

Abstract
Besides inhalation, a few studies have indicated that the uptake of nicotine through air or clothing may be a significant pathway of its exposure among passive smokers. Nicotine is well known to exert various physiological impacts, including stimulating sympathetic nervous system, causing vascular disturbances, and inducing cell death. Therefore, we aimed to establish whether exposure of nicotine could induce articular cartilage degeneration in a mouse model of osteoarthritis (OA). We specifically assessed dose-dependent effect of nicotine in vitro to mimic its accumulation. Further, during the in vivo studies, mice subcutaneously administered with nicotine was examined for OA-associated pathologic changes. We found that nicotine significantly suppressed chondrocytes and chondrogenic markers (Sox, Col II, and aggrecan). Nicotine-treated mice also showed altered knee joint ultrastructure with reduced Col II and proteoglycans. After corroborating nicotine-induced OA characteristics, we treated this pathologic condition through employing platelet-derived biomaterial (PDB)-based regenerative therapy. The PDB significantly suppressed OA-like pathophysiological characteristics by 4 weeks. The mechanistic insight underlying this therapy demonstrated that PDB significantly restored levels of insulin-like growth factor 1 (IGF-1) signaling pathway proteins, especially pIGF-1 R, pAKT, and IRS-1, regulating extracellular matrix synthesis by chondrocytes. Taken together, the PDB exerts regenerative and reparative activities in nicotine-mediated initiation and progression of OA, through modulating IGF-1/AKT/IRS-1 signaling axis.
Keywords: nicotine osteoarthritis, platelet-derived biomaterials, IGF-1
Introduction
Osteoarthritis (OA) is a chronic disease, which is characterized by deteriorative changes in the osteochondral unit of knee joint, comprising of cartilage, meniscus, and subchondral bone1,2. The etiology of OA is multifactorial, which includes aging, heredity, obesity, smoking, and mechanical stress, such as joint overuse or injury3. Nicotine, an addictive component of cigarette smoke, has various physiological impacts, including stimulating sympathetic nervous system, causing vascular disturbances, and inducing cell death4. It is widely known that the nicotine exhibits the deteriorating impacts through smoking as a major pathway. However, recent studies suggest that nicotine has also been identified as dermal permeant and could be absorbed from transdermal nicotine patches, during harvesting tobacco leaves (green tobacco sickness), vaping, or nicotine-exposed cloths, thereby may contribute its substantial accumulation over a period of time5,6. A case report also evidenced an acute nicotine poisoning associated with a traditional remedy for eczema7, while other previous reports have documented the adverse impacts of nicotine such as reduced bone metabolic activity healing, which also indicate to influence knee joint, causing OA8,9. Therefore, this study initially aimed to establish a mouse model of nicotine-induced OA as described previously10. In the animal models sometimes it seems difficult to induce the chronic accumulation of toxic substances such as nicotine to observe its dose-dependent effect, and therefore, its higher doses are preferred in vitro. To mimic chronic accumulation-induced toxicity by employing higher doses, even previous studies have attempted to understand its effect on functional properties of human umbilical vein endothelial cells (HUVECs)11, playing key role in the progression of both periodontal disease and cardiovascular disease.
Further, the goals of current therapies of OA only offer temporary and limited benefits, such as nonsteroidal anti-inflammatory drug, corticosteroids injections, hyaluronic acid (HA)–based viscosupplementation, autologous chondrocyte implantation12, merely relieving pain with partial recovery from pathologic symptoms, but with respect to potential safety concerns, and no reliable therapy has completely restored it. Hence, the novel methods are urgently needed to effectively address the OA treatment.
Platelet-rich plasma is isolated from whole blood and has been more specifically designated as platelet-derived biomaterial (PDB) in the recent studies13,14, due to their mediated therapeutic effects through released biomaterials from platelets. PDB has been employed for regenerating cartilage, owing to its secretion of multiple growth factors, in particular, transforming growth factor-β1 (TGF-β1) and platelet-derived growth factor (PDGF) [2]. Previously, we have demonstrated the regenerative potential of PDB through inhibition of inflammatory cytokines and promotion of chondrogenesis15. Therefore, we for the first time aimed to determine if PDB, at optimized concentrations of human plasmatic fractions, could rescue nicotine-induced OA.
Furthermore, as the intracellular signaling pathways in chondrocytes are regulated by soluble mediators and changes in cartilaginous extracellular matrix16, a clear understanding of OA-associated specific pathway is very important to identify therapeutic targets. Previous studies have shown that insulin-like growth factor 1 (IGF-1) stimulates PI-3 kinase-Akt pathway in OA, promote not only chondrocyte survival but also proteoglycan (PG) and collagen synthesis17. However, contradictory findings also showed that PI-3 kinase-Akt pathway is also stimulated by inflammatory cytokines, such as interleukin-1β (IL-1β), consequently increase production of matrix metalloproteinases (MMPs) in cartilaginous matrix18. Therefore, this study aimed to identify nicotine-induced initiation and progression of OA and its therapy through PDB in vitro and in vivo. We further investigated whether this therapeutic effect of PDB is associated with modulation of IGF-1/AKT/IRS-1 signaling axis.
Materials and Methods
Chondrocyte Culture, its Maintenance, and Ethics
The human chondrocytes cultures were obtained from patients who underwent joint replacement therapy. The osteoarthritic cartilage harvested from patients was turned into pieces and digested with an enzymatic solution [8 mg/ml hyaluronidase (Sigma-Aldrich, St. Louis, MO, USA), 8 mg/ml collagenase (Sigma-Aldrich), and 2.5 mg/ml trypsin (Sigma-Aldrich)] for 6 h at 37°C. The cellular suspension was centrifuged at 1,500 rpm for 5 min and resuspended into Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Gibco BRL, New York, NY, USA) with 10% fetal bovine serum (FBS; Gibco BRL), and 1% Penicillin-Streptomycin-Amphotericin B (PSA) (Biological Industries, Beit Haemek, Israel) in a humidified atmosphere containing 5% CO2. Thereafter, the primary OA chondrocytes were cultured, passaged, and maintained in DMEM/F12 medium.
Based on previous reports showing various inhibitory activities of nicotine against bone density19, human renal proximal tubular epithelial cells20, and myoblast differentiation21, we firstly established in vitro model of nicotine (Sigma 36733) OA, by treatment of various doses of nicotine (100, 500, and 1,000 µM) to chondrocytes (passage 2) in the culture medium. After validating OA properties in nicotine-treated chondrocytes, we further treated them with PDB for 7 days.
PDB Preparations
The PDB was prepared and quantified as described in a previous study22. Various PDB concentrations according to enzyme-linked immunosorbent assay results of TGF-β1 were dissolved in DMEM/F12 1% FBS medium. Chondrocytes were seeded into six-well plate at a density of 5 × 105 cells/ml and treated with PDB (TGF-β1 = 1 ng/ml)-conditioned medium, while 1% FBS was used as experimental control. The optical density (OD) values were noted through Multiskan RC (Labsystems, Helsinki, Finland). Further, after 7 days of nicotine treatment, the PDB was employed to assess their effect on cellular proliferation and cell numbers were evaluated with automated cell counter Countess™ (Life Technologies, Carlsbad, CA, USA).
Proliferation Assay
For determining therapeutic effects of PDB, cell proliferation was assessed after 7-day treatment with nicotine in the presence of PDB, and cell numbers were evaluated with automated cell counter Countess™ (Life Technologies).
Real-time Polymerase Chain Reaction–based Expression of Cytokines
Total RNA was extracted from cells using High Pure RNA Isolation Kit (Roche, Mannheim, Germany) according to manufacturer’s instructions. Reverse transcription (RT) was performed as previously described23. Quantitative real-time polymerase chain reaction (PCR) was performed using an ABI 7300 real-time PCR system (Applied Biosystems, Foster, CA, USA), and gene expression was calculated by the 2−ΔCt or 2−ΔΔCt method with calibration samples included in each experiment. The primers used are shown in Table 1.
Table 1.
List of Genes and Their Primer Sequences Used in RT-PCR.
| Gene | Primer direction | Sequence |
|---|---|---|
| Sox9 | Forward | AGACCTTTGGGCTGCCTTAT TAGCCTCCCTCACTCCAAGA |
| Reverse | ||
| Col ll | Forward | CCTTCCTGCGCCTGCTGTC GGCCCGGATCTCCACGTC |
| Reverse | ||
| Aggrecan | Forward | CCGCTACGACGCCATCTG CCCCCACTCCAAAGAAGTTTT |
| Reverse | ||
| MMP-1 | Forward | AGCTAGCTCAGGATGACATTGATG GCCGATGGGCTGGACAG |
| Reverse | ||
| MMP-3 | Forward | TGGCATTCAGTCCCTCTATGG AGGACAAAGCAGGATCACAGTT |
| Reverse | ||
| MMP-9 | Forward | CCTGGAGACCTGAGAACCAATC CCACCCGAGTGTAACCATAGC |
| Reverse | ||
| β-Actin | Forward | AGAGCTACGAGCTGCCTGAC AGCACTGTGTTGGCGTACAG |
| Reverse |
MMP: matrix metalloproteinase; RT-PCR: reverse transcription polymerase chain reaction.
Western Blotting
Cell lysis was performed in radioimmunoprecipitation assay buffer (50 mM Tris, 150 mM NaCl, 0.5% deoxycholate (DOC), 1% NP-40, and 0.1% sodium dodecyl sulfate); thereafter, total protein was extracted, denatured for 5 min at 95°C, and separated on a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis. Further, the proteins were transferred on to the polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA) and blocked with 4% bovine serum albumin (BSA) blocking buffer. The membrane was then reacted with Sox9 (ab59252, 1:500, Abcam, Cambridge, UK), Col II (ab34712, 1:1000, Abcam), IGF-1 R (9750 S, 1:500, Cell Signaling, Danvers, MA, USA), pIGF-1 R (3024 S, 1:500, Cell Signaling), AKT (GTX121937, 1:2,500, GeneTex, Irvine, CA, USA), pAKT (GTX59559, 1:1,000, GeneTex), IRS-1 (GTX78916, 1:500, GeneTex), and β-actin (GTX109639, 1:1,000, GeneTex) polyclonal antibodies. Membranes were then incubated with anti-rabbit secondary peroxidase-conjugated antibody (111-035-003; Jackson ImmunoResearch, Newmarket, UK). Additionally, the membranes reacted with AGN (MABT83, 1:500, Millipore, Billerica, MA, USA) monoclonal antibodies were then incubated with anti-mouse secondary peroxidase-conjugated antibody (115-035-003, Jackson ImmunoResearch). The bands were visualized by using enhanced chemiluminescence detection kit (WBKLS0500, Millipore, Billerica, MA, USA) and images were analyzed using Mutigel-21 (Top Bio, Taipei, Taiwan).
In Vitro Osteoarthritic Neo-cartilage Formation and Histological Examination
The neo-cartilage formation of articular chondrocyte embedded in collagen has been described in our previous study15. Neo-cartilages were then subjected to rotatory cell culture system (RCCS-4D®, Synthecon, Houston, TX, USA) and cultured in DMEM/F12 (for control group), nicotine (1,000 µM) (for OA neo-cartilage formation), nicotine/PDB (treatment group) containing medium in a 37°C, 5% CO2 incubator, and medium were changed every 2 days. Neo-cartilages were processed for histological analysis after 4-week culture period. Samples were first counterstained with hematoxylin and eosin (H&E) for identifying cellular distribution. Additionally, immunohistochemical (IHC) staining of Col II (Millipore, Temecula, CA, USA) and alcian blue stain for PG were conducted to examine accumulation of cartilaginous extracellular matrix (ECM).
Nicotine-induced OA Animal Model
In 8-week-old male C57BL/6 J mice, the OA was induced through subcutaneous injection of 1.5 mg/kg nicotine (n = 5) as followed in previous study10, while mice with no treatment were referred as the control (n = 3). After 1 month of nicotine induction, 10 µl PDB (1 ng/ml) was intra-articularly administered into knee of mice (n = 5) once per week for 4 weeks. After another 1 month of PDB treatment, the animals were euthanized and knee joints from all groups were then isolated for histologic and IHC studies.
Histologic and Immunohistological Staining
IHC staining was performed on fixed tissue sections using avidin–biotin peroxidase technique. Briefly, unstained sections were deparaffinized with xylene and rehydrated with decreasing concentrations of ethanol. Nonspecific binding was blocked with 4% BSA. Avidin and biotin binding sites contained in tissue samples were blocked using a commercial avidin–biotin blocking kit (Vector Laboratories, Burlingame, CA, USA). Sections were then incubated for 30 min at room temperature with following antibodies diluted in phosphate buffered saline containing BSA and incubated at 4°C for overnight. Sections were washed in ice-cold saline and incubated with a secondary biotinylated anti-mouse immunoglobulin G. The activity of endogenous peroxidase was blocked using 0.3% H2O2 in horseradish peroxidase (Vector Laboratories). Peroxidase activity was visualized using diaminobenzidine (Vector Laboratories). This technique uses unlabeled primary antibody, biotinylated secondary antibody, and a preformed avidin and biotinylated horseradish peroxidase macromolecular complex. The slides were further rinsed in water and lightly counterstained with hematoxylin. Besides, knee joint tissue sections were also stained with Col II and alcian blue to determine the content of cartilage and PG, respectively, in the ECM of knee joint, and OA grade was assessed using scoring system as described previously24.
Statistical Analysis
Data are represented as mean ± standard deviation for each group. The experiments were performed in triplicates, and differences between groups were estimated with Student’s t-test and one-way analysis of variance (Sigma Plot Version 10.0 and GraphPad Prism 7). Symbols with *, **, and *** indicate P < 0.05, P < 0.01, and P < 0.001, respectively.
Results
Characterizing Nicotine-altered Chondrocyte Physiology
Cartilage degeneration is accompanied by various etiological factors leading to chondrocytic death. Therefore, to mimic the accumulation of nicotine during chronic exposure, higher doses of nicotine were chosen for its detrimental effect on chondrocytes. Specifically, the degenerative characteristics of cellular state were assessed after treatment with varying doses of nicotine (100, 500, and 1,000 µM) for 7 days (Fig. 1A). Compared to control, nicotine-treated chondrocytes demonstrated a significantly reduced cell numbers at 500 µM, which was further reduced in 1,000 µM concentration. Therefore, to observe higher damaging effects, the optimized dose of 1,000 µM was used in further experiments. The degenerative effect at dose of 1,000 µM was validated by western blot analysis (Fig. 1B), which revealed significantly suppressed levels of cartilage-specific proteins. Furthermore, as loss of PGs from articular cartilage is a hallmark of the osteoarthritic process25, we conducted alcian blue staining, which also showed relatively feeble staining compared to control (Fig. 1C). These above results imply that nicotine adversely altered chondrocyte physiological characteristics leading to OA.
Figure 1.

Effects of nicotine on cellular activity and chondrogenic markers of chondrocytes. Initially, chondrocytes were treated with nicotine doses of 100, 500, and 1,000 µM for 7 days to determine their in vitro proliferation ability (A). Further, due to higher damaging effect, 1,000 µM nicotine was employed in further experiments. (B) Western blotting dependent protein expressions of chondrocyte-specific markers including Sox9, Col II, and aggrecan. Further, the relative quantification of alcian blue staining (C) of control and nicotine-treated chondrocytes was done. The results are presented as mean ± SD (n = 3; **P < 0.05 and ***P < 0.001, respectively). SD: standard deviation.
Effect of PDB on Nicotine-accelerated OA-associated Gene/Proteins
Our results showed that nicotine strongly inhibited chondrocyte cell number; however, the inhibitory effect was reversed by PDB treatment (Fig. 2A). RT-PCR assay revealed an increased expression levels of cartilage-specific genes, including Sox9, Col II, and aggrecan (AGN) (Fig. 2B). Western blot analysis of Sox9, Col II, and AGN proteins (Fig. 2C) also followed the similar trend as of cell number and RT-PCR. These results were further supported by enhanced alcian blue staining and its quantified results in PDB-treated group (Fig. 2D).
Figure 2.

Influence of PDB on cellular activity and chondrogenic markers of nicotine-treated chondrocytes. After treatment with PDB for 7 days, the characteristics of chondrocytes were examined for their (A) proliferation ability, (B and C) reverse transcription and western blotting-dependent gene and protein expressions, respectively, of chondrocyte-specific markers including Sox9, Col II, and aggrecan. (D) Relative quantification of alcian blue staining of control and nicotine-treated chondrocytes. The results are presented as mean ± SD (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001). PDB: platelet-derived biomaterial; SD: standard deviation.
Therapeutic Potential of PDB on Nicotine-treated Three-dimensional Neo-cartilage OA Model
In order to assess the therapeutic effect of PDB on osteoarthritic chondrocytes, a three-dimensional (3D) neo-cartilage OA model was established. H&E staining showed that compared to control group (Fig. 3A-a), the neo-cartilage treated with nicotine revealed an altered chondrocytic morphology (Fig. 3A-b) along with increased cell death, indicating osteoarthritic characteristics. However, PDB treatment not only suppressed the cell death in OA-like 3D neo-cartilage but also restored normal morphology of chondrocytes (Fig. 3A-c). Further, nicotine-treated neo-cartilage group showed a highly reduced cartilaginous ECM and glycosaminoglycans (GAGs), as demonstrated by IHC Col II (Fig. 3A-e) and alcian blue staining (Fig. 3A-h), when compared to their respective controls (Fig. 3A-d, g). However, the intense positive signals of Col II and alcian blue implied an enhanced ECM and GAGs synthesis in PDB-treated group (Fig. 3A-f, i, respectively). These results were also confirmed through quantification of their positive signals (Fig. 4B, C, respectively).
Figure 3.

Determination of chondro-regenerative effects of PDB on nicotine-treated 3D neo-cartilage OA model. (A) H&E staining (a–c, upper panel), immunocytochemical staining of Col II (ICC Col II) (d–f, middle panel), and alcian blue staining (g–i, lower panel) were conducted to determine the morphologic impairments, collagen, and proteoglycan content, respectively, in synthetic cartilaginous matrices of control, nicotine-treated, and PDB-treated nicotine-OA group. All the images were obtained at ×20 magnification (scale bar: 200 µm). Further, based on these stainings, the relative quantifications of (B) Col II and (C) proteoglycan were done. The results are presented as mean ± SD (n = 3; **P < 0.01 and *** P < 0.001, respectively). H&E: hematoxylin & eosin; OA: osteoarthritis; PDB: platelet-derived biomaterial; SD: standard deviation.
Figure 4.

PDB administration and assessment of OA-associated histologic improvements. (A) Experimental protocol for PDB therapy in nicotine-induced OA mice. Following 2 weeks of lab adaptation, mice knee joint (n = 5) was subcutaneously injected with nicotine (1.5 mg/kg) for 4 weeks. Thereafter, PDB was intra-articularly injected in the animals (n = 5) once per week for 4 weeks, and the improvement in OA status was examined after further 4 weeks of treatment time. (B) Representative H&E staining (a–c, upper panel), immunohistochemical staining of Col II (IHC Col II) (d–f, middle panel), and alcian blue staining (g–i, lower panel) were conducted to determine the ultrastructural changes, collagen and proteoglycan content, respectively, in the extracellular matrix of articular cartilage of control, nicotine-treated, and PDB-treated nicotine-OA group. All the images were obtained at ×100 magnification (scale bar: 50 µm). Further, based on these histologic analyses, the severity of OA score (C) was determined. The results are presented as mean ± SD [n = 3 (control); n = 5 (nicotine and nicotine + PDB); *** P < 0.001]. H&E: hematoxylin & eosin; OA: osteoarthritis; PDB: platelet-derived biomaterial; SD: standard deviation.
Intra-articular Administration of PDB in Nicotine-induced OA Mouse Model
Eventually, the therapeutic potential of PDB was investigated in the in vivo model of nicotine-induced OA mice, as described in Fig. 4A. After 4 weeks of OA induction, the PDB was intra-articularly administered into nicotine-induced OA knee joint for further 4 weeks. H&E staining demonstrated that compared to control (Fig. 4B-a), highly reduced number of chondrocytes and lacunae appeared in wavy and disrupted collagen network in nicotine-treated knee joint (Fig. 4B-b), which were recovered through PDB treatment (Fig. 4B-c). The IHC staining for Col II in control group showed intense positive brown signals (Fig. 4B-d), whereas nicotine-treated group revealed a feeble staining (Fig. 4B-e), which was later reappeared in PDB-treated group (Fig. 4B-f). Similarly, much reduced blue staining in nicotine-treated group indicated huge loss of GAGs (Fig. 4B-h), when compared to control (Fig. 4B-g). However, an intensive blue staining signal was present in cartilaginous structure of PDB-treated group (Fig. 4B-i). Furthermore, compared to control, the severity of OA after scoring was much higher in nicotine-treated group, which was significantly suppressed through PDB treatment (Fig. 4C).
Efficacy of PDB on IGF-1 R Signaling Pathway in Nicotine-induced OA Knee Joint
The IGF-1/IRS pathway has been documented to modulate PI3K/AKT phosphorylation and regulate synthesis of ECM proteins during the early development of chondrocytes17. Further, IGF-1 also functions systemically as well as locally to exhibit endocrine, paracrine, and autocrine response. Our results demonstrated that exposure of nicotine for 4 weeks suppressed protein levels of IGF-1 signaling pathway, especially pIGF-1 R (Fig. 5A), pAKT (Fig. 5B), and IRS-1 (Fig. 5C) in knee joint compared to control. However, the administration of PDB significantly restored levels of these proteins, indicating its positive effect on IGF-1 signaling pathway leading to enhanced synthesis of ECM.
Figure 5.

Efficacy of PDB on IGF-1 signaling, inflammatory status, and chondrogenic proteins in knee joint. Assessment of expression levels of (A) pIGF-1, (B) pAKT, and (C) IRS-1 in control, nicotine-treated, and PDB-treated nicotine-OA group. Further, in these groups, the gene expression of inflammatory mediators including IL-1β, MMP-1, MMP-3, and MMP-9 (D) and chondrogenic genes (E) was investigated. The results are presented as mean ± SD (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001). IGF-1: insulin-like growth factor 1; IL-1β: interleukin-1β; MMP: matrix metalloproteinase; OA: osteoarthritis; PDB: platelet-derived biomaterial; SD: standard deviation.
PDB Intervention and Levels of Inflammatory Biomarkers in Nicotine-mediated OA Knee Joint
PI3K/AKT signaling axis has been reported to play a stimulating role in production of MMPs26. Therefore, to address role of this signaling pathway in inducing synthesis of catabolic proteases IL-1β, MMP-1, 3, and 9 in cartilaginous matrix of nicotine-induced OA knee joint, we conducted RT-PCR analysis, which revealed increased expression levels of these inflammatory molecules compared to control group (Fig. 5D). However, the reduced expression of all MMPs was exhibited in PDB-treated group, implying its anti-inflammatory activities.
Effect of PDB on Chondrocyte-specific Markers in Nicotine-induced OA Knee Joint
Our RT-PCR results demonstrated an inhibited expression of chondrocyte-lineage-specific genes, including SOX9, Col II, and AGN in nicotine-treated group when compared to control (Fig. 5E). However, a notable increase in the levels of these genes was evidenced in PDB-administered group. These results suggest that nicotine microenvironment adversely impacted cartilage health, specifically in the form of collagen and PG loss, which were lessened through intervention of PDB through inhibiting MMPs.
Discussion
This is the first report revealing PDB efficacy on nicotine-induced OA in mouse model. Our results clearly demonstrated reduced chondrocyte numbers with increasing doses of nicotine, which was employed to mimic the accumulation of nicotine during chronic exposure. The RT-PCR and western blot analysis showed a suppressed expression of chondrogenic genes and proteins, respectively, including SOX9, Col II, and AGN. Additionally, the alcian blue staining showed decreased PG content, which implied nicotine-suppressed chondrogenesis. These deteriorating outcomes might also be ascribed to different isomers of nicotine. This is in line with an important study that documented that (-)-nicotine isomeric form is more active and twice toxic than (+)-nicotine27. Our results are supported by study of Yang et al. showing inhibited proliferation and suppressed expression of mRNA expression of Sox9, type II collagen, and AGN in nicotine-treated group compared to control28. These pathophysiological impacts strongly portend the risk of nicotine to compromise knee joint integrity and cause OA.
Further, after confirming nicotine-induced OA, we administered PDB, which is a cocktail of growth factors released from concentrated platelets, and entails various clinical applications owing to their stronger anti-inflammatory and regenerative activity29. We showed that PDB-treated nicotine-induced OA group had an increased expression of chondrogenic proteins and PG content, indicating therapeutic behavior of PDB against inhibitory effect of nicotine. Interestingly, it has been documented that the nicotine as a cholinergic alkaloids act on central and peripheral nicotinic and muscarinic receptors causing central nervous system, sympathetic autonomic, parasympathetic autonomic, and neuromuscular effects in varying combinations, relying on dose of ingested substance7. Nicotine initially functions as an agonist at nicotinic receptor causing effects consistent with sympathetic stimulation, but then blocks the receptor producing late parasympathetic effects and neuromuscular blockade. This is an indicative of nicotine-mediated actions in two phases, with the first being stimulatory, while second the inhibitory. Therefore, our in vivo and in vitro studies revealing pathological influence of nicotine, in terms of initiation as well progression of knee OA, might be attributed to predominance of inhibitory response of chronic accumulation of nicotine.
Further, it has been suggested that nicotine exposure delays chondrogenesis through downregulation of IGF-1 signaling, leading to inhibited matrix synthesis by growth plate chondrocytes30. In our previous studies, we have demonstrated that 3D cultures are critical for chondrocyte survival in tissue-engineered constructs, which mimic in vivo microenvironment maintaining chondrocytic physiology31. Whereas, this study revealed nicotine-induced chondrocyte apoptosis and inhibited ECM synthesis, leading to deformed morphology of neo-cartilage. After treatment with PDB, strong Col II and Alcian blue stains indicated resynthesized and reaccumulated mature chondrogenic ECM in arthritic neo-cartilage. Collectively, PDB may overcome nicotine-induced neo-cartilage deformation through ECM reformation, leading to recovery of chondrocytes.
Beside in vitro studies, the establishment of various diseases/trauma-induced OA animal model plays a key role in understanding human OA pathophysiology, which helps to develop disease-modifying therapies from preclinical to clinical trials. Therefore, we established nicotine-induced OA mouse model, which was later treated with PDB. Our H&E-stained tissue sections showed the deformed joint, degraded cartilage surfaces, collagen fibrils, and PGs in nicotine-treated group, while significant recovery through PDB treatment in this group indicated its marked healing efficacies in terms of enhanced IHC collagen type II and alcian blue stainings. Further, the huge loss of chondrocytes and PG in wavy fibrils of collagen fibers of extracellular matrix, as revealed by H&E, IHC, and alcian blue staining in nicotine-induced OA group, accounts for cartilage destruction score of over 3, which was decreased to 50% after PDB intervention. These therapeutic efficacies of PDB might also be attributed to the anti-inflammatory activities of TGF-β132 and lipoxins33, regulating repair of inflamed tissues leading to matrix restoration34 and regeneration35. Further, the therapeutic efficacies of PDB may also be attributed to growth factors such as hepatocyte growth factor (HGF), epidermal growth factor (EGF), and fibroblast growth factor, which are present as a cocktail.36 It is further well known that the viscosity of synovial fluid is ascribed to HA, which acts as a lubricant for joint movements, leading to nearly zero coefficient of friction in joint cartilage37. It has been evidenced that the PDGF primarily activates HA synthase isoform 238; therefore, through regulating the endogenous HA synthesis, PDB would restore HA levels leading to elevated cartilage protection and joint lubrication39. Previous studies have also underlined the synergy between HGF and VEGF, which may likely to act cooperatively in normal physiology40,41, implying a role in non-inflammatory instead of inflammatory angiogenesis42,43. Hence it is possible that PDB could modify the angiogenic balance through triggering the secretion of HGF44. In similar trend, IGF-1 and PDGF could also inhibit IL-1β-induced cartilage degradation via downregulating NF-κB signaling45. In addition, PDB-contained PDGF, TGF-β, IGF-1, and EGF, which are regulators of cartilage growth, may also improve chondrocyte metabolism46,47.
Further, it has already been demonstrated that a key anabolic factor, IGF-1 in adult articular cartilage, stimulates phosphorylation of cell signaling protein AKT, required for PG synthesis in healthy chondrocytes48. In our study, we found that phosphorylated level of IGF-1, AKT, and IRS was suppressed in nicotine-treated group, which was later restored through PDB intervention. Further, the PI3K/AKT signaling axis has been reported to stimulate the production of MMPs26. The secretome analysis of nicotine-treated human articular chondrocytes has been demonstrated with its pathologic effects by increased synthesis of catabolic molecules such as other interleukins and MMPs (MMP-1, -2, and -3), which are critical mediators of OA pathophysiology49. This study also suggested that nicotine at physiological levels is not only unable to diminish this catabolic effect of IL-1, but even increases the secretion. Our previous study has also documented the higher expression of various matrix-associated inflammatory markers, including IL-1β, MMP-1, MMP-3, and MMP-9, which were found to be significantly suppressed in nicotine-treated groups by PDB therapy.
Besides various significant therapeutic outcomes, this study includes a few limitations, such as use of higher doses of nicotine during in vitro assessment of its toxicity to mimic chronic accumulation in human body. However, compared to our study, even high range of doses of nicotine (10 µM–10 mM) had been tested in the previous study to determine its impact on functional properties of HUVECs11, which play an important role in the progression of both periodontal disease and cardiovascular disease. Interestingly, the HUVECs were inhibited by higher dose (10 mM) and not the lower one (10 µM), implying chronic accumulation-mimicking ability of higher doses. Further, since skin is exposed to nicotine in daily life through dermal mode and uptake transdermally, we established nicotine-OA model in mice model to mimic toxicity through subcutaneous injection. Our future studies will also focus on confirming roles of other specific PDB-contained biomaterials on nicotine-induced OA. Additionally, other signaling axes will also be targeted.
Taken together, it could be inferred that PDB could efficiently inhibit nicotine-induced inflammatory profile; as a consequence, the levels of cartilage-specific genes as well proteins (SOX9, Col II, and AGN) were enhanced (Fig. 6).
Figure 6.
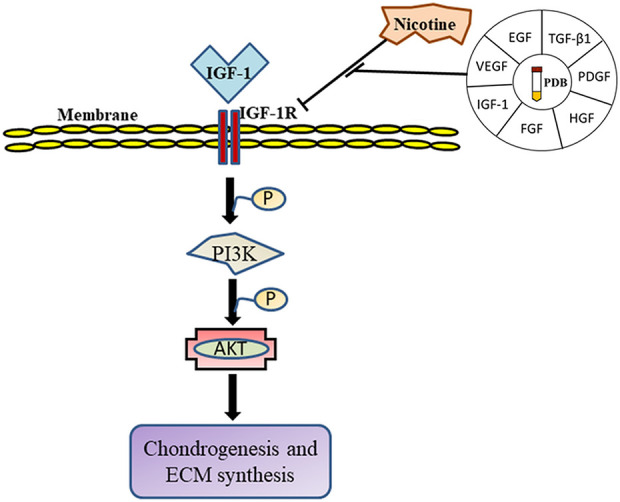
Schematic summary of mechanistic insight of PDB therapy in nicotine-induced OA. ECM: extracellular matrix; EGF: epidermal growth factor; FGF: fibroblast growth factor; HGF: hepatocyte growth factor; IGF-1: insulin-derived growth factor-1; OA: osteoarthritis; PDB: platelet-derived biomaterial; PDGF: platelet-derived growth factor; TGF-β1: transforming growth factor-β1; VEGF: vascular endothelial growth factor.
Footnotes
Ethical Approval: All the animal care and used protocols were approved by the Institutional Animal Care and Use Committee (IACUC), Taipei Medical University, Taiwan.
Statement of Human and Animal Rights: A prior informed consent was obtained from patients and all the protocols were followed as per the Institutional Review Board (IRB No. 201305026).
Statement of Informed Consent: Verbal informed consent was obtained from the patients for their anonymized information to be published in this article.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the following grants and agencies: Ministry of Science and Technology Taiwan (MOST 108-2221-E-038-014), Taipei Medical University (TMU 108-5601-004-111), and Stem Cell Research Center, College of Oral Medicine, Taipei Medical University, Taiwan.
ORCID iD: Win-Ping Deng  https://orcid.org/0000-0003-2180-0472
https://orcid.org/0000-0003-2180-0472
References
- 1. Dubey NK, Wei H-J, Yu S-H, Williams DF, Wang JR, Deng Y-H, Tsai F-C, Wang PD, Deng W-P. Adipose-derived stem cells attenuates diabetic osteoarthritis via inhibition of glycation-mediated inflammatory cascade. Aging Dis. 2019;10(3):483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dubey NK, Mishra VK, Dubey R, Syed-Abdul S, Wang JR, Wang PD, Deng WP. Combating osteoarthritis through stem cell therapies by rejuvenating cartilage: a review. Stem Cells Int. 2018;2018:5421019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Johnson VL, Hunter DJ. The epidemiology of osteoarthritis. Best Pract Res Clin Rheumatol. 2014;28(1):5–15. [DOI] [PubMed] [Google Scholar]
- 4. Zevin S, Gourlay SG, Benowitz NL. Clinical pharmacology of nicotine. Clin Dermatol. 1998;16(5):557–564. [DOI] [PubMed] [Google Scholar]
- 5. Faulkner JM. Nicotine poisoning by absorption through the skin. J Am Med Assoc. 1933;100(21):1664–1665. [Google Scholar]
- 6. Benowitz NL, Lake T, Keller KH, Lee BL. Prolonged absorption with development of tolerance to toxic effects after cutaneous exposure to nicotine. Clin Pharmacol Ther. 1987;42(1):119–120. [DOI] [PubMed] [Google Scholar]
- 7. Davies P, Levy S, Pahari A, Martinez D. Acute nicotine poisoning associated with a traditional remedy for eczema. Arch Dis Child. 2001;85(6):500–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porter SE, Hanley EN., Jr The musculoskeletal effects of smoking. J Am Acad Orthop Surg. 2001;9(1):9–17. [DOI] [PubMed] [Google Scholar]
- 9. Berman D, Oren JH, Bendo J, Spivak J. The effect of smoking on spinal fusion. Int J Spine Surg. 2017;11(4):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mangubat M, Lutfy K, Lee ML, Pulido L, Stout D, Davis R, Shin CS, Shahbazian M, Seasholtz S, Sinha-Hikim A, Sinha-Hikim I, et al. Effect of nicotine on body composition in mice. J Endocrinol. 2012;212(3):317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. An N, Andrukhov O, Tang Y, Falkensammer F, Bantleon HP, Ouyang X, Rausch-Fan X. Effect of nicotine and porphyromonas gingivalis lipopolysaccharide on endothelial cells in vitro. PLoS One. 2014;9(5):e96942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mora JC, Przkora R, Cruz-Almeida Y. Knee osteoarthritis: pathophysiology and current treatment modalities. J Pain Res. 2018;11:2189–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arora S, Agnihotri N. Platelet derived biomaterials for therapeutic use: review of technical aspects. Indian J Hematol Blood Transfus. 2017;33(2):159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yip HK, Chen KH, Dubey NK, Sun CK, Deng YH, Su CW, Lo WC, Cheng HC, Deng WP. Cerebro- and renoprotective activities through platelet-derived biomaterials against cerebrorenal syndrome in rat model. Biomaterials. 2019;214:119227. [DOI] [PubMed] [Google Scholar]
- 15. Wu CC, Chen WH, Zao B, Lai PL, Lin TC, Lo HY, Shieh YH, Wu CH, Deng WP. Regenerative potentials of platelet-rich plasma enhanced by collagen in retrieving pro-inflammatory cytokine-inhibited chondrogenesis. Biomaterials. 2011;32(25):5847–5854. [DOI] [PubMed] [Google Scholar]
- 16. Houard X, Goldring MB, Berenbaum F. Homeostatic mechanisms in articular cartilage and role of inflammation in osteoarthritis. Curr Rheumatol Rep. 2013;15(11):375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Starkman BG, Cravero JD, Delcarlo M, Loeser RF. IGF-I stimulation of proteoglycan synthesis by chondrocytes requires activation of the PI 3-kinase pathway but not ERK MAPK. Biochem J. 2005;389(Pt 3):723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Litherland GJ, Dixon C, Lakey RL, Robson T, Jones D, Young DA, Cawston TE, Rowan AD. Synergistic collagenase expression and cartilage collagenolysis are phosphatidylinositol 3-kinase/Akt signaling-dependent. J Biol Chem. 2008;283(21):14221–14229. [DOI] [PubMed] [Google Scholar]
- 19. Broulik PD, Rosenkrancova J, Ruzicka P, Sedlacek R, Kurcova I. The effect of chronic nicotine administration on bone mineral content and bone strength in normal and castrated male rats. Horm Metab Res. 2007;39(1):20–24. [DOI] [PubMed] [Google Scholar]
- 20. Kim CS, Choi JS, Joo SY, Bae EH, Ma SK, Lee J, Kim SW. Nicotine-induced apoptosis in human renal proximal tubular epithelial cells. PLoS One. 2016;11(3):e0152591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lei W, Lerner C, Sundar IK, Rahman I. Myofibroblast differentiation and its functional properties are inhibited by nicotine and e-cigarette via mitochondrial OXPHOS complex III. Sci Rep. 2017;7:43213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen WH, Lo WC, Lee JJ, Su CH, Lin CT, Liu HY, Lin TW, Lin WC, Huang TY, Deng WP. Tissue-engineered intervertebral disc and chondrogenesis using human nucleus pulposus regulated through TGF-beta1 in platelet-rich plasma. J Cell Physiol. 2006;209(3):744–754. [DOI] [PubMed] [Google Scholar]
- 23. Wei HJ, Wu AT, Hsu CH, Lin YP, Cheng WF, Su CH, Chiu WT, Whang-Peng J, Douglas FL, Deng WP. The development of a novel cancer immunotherapeutic platform using tumor-targeting mesenchymal stem cells and a protein vaccine. Mol Ther. 2011;19(12):2249–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kamekura S, Hoshi K, Shimoaka T, Chung U, Chikuda H, Yamada T, Uchida M, Ogata N, Seichi A, Nakamura K, Kawaguchi H. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthritis Cartilage. 2005;13(7):632–641. [DOI] [PubMed] [Google Scholar]
- 25. Fosang AJ, Last K, Maciewicz RA. Aggrecan is degraded by matrix metalloproteinases in human arthritis. Evidence that matrix metalloproteinase and aggrecanase activities can be independent. J Clin Invest. 1996;98(10):2292–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Greene MA, Loeser RF. Function of the chondrocyte PI-3 kinase-Akt signaling pathway is stimulus dependent. Osteoarthritis Cartilage. 2015;23(6):949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barlow RB, Hamilton JT. The stereospecificity of nicotine. Br J Pharmacol Chemother. 1965;25(1):206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang X, Qi Y, Avercenc-Leger L, Vincourt JB, Hupont S, Huselstein C, Wang H, Chen L, Magdalou J. Effect of nicotine on the proliferation and chondrogenic differentiation of the human Wharton’s jelly mesenchymal stem cells. Biomed Mater Eng. 2017;28(s1): S217–S228. [DOI] [PubMed] [Google Scholar]
- 29. Chen WH, Lin CM, Huang CF, Hsu WC, Lee CH, Ou KL, Dubey NK, Deng WP. Functional recovery in osteoarthritic chondrocytes through hyaluronic acid and platelet-rich plasma-inhibited infrapatellar fat pad adipocytes. Am J Sports Med. 2016;44(10):2696–2705. [DOI] [PubMed] [Google Scholar]
- 30. Deng Y, Cao H, Cu F, Xu D, Lei Y, Tan Y, Magdalou J, Wang H, Chen L. Nicotine-induced retardation of chondrogenesis through down-regulation of IGF-1 signaling pathway to inhibit matrix synthesis of growth plate chondrocytes in fetal rats. Toxicol Appl Pharmacol. 2013;269(1):25–33. [DOI] [PubMed] [Google Scholar]
- 31. Chen WH, Lo WC, Hsu WC, Wei HJ, Liu HY, Lee CH, Tina Chen SY, Shieh YH, Williams DF, Deng WP. Synergistic anabolic actions of hyaluronic acid and platelet-rich plasma on cartilage regeneration in osteoarthritis therapy. Biomaterials. 2014;35(36):9599–9607. [DOI] [PubMed] [Google Scholar]
- 32. Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109(1):41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abdul Ameer LA, Raheem ZJ, Abdulrazaq SS, Ali BG, Nasser MM, Aldeen Khairi AW. The anti-inflammatory effect of the platelet-rich plasma in the periodontal pocket. Eur J Dent. 2018;12(4):528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sundman EA, Cole BJ, Karas V, Della Valle C, Tetreault MW, Mohammed HO, Fortier LA. The anti-inflammatory and matrix restorative mechanisms of platelet-rich plasma in osteoarthritis. Am J Sports Med. 2014;42(1):35–41. [DOI] [PubMed] [Google Scholar]
- 35. Shoeib HM, Keshk WA, Foda AM, Abo El Noeman S. A study on the regenerative effect of platelet-rich plasma on experimentally induced hepatic damage in albino rats. Can J Physiol Pharmacol. 2018;96(6):630–636. [DOI] [PubMed] [Google Scholar]
- 36. Murata S, Maruyama T, Nowatari T, Takahashi K, Ohkohchi N. Signal transduction of platelet-induced liver regeneration and decrease of liver fibrosis. Int J Mol Sci. 2014;15(4):5412–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seror J, Merkher Y, Kampf N, Collinson L, Day AJ, Maroudas A, Klein J. Articular cartilage proteoglycans as boundary lubricants: structure and frictional interaction of surface-attached hyaluronan and hyaluronan--aggrecan complexes. Biomacromolecules. 2011;12(10):3432–3443. [DOI] [PubMed] [Google Scholar]
- 38. Jacobson A, Brinck J, Briskin MJ, Spicer AP, Heldin P. Expression of human hyaluronan synthases in response to external stimuli. Biochem J. 2000;348(Pt 1):29–35. [PMC free article] [PubMed] [Google Scholar]
- 39. Goldring SR, Goldring MB. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res. 2004;(427 Suppl):S27–S36. [DOI] [PubMed] [Google Scholar]
- 40. Xin X, Yang S, Ingle G, Zlot C, Rangell L, Kowalski J, Schwall R, Ferrara N, Gerritsen ME. Hepatocyte growth factor enhances vascular endothelial growth factor-induced angiogenesis in vitro and in vivo. Am J Pathol. 2001;158(3):1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Beilmann M, Birk G, Lenter MC. Human primary co-culture angiogenesis assay reveals additive stimulation and different angiogenic properties of VEGF and HGF. Cytokine. 2004;26(4):178–185. [DOI] [PubMed] [Google Scholar]
- 42. Min JK, Lee YM, Kim JH, Kim YM, Kim SW, Lee SY, Gho YS, Oh GT, Kwon YG. Hepatocyte growth factor suppresses vascular endothelial growth factor-induced expression of endothelial ICAM-1 and VCAM-1 by inhibiting the nuclear factor-kappaB pathway. Circ Res. 2005;96(3):300–307. [DOI] [PubMed] [Google Scholar]
- 43. Gerritsen ME, Tomlinson JE, Zlot C, Ziman M, Hwang S. Using gene expression profiling to identify the molecular basis of the synergistic actions of hepatocyte growth factor and vascular endothelial growth factor in human endothelial cells. Br J Pharmacol. 2003;140(4):595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Anitua E, Sanchez M, Nurden AT, Zalduendo M, de la Fuente M, Azofra J, Andia I. Reciprocal actions of platelet-secreted TGF-beta1 on the production of VEGF and HGF by human tendon cells. Plast Reconstr Surg. 2007;119(3):950–959. [DOI] [PubMed] [Google Scholar]
- 45. Montaseri A, Busch F, Mobasheri A, Buhrmann C, Aldinger C, Rad JS, Shakibaei M. IGF-1 and PDGF-bb suppress IL-1beta-induced cartilage degradation through down-regulation of NF-kappaB signaling: involvement of Src/PI-3K/AKT pathway. PLoS One. 2011;6(12):e28663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Makower AM, Wroblewski J, Pawlowski A. Effects of IGF-I, rGH, FGF, EGF and NCS on DNA-synthesis, cell proliferation and morphology of chondrocytes isolated from rat rib growth cartilage. Cell Biol Int Rep. 1989;13(3):259–270. [DOI] [PubMed] [Google Scholar]
- 47. Asanbaeva A, Masuda K, Thonar EJ, Klisch SM, Sah RL. Regulation of immature cartilage growth by IGF-I, TGF-beta1, BMP-7, and PDGF-AB: role of metabolic balance between fixed charge and collagen network. Biomech Model Mechanobiol. 2008;7(4):263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Loeser RF, Gandhi U, Long DL, Yin W, Chubinskaya S. Aging and oxidative stress reduce the response of human articular chondrocytes to insulin-like growth factor 1 and osteogenic protein 1. Arthritis Rheumatol. 2014;66(8):2201–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lourido L, Calamia V, Fernandez-Puente P, Mateos J, Oreiro N, Blanco FJ, Ruiz-Romero C. Secretome analysis of human articular chondrocytes unravels catabolic effects of nicotine on the joint. Proteomics Clin Appl. 2016;10(6):671–680. [DOI] [PubMed] [Google Scholar]