IFN responses35. Like TRIF, it also contains the RHIM motif which can link RIPK1 and RIPK3, and induce necroptosis35. Radiation was supposed to be an invitation to necroptosis in endocrine cancers, but the mechanism remained to be clarified26.
2.2. Execution of necroptosis
The functional necrosome consists of RIPK1, RIPK3 and MLKL. Inactivated MLKL is a monomer in the cytoplasm36. It turns into the form of trimer once phosphorylated by RIPK3 at the kinase-like domain of MLK. Afterwards, the trimers bind to phosphatidylinositol phosphates (PIPs) and cardiolipin (CL) which are mitochondria-specific, leading to their translocation to membrane rich in PIPs and CL36. There is evidence that MLKL in the plasma membrane binds to the ion channel transient receptor potential melastatin related 7 (TRMP7) and then leads to the influx of Ca2+ ions and subsequent cell death36. Another study revealed that MLKL in the membrane promoted the influx of Na+ and then increased the osmotic pressure, causing the rupture of plasma membrane37. Papatriantafyllou et al.38 proposed that phosphoglycerte mutase family member 5 (PAGM5) was another target of necrosome. PGAM5 recruited the mitochondrial fission factor dynamin-related protein 1 (Drp1) and activated its GTP-binding proteins (GTPase) activity, leading to mitochondrial fragmentation which is an early and indispensible step for necrosis execution38. Lysosomal permeability transition (LMT) involved the increase of Ca2+ concentration in response of FAS and TNF, lysosome enlargement and lysosome membrane permeabilization39. Then, the leakage of proteolytic enzyme into the cytoplasm degraded proteins and activated other hydrolases participated in the necroptosis39.
Reactive oxygen species (ROS)40, adenine nucleotide translocase (ANT)41, reactive nitrogen species (RNS)42 are important regulators in the execution of necroptosis. RIPK3 activates the activity of glycogen phosphorylase (PYLG), glutamate ammonia ligase (GLUL) and glutamate dehydrogenase (GLUD1), which promotes the production of ROS. Then the increasing amount of ROS activates the pro-necrotic effect of c-Jun NH2-terminal kinase (JNK), which can suppress TNF-α-induced apoptosis40,43, leading cells to undergo necrotic or necroptotic pathways. ANT, an ADP/ATP carrier located in the inner mitochondrial membrane, is essential for the synthesis of mitochondrial ATP41. In case that the ability of ANT to transport cytoplasmic ADP is blocked, mitochondrial ATP will be in shortage, and then necrotic or necroptotic pathways are triggered41. Another study indicated that excessive nitric oxide induced the production of RNS which played an important role in lipid and protein oxidation and peroxidation42. In addition, nitration also could induce RIPK1- and RIPK3-dependent necroptosis44. Nitration of NDUFB8, a vital component of mitochondrial supercomplex, resulted in the dissociation of mitochondrial supercomplex and mitochondrial depolarization, which were associated with necroptosis.
2.3. Propagation of necroptosis
Previous studies confirmed that necroptosis was an invitation to inflammation33. Damage-associated molecule pattern (DAMP), including cytokines and other chemo-attractants, are capable of recruiting primary immune cells to the necroptotic cells, followed by phagocytosis and termination of cell death signaling45. Furthermore, inflammation is inclined to tumorgenesis46 and promotes cancer progression and metastasis, attenuating adaptive immune responses and increasing drug resistance47.
There is another assumption that poly ADP-ribose polymerase 1 (PARP-1) participates in the disposal of necroptotic cells48. PARP-1 is a nuclear enzyme that is essential in repairing DNA damage49. PARP-1 overexpression induced by DNA-strand breakage results in NAD+ and ATP consumption, and the release of apoptosis-inducing factor (AIF) that binds to DNA from mitochondria to nucleus, causes chromatin condensation and DNA fragmentation, and induces necrosis50. Compound 2 is able to inhibit the effects of PARP-1 and block the release of AIF from mitochondria to nucleus, whereas direct inhibition of PARP-1 fails to protect cells from necroptosis48. Thus, PARP-1-induced necrosis may be associated with the downstream pathway of necroptosis.
Necroptotic cells are cleared from the immune system through pinocytosis, or cellular drinking, which is mediated by macropinosome, a subcellular component of macrophages51. The process of propagation of necroptosis is irreversible; therefore, more attention should be paid to discover inhibitors to block the process of initiation and execution of necrosis.
3. Detection of necroptosis
Since the mechanism of necroptosis has been clarified quite clearly, several methods have been developed to detect necroptosis based on the morphological and biochemical characteristics (Fig. 3). It should be noted that these methods were often used in combination rather than alone since there are many interference factors in the experiments.
Figure 3.

Methods of detection of necroptosis.
3.1. Direct observation of necroptosis cells by morphology change
Direct observation of cell morphology by transmission electron microscopy (TEM) provides a preliminary judgment to distinguish apoptosis, necrosis and necroptosis52, 53, 54. Apoptosis is characterized by cell shrinkage, nuclear condensation and organelle fragmentation52,55, which is apparently distinct from necrosis and necroptosis because they are characterized by cell swelling and membrane rupture53. For necroptosis, it can be distinguished from necrosis that necroptotic cells retain integral nucleus while necrotic cells do not54. Chen et al.56 clearly observed necroptotic cell morphology including the changes of endoplasmic reticula, mitochondria, cytomembrane and cell nucleus compared with normal cells by TEM.
3.2. Identifying necroptotic cells by flow cytometry
Flow cytometry is able to distinguish necroptotic cells. Compared with the control group, an increase in annexin V–/propidium iodide+ or annexin V+/propidium iodide+ in treated cells is the indicative of primary necrosis or secondary necrosis, respectively, while a decrease in annexin V–/propidium iodide+ is the indicative of apoptosis57. Notably, this method is not enough to accurately identify necroptosis, which should be applied in combination with other methods.
3.3. Detecting necroptotic cells by content variation and phosphorylation status of proteins
Expression of important proteins involved in necroptosis, such as RIPK1, RIPK3, and MLKL, can be detected by Western blot or immunofluorescence. Their overexpression and phosphorylation are commonly the indicative of necroptosis. There are several antibodies that can be applied to verify whether the proteins are activated at the phosphorylation sites. For example, anti-phospho-S14/15 RIPK1 antibody and anti-phospho-S166 hRIPKl antibody were used to detect the activation of RIPK158,59. Anti-phospho-S227 hRIPK3 antibody60 and anti-phospho-S232 mRIPK3 antibody61 were used to detect the activation of RIPK3, while anti-phospho-T357/S358 hMLKL antibody62 was often used in detecting phosphorylation of MLKL.
3.4. Detection of necrosome formation
Necrosome formation can be detected by RIPK1/RIPK3 interactions in FADD or caspase-8 pull-down experiments. For RIPK3/MLKL or complex formations, immunoprecipitation or other methods are commonly used62, 63, 64.
3.5. Cell death inhibition in RIPK1, RIPK3 or MLKL deficient transgenic models
If cells are killed by necroptosis, knockdown of RIPK1, RIPK3 or MLKL can rescue cells, leading to a higher IC50 value or higher cell viability compared to the normal models. The mechanism is similar to that of necroptosis inhibitors. For example, knockdown of RIPK3 or MLKL through siRNA in NP cells showed decreased cytotoxicity compared with untransfected cells under the same circumstances, indicating the untransfected cells were killed through necroptosis56.
3.6. Rescuing cells from necroptosis by chemical inhibitors
Recently, the development of the new screening methods facilitated the discovery of a number of necroptosis inhibitors65. Combination use with necroptosis inhibitors would rescue necroptotic cells from the single treatment, leading to higher IC50 values or higher cell viability66. Classification and activity of necroptosis inhibitors will be introduced in the following sections (Figure 4, Figure 5, Figure 6, Figure 7).
Figure 4.

Chemical structures of RIPK1 inhibitors.
Figure 5.

Chemical structures of RIPK3 inhibitors and caspase inhibitor 17.
Figure 6.

Chemical structures of MLKL inhibitors and caspase inhibitor 24.
Figure 7.

Chemical structures of multi-targeting necroptosis inhibitors.
4. Inhibitors of necroptosis
Release of DMAPs during necroptosis could induce inflammation, which is known to promote tumorgenesis and cancer metastasis67. The mechanism will be introduced in the following sections. Therefore, inhibiting necroptosis may be helpful in cancer treatment.
4.1. RIPK1 inhibitors
Compound 2 was a RIPK1 inhibitor (RIPK1 IC50 = 1.98 μmol/L) and has been widely used as a tool compound in the mechanism studies of necroptosis68. However, it was relatively toxic because it also inhibited indoleamine 2,3-dioxygenase (IDO), resulting in the interference in inflammation-associated tumorgenesis to break tumor immune-tolerance and sensitize tumors to cell death68. In contrast, its analog 7-Cl-O-Nec-1 (3, RIPK1 IC50 = 0.91 μmol/L, Fig. 4) is less toxic because it did not show any inhibition towards IDO68.
GSK′963 (4, RIPK1 IC50 = 29 nmol/L) was discovered by screening GSK compound collections and exhibited higher inhibitory activity than compound 2 at molecular and cellular levels69. It was verified as a selective RIPK1 inhibitor because it was inactive against more than 100 kinases69. At the dose of 2 mg/kg, compound 4 could inhibit 90% RIPK1 activity and protect mice from shock even at a lower dosage (0.2 mg/kg), while its enantiomer was inactive so that it can be used to validate on-target effects69.
GSK2982772 (5, RIPK1 IC50 = 16 nmol/L) is the first orally-active RIPK1 inhibitor and is currently undergoing phase II clinical studies for treatment of ulcerative colitis, psoriasis and rheumatoid arthritis70. Compound 5 showed excellent activity in rats and monkeys with a wide therapeutic window (dosage range: 2–1000 mg/kg)70,71. It also demonstrated low toxicity such as no inhibition towards recombinant human cytochrome P450 (CYP450s), weak inhibition towards human Ether-a-go-go-related gene (hERG, IC50 = 195 μmol/L) and weak activation of human Pregnane X receptor (hPXR, EC50 = 13 μmol/L)70,71.
GSK′481 (6, RIPK1 IC50 = 10 nmol/L) was identified by screening GSK's DNA-encoded small-molecule libraries, and was considered as a promising lead compound due to its high selectivity and potency72. GSK′547 (7) is also a highly selective RIPK1 inhibitor against other kinases with favorable pharmacokinetic properties in mice, whose oral exposure was 400 times higher than that of compound 473. In the model of type C1 Niemann-Pick disease (NPC1), treatment with compound 7 could significantly extend the life span of mice74.
Robert's group75 discovered DHP77 (8, RIPK1 IC50 = 2.0 nmol/L) as an orally active RIPK1 inhibitor through high-throughput screening and hit optimization. Compound 8 is a potent and selective dihydropyrazole RIPK1 inhibitor and with good pharmacokinetic and pharmacodynamic profiles in several animal models, such as sclerosis and retinitis pigmentosa75.
Natural product derivative 6E11 (9, RIPK1 IC50 = 90 nmol/L) inhibited necroptosis induced by TNF-α or TRAIL, but failed to inhibit TRAIL-induced apoptosis, thus providing a novel structural type for RIPK1 inhibitors76.
Derived from pharmacophore fusion of ponatinib (29, dual RIPK1 and RIPK3 inhibitor) and compound 2, PN10 (10, RIPK1 IC50 = 90 nmol/L) was discovered as a potent and specific RIPK1 inhibitor and showed excellent in vivo activity in treatment with TNF-α77. LY3009120 (11), a pan-RAF inhibitor, was able to inhibit necroptosis by blocking phosphorylation of RIPK1 and subsequent phosphorylation of RIPK3 and MLKL78. NTB451 (12) was confirmed to be a specific RIPK1 inhibitor (IC50 = 7.9 μmol/L) through molecular docking, molecular dynamics simulation and drug affinity-responsive target stability (DARTS).
Furthermore, DNL747 and DNL758 were validated as highly efficacious RIPK1 inhibitors, whose chemical structures remain undisclosed. DNL747 is a blood–brain barrier (BBB) penetrable compound and has entered clinical trials for the treatment of Alzheimer's disease (AD)79, while DNL758 was unable to penetrate BBB and was supposed to be used in the treatment of systemic inflammatory79.
4.2. RIPK3 inhibitors
GSK′840 (13, RIPK3 IC50 = 0.3 nmol/L), GSK′843 (14, RIPK3 IC50 = 6.5 nmol/L) and GSK′872 (15, RIPK3 IC50 = 1.3 nmol/L) are classic and potent RIPK3 inhibitors identified by screening compound libraries (Fig. 5)80. Dabrafenib (16, RIPK3 IC50 = 250 nmol/L), a clinical drug for the treatment of BrafV600E melanoma, was also validated to target RIPK3, which showed excellent selectivity compared to its inhibition against RIPK1, RIPK2 and RIPK581. TNF-α, Smac mimetic and caspase inhibitor z-VAD-fmk (17, TSZ) are widely accepted to be able to induce RIPK3-mediated necroptosis82. Dabrafenib rescued cells from TSZ-induced cell death in RIPK3-expressed HT29 cells, in which morphology changes were apparently observed81. Dabrafenib had no influence on the apoptosis induced by TNF-α and Smac mimetic in HT29 cells, suggesting that it had little impact on apoptosis81. Dabrafenib was proven to be able to bind to RIPK3 through molecular docking and the surface plasma resonance assay81. Further analysis indicated that it disrupted the RIPK3–MLKL interaction rather than RIPK1 and RIPK1–RIPK3 interaction81.
HS-1371 (18) was discovered to be a RIPK3 inhibitor (IC50 = 20.8 nmol/L) by screening kinase-targeted chemical libraries83. Molecular docking studies demonstrated that HS-1371 directly bound to the ATP binding pocket of RIPK383. ATP competition assay showed that HS-1371 inhibited RIPK3 in a time-dependent and ATP-competitive manner83. HS-1371 declined phosphorylation of RIPK3, thus blocking the interaction of RIPK3 and MLKL83. Furthermore, HS-1371 was able to rescue cells from TNF-induced necroptosis rather than apoptosis83. Pretreatment or post-treatment of cells with HS-1731 both inhibited RIPK3-mediated necroptosis83.
Structural optimization of dual RIPK1 and RIPK3 inhibitor TAK632 (19) led to a selective RIPK3 inhibitor (20, RIPK3 Kd = 81 nmol/L), which showed more than 60-fold selectivity against RIPK3 than RIPK184. Molecular docking studies showed that compound 20 could form π–π stacking and hydrogen bonding interactions with RIPK3 rather than RIPK1, accounting for the high selectivity84. Compound 20 rescued cells from necroptosis rather than apoptosis, indicating it is a selective necroptosis inhibitor84. It blocked the phosphorylation of RIPK3 and MLKL while having no influence on the phosphorylation of RIPK184. Compound 20 also displayed favorable pharmacokinetic properties in rats with an oral bioavailability of 25.2% without obvious side effects84.
Green's group85 previously confirmed the significant role of the phosphorylation of Ser345 in necroptosis, and then designed a selective monoclonal antibody for detection of necroptosis. Using this antibody to detect their capability to inhibit necroptosis, GW4401398 (21, EC50 = 73.6 nmol/L) was identified by screening 8904 compounds85. GW4401398 was proven to inhibit TNF- and TRIF-mediated necroptosis independent of RIPK1–RIPK3 interaction85. It directly inhibited RIPK3-mediated MLKL phosphorylation, thus inhibiting necroptosis85.
4.3. MLKL inhibitors
Necrosulfonamide (NSA, 22, Fig. 6) was identified by screening compound libraries and blocked necroptosis induced by different stimulus86. Target validation investigations by synthesizing biotin-based probes and pull-down experiments further confirmed that MLKL was a specific target for NSA86.
By screening a library of 367 compounds using thermal shift assay, GW806742X (23, RIPK3 Kd = 9.3 μmol/L) was validated to bind to the MLKL pseudokinase domain in an ADP or ATP dependent manner87. It dose-dependently inhibited necroptosis of wild-type mouse dermal fibroblasts stimulated with TSQ consisting of TNF, Smac-mimetic, and caspase inhibitor Q-VD-OPh (24)87.
Yan et al.88 identified TC13172 (25) as a novel MLKL inhibitor with an EC50 value of 2 nmol/L through phenotype-based screen and subsequent structure–activity relationship study, which was inactive against RIPK1 and RIPK3. It was confirmed to bind to MLKL in cell lysates at the site of Cys-86 by a biotin-tag and pull-down experiments88. Further mechanism studies indicated that compound 25 inhibited the translocation of MLKL to cell membrane rather than the phosphorylation of MLKL to block cell necroptosis88.
4.4. Multi-targeting necroptosis inhibitors
Ligustroflavone (26, Fig. 7), a natural product extracted from privet, was verified to possess anti-inflammation activity89. Upon the treatment of ligustroflavone, RIPK3 and MLKL were significantly decreased and the interaction between RIPK3 and RIPK1 or MLKL was reduced, while RIPK1 remained unaffected89. Thus, ligustroflavone was considered as a triple RIPK1/RIPK3/MLKL inhibitor and blocked necroptosis89.
GSK′067 (27) and GSK′074 (28) were RIPK1 and RIPK3 dual inhibitors identified by screening compound libraries90. Further mechanism studies indicated that compound 28 blocked the formation of RIPK1–RIPK3 complex and phosphorylation of MLKL without affecting the expression levels of RIPK1 and RIPK390. It blocked necroptosis with an IC50 value of 3 nmol/L and showed high binding affinity to RIPK1 (Kd = 12 nmol/L) and RIPK3 (Kd = 130 nmol/L) without activity to other kinases, indicating its selectivity to RIPK1 and RIPK390.
Fauster et al.91 screened 268 U.S. Food and Drug Adminstration (FDA) approved drugs for their ability to inhibit TNF-α-induced necroptosis, and identified ponatinib (29) and pazopanib (30) as necroptosis inhibitors. Ponatinib is a potent and orally available antitumor agent targeting BCR-ABL, VEGFR2 and FGFR1, while pazopanib is also a multi-targeting kinase inhibitor with inhibitory activity against VEGFR1, VEGFR2, VEGFR3, PDGFR, FGFR, c-Kit and c-Fms91. Ponatinib and pazopanib both displayed potent inhibitory activity against necroptosis with an EC50 of 89 and 254 nmol/L, respectively91. Further mechanism studies demonstrated that they blocked necroptosis induced by TNF-α, TRAIL and FasLs91. Ponatinib declined the phosphorylation of RIPK1, RIPK3 and MLKL, and inhibited the interaction between RIPK3 and MLKL91. Furthermore, thermal shift assay confirmed that RIPK3 was a target of ponatinib91. These data indicated that ponatinib directly targeted RIPK1 and RIPK391. Similarly, pazopanib showed relatively lower affinity towards RIPK1 and RIPK3. However, it had no influence on the interaction between RIPK3 and MLKL91.
5. Necroptosis in cancer therapy
5.1. Necroptosis is important in cancer treatment
5.1.1. Cancerous cells undergoing necroptosis is immunogenic
When the necroptotic cells are co-cultured with BMDCs (mouse bone marrow-derived dendritic cells), they can be phagocytized, resulting in the phenotypic maturation of BMDCs92. Meanwhile, necroptotic cancer cells in the form of vaccination effectively cross-primed cytotoxic CD8a+ T cells in vivo and induced strong CT26 tumor antigen-specific production of IFN-γ ex vivo, which was independent of the extent of NF-κB activation93. Thus, necroptotic cancer cells were considered as potent inducers of adaptive immune response and could mediate efficient antitumor immunity92. Additionally, there was evidence that compound 2 could prolong the life span of activated primary macrophages, suggesting that necroptosis could modulate the amount of active macrophages during infection92. Thus, necroptosis may serve as a cellular defense mechanism against the invasion of foreign organisms.
5.1.2. Necroptosis influences the metastasis of cancer cells
Metastasis is the primary cause of morbidity and mortality in cancer patients, which includes dissociation of malignant cells in the primary tumor, local invasion, angiogenesis, intravasation of invading cells into the vasculature or lymphatic systems, survival in these channels, extravasation, and proliferation at a distant site94. It was reported that shikonin (37) dramatically reduced the metastasis of osteosarcoma C6 and U87 glioma cells due to its induction of necroptosis95,96. Besides, ROS plays a significant role in the migration and invasion of cells through regulation of cytoskeleton dynamics and adhesion molecules, such as controlling expression levels of essential extracellular matrix-degrading enzymes (e.g., uPA and uPAR) in cell metastasis96,97. As mentioned above, overproduction of ROS promotes the execution of necroptosis. In other studies, RIPK3 was supposed to modulate the ROS level in case of necroptosis97. Thus, necroptosis has a close relationship with the metastasis of cancer cells.
5.1.3. Necroptosis serves as a back-up way for apoptosis
Before the concept of necroptosis was suggested, a prevailing trend in cancer chemotherapy is to discover molecules that can induce apoptosis98. However, apoptosis inducers cannot work in some circumstances, such as drug resistance due to the activation of alternative compensatory pathways, upregulation of drug transporters and multidrug resistance98. For example, proapoptotic agents, such a TRAIL and BCL-2 inhibitors, can induce necroptosis in cancer cells when apoptosis is blocked99. When a diphtheria-based fusion toxin or Smac mimetic applied with demethylating agents (e.g., 5-azacytidine or 5-aza-20-deoxycytidine), it can synergistically trigger cancer cell death and overcome apoptosis resistance by inducing necroptosis98. Trastuzumab, a humanized recombination monoclonal antibody, induces necroptosis to overcome the resistance of tumor cells against antibody treatment100. Treatment of L929 cells with caspase-inhibitor 17 (Fig. 4) mimicked the invasion of virus involved in innate immunity. As a result, such cells were proven to commit suicide through necroptosis92. The underlying mechanism was associated with the fact that apoptosis relied on the activation of caspases whereas necroptosis is independent of the activation of caspases92. Therefore, necroptosis can serve as an alternative way in cancer therapy in replace of various pathways of apoptosis.
5.2. Necroptosis may be conducive to cancer progression
Necroptotic cells are characterized with cell membrane leakage and release of intracellular damage-associated molecular patterns (e.g., IL-1 family cytokines, nucleic acids, ribonucleoproteins, histones, HMGB family members, and heat-shock proteins) which result in inflammatory responses and related side effects45. Subsequently, they might facilitate tumor progression, promote tumor cell proliferation and survival, as well as tumor angiogenesis, invasion and metastasis, leaving a detrimental effect on the tissue which is not favorable for the treatment101. In turn, the damaged tissue may also promote the metastasis of cancer cells101.
5.3. Necroptosis is not universally sensitive in tumor cell lines
Different cancer cell lines vary in terms of their sensitivity to necroptosis due to different environments such as the availability of oxygen or nutrients102. For example, human colorectal carcinoma cells are more sensitive to necroptosis under the hypoxic circumstances or in the absence of glucose compared to that in presence of abundant oxygen or glucose, possibly because of pyruvate scavenging of mitochondrial free radicals103. Also, the expression level of RIPK3 is fundamental for inducing necroptosis102. Numerous cancer cell lines, such as human breast adenocarcinoma cell line MCF-7, human hypotriploid cell line Hek293 and human cervix adenocarcinoma cell line HeLa, are unsuitable for necroptosis-based therapy due to low level of RIPK3 expression102. Therefore, necroptosis-based chemotherapy is only effective in limited tumor cell lines.
6. Necroptosis inducers
Recently, a series of compounds were reported to be capable of inducing tumor necroptosis in different pathways (Table 1)95,104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129. Several of them have been previously known to act by typical antitumor mechanisms, whose new role in necroptosis aid to draw a comprehensive mechanism diagram of these antitumor agents.
Table 1.
Necroptosis-inducing agents and their therapeutic applications.
| Necroptosis-inducing agent | Type of cells | Dosage | Ref. |
|---|---|---|---|
| Inorganic salts and metal complexes | |||
| CoCl2 31 (Cobalt chloride) |
HT-29 (human colon carcinoma cells) | 300 μmol/L | 104 |
| LiCl 32 (Lithium chloride) 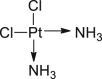
|
Human primary Schwannoma cells | 20 mmol/L | 105 |
33 (Cisplatin)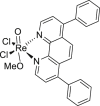
|
L929 cells (mouse fibroblast cells) | 100 μmol/L | 106 |
|
34 (Rhenium(V) oxo complexes) Natural products 
|
A549 cells (human lung cancer cells) | 20 μmol/L | 107 |
35 (Matrine)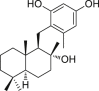
|
Mz-ChA-1 and QBC939 (human cholangiocarcinoma cells) | 1.5 mg/mL | 108 |
36 (Neoalbaconol)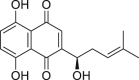
|
C666-1 and HK1 cells (human nasopharyngeal carcinoma cells) | 40 μmol/L | 109 |
37 (Shikonin)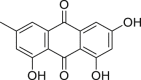
|
C6 and U87 (human glioma cells) | 3 μmol/L | 95 |
38 (Emodin)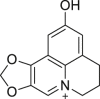
|
U251 (human glioblastoma cells) | 20 μmol/L | 110 |
|
39 (Ungeremine) Antitumor agents 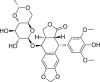
|
CCRF-CEM (human leukemia cells) | 4.89 μmol/L | 111 |
40 (Etoposide)
|
MDA-MB-231 (human breast cancer cells) HT1080 (human sarcoma cells) |
20 mmol/L | 112 |
41 (Givinostat)
|
HDLM-2 and L-540 HL cells (human Hodgkin lymphoma cells) | 100 μmol/L 5 μmol/L |
113 |
42 (Sorafenib)
|
MDA-MB-435 cells (human breast cancer cells) | 40 μmol/L | 114 |
43 (3-Bromopyruvate)
|
TE671 and RMS13 cells (human bone marrow transverse muscle carcinoma cells) | 200 nmol/L | 115 |
44 (Obatoclax)
|
CRC cells (human colorectal cancer cells) | 50 μg/mL | 116 |
45 (5-Fluorouracil)
|
|||
46 (IDN-7314)
|
A549 cells (human lung cancer cells) | 100 μmol/L | 117 |
47 (Methyl methanesulfonate)
|
MiaPaCa-2 (human pancreatic cells) | 15 μmol/L | 118 |
48 (ZZW-115)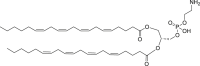
|
NCI–H28 cells (human malignant pleural mesothelioma cells) | 100 μmol/L | 119 |
49 (Diarachidonoylphosphoethanolamine)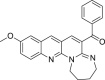
|
A375 Cells (human melanoma cells) | 8 μmol/L | 120 |
50 (3u)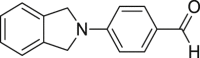
|
SKOV3 (human ovarian cells) | 12.5 μmol/L | 121 |
|
51 (673 A) Non-antitumor agents 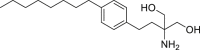
|
U251MG, U87MG cells (human glioblastoma cells) | 15 μmol/L | 122 |
52 (FTY720)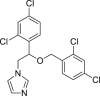
|
MDA-MB-231 (human breast cancer cells) | 40 μmol/L | 123 |
53 (Miconazole)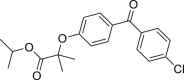
|
Hep3B (human hepatoma cells) | 50 μmol/L | 124 |
|
54 (Fenofibrate) Other necroptosis inducers 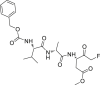 17 (Z-VAD-fmk) |
Classically activated macrophages | 20 μmol/L | 125 |
55 (Furosine)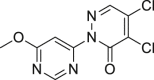
|
Primary hepatocytes | 100 μg/mL | 126 |
56 (LGH00168)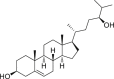
|
A549 cells (human lung cancer cells) | 3.26 μmol/L | 127 |
57 (24(S)-hydroxycholesterol)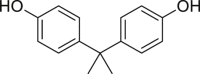
|
SH-SY5Y cells (human neuroblastoma cells) | 50 μmol/L | 128 |
| 58 (Bisphenol A) | SH-SY5Y (human neuroblastoma cells) | 1 nmol/L | 129 |
6.1. Inorganic salts and metal complexes as necroptosis inducers
Cobalt chloride (CoCl2, 31) was able to induce necrosis through direct observation by TEM. While it was verified to elevate the level of hypoxia-inducible factor-1α (HIF-1α) and induce necroptosis at 300 μmol/L with increased levels of RIPK1, RIPK3 and MLKL proteins when caspase activity was suppressed by compound 17 in HT-29 cells104.
Lithium chloride (LiCl, 32) was confirmed to induce cell death through activating TNF-α and inducing necroptosis105. The effect could be reversed by the treatment of necroptosis inhibitor 2, ROS inhibitor N-acetyl-l-cysteine (NAC), and inhibitors of the AKT/mTOR pathway in schwannoma cells, suggesting that LiCl-induced necroptosis was associated with ROS production105.
Cisplatin (33) is conventionally considered to induce cell apoptosis130. Chen et al.106 found that combinational treatment of cisplatin and pan-caspase inhibitor 17 increased the death of L929 cells compared to single treatment with cisplatin, suggesting there is additional pathway in cisplatin-induced cell death. Moreover, the addition of necroptosis inhibitor 2 dramatically rescued almost all of the cells. Further studies revealed that L929 cells treated with cisplatin secreted TNF-α which promoted the formation and translocation of RIPK1/RIPK3/MLKL complex, followed by the mitochondrial permeability transition and release of ROS, which finally led to the necroptosis106.
Rhenium(V) oxo complexes (34) effectively killed cancer cells by triggering necroptosis observed by fluorescence microscopy with cell membrane disintegration and PI uptake107. They also induced mitochondrial membrane rupture which is a downstream of ROS generation107. ROS generation induced by Rhenium(V) oxo complexes could be downregulated by co-treatment with necroptosis inhibitor 2, suggesting that RIPK1 is essential in ROS production, which is indicative of necroptotic prosperities107.
6.2. Natural products as necroptosis inducers
Matrine (35), an alkaloid extracted from traditional Chinese herb Sophora flavescens, was reported to induce necroptosis in cholangiocarcinoma cells, which was different from its classic apoptosis-inducing effects in other cancer cell lines108. Mz-ChA-1 and QBC939 cells (cholangiocarcinoma cells) treated with matrine were characterized with extensive organelle, plasma membrane rupture and integral nuclei which coincided with the morphology of necroptotic cells108. Additionally, cells died due to the treatment of matrine could not be rescued by the addition of pan-caspase inhibitor 17, but they could be rescued by necroptosis inhibitor 2108. Further study verified that matrine induced necroptosis through the formation of RIPK1/RIPK3/MLKL complex108. Osmotic pressure and release of ROS also facilitated the necroptotic process108. More interestingly, matrine was able to upregulate the expression of RIPK3 in Mz-ChA-1 cells, making it possible to treat cholangiocarcinoma with a low expression of RIPK3108. This work potentially provided a new strategy to deal with the apoptosis-resistance in cholangiocarcinoma therapy108.
Neoalbaconol (NA, 36), extracted from Albatrellus confluens, was reported to trigger several kinds of cell death109. NA down-regulated E3 ubiquitin ligases, resulting in reduced ubiquitination of RIPK1. Thus, an elevated expression level of RIPK1 was observed, which also activated the transcription of TNF-α109. Moreover, NA caused RIPK3-mediated ROS generation which contributed to cell death109. Thus, it can be concluded that NA was able to induce necroptosis through activation of TNF-α and RIPK3-dependent production of ROS109.
Shikonin (37), extracted from the traditional Chinese herb “Zicao”, was originally used to treat wound healing because of its anti-inflammatory and antimicrobial properties131. It was reported to kill tumor cells by inducing apoptosis131. Huang et al.95 found that lost in plasma membrane integrity and intact nuclear membrane in glioma cells were observed in cells treated with shikonin directly by electronic transmission microscopy. Shikonin-induced C6 and u87 glioma cells can be rescued by necroptosis inhibitor 2 but unaffected by treatment with the caspase inhibitor 17. Also, an increased expression of RIPK1 was observed after treatment with shikonin95. All these facts indicated that shikonin-induced necroptosis through RIPK1 activation.
Emodin (38), an anthraquinone derivative extracted from traditional Chinese medicine Rheum palmatum, has been used to treat various diseases due to its antitumor, anti-inflammation, anti-metastasis and immunosuppressive effects, but its mechanism of action remained unclear132. Zhou et al.110 found that cells treated with emodin showed increased levels of RIPK1, RIPK3 and TNF-α. Moreover, combinational use of emodin with necroptosis inhibitor 2 or 15 reduced the release of lactate dehydrogenase (LDH)110. Hematoxylin-eosin (H&E) staining of tumor tissues separated from mice treated with emodin demonstrated obvious necrosis effect110. Further studies indicated that TNF-α, RIPK1, RIPK3 and MLKL in tumor tissues were upregulated when treated with emodin, indicating that it could inhibit glioma growth in vivo through necroptosis110.
Ungeremine (39), an alkaloid extracted from Ungernia minor, showed potent cytotoxicity in drug-resistant cancer cell lines and upregulated RIPK3 level111. Co-treatment with necroptosis inhibitor 2 decreased the cytotoxicity of ungeremine, and flow cytometry analysis indicated that 13.1% cells were necoptotic111.
6.3. Antitumor agents as necroptosis inducers
Etoposide (40), a topoisomerase II inhibitor, has been used for the treatment of several various cancers, such as small cell lung cancer, lymphomas and testicular cancer133. Meier's group112 reported that etoposide (20 mmol/L) promoted the binding of caspase-8 to RIPK1 and FADD in cancer cells independent of TNF, TRAIL death receptors and mitochondria, while the effect could not be observed in normal HUVEC cells.
Combinational treatment of HDAC inhibitor givinostat (41) and kinase inhibitor sorafenib (42) showed synergistic activity in triggering cell death, ROS generation and mitochondrion disruption113. Further studies indicated that the combinational use induced cell necrosis three times higher than the single dosage113. Treatments with necroptosis inhibitor 2 or the ROS inhibitor resulted in cell survival, which indicated combinational use of givinostat and sorafenib-induced necroptotic cell death113.
3-Bromopyruvate (3-BrPA, 43), an alkylating agent, was reported to induce cell death in various ways through inhibiting hexokinase II activity which is a key enzyme of glycolysis overexpressed in cancer cells134. The addition of NSA reversed T24 cell (bladder carcinoma cell line) death caused by 3-BrPA, however radically treatment combined with necroptosis inhibitor 2 could rescue cells from death, indicating that 3-BrPA induced necroptosis in a RIPK1-independent but RIPK3-dependent pathway114.
Obatoclax (44), a small-molecule inhibitor of BCL-2 proteins, was verified to trigger cell death via autophagy115. Recently, it was reported to stimulate the interaction between Atg 5 (a constituent of autophagosomal membranes) and components of the necrosome (e.g., FADD, RIPK1 and RIPK3) through co-immunoprecipitation assay115. Knockdown of RIPK1 or treatment of necroptosis inhibitor 2 both suppressed obatoclax-induced cell death115. Moreover, knockdown of RIPK3 also partially reversed cell death115. Knockdown of RIPK1 and RIPK3 failed to affect the cells undergoing autography, indicating that necroptosis induced by obatoclax is the downstream of autography115. In vivo study was performed in RIPK1 wide-type and RIPK1 knockdown models, and the results showed that RIPK1 knockdown significantly suppressed the tumor inhibition caused by obatoclax115.
5-Fluorouracil (5-FU, 45) is a thymidylate synthetase inhibitor and is used as the standard chemotherapy for colorectal cancer135. However, drug resistance of 5-FU is a huge challenge in clinical studies116. Recently, it was reported that pan-caspase inhibitors were able to sensitize 5-FU to cell death in drug resistant cells116. In vivo studies showed that the combination of pan-caspase inhibitor IDN-7314 (46) and 5-FU showed synergistic effects in inhibiting tumor growth of HT29 xenografts116. Further mechanism study showed that the combinational use could suppress caspase-8 mediated cleavage of RIPK1 and stabilize RIPK1-containg complexes and the component of necrosome was observed. Thus, 5-FU induced necroptosis in the presence of pan-caspase inhibitors in drug-resistant models116.
Methyl methanesulfonate (47), an alkylating agent, was reported to induce cell death through apoptosis136. Recently, Jiang et al.117 found that high dosage (400 and 800 μmol/L) of compound 47 induced cell death through necroptosis rather than apoptosis. Co-treatment of compound 47 with necroptosis inhibitor 2 could reverse cell death, while co-treatment with caspase inhibitor 17 failed117. Necroptosis biomarkers, such as leakage of LDH, HMGB1, ROS and RIPKs, were upregulated, while apoptosis biomarkers caspase-3 and caspase-9 were not influenced, suggesting that compound 47 was able to induce necroptosis in A549 cells (human lung cancer cells)117.
Trifluoperazine, an antipsychotic agent, possesses antitumor activity137. However, its application in cancer therapy is limited by significant side effects on central nervous system137. Based on the structure–activity relationship study, trifluoperazine derivative ZZW-115 (48) was found to induce cell death mainly by necroptosis without neurological effects118. Co-treatment with necroptosis inhibitor 2 or caspase inhibitor 19 could partially reverse cell death induced by compound 48, indicating that it induced necroptosis and apoptosis through independent mechanisms118.
Diarachidonoylphosphoethanolamine (DAPE, 49) was verified to induce apoptosis and necrosis through flow cytomety. Later, it was found that the cell death induced by NCI-H28 cells could be partially reversed by necroptosis inhibitor 2 or knockdown of RIPK1, indicating that DAPE could induce necroptosis119. Moreover, mitochondrial membranes were disrupted and ROS was released, which could be reversed by cyclophilin D inhibitors. Therefore, DAPE induced necroptosis through activation of RIPK1- and RIPK3-dependent production of ROS119.
Naphthyridine derivatives are heterocycles widely used in anti-tumour therapy138. Kong et al.120 discovered a potent naphthyridine derivative (50) through screening 1,3-diazaheterocycle fused naphthyridine derivatives. Low dosage (4 and 8 μmol/L) of compound 50-induced cell death could not be clearly rescued by caspase inhibitor 17, while rescue effects were observed at high dosage (12, 16 and 20 μmol/L), suggesting that there was another pathway to induce cell death when compound 50 was used at low dosages. Western blot studies showed that RIPK1 and MLKL were upregulated at low concentrations while cleaved by caspase-8 at high concentrations, suggesting that necroptosis was induced at low concentration and inhibited at high concentrations120.
Aldehyde dehydrogenase (ALDH) is associated with antitumor drug resistance and is a potential target for cancer therapy139. Pre-treatment of cells with the caspase inhibitor 17 before ALDH inhibitor 673 A (51) did not promote cell viability, indicating that compound 51 induces cell death in a non-apoptotic way121. Compound 51-treated cells stained with DAPI led to nuclear swelling and loss of nuclear content, which was consistent with TEM, indicating that cells are necroptotic121. Downregulation of RIPK1 levels failed to rescue necroptotic cells trigger by compound 51, and similar results were observed for the necroptosis inhibitor 2 treatment121. Low dose of MLKL inhibitor NSA could protect cells from necroptotic, suggesting that compound 51 induced necroptosis is RIPK3 dependent121.
6.4. Non-antitumor agents as necroptosis inducers
FTY720 (52) is an immunosuppressive drug that was approved for the treatment of multiple sclerosis140. The levels of RIPK1 and RIPK3 were upregulated in human glioblastoma cells when treated with compound 52, indicating that it induced RIPK1/RIPK3-mediated necroptosis122. Cells treated with compound 52 and necroptosis inhibitor 2 rescued most of the cells, while knockdown of RIPK3 also resulted in rescuing cells from death122.
Antifungal agent miconazole (53) induced both apoptosis and necroptosis in MDA-MB-231 cells, which could be directly observed by TEM123. Treatment with caspase inhibitor 17 or necroptosis inhibitor 2 could partially rescue cells, indicating that micronazole induced both apoptosis and necroptosis123. Furthermore, upregulated RIPK3 levels, MLKL levels and ROS generation were also observed as necroptotic properties123.
Fenofibrate (54) was widely used to reduce high cholesterol and high triglyceride levels in blood141. Hep3B cells (human hematoma cells) treated with fenofibrate for 24 h underwent nuclear morphology changes such as chromatin condensation, nuclear fragmentation and irregularly shaped nucleus124. Analysis of the effect of fenofibrate on the expression level of caspases and necroptotic markers showed that active form of caspase-3 and caspase-8 were decreased while RIPK1, RIPK3 and MLKL were upregulated, indicating that fenofibrate induced necroptosis in Hep3B cells124.
6.5. Other necroptosis inducers
As described above, compound 17 is a pan-caspase inhibitor and is commonly used to sensitize resistant cells through inducing necroptosis125. When it was used alone, the expression level of phosphorylated MLKL was upregulated in LPS-activated macrophages (CAMs)125. Furthermore, co-treatment with necroptosis inhibitor 2 partially inhibited LDH release and phosphorylation of MLKL in CAMs, while co-treatment with TNF-α antagonist had no influence on compound 17 induced cell death, indicating that it induced necroptosis is TNF-α independent125.
Furosine (55) was reported to activate the expression of RIPK1, RIPK3, MLKL and TNF-α in hepatocytes, suggesting that it induced the necroptosis pathway and subsequent inflammatory reactions126.
C/EBP homologous protein (CHOP) played a significant role in endoplasmic reticulum (ER) stress-induced apoptosis142. A549 cells treated with CHOP activator LGH00168 (56) were regarded as necroptotic due to rupture of plasma membrane and lysosomal membrane permeabilization and caspase-8 inhibition127. Moreover, it could generate ROS in a dose-dependent manner and led to rupture of the plasma membrane, which was a symbol of necrosis. However, it can be effectively attenuated by ROS scavenger NAC, which indicated that necroptosis rather than necrosis played an essential role in inducing cell death127. Co-treatment with necroptosis inhibitor 2 rescued cells from death and reduced PI uptake, suggesting that the induced necroptosis was in a RIPK1-dependent manner127.
24(S)-Hydroxycholesterol (24S–OHC, 57) is enzymatically produced in the brain to maintain cholesterol homeostasis143. In T-lymphoma Jurkat cells, it triggered caspase-independent cell death, which was suppressed by additional treatment of necroptosis inhibitor 2 or knockdown of RIPK3, suggesting its necroptosis inducing roles128.
Bisphenol A (BPA, 58) is an endocrine disruptor with estrogenic and obesogenic activity42. SH-SY5Y cells (human neuroblastoma cell line) treated with compound 58 underwent apoptosis within 48 h, then necrosis was occurred and increased gradually129. When necroptosis inhibitor 2 was added in combination treatment with compound 58, necroptosis was replaced by apoptosis, indicating that it induced necroptosis129.
7. Necroptosis and other diseases
Apart from the relationship with tumor therapy, necroptosis has been reported to be related to cerebral ischemia, neurodegenerative diseases and immune diseases. Dabrafenib, a potent RIPK3 inhibitor, was proven to be able to block LPS-induced activation of TNF-α and significantly reduce infarct lesion size, indicating that inhibiting necroptosis could be neuroprotective and attenuate cerebral ischemia144. Necroptosis could mediate neuronal loss in neurogenerative diseases. Either inhibition of RIPK1 or the knockdown of RIPK3 and MLKL was able to decrease degeneration, suggesting inhibiting necroptosis was favorable in treating neurodegenerative diseases145. Overexpression of RIPK3 was observed in human Paneth cells and increased necroptosis in Crohn's disease, suggesting potential therapeutic effect of necroptosis inhibition146.
8. Conclusions and perspectives
In summary, the mechanism of necroptosis and its applications in cancer therapy were reviewed. Necroptosis of cancer cells could be initiated by various stimuli and through different pathways. RIPK1, RIPK3 and MLKL played an important role in the process of necroptosis, resulting in membrane leakage and release of cytokines. Necroptosis is associated with tumor metastasis which is the major cause of morbidity and mortality in cancer patients in clinic. Therefore, induction of necroptosis is an effective strategy in clinical cancer therapy. Necroptotic cells could potentially induce intrinsic and adaptive immune response and thus mediate efficient antitumor immunity. In addition, necroptosis can serve as a back-up way for apoptosis-resistant circumstances, which are capable of overcoming the obstacle of drug-resistance which is common but intricate in clinical cancer therapy. For those apoptosis-resistant cases, necroptosis inducers could sensitize tumor cells to death, which provides another way to overcome drug resistance when common therapy is failed. Thus, drug development of necroptosis inducers deserves to be paid more attention. However, the release of cytokines also induces inflammation which would be harmful to the tissue and in turn tissue damage could facilitate the metastasis of tumor cells. However, necroptosis is accompanied by the release of cytokines and induce inflammation, which is harmful to the tissue and in turn tissue damage can facilitate the metastasis of tumor cells. Also, it should be noted that necroptosis is not widely sensitive in cancer cells in that apoptosis is the main cause of cancer death. Therefore, the necroptosis inducers are mostly favorable in apoptosis-resistant cases in clinical cancer therapy. Currently, several antitumor agents have been verified to act as necroptosis inducers while more medicinal chemistry efforts are required to discover new inducers with drug-like properties.
So far, a series of necroptotic inducers have been identified, paving the pathway for investigating the new mode of cancer death and providing novel therapeutic tools. However, most studies are carried out by in vitro experiments, and the in vivo efficacy of necroptosis inducer and their selectivity in killing tumors remain to be further explored. Also, there is still lack of in vivo necroptotic markers. Thus, it is highly desirable to discover new necroptotic markers and investigate their effects on the selectivity of cancer cells. With better understanding of the mechanism of necroptosis in cancer cells, targeting necroptosis will be an effective strategy for cancer therapy.
Acknowledgments
This work was supported by the National Key R&D Program of China (Grant 2017YFA0506000 to Chunquan Sheng), National Natural Science Foundation of China (Grants 81725020 and 21738002 to Chunquan Sheng and 81872742 to Guoqiang Dong) and the Innovation Program of Shanghai Municipal Education Commission (Grant 2019-01-07-00-07-E00073 to Chunquan Sheng, China).
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Guoqiang Dong, Email: dgq-81@163.com.
Chunquan Sheng, Email: shengcq@smmu.edu.cn.
Author contributions
Chunquan Sheng and Guoqiang Dong proposed the idea and revised the manuscript. Ying Wu and Guoqiang Dong wrote the manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- 1.Hardy K., Handyside A.H., Winston R.M. The human blastocyst: cell number, death and allocation during late preimplantation development in vitro. Development. 1989;107:597–604. doi: 10.1242/dev.107.3.597. [DOI] [PubMed] [Google Scholar]
- 2.Lockshin R.A., Zakeri Z. Cell death in health and disease. J Cell Mol Med. 2007;11:1214–1224. doi: 10.1111/j.1582-4934.2007.00150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kerr J.F., Wyllie A.H., Currie A.R. Apoptosis: a basic biological phenomenon with wide ranging implications in tissue kinetics. Br J Canc. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuchs Y., Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Festjens N., Vanden Berghe T., Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta. 2006;1757:1371–1387. doi: 10.1016/j.bbabio.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 6.Golstein P., Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Krantic S. Programmed cell death: apoptotic versus necrotic. 3rd mediterranean conference of neuroscience, Alexandria, Egypt. Front Neurosci. 13 Dec–16 Dec, 2009 doi: 10.3389/conf.neuro.01.2009.16.076. Available from: [DOI] [Google Scholar]
- 8.Laster S.M., Wood J.G., Gooding L.R. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141:2629–2634. [PubMed] [Google Scholar]
- 9.Zong W.X., Ditsworth D., Bauer D.E., Wang Z.Q., Thompson C.B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 11.Brown J.M., Wilson G. Apoptosis genes and resistance to cancer therapy: what does the experimental and clinical data tell us? Cancer Biol Ther. 2003;2:477–490. doi: 10.4161/cbt.2.5.450. [DOI] [PubMed] [Google Scholar]
- 12.Jin G.S., Liu Y.H., Xu P.Q., Jin G.S. Induction of necroptosis in human breast cancer drug-resistant cells by SMAC analog LCL161 after caspase inhibition requires RIP3. Die Pharmazie. 2019;74:363–368. doi: 10.1691/ph.2019.9307. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y.G., Hao F.S., Nan Y.H., Qu L.C., Na W.L., Jia C.S. PKM2 inhibitor shikonin overcomes the cisplatin eesistance in bladder cancer by inducing necroptosis. Int J Biol Sci. 2018;14:1883–1891. doi: 10.7150/ijbs.27854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi M.E., Price D.R., Ryter S.W., Choi A.M.K. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. 2019;4 doi: 10.1172/jci.insight.128834. pii 128834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conrad M., Angeli J.P., Vandenabeele P., Stockwell B.R. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–366. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weinlich R., Oberst A., Beere H.M., Green D.R. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18:127–136. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 17.Bertrand M.J., Milutinovic S., Dickson K.M., Ho W.C., Boudreault A., Durkin J. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 18.Hsu H., Xiong J., Goeddel D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 19.Wang L., Du F.H., Wang X.D. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 20.He S., Liang Y., Shao F., Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holler N., Zaru R., Micheau O., Thome M., Attinger A., Valitutti S. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 22.Seya T., Shime H., Takaki H., Azuma M., Oshiumi H., Matsumoto M. TLR3/TICAM-1 signaling in tumor cell RIP3-dependent necroptosis. OncoImmunology. 2012;1:917–923. doi: 10.4161/onci.21244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen D.S., Yu J., Zhang L. Necroptosis: an alternative cell death program defending against cancer. Biochim Biophys Acta. 2016;1865:228–236. doi: 10.1016/j.bbcan.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong Y.T., Fan Z.Y., Luo G.P., Yang C., Huang Q.Y., Fan K. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019;18:100. doi: 10.1186/s12943-019-1029-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philipp S., Sosna J., Adam D. Cancer and necroptosis: friend or foe? Cell Mol Life Sci. 2016;73:2183–2193. doi: 10.1007/s00018-016-2193-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nehs M.A., Lin C.I., Kozono D.E., Whang E.E., Cho N.L., Zhu K. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery. 2011;150:1032–1039. doi: 10.1016/j.surg.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 27.Wu X., Wu M.Y., Jiang M., Zhi Q.M., Bian X.J., Xu M.D. TNF-alpha sensitizes chemotherapy and radiotherapy against breast cancer cells. Cancer Cell Int. 2017;17:13. doi: 10.1186/s12935-017-0382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H.X., Lin X. Positive and negative signaling components involved in TNFα-induced NF-κB activation. Cytokine. 2008;41:1–8. doi: 10.1016/j.cyto.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 29.Boatright K.M., Salvesen G.S. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 30.Cook W.D., Moujalled D.M., Ralph T.J., Lock P., Young S.N., Murphy J.M. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ. 2014;21:1600–1612. doi: 10.1038/cdd.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie T., Peng W., Yan C., Wu J., Gong X., Shi Y. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 2013;5:70–78. doi: 10.1016/j.celrep.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 32.Dickens L.S., Powley I.R., Hughes M.A., MacFarlane M. The ‘complexities’ of life and death: death receptor signalling platforms. Exp Cell Res. 2012;318:1269–1277. doi: 10.1016/j.yexcr.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Thomas L.R., Henson A., Reed J.C., Salsbury F.R., Thorburn A. Direct binding of Fas-associated death domain (FADD) to the tumor necrosis factor-related apoptosis-inducing ligand receptor DR5 is regulated by the death effector domain of FADD. J Biol Chem. 2004;279:32780–32785. doi: 10.1074/jbc.M401680200. [DOI] [PubMed] [Google Scholar]
- 34.Pasparakis M., Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 35.Sun L.M., Wang X.D. A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends Biochem Sci. 2014;39:587–593. doi: 10.1016/j.tibs.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 36.Cai Z.Y., Jitkaew S., Zhao J., Chiang H.C., Choksi S., Liu J. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X., Li W.J., Ren J.M., Huang D.L., He W.T., Song Y.L. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–121. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papatriantafyllou M. Cell death: programmed necrosis: putting the pieces together. Nat Rev Mol Cell Biol. 2012;13:135. doi: 10.1038/nrm3292. [DOI] [PubMed] [Google Scholar]
- 39.Yashin D.V., Romanova E.A., Ivanova O.K., Sashchenko L.P. The Tag7-Hsp70 cytotoxic complex induces tumor cell necroptosis via permeabilisation of lysosomes and mitochondria. Biochimie. 2016;123:32–36. doi: 10.1016/j.biochi.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 40.Wu T., Chen W.Z., Han J.H. Role of RIP3 in necrotic cell death. In: Shen H.M., Vandenabeele P., editors. Necrotic cell death. Cell death in biology and diseases. Springer (Humana Press); New York: 2014. pp. 44–45. [Google Scholar]
- 41.Christofferson D.E., Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Legeay S., Faure S. Is bisphenol A an environmental obesogen? Fundam Clin Pharmacol. 2017;31:594–609. doi: 10.1111/fcp.12300. [DOI] [PubMed] [Google Scholar]
- 43.Fulda S. Regulation of necroptosis signaling and cell death by reactive oxygen species. Biol Chem. 2016;397:657–660. doi: 10.1515/hsz-2016-0102. [DOI] [PubMed] [Google Scholar]
- 44.Davis C.W., Hawkins B.J., Ramasamy S., Irrinki K.M., Cameron B.A., Islam K. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic Biol Med. 2010;48:306–317. doi: 10.1016/j.freeradbiomed.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaczmarek A., Vandenabeele P., Krysko D.V. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:20923. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Greten F.R., Eckmann L., Greten T.F., Park J.M., Li Z.W., Egan L.J. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 47.Grivennikov S.I., Greten F.R., Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu X.S., Chua C.C., Zhang M., Geng D.Q., Liu C.F., Hamdy R.C. The role of PARP activation in glutamate-induced necroptosis in HT-22 cells. Brain Res. 2010;1343:206–212. doi: 10.1016/j.brainres.2010.04.080. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y.J., Luo W.B., Wang Y.F. PARP-1 and its associated nucleases in DNA damage response. DNA Repair (Amst) 2019;81:102651. doi: 10.1016/j.dnarep.2019.102651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moubarak R.S., Yuste V.J., Artus C., Bouharrour A., Greer P.A., Murcia M.D. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol. 2007;27:4844–4862. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Q., Ju X.L., Yang Z., Chen K.P. Necroptotic cells release find-me signal and are engulfed without proinflammatory cytokine production. In Vitro Cell Dev Biol Anim. 2015;51:1033–1039. doi: 10.1007/s11626-015-9926-7. [DOI] [PubMed] [Google Scholar]
- 52.Häcker G., Hacker G. The morphology of apoptosis. Cell Tissue Res. 2000;301:5–17. doi: 10.1007/s004410000193. [DOI] [PubMed] [Google Scholar]
- 53.Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 54.Gizycka A., Chorostowska-Wynimko J. Programmed necrosis and necroptosis—molecular mechanisms. Postepy Hig Med Dosw. 2015;69:1353–1363. doi: 10.5604/17322693.1186337. [DOI] [PubMed] [Google Scholar]
- 55.Qi S.S., Guo L.Y., Yan S.Z., Lee R.J., Yu S.Q. Hypocrellin A-based photodynamic action induces apoptosis in A549 cells through ROS-mediated mitochondrial signaling pathway. Acta Pharm Sin B. 2019;9:279–293. doi: 10.1016/j.apsb.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen S.F., Lv X., Hu B.W., Shao Z.W., Wang B.C., Ma K.G. RIPK1/RIPK3/MLKL-mediated necroptosis contributes to compression-induced rat nucleus pulposus cells death. Apoptosis. 2017;22:626–638. doi: 10.1007/s10495-017-1358-2. [DOI] [PubMed] [Google Scholar]
- 57.Bailey L.J., Alahari S., Tagliaferro A., Post M., Caniggia I. Augmented trophoblast cell death in preeclampsia can proceed via ceramide-mediated necroptosis. Cell Death Dis. 2017;8 doi: 10.1038/cddis.2016.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berger S.B., Kasparcova V., Hoffman S., Swift B., Dare L., Schaeffer M. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192:5476–5480. doi: 10.4049/jimmunol.1400499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ofengeim D., Ito Y., Najafov A., Zhang Y., Shan B., DeWitt J.P. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10:1836–1849. doi: 10.1016/j.celrep.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li D.R., Xu T., Cao Y., Wang H.Y., Li L., Chen S. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci U S A. 2015;112:5017–5022. doi: 10.1073/pnas.1505244112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meng L.J., Jin W., Wang X.D. RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc Natl Acad Sci U S A. 2015;112:11007–11012. doi: 10.1073/pnas.1514730112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang H.Y., Sun L.M., Su L.J., Rizo J., Liu L., Wang L.F. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 63.Mcquade T., Cho Y.S., Chan K.M. Positive and negative phosphorylation regulates RIP1 and RIP3-induced programmed necrosis. Biochem J. 2013;456:409–415. doi: 10.1042/BJ20130860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cho Y.S., Challa S., Moquin D., Genga R., Ray T.D., Guildford M. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1113. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pan P.C., Cai Z.Y., Zhuang C.L., Chen X.F., Chai Y.F. Methodology of drug screening and target identification for new necroptosis inhibitors. J Pharm Anal. 2019;9:71–76. doi: 10.1016/j.jpha.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suntharalingam K., Awuah S.G., Bruno P.M., Johnstone T.C., Wang F., Lin W. Necroptosis-inducing Rhenium(V) oxo complexes. J Am Chem Soc. 2015;137:2967–2974. doi: 10.1021/ja511978y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Z.Y., Wu B., Guo Y.S., Zhou Y.H., Fu Z.G., Xu B.Q. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am J Cancer Res. 2015;5:3174–3185. [PMC free article] [PubMed] [Google Scholar]
- 68.Degterev A., Maki J.L., Yuan J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2013;20:366. doi: 10.1038/cdd.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berger S.B., Harris P., Nagilla R., Kasparcova V., Hoffman S., Swift B. Characterization of GSK'963: a structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Dis. 2015;1:15009. doi: 10.1038/cddiscovery.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harris P.A., Berger S.B., Jeong J.U., Nagilla R., Bandyopadhyay D., Campobasso N. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase aspecific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J Med Chem. 2017;60:1247–1261. doi: 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- 71.Weisel K., Scott N.E., Tompson D.J., Votta B.J., Madhavan S., Povey K. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol Res Perspect. 2017;5 doi: 10.1002/prp2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harris P.A., King B.W., Bandyopadhyay D., Berger S.B., Campobasso N., Capriotti C.A. DNA-encoded library screening identifies benzo[b][1,4]oxazepin-4-ones as highly potent and monoselective receptor interacting protein 1 kinase inhibitors. J Med Chem. 2016;59:2163–2178. doi: 10.1021/acs.jmedchem.5b01898. [DOI] [PubMed] [Google Scholar]
- 73.Wang W., Marinis J.M., Beal A.M., Savadkar S., Wu Y., Khan M. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell. 2018;34:757–774. doi: 10.1016/j.ccell.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cougnoux A., Clifford S., Salman A., Ng S.L., Bertin J., Porter F.D. Necroptosis inhibition as a therapy for Niemann-Pick disease, type C1: inhibition of RIP kinases and combination therapy with 2-hydroxypropyl-beta-cyclodextrin. Mol Genet Metab. 2018;125:345–350. doi: 10.1016/j.ymgme.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harris P.A., Faucher N., George N., Eidam P.M., King B.W., White G.V. Discovery and lead-optimization of 4,5-dihydropyrazoles as mono-kinase selective, orally vioavailable and efficacious inhibitors of receptor interacting protein 1 (RIP1) kinase. J Med Chem. 2019;62:5096–5110. doi: 10.1021/acs.jmedchem.9b00318. [DOI] [PubMed] [Google Scholar]
- 76.Delehouze C., Leverrier-Penna S., Le Cann F., Comte A., Jacquard-Fevai M., Delalande O. 6E11, a highly selective inhibitor of receptor-interacting protein kinase 1, protects cells against cold hypoxia-reoxygenation injury. Sci Rep. 2017;7:12931. doi: 10.1038/s41598-017-12788-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Najjar M., Suebsuwong C., Ray S.S., Thapa R.J., Maki J.L., Nogusa S. Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 2015;10:1850–1860. doi: 10.1016/j.celrep.2015.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang C., Luo Y.Q., He Q.L., Liu S., He A.D., Yan J. A pan-RAF inhibitor LY3009120 inhibits necroptosis by preventing phosphorylation of RIPK1 and alleviates dextran sulfate sodium-induced colitis. Clin Sci (Lond) 2019;133:919–932. doi: 10.1042/CS20181081. [DOI] [PubMed] [Google Scholar]
- 79.von Schaper E. Everything but amyloid: new thinking prompts FDA revamp. Nat Biotechnol. 2018;36:4834. doi: 10.1038/nbt0618-483. [DOI] [PubMed] [Google Scholar]
- 80.Mandal P., Berger S.B., Pillay S., Moriwaki K., Huang C., Guo H. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li J.X., Feng J.M., Wang Y., Li X.H., Chen X.X., Su Y. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 2014;5:e1278. doi: 10.1038/cddis.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hussain M., Zimmermann V., van Wijk S.J.L., Fulda S. Mouse lung fibroblasts are highly susceptible to necroptosis in a reactive oxygen species-dependent manner. Biochem Pharmacol. 2018;153:242–247. doi: 10.1016/j.bcp.2018.01.025. [DOI] [PubMed] [Google Scholar]
- 83.Park H.H., Park S.Y., Mah S., Park J.H., Hong S.S., Hong S. HS-1371, a novel kinase inhibitor of RIP3-mediated necroptosis. Exp Mol Med. 2018;50:125. doi: 10.1038/s12276-018-0152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang H., Xu L.J., Qin X., Chen X.F., Cong H., Hu L.M. N-(7-Cyano-6-(4-fluoro-3-(2-(3-(trifluoromethyl)phenyl)acetamido)phenoxy)benzo[d]thiazol-2-yl)cyclopropanecarboxamide (TAK-632) analogues as novel necroptosis inhibitors by targeting receptor-interacting protein kinase 3 (RIPK3): synthesis, structure–activity relationships, and in vivo efficacy. J Med Chem. 2019;62:6665–6681. doi: 10.1021/acs.jmedchem.9b00611. [DOI] [PubMed] [Google Scholar]
- 85.Rodriguez D.A., Weinlich R., Brown S., Guy C., Fitzgerald P., Dillon C.P. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016;23:76–88. doi: 10.1038/cdd.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liao D.H., Sun L.M., Liu W.L., He S.D., Wang X.D., Lei X.G. Necrosulfonamide inhibits necroptosis by selectively targeting the mixed lineage kinase domain-like protein. Med Chem Comm. 2014;5:333–337. [Google Scholar]
- 87.Hildebrand J.M., Tanzer M.C., Lucet I.S., Young S.N., Spall S.K., Sharma P. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;11:15072–15077. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yan B., Liu L., Huang S.Q., Ren Y., Wang H.Y., Yao Z.L. Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem Commun. 2017;53:3637–3640. doi: 10.1039/c7cc00667e. [DOI] [PubMed] [Google Scholar]
- 89.Zhang Y.Y., Liu W.N., Li Y.Q., Zhang X.J., Yang J., Luo X.J. Ligustroflavone reduces necroptosis in rat brain after ischemic stroke through targeting RIPK1/RIPK3/MLKL pathway. Naunyn-Schmiedebergs Arch Pharmakol. 2019;392:1085–1095. doi: 10.1007/s00210-019-01656-9. [DOI] [PubMed] [Google Scholar]
- 90.Zhou T., Wang Q.W., Phan N., Ren J., Yang H., Feldman C.C. Identification of a novel class of RIP1/RIP3 dual inhibitors that impede cell death and inflammation in mouse abdominal aortic aneurysm models. Cell Death Dis. 2019;10:226. doi: 10.1038/s41419-019-1468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fauster A., Rebsamen M., Huber K.V., Bigenzahn J.W., Stukalov A., Lardeau C.H. A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis. 2015;6:e1767. doi: 10.1038/cddis.2015.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hitomi J., Christofferson D.E., Ng A., Yao J., Degterev A., Xavier R.J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aaes T.L., Kaczmarek A., Delvaeye T., de Craene B., de Koker S., Heyndrickx L. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep. 2016;15:274–287. doi: 10.1016/j.celrep.2016.03.037. [DOI] [PubMed] [Google Scholar]
- 94.Duffy M., Mcgowan P., Gallagher W. Cancer invasion and metastasis: changing views. J Pathol. 2008;214:283–293. doi: 10.1002/path.2282. [DOI] [PubMed] [Google Scholar]
- 95.Huang C.J., Luo Y.N., Zhao J.W., Yang F.W., Zhao H.W., Fan W.H. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One. 2013;8 doi: 10.1371/journal.pone.0066326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fu Z.Z., Deng B.Y., Liao Y.X., Shan L.C., Yin F., Wang Z.Y. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer. 2013;13:580. doi: 10.1186/1471-2407-13-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim Y.S., Morgan M.J., Choksi S., Liu Z.G. TNF-induced activation of the Nox 1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell. 2007;26:675–687. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 98.Steinhart L., Belz K., Fulda S. Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell Death Dis. 2013;4 doi: 10.1038/cddis.2013.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yan C., Oh J.S., Yoo S.H., Lee J.S., Yoon Y.G., Oh Y.J. The targeted inhibition of mitochondrial Hsp 90 overcomes the apoptosis resistance conferred by Bcl-2 in Hep3B cells via necroptosis. Toxicol Appl Pharmacol. 2013;266:9–18. doi: 10.1016/j.taap.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 100.Hu J.M., Liu X.L., Hughes D., Esteva F.J., Liu B.L., Chandra J. Herceptin conjugates linked by EDC boost direct tumor cell death via programmed tumor cell necrosis. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Smith C.C., Yellon D.M. Necroptosis, necrostatins and tissue injury. J Cell Mol Med. 2011;15:1797–1806. doi: 10.1111/j.1582-4934.2011.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen W.Z., Wu J.F., Li L.S., Zhang Z.M., Ren J.M., Liang Y.J. Ppm1b negatively regulates necroptosis through dephosphorylating Rip 3. Nat Cell Biol. 2015;17:434–444. doi: 10.1038/ncb3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang C.Y., Kuo W.T., Huang Y.C., Lee T.C., Yu L.C. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013;4:e622. doi: 10.1038/cddis.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang H.Y., Zhang B. Cobalt chloride induces necroptosis in human colon cancer HT-29 cells. Asian Pac J Cancer Prev APJCP. 2015;16:2569–2574. doi: 10.7314/apjcp.2015.16.6.2569. [DOI] [PubMed] [Google Scholar]
- 105.Wang Y., Zhang Q., Wang B., Li P., Liu P.N. LiCl Treatment induces programmed cell death of Schwannoma cells through AKT- and MTOR-mediated necroptosis. Neurochem Res. 2017;42:2363–2371. doi: 10.1007/s11064-017-2256-2. [DOI] [PubMed] [Google Scholar]
- 106.Xu Y.F., Ma H.B., Fang Y.L., Zhang Z.R., Shao J., Hong M. Cisplatin-induced necroptosis in TNFalpha dependent and independent pathways. Cell Signal. 2017;31:112–123. doi: 10.1016/j.cellsig.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 107.Suntharalingam K., Awuah S.G., Bruno P.M., Johnstone T.C., Wang F., Lin W. Necroptosis-inducing rhenium(V) oxo complexes. J Am Chem Soc. 2015;137:2967–2974. doi: 10.1021/ja511978y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu B.B., Xu M.Y., Tian Y., Yu Q., Zhao Y.J., Chen X. Matrine induces RIP3-dependent necroptosis in cholangiocarcinoma cells. Cell Death Dis. 2017;3:16096. doi: 10.1038/cddiscovery.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yu X.F., Deng Q.P., Li W., Xiao L.B., Luo X.J., Liu X.L. Neoalbaconol induces cell death through necroptosis by regulating RIPK-dependent autocrine TNFalpha and ROS production. Oncotarget. 2015;6:1995–2008. doi: 10.18632/oncotarget.3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou J.B., Li G.H., Han G.K., Feng S., Liu Y.H., Chen J. Emodin induced necroptosis in the glioma cell line U251 via the TNF-alpha/RIP1/RIP3 pathway. Investig New Drugs. 2020;38:50–59. doi: 10.1007/s10637-019-00764-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mbaveng A.T., Bitchagno G.T.M., Kuete V., Tane P., Efferth T. Cytotoxicity of ungeremine towards multi-factorial drug resistant cancer cells and induction of apoptosis, ferroptosis, necroptosis and autophagy. Phytomedicine. 2019:152832. doi: 10.1016/j.phymed.2019.152832. [DOI] [PubMed] [Google Scholar]
- 112.Tenev T., Bianchi K., Darding M., Broemer M., Langlais C., Wallberg F. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 113.Locatelli S.L., Cleris L., Stirparo G.G., Tartari S., Saba E., Pierdominici M. BIM upregulation and ROS-dependent necroptosis mediate the antitumor effects of the HDACi givinostat and sorafenib in Hodgkin lymphoma cell line xenografts. Leukemia. 2014;28:1861–1871. doi: 10.1038/leu.2014.81. [DOI] [PubMed] [Google Scholar]
- 114.Konstantakou E.G., Voutsinas G.E., Velentzas A.D., Basogianni A.S., Paronis E., Balafas E. 3-BrPA eliminates human bladder cancer cells with highly oncogenic signatures via engagement of specific death programs and perturbation of multiple signaling and metabolic determinants. Mol Cancer. 2015;14:135. doi: 10.1186/s12943-015-0399-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Basit F., Cristofanon S., Fulda S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013;20:1161–1173. doi: 10.1038/cdd.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Oliver Metzig M., Fuchs D., Tagscherer K.E., Gröne H.J., Schirmacher P., Roth W. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-alpha-dependent necroptosis driven by RIP1 kinase and NF-kappaB. Oncogene. 2016;35:3399–3409. doi: 10.1038/onc.2015.398. [DOI] [PubMed] [Google Scholar]
- 117.Jiang Y., Shan S.G., Chi L.F., Zhang G.L., Gao X.J., Li H.J. Methyl methanesulfonate induces necroptosis in human lung adenoma A549 cells through the PIG-3-reactive oxygen species pathway. Tumour Biol. 2016;37:3785–3795. doi: 10.1007/s13277-015-3531-y. [DOI] [PubMed] [Google Scholar]
- 118.Santofimia-Castaño P., Xia Y., Lan W., Zhou Z., Huang C., Peng L. Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J Clin Investig. 2019;129:2500–2513. doi: 10.1172/JCI127223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kaku Y., Tsuchiya A., Kanno T., Nakano T., Nishizaki T. Diarachidonoylphosphoethanolamine induces necrosis/necroptosis of malignant pleural mesothelioma cells. Cell Signal. 2015;27:1713–1719. doi: 10.1016/j.cellsig.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 120.Kong Q.H., Lv J.X., Yan S.J., Chang K.J., Wang G.L. A novel naphthyridine derivative, 3u, induces necroptosis at low concentrations and apoptosis at high concentrations in human melanoma A375 cells. Int J Mol Sci. 2018;19:e2975. doi: 10.3390/ijms19102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chefetz I., Grimley E., Yang K., Hong L., Vinogradova E.V., Suciu R. A Pan-ALDH1A inhibitor induces necroptosis in ovarian cancer stem-like cells. Cell Rep. 2019;26:3061–3075. doi: 10.1016/j.celrep.2019.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang L., Wang H.D., Ding K., Xu J.G. FTY720 induces autophagy-related apoptosis and necroptosis in human glioblastoma cells. Toxicol Lett. 2015;236:43–59. doi: 10.1016/j.toxlet.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 123.Wu C.Z., Gao M.J., Shen L., Li B.H., Bai X.J., Gui J.H. Miconazole triggers various forms of cell death in human breast cancer MDA-MB-231 cells. Die Pharmazie. 2019;74:290–294. doi: 10.1691/ph.2019.8812. [DOI] [PubMed] [Google Scholar]
- 124.You B.J., Hour M.J., Chen L.Y., Luo S.C., Hsu P.H., Lee H.Z. Fenofibrate induces human hepatoma Hep3B cells apoptosis and necroptosis through inhibition of thioesterase domain of fatty acid synthase. Sci Rep. 2019;9:3306. doi: 10.1038/s41598-019-39778-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koike A., Hanatani M., Fujimori K. Pan-caspase inhibitors induce necroptosis via ROS-mediated activation of mixed lineage kinase domain-like protein and p38 in classically activated macrophages. Exp Cell Res. 2019;380:171–179. doi: 10.1016/j.yexcr.2019.04.027. [DOI] [PubMed] [Google Scholar]
- 126.Li H.Y., Wang Y.Z., Yang H.G., Zhang Y.D., Xing L., Wang J.Q. Furosine, a maillard reaction product, triggers necroptosis in hepatocytes by regulating the RIPK1/RIPK3/MLKL pathway. Int J Mol Sci. 2019;20:e2388. doi: 10.3390/ijms20102388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ma Y.M., Peng Y.M., Zhu Q.H., Gao A.H., Chao B., He Q.J. Novel CHOP activator LGH00168 induces necroptosis in A549 human lung cancer cells via ROS-mediated ER stress and NF-kappaB inhibition. Acta Pharmacol Sin. 2016;37:1381–1390. doi: 10.1038/aps.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yamanaka K., Urano Y., Takabe W., Saito Y., Noguchi N. Induction of apoptosis and necroptosis by 24(S)-hydroxycholesterol is dependent on activity of acyl-CoA:cholesterol acyltransferase 1. Cell Death Dis. 2014;5:e990. doi: 10.1038/cddis.2013.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ayazgök B., Tüylü Küçükkılınç T. Low-dose bisphenol A induces RIPK1-mediated necroptosis in SH-SY5Y cells: effects on TNF-alpha and acetylcholinesterase. J Biochem Mol Toxicol. 2018:e22233. doi: 10.1002/jbt.22233. [DOI] [PubMed] [Google Scholar]
- 130.Gill J.S., Windebank A.J. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Investig. 1998;101:2842–2850. doi: 10.1172/JCI1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Andújar I., Ríos J.L., Giner R.M., Recio M.C. Pharmacological properties of shikonin—a review of literature since 2002. Planta Med. 2013;79:1685–1697. doi: 10.1055/s-0033-1350934. [DOI] [PubMed] [Google Scholar]
- 132.Dong X.X., Fu J., Yin X.B., Cao S.L., Li X.C., Lin L.F. Emodin: a review of its pharmacology, toxicity and pharmacokinetics. Phytother Res. 2016;30:1207–1218. doi: 10.1002/ptr.5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hande K.R. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34:151421. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 134.Cardaci S., Desideri E., Ciriolo M.R. Targeting aerobic glycolysis: 3-bromopyruvate as a promising anticancer drug. J Bioenerg Biomembr. 2012;44:17–29. doi: 10.1007/s10863-012-9422-7. [DOI] [PubMed] [Google Scholar]
- 135.Tournigand C., André T., Achille E., Lledo G., Flesh M., Mery-Mignard D. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22:229–237. doi: 10.1200/JCO.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 136.Kuo M.L., Chou Y.W., Chau Y.P., Meng T.C. Differential induction of apoptosis in oncogene-transformed NIH 3T3 cells by methylmethanesulfonate. Biochem Pharmacol. 1996;52:481–488. doi: 10.1016/0006-2952(96)00251-1. [DOI] [PubMed] [Google Scholar]
- 137.Budd G.T., Bukowski R.M., Lichtin A., Bauer L., van Kirk P., Ganapathi R. Phase II trial of doxorubicin and trifluoperazine in metastatic breast cancer. Investig New Drugs. 1993;11:75–79. doi: 10.1007/BF00873916. [DOI] [PubMed] [Google Scholar]
- 138.Srivastava S.K., Jha A., Agarwal S.K., Mukherjee R., Burman A.C. Synthesis and structure–activity relationships of potent antitumor active quinoline and naphthyridine derivatives. Anti Cancer Agents Med Chem. 2007;7:685–709. doi: 10.2174/187152007784111313. [DOI] [PubMed] [Google Scholar]
- 139.Moreb J.S., Ucar-Bilyeu D.A., Khan A. Use of retinoic acid/aldehyde dehydrogenase pathway as potential targeted therapy against cancer stem cells. Cancer Chemother Pharmacol. 2017;79:295–301. doi: 10.1007/s00280-016-3213-5. [DOI] [PubMed] [Google Scholar]
- 140.Wallington-Beddoe C.T., Don A.S., Hewson J., Qiao Q., Papa R.A., Lock R.B. Disparate in vivo efficacy of FTY720 in xenograft models of Philadelphia positive and negative B-lineage acute lymphoblastic leukemia. PLoS One. 2012;7 doi: 10.1371/journal.pone.0036429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Arai H., Yamashita S., Yokote K., Araki E., Suganami H., Ishibashi S. Efficacy and safety of pemafibrate versus fenofibrate in patients with high triglyceride and low HDL cholesterol levels: a multicenter, placebo-controlled, double-blind, randomized trial. J Atheroscler Thromb. 2018;25:521–538. doi: 10.5551/jat.44412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shimodaira Y., Takahashi S., Kinouchi Y., Endo K., Shiga H., Kakuta Y. Modulation of endoplasmic reticulum ER. stress-induced autophagy by C/EBP homologous protein (CHOP) and inositol-requiring enzyme 1 alpha (IRE1alpha) in human colon cancer cells. Biochem Biophys Res Commun. 2014;445:524–533. doi: 10.1016/j.bbrc.2014.02.054. [DOI] [PubMed] [Google Scholar]
- 143.Noguchi N., Saito Y., Urano Y. Diverse functions of 24(S)-hydroxycholesterol in the brain. Biochem Biophys Res Commun. 2014;446:692–696. doi: 10.1016/j.bbrc.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 144.Cruz S.A., Qin Z., Stewart A.F.R., Chen H.H. Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen Res. 2018;13:252–256. doi: 10.4103/1673-5374.226394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Oñate M., Catenaccio A., Salvadores N., Saquel C., Martinez A., Moreno-Gonzalez The necroptosis machinery mediates axonal degeneration in a model of Parkinson disease. Cell Death Differ. 2019:1–17. doi: 10.1038/s41418-019-0408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Günther C., Martini E., Wittkopf N., Amann K., Weigmann B., Neumann H. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]