

Key Points
Question
What are the clinical features and course of retinitis pigmentosa associated with biallelic sequence variations in the PDE6A gene?
Findings
In this longitudinal cohort study of 57 adults, 17 of the PDE6A variants appeared to be novel. Disease was highly symmetrical between right and left eyes, and visual impairment was mild or moderate in 90% of patients.
Meaning
These data suggest that PDE6A–retinitis pigmentosa may be amenable to gene therapy.
In this cohort study, 57 patients with biallelic sequence variations in the PDE6A gene and retinitis pigmentosa were followed up to assess clinical features, genetic findings, and genotype-phenotype correlations of the disease.
Abstract
Importance
Treatment trials require sound knowledge on the natural course of disease.
Objective
To assess clinical features, genetic findings, and genotype-phenotype correlations in patients with retinitis pigmentosa (RP) associated with biallelic sequence variations in the PDE6A gene in preparation for a gene supplementation trial.
Design, Setting, and Participants
This prospective, longitudinal, observational cohort study was conducted from January 2001 to December 2019 in a single center (Centre for Ophthalmology of the University of Tübingen, Germany) with patients recruited multinationally from 12 collaborating European tertiary referral centers. Patients with retinitis pigmentosa, sequence variants in PDE6A, and the ability to provide informed consent were included.
Exposures
Comprehensive ophthalmological examinations; validation of compound heterozygosity and biallelism by familial segregation analysis, allelic cloning, or assessment of next-generation sequencing–read data, where possible.
Main Outcomes and Measures
Genetic findings and clinical features describing the entire cohort and comparing patients harboring the 2 most common disease-causing variants in a homozygous state (c.304C>A;p.(R102S) and c.998 + 1G>A;p.?).
Results
Fifty-seven patients (32 female patients [56%]; mean [SD], 40 [14] years) from 44 families were included. All patients completed the study. Thirty patients were homozygous for disease-causing alleles. Twenty-seven patients were heterozygous for 2 different PDE6A variants each. The most frequently observed alleles were c.304C>A;p.(R102S), c.998 + 1G>A;p.?, and c.2053G>A;p.(V685M). The mean (SD) best-corrected visual acuity was 0.43 (0.48) logMAR (Snellen equivalent, 20/50). The median visual field area with object III4e was 660 square degrees (5th and 95th percentiles, 76 and 11 019 square degrees; 25th and 75th percentiles, 255 and 3923 square degrees). Dark-adapted and light-adapted full-field electroretinography showed no responses in 88 of 108 eyes (81.5%). Sixty-nine of 108 eyes (62.9%) showed additional findings on optical coherence tomography imaging (eg, cystoid macular edema or macular atrophy). The variant c.998 + 1G>A;p.? led to a more severe phenotype when compared with the variant c.304C>A;p.(R102S).
Conclusions and Relevance
Seventeen of the PDE6A variants found in these patients appeared to be novel. Regarding the clinical findings, disease was highly symmetrical between the right and left eyes and visual impairment was mild or moderate in 90% of patients, providing a window of opportunity for gene therapy.
Introduction
Retinitis pigmentosa (RP) is a hereditary, degenerative retinal disease that causes severe visual impairment and visual acuity loss because of progressive degeneration of primarily the rod and secondarily the cone photoreceptors. The disease manifests with early-onset nyctalopia, followed by daytime visual field defects progressing from the midperiphery to the periphery and the center. Best-corrected visual acuity typically remains relatively well preserved until macular involvement by macular edema and/or photoreceptor atrophy causes central visual acuity loss.
To date, sequence variations in 89 genes are known to be associated with RP.1 In 1995, the gene encoding for the α subunit of the rod photoreceptor cyclic guanosine monophosphate (cGMP) phosphodiesterase (PDE6A) was identified as the seventh RP locus, on chromosome 5q31.2-q34 (OMIM, 180071 and 613810).2,3 It includes 22 coding exons and encodes 859 amino acid residues. Rod photoreceptor cGMP phosphodiesterase is made of 4 subunits, the catalytic α subunit (PDE6A), the catalytic β subunit (PDE6B), and 2 inhibitory γ subunits (PDE6G). The enzyme functions to hydrolize the intracellular cytoplasmic cGMP level, which causes closure of cyclic nucleotide–gated channels, an essential step in vertebrate phototransduction.4 The PDE6A gene appears to account for disease in less than 4% of families with autosomal recessive RP in North America, approximately 2% of the cases in cohorts of French and Pakistani patients, and approximately 1% of families with inherited retinal diseases in Israel.5,6,7,8
Sequence variations in PDE6A do not cause retinal dystrophy in only humans. Biallelic sequence variations in the PDE6A gene have been identified to cause autosomal recessive progressive retinal atrophy in the Cardigan Welsh Corgi dog.9,10 Additionally, induced and natural mouse models have been studied for variations of the disease phenotype (eg, pace of photoreceptor degeneration) associated with different PDE6A sequence variations.10,11 In preclinical gene supplementation trials, mice showed more effective photoreceptor cell rescue when the rod-specific transgene AAV2/8(Y733F)-Rho-Pde6α was delivered before the onset of disease. It was thus concluded that the success of therapeutic clinical trials will depend on identifying patients as early as possible to maximize the number of rods still viable and treatable with gene therapy.12,13,14
In 2012, the RD-CURE Consortium started working on a collaborative project for the clinical translation of gene therapy for patients with inherited retinal dystrophies; PDE6A-associated RP is one of the diseases for which a therapeutic transgene is currently evaluated in a human gene therapy trial, using recombinant adeno-associated viral vectors. The aim of the present study was to assess clinical features and genetic findings of patients with RP associated with biallelic sequence variations in the PDE6A gene in preparation for this gene supplementation trial.
Methods
The study (ClinicalTrials.gov Identifier NCT02759952) was conducted in accordance with the Declaration of Helsinki, with approval from the ethics committee of the University of Tübingen. Written informed consent was obtained from all participants. Patients were recruited from the clinics for hereditary retinal degenerations at the Centre of Ophthalmology of the University of Tübingen and 12 collaborating European tertiary referral centers. Patient travel and accommodation costs were compensated.
Ophthalmological Testing
All patients were examined at the Centre for Ophthalmology of the University of Tübingen, Germany, between January 2001 and December 2019. A comprehensive ophthalmological examination was performed, including best-corrected visual acuity (BCVA) with Early Treatment Diabetic Retinopathy Study charts, a semiautomated 90° kinetic visual field (VF) exam with objects lll4e and I4e (Octopus 900 [Haag-Streit]), full-field electroretinography (ff-ERG) and multifocal electroretinography testing according to International Society for Clinical Electrophysiology of Vision standards (Espion [Diagnosys]), spectral-domain optical coherence tomography imaging (Spectralis HRA+OCT [Heidelberg Engineering]), and slitlamp and dilated fundus examinations and photography. Results of BCVA testing were converted to logarithm of the minimum angle of resolution (logMAR) visual acuity values.15 Visual field parameters were assessed as total VF area in square degrees for both objects III4e and I4e, using the built-in software of the testing device.
Genetic Testing and Variant Classification
The patients enrolled in this study had a confirmed genetic diagnosis of PDE6A-associated retinitis pigmentosa. If possible, validation of homozygosity, compound heterozygosity, and biallelism was performed by familial segregation analysis, allelic cloning, or assessment of next-generation sequencing–read data. Whole-exome sequencing to exclude the presence of pathogenic variants in other genes associated with inherited retinal degeneration was performed for 23 cases. Standards and guidelines provided by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology were applied to classify the identified variants.16 The potential pathogenicity of missense changes was assessed using 5 online prediction software tools, namely SIFT, PolyPhen-2, Mutation Taster, Mutation Assessor, and Provean.17,18,19,20,21 Assessment of variants potentially affecting splicing was performed with the Alamut Genova software version 1.4 (Sophia Genetics) using default parameters. Variant designation is based on the National Center for Biotechnology Information reference sequence for PDE6A (NM_000440.3; GRCh38) involving 22 coding exons.
Statistical Analysis
Statistical analyses were performed using SPSS Statistics for Windows, version 26.0 (IBM). Normal distribution was tested using the Shapiro-Wilk test. Differences between genetic subgroups were tested using regression analysis.
Results
Fifty-seven patients from across Europe were included. Of the 57 patients (114 eyes) in this analysis, 25 (44%) were male and 32 (56%) were female. The mean (SD) age at baseline was 40 (14) years (range, 12-78 years). The mean (SD) follow-up time was 2.9 (2.1) years. Forty of the 57 patients (70%) were followed up as part of the study. The mean (SD) follow-up time was 2.9 (2.1) years.
Genetic Findings
All 57 affected individuals harbored rare and potentially disease-causing variants compatible with autosomal recessive inheritance in the PDE6A gene (Table 1). Thirty patients were homozygous for disease-causing alleles. Validation of true homozygosity by parental segregation analysis was possible in 9 cases. Twenty-seven patients were heterozygous for 2 different PDE6A variants each. In these, compound heterozygosity of variants could be validated in 19 cases by segregation analysis. In addition, in 2 cases, transconfiguration of variants was established by assessing independent next-generation sequencing–read data or allelic cloning, respectively.
Table 1. Genetic and Clinical Findingsa.
| Patient No. | Key | Decade of life | Variant 1 | Variant 2 | Segregation | Visual acuity, logMAR | Visual acuity, Snellen | Visual field object III4e, square degrees | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sequence variation | HGMDb | Sequence variation | HGMDb | ||||||||||
| Right eye | Left eye | Right eye | Left eye | Right eye | Left eye | ||||||||
| 34 | ARRP 322 | 5 | c.304C>A/p.R102S | CM994742 | c.304C>A/p.R102S | CM994742 | No | −0.1 | 0 | 20/16 | 20/20 | 1520 | 3249 |
| 50 | SRP 1101 | 2 | c.1957C>T/p.R653* | CM161562 | c.998 + 2T>G/p.? | Novel | Yes | 0 | 0 | 20/20 | 20/20 | 3538 | 3540 |
| 110 | ARRP 415 | 3 | c.304C>A/p.R102S | CM994742 | c.1689C>A/p.H563Q | CM1825637 | No | 0 | 0 | 20/20 | 20/20 | 7522 | 6252 |
| 102 | ARRP 150 | 3 | c.304C>A/p.R102S | CM994742 | c.304C>A/p.R102S | CM994742 | No | 0 | 0 | 20/20 | 20/20 | 13 123 | 12 422 |
| 49 | SRP 1064 | 3 | c.304C>A/p.R102S | CM994742 | c.2053G>A/p.V685M | CM106644 | No | 0 | 0 | 20/20 | 20/20 | 10 889 | 10 754 |
| 101 | ARRP 150 | 3 | c.304C>A/p.R102S | CM994742 | c.304C>A/p.R102S | CM994742 | No | 0 | 0 | 20/20 | 20/20 | 12 569 | 11 105 |
| 36 | ARRP 324 | 4 | c.1705C>A/p.Q569K | CM994743 | c.1705C>A/p.Q569K | CM994743 | No | 0 | 0 | 20/20 | 20/20 | 3914 | 6895 |
| 104 | SRP 578 | 4 | c.1684C>T/p.R562W | CM1510052 | c.2053G>A/p.V685M | CM106644 | Yes | 0 | 0 | 20/20 | 20/20 | NA | NA |
| 111 | ARRP 410 | 2 | c.1927-1G>T/p.? | Novel | c.2332_2335del/ p.D778Lfs*42 | Novel | No | 0.1 | 0 | 20/25 | 20/20 | 6074 | 8234 |
| 48 | SRP 1024 | 4 | c.1683G>A/p.W561* | CM950913 | c.1263 + 1G>A/p.? | Novel | Yes | 0.1 | 0 | 20/25 | 20/20 | 2070 | 2599 |
| 23 | ARRP 291 | 3 | c.998 + 1G>A/p.? | CS994749 | c.998 + 1G>A/p.? | CS994749 | Yes | 0.6 | 0 | 20/80 | 20/20 | 3759 | 4526 |
| 6 | SRP 976 | 4 | c.1620 + 1G>A/p.? | Novel | c.1705C>A/p.Q569K | CM994743 | Yes | 0 | 0.1 | 20/20 | 20/25 | 4246 | 5036 |
| 3 | SRP 150 | 5 | c.304C>A/p.R102S | CM994742 | c.1689C>A/p.H563Q | CM1825637 | Yes | 0 | 0.1 | 20/20 | 20/25 | 375 | 220 |
| 17 | ARRP 128 | 2 | c.769C>T/p.R257* | CM066961 | c.769C>T/p.R257* | CM066961 | Yes | 0.1 | 0.1 | 20/25 | 20/25 | 11 132 | 10 740 |
| 8 | SRP 977 | 4 | c.998 + 1G>A/p.? | CS994749 | c.998 + 1G>A/p.? | CS994749 | No | 0.1 | 0.1 | 20/25 | 20/25 | 795 | 777 |
| 105 | SRP 125 | 4 | c.304C>A/p.R102S | CM994742 | c.1966G>T/p.E656* | CM1717824 | No | 0.1 | 0.1 | 20/25 | 20/25 | NA | NA |
| 47 | ARRP 322 | 5 | c.304C>A/p.R102S | CM994742 | c.304C>A/p.R102S | CM994742 | No | 0.1 | 0.1 | 20/25 | 20/25 | 336 | 355 |
| 29 | ARRP 316 | 4 | c.304C>A/p.R102S | CM994742 | c.2053G>A/p.V685M | CM106644 | Yes | 0.2 | 0.1 | 20/30 | 20/25 | 244 | 203 |
| 46 | SRP 1009 | 5 | c.304C>A/p.R102S | CM994742 | c.1705C>A/p.Q569K | CM994743 | Yes | 0.2 | 0.1 | 20/30 | 20/25 | 440 | 464 |
| 109 | SRP 1080 | 6 | c.1610T>C/p.I537T | Novel | c.743T>A/p.V248D | Novel | No | 0.4 | 0.1 | 20/50 | 20/25 | 171 | 142 |
| 14 | SRP 159 | 4 | c.1862T>G/p.L621R | Novel | c.2053G>A/p.V685M | CM106644 | Yes | 0.1 | 0.2 | 20/25 | 20/30 | 1008 | 1528 |
| 15 | SRP 894 | 4 | c.304C>A/p.R102S | CM994742 | c.2053G>A/p.V685M | CM106644 | Yes | 0.2 | 0.2 | 20/30 | 20/30 | 1728 | 2226 |
| 21 | ARRP 290 | 4 | c.1957C>T/p.R653* | CM161562 | c.1957C>T/p.R653* | CM161562 | No | 0.2 | 0.2 | 20/30 | 20/30 | 434 | 426 |
| 12 | ARRP 291 | 5 | c.998 + 1G>A/p.? | CS994749 | c.998 + 1G>A/p.? | CS994749 | Yes | 0.2 | 0.2 | 20/30 | 20/30 | 163 | 187 |
| 16 | SRP 552 | 5 | c.304C>A/p.R102S | CM994742 | c.2053G>A/p.V685M | CM106644 | Yes | 0.2 | 0.2 | 20/30 | 20/30 | 2383 | 2593 |
| 30 | ARRP 316 | 3 | c.304C>A/p.R102S | CM994742 | c.2053G>A/p.V685M | CM106644 | Yes | 0.3 | 0.2 | 20/40 | 20/30 | 86 | 107 |
| 26 | ARRP 291 | 4 | c.998 + 1G>A/p.? | CS994749 | c.998 + 1G>A/p.? | CS994749 | Yes | 0.3 | 0.2 | 20/40 | 20/30 | 1837 | 1585 |
| 27 | SRP 829 | 7 | c.304C>A/p.R102S | CM994742 | c.304C>A/p.R102S | CM994742 | No | 0.3 | 0.2 | 20/40 | 20/30 | 1680 | 3923 |
| 41 | SRP 986 | 6 | c.612del/p.K205Rfs*16 | CD192430 | c.612del/p.K205Rfs*16 | CD192430 | No | 0.5 | 0.2 | 20/60 | 20/30 | 187 | 117 |
| 33 | ARRP 307 | 7 | c.1705C>A/p.Q569K | CM994743 | c.1065 + 2T>A/p.? | Novel | Yes | 0.6 | 0.2 | 20/80 | 20/30 | 70 | 124 |
| 42 | SRP 1089 | 2 | c.1926 + 1G>A/p.? | Novel | c.1926 + 1G>A/p.? | Novel | No | 0.3 | 0.3 | 20/40 | 20/40 | 1967 | 2961 |
| 5 | ARRP 399 | 3 | c.769C>T/p.R257* | CM066961 | c.769C>T/p.R257* | CM066961 | No | 0.3 | 0.3 | 20/40 | 20/40 | 6737 | 7446 |
| 19 | SRP 905 | 5 | c.1957C>T/p.R653* | CM161562 | c.2332_2335del/ p.D778Lfs*42 | Novel | Yes | 0.3 | 0.3 | 20/40 | 20/40 | 562 | 1299 |
| 4 | ARRP 399 | 3 | c.769C>T/p.R257* | CM066961 | c.769C>T/p.R257* | CM066961 | No | 0.4 | 0.3 | 20/50 | 20/40 | 8937 | 7597 |
| 45 | ARRP 341 | 7 | c.959A>G/p.D320G | Novel | c.1749C>G/p.Y583* | CM950914 | No | 0.4 | 0.3 | 20/50 | 20/40 | 660 | 523 |
| 1 | ARRP 254 | 8 | c.304C>A/p.R102S | CM994742 | c.304C>A/p.R102S | CM994742 | No | 1 | 0.3 | 20/200 | 20/40 | 140 | 107 |
| 39 | SRP 928 | 5 | c.1957C>T/p.R653* | CM161562 | c.1956_1957ins20/ p.R653* | Novel | Yes | 0.4 | 0.4 | 20/50 | 20/50 | 702 | 683 |
| 20 | ARRP 290 | 4 | c.1957C>T/p.R653* | CM161562 | c.1957C>T/p.R653* | CM161562 | No | 0.5 | 0.4 | 20/60 | 20/50 | 374 | 267 |
| 108 | SRP 973 | 4 | c.2083C>T/p.Q695* | Novel | c.1065 + 2T>A/p.? | Novel | No | 1 | 0.4 | 20/200 | 20/50 | 444 | 751 |
| 28 | ARRP 314 | 4 | c.305G>A/p.R102H | CM994741 | c.305G>A/p.R102H | CM994741 | No | 0.2 | 0.5 | 20/30 | 20/60 | 753 | 576 |
| 7 | ARRP 219 | 6 | c.304C>A/p.R102S | CM994742 | c.304C>A/p.R102S | CM994742 | No | 0.2 | 0.5 | 20/30 | 20/60 | 8076 | 9060 |
| 103 | ARRP 223 | 3 | c.1166C>T/p.P389L | CM112500 | c.1166C>T/p.P389L | CM112500 | No | 0.3 | 0.5 | 20/40 | 20/60 | 7812 | 9453 |
| 31 | ARRP 307 | 6 | c.1705C>A/p.Q569K | CM994743 | c.1065 + 2T>A/p.? | Novel | Yes | 0.5 | 0.5 | 20/60 | 20/60 | 221 | 167 |
| 2 | SRP 564 | 4 | c.63_68del/ p.K21_Y23delinsN | Novel | c.1926 + 1G>A/p.? | Novel | Yes | 0.3 | 0.7 | 20/40 | 20/100 | 324 | 305 |
| 107 | ARRP 320 | 3 | c.769C>T/p.R257* | CM066961 | c.769C>T/p.R257* | CM066961 | No | 0.7 | 0.7 | 20/100 | 20/100 | 6644 | 6108 |
| 106 | SRP 764 | 7 | c.676del/p.H226Tfs*2 | CD140529 | c.2053G>A/p.V685M | CM106644 | No | 0.9 | 0.7 | 20/160 | 20/100 | 255 | 322 |
| 24 | ARRP 291 | 5 | c.998 + 1G>A/p.? | CS994749 | c.998 + 1G>A/p.? | CS994749 | Yes | 1 | 0.7 | 20/200 | 20/100 | 496 | 365 |
| 44 | ARRP 317 | 6 | c.627 + 2T>G/p.? | Novel | c.627 + 2T>G/p.? | Novel | No | 0.5 | 0.8 | 20/60 | 20/125 | 318 | 292 |
| 11 | ARRP 60 | 5 | c.2053G>A/p.V685M | CM106644 | c.2053G>A/p.V685M | CM106644 | Yes | 0.7 | 0.9 | 20/100 | 20/160 | 163 | 204 |
| 22 | ARRP 291 | 5 | c.998 + 1G>A/p.? | CS994749 | c.998 + 1G>A/p.? | CS994749 | Yes | 0.7 | 0.9 | 20/100 | 20/160 | 592 | 568 |
| 25 | ARRP 291 | 4 | c.998 + 1G>A/p.? | CS994749 | c.998 + 1G>A/p.? | CS994749 | Yes | 1 | 1 | 20/200 | 20/200 | 394 | 441 |
| 10 | ARRP 60 | 5 | c.2053G>A/p.V685M | CM106644 | c.2053G>A/p.V685M | CM106644 | Yes | 1.2 | 1 | 20/300 | 20/200 | 129 | 90 |
| 32 | ARRP 307 | 7 | c.1705C>A/p.Q569K | CM994743 | c.1065 + 2T>A/p.? | Novel | Yes | 1.1 | 1.1 | 20/250 | 20/250 | 49 | 33 |
| 35 | ARRP 323 | 5 | c.84C>G/p.Y28* | Novel | c.84C>G/p.Y28* | Novel | No | 1.2 | 1.2 | 20/300 | 20/300 | 436 | 583 |
| 40 | SRP 929 | 6 | c.305G>A/p.R102H | CM994741 | c.1359_1361delinsCC/ p.V454Qfs*5 | Novel | Yes | 1.3 | 1.3 | 20/400 | 20/400 | NA | 9 |
| 18 | SRP 250 | 6 | c.1235T>C/p.F412S | Novel | c.1966G>T/p.E656* | CM1717824 | Yes | 1/35 at 1 m | HM | 1/35 at 1 m | HM | 218 | 20 |
| 38 | ARRP 306 | 7 | c.1705C>A/p.Q569K | CM994743 | c.1705C>A/p.Q569K | CM994743 | No | HM | HM | HM | HM | NA | NA |
Abbreviations: ARRP, autosomal recessive retinitis pigmentosa; CD, deletion, coding; CM, substitution, coding; CS, splicing; HGMD, Human Gene Mutation Database; HM, hand movements; NA, not applicable; SRP, sporadic retinitis pigmentosa.
Patients are sorted by age.
The codes CD, CM, and CS refer to variants in the HGMD Accession number definitions.
The sequence variation spectrum included 33 different alleles, 17 of which have not been reported to date, to our knowledge. They included 12 missense variants, 8 canonical splice-site variants, 7 nonsense variants, 5 frameshift variants, and 1 in-frame deletion or insertion variants. Recurrent alleles were frequently observed: 16 patients carried the c.304C>A;p.(R102S) variant on 1 or both alleles, making it the most common allele in the cohort (23 alleles), followed by c.2053G>A;p./(V685M) (12 alleles). The splice-site variant c.998 + 1G>A;p.? accounted for 14 alleles in the cohort. However, this high number is associated with 6 siblings from the same family having homozygosity for this variant (Figure 1). All variants were classified according to their pathogenicity based on the ACMG guidelines (Table 2).16
Figure 1. Genomic and Protein Structure of PDE6A and Location of Variants.

The exon and intron organization is shown to scale at the top; the polypeptide and its functional domains, below. Blue indicates splice-site variants; black, frameshift variants; red, nonsense variants; gray, in-frame deletion or insertion; green, missense variants. cGMP indicates cyclic guanosine monophosphate; GAF, cGMP-specific phosphodiesterases, adenylyl cyclases and FhlA; PDE and PDEase, phosphodiesterase.
Table 2. PDE6A Sequence Variants Identified in This Study.
| Nucleotide (NM_000440.3) | PDE6A protein (NP_000431.2) | Initial known description | Localization of missense variants | Consensus prediction for missense variantsa | American College of Medical Genetics and Genomics | gnomAD minor allele frequency | |
|---|---|---|---|---|---|---|---|
| Categoriesb | Prediction, final classification | ||||||
| Splice site variants | |||||||
| c.627 + 2T>G | NA (p.D159_E209del)c | This study | NA | NA | PVS1; PM2 | Likely pathogenic | None |
| c.998 + 1G>A | NA (p.I313Sfs*4)c | Dryja et al,5 1999 | NA | NA | PVS1; PM2; PP1 | Pathogenic | 0.00002830 |
| c.998 + 2T>G | NA (p.I313Sfs*4)c | This study | NA | NA | PVS1; PM2; PM3 | Pathogenic | None |
| c.1065 + 2T>A | NA (p.N334Ffs*5)c | Khateb et al,6 2019 | NA | NA | PVS1; PM2; PM3 | Pathogenic | None |
| c.1263 + 1G>A | NA (p.K372_E421del)c | This study | NA | NA | PVS1; PM2; PM3 | Pathogenic | None |
| c.1620 + 1G>A | NA (p.Q492_E540del)c | This study | NA | NA | PVS1; PM2; PM3 | Pathogenic | 0.000007958 |
| c.1926 + 1G>A | NA (p.S614Afs*2)c | This study | NA | NA | PVS1; PM2; PM3 | Pathogenic | 0.000003978 |
| c.1927-1G>T | NA (p.S643Efs*13)c | This study | NA | NA | PVS1; PM2 | Likely pathogenic | None |
| Frameshift deletions and insertions | |||||||
| c.612del | p.K205Rfs*16 | Jespersgaard et al,22 2019 | NA | NA | PM2; PVS1 | Likely pathogenic | None |
| c.676del | p.H226Tfs*2 | Glöckle et al,23 2014d | NA | NA | PM2; PVS1 | Likely pathogenic | None |
| c.2332_2335del | p.D778Lfs*42 | This study | NA | NA | PM2; PVS1; PM3 | Pathogenic | 0.00002475 |
| c.1359_1361 delinsCC |
p.V454Qfs*5 | This study | NA | NA | PM2; PVS1; PM3 | Pathogenic | None |
| c.1956_1957ins20 | p.R653* | This study | PM2; PVS1; PM3 | Pathogenic | None | ||
| Nonsense variants | |||||||
| c.84C>G | p.Y28* | This study | NA | NA | PM2; PVS1 | Likely pathogenic | 0.000003980 |
| c.769C>T | p.R257* | Riazuddin et al,24 2006 | NA | NA | PM2; PVS1; PM3 | Pathogenic | 0.00003890 |
| c.1683G>A | p.W561* | Huang et al,2 1995 | NA | NA | PM2; PVS1; PM3 | Pathogenic | 0.000003977 |
| c.1749C>G | p.Y583* | Huang et al,2 1995 | NA | NA | PM2; PVS1 | Likely pathogenic | None |
| c.1957C>T | p.R653* | Perez-Carro et al,25 2016 | NA | NA | PM2; PVS1; PM3 | Pathogenic | 0.00002787 |
| c.1966G>T | p.E656* | Soens et al,26 2017 | NA | NA | PM2; PVS1; PM3 | Pathogenic | 0.000007079 |
| c.2083C>T | p.Q695* | This study | NA | NA | PM2; PVS1 | Likely pathogenic | 0.000003981 |
| In-frame deletion and insertion | |||||||
| c.63_68del | p.K21_Y23delinsN | This study | NA | NA | PM2; PM3; PM4 | Likely pathogenic | None |
| Missense variants | |||||||
| c.304C>A | p.R102S | Dryja et al,5 1999 | GAF domain | Damaging | PM2; PM3; PM5; PS3 | Pathogenic | 0.0001556 |
| c.305G>A | p.R102H | Dryja et al,5 1999 | GAF domain | Damaging | PM2; PM3; PM5; PS3 | Pathogenic | 0.00002122 |
| c.743T>A | p.V248D | This study | GAF domain | Damaging | PM2 | VUS | None |
| c.959A>G | p.D320G | This study | GAF domain | Damaging | PM2 | VUS | None |
| c.1166C>T | p.P389L | Collin et al,27 2011 | GAF domain | Damaging | PM2; PS3 | Likely pathogenic | 0.000007954 |
| c.1235T>C | p.F412S | This study | GAF domain | Damaging | PM2; PM3 | VUS | None |
| c.1610T>C | p.I537T | This study | Catalytic domain | Damaging | PM1; PM2 | VUS | None |
| c.1684C>T | p.R562W | Sothilingam et al,11 2015d | Catalytic domain | Damaging | PM1; PM2; PM3; PS3 | Pathogenic | 0.00001193 |
| c.1689C>A | p.H563Q | Birtel et al,28 2018 | Catalytic domain | Damaging | PM1; PM2; PM3 | Likely pathogenic | 0.000007954 |
| c.1705C>A | p.Q569K | Dryja et al,5 1999 | Catalytic domain | Damaging | PM1; PM2; PM3 | Likely pathogenic | 0.0001344 |
| c.1862T>G | p.L621R | This study | Catalytic domain | Damaging | PM1; PM2; PM3 | Likely pathogenic | None |
| c.2053G>A | p.V685M | Corton et al,29 2010 | Catalytic domain | Damaging | PM1; PM2; PM3; PS3 | Pathogenic | 0.00004957 |
Abbreviations: NA, not applicable; GAF, cGMP-specific phosphodiesterases, adenylyl cyclases and FhlA; gnomAD, Genome Aggregation Database; PM, pathogenic, moderate; PP, pathogenic, supporting; PS, pathogenic, strong; PVS, pathogenic, very strong; VUS, variant of uncertain significance.
The American College of Medical Genetics and Genomics categories PM, PP, PS, and PVS are further subdivided and ranked from 1 to 6 for PM, with lower numbers higher in influence. It is the state-of-the-art standard classification of genetic variants.16
Consensus of 4 or more prediction algorithms.
Consequence of putative exon skipping.
The patient described is the same as in the present study.
Ophthalmological Findings
The mean (SD) BCVA was 0.43 (0.48) logMAR (range, −0.10 to 2.30; 114 eyes; Snellen equivalent, 20/50; range, hand movement to 20/16). Visual impairment was mild in 15 of 114 eyes (13.2%), moderate in 19 of 114 eyes (16.7%), and severe in 7 of 114 eyes (6.1%), and 4 of 114 eyes (3.5%) met the criteria for legal blindness as defined by the World Health Organization with respect to BCVA. The BCVA findings were highly symmetrical in right and left eyes (R2, 0.786).
Kinetic visual field testing with object III4e of Goldmann allowed for evaluation in 107 of 114 eyes (93.9%). The median visual field area in these 107 eyes was 660 (5th-95th percentiles, 76-11 019; 25th-75th percentiles, 255-3923) square degrees (Figure 2). In 3 eyes, object III4e was not recognized. Kinetic visual field testing obtained with object I4e allowed for evaluation in 92 of 114 eyes (80.7%). The median visual field area in these 92 eyes was 150 (range, 2-2833) square degrees (Figure 2). In 16 eyes, object I4e was not recognized. Kinetic visual field measurements were highly symmetrical in right and left eyes (R2, 0.951). Progression of visual field defects with increasing age or disease duration is shown in Figure 2.
Figure 2. Visual Field Data.
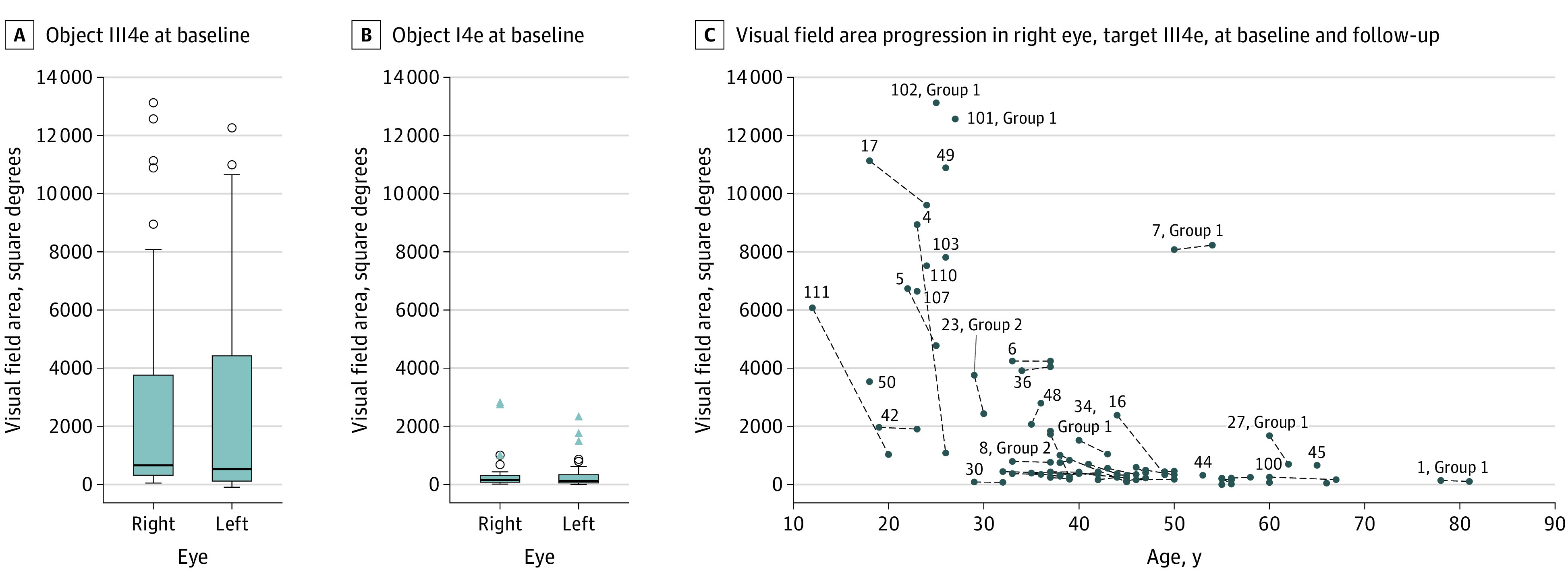
A and B, Visual field area at baseline for objects III4e and I4e. C, Visual field area of right eyes at baseline, and, if available, at last follow-up. Note the drop in visual field area that is most rapid in the second and third decades of life.
Optical coherence tomography (OCT) images were available in 108 of 114 eyes (94.7%). As is typical for RP, OCT imaging revealed thinning of the outer retinal layers from the periphery to the center of the retina (macula or fovea), with disruption or loss of the ellipsoid zone in advanced disease. In the cohort, 69 of 108 eyes (63.9%) showed additional findings on OCT: cystoid macular edema (CME) in 27 of 108 eyes (25.0%), CME and macular atrophy in 2 of 108 (1.9%), macular atrophy in 18 of 108 (16.7%), epiretinal membrane (ERM) in 8 of 108 (7.4%), ERM with traction in 6 of 108 (5.6%), ERM with a lamellar hole in 2 of 108 (1.9%), an isolated lamellar hole in 1 of 108 (1%), a full-thickness macular hole in 1 of 108 (0.9%), and posterior staphyloma in 4 of 108 (3.7%) (eFigure 1 in the Supplement).
Full-field ERG findings were available in 108 of 114 eyes (94.7%). In 4 of 108 eyes (3.7%), dark-adapted and light-adapted ff-ERG responses were within normal limits. In 8 of 108 (7.4%) each, light-adapted ff-ERG showed some measurable, albeit subnormal responses, whereas dark-adapted ff-ERG showed either residual or no responses. In 88 of 108 (81.5%), responses for both dark-adapted and light-adapted ff-ERGs were absent. Multifocal ERG findings were available in 90 of 114 eyes (78.9%). In 53 of 90 eyes (88%), multifocal electroretinography showed no responses.
We also evaluated whether the clinical findings differed between patients harboring 1 of the common disease-causing PDE6A variants in a homozygous state, comparing the c.304C>A;p.(R102S) (group 1), and c.998 + 1G>A;p.? (group 2). Seven patients each were homozygous for either the variant c.304C>A;p.R(102S) or the variant c.998 + 1G>A;p.?. Visual field maps, OCT, and fundus autofluorescence findings of right eyes are shown in eFigure 1 in the Supplement. The respective groups (1 and 2) differed in their mean (SD) ages at baseline (46 [17] vs 38 [6] years). The mean (SD) visual acuity was worse in group 2 when compared with group 1 (0.50 [0.37] vs 0.19 [0.27] logMAR; Snellen equivalent: 20/60 vs 20/30). Similarly, the mean (SD) VF area was worse in group 2 when compared with group 1 (1178 [1304] vs 5548 [5040] square degrees). When corrected for age, these differences were statistically significant. The BCVA mean difference was −0.31 (95% CI, −0.70 to 0.07; P = .01), and the VF area mean difference was 4370 (95% CI, −644 to 9384; P = .003).
Discussion
In this study, we examined 57 patients from across Europe with RP associated with pathogenic, biallelic variants in the PDE6A gene. Given that PDE6A-associated RP is such a rare genetic subtype of RP, the size of this cohort is uniquely large.6 From comprehensive genetic analyses in association with this study of PDE6A in patients with a clinical diagnosis of RP, we conclude that there is a frequency of PDE6A-associated RP of 1.6% in Germany (1100 patients residing in Germany, of whom 17 were found to harbor biallelic variants in PDE6A [data not shown]).
Clinical Findings
All patients with PDE6A-associated RP in our study exhibited highly symmetrical findings typical for RP with respect to visual acuity, visual field, and other modalities. When comparing BCVA with VF area in terms of symmetry, the VF area was more symmetrical in right and left eyes. Disease symmetry presents a strong case for using the untreated eye as the control eye in an interventional study with small patient numbers. The untreated eye may serve as a control to assess possible negative adverse effects as well as positive outcomes of therapy (such as gene supplementation therapy; ie, the preservation of VF area or the deceleration of VF area loss).
The greatest (numberwise) loss in VF area was approximately −7900 square degrees in a 23-year-old patient with a VF area of 8900 square degrees at baseline 2 years earlier. This change reflects the natural course of disease, characterized by a slow progression of VF defects before young adulthood, typically followed by a sudden loss of large or larger VF areas in young adulthood and subsequent slow progression of VF defects the smaller the remaining VF area gets.
Kinetic visual field testing with object III4e allowed for evaluation in more than 90% of eyes. However, kinetic visual field testing is a set of psychophysical examinations with advantages but also limitations. The limitations are that the condition of the patient affects the results of the examination, in that a patient who is alert will achieve better results than a patient with fatigue, and the same is true for the technician performing the examination and encouraging the patient. Thus, VF findings can vary between visits, and comparisons between patients, as well as within the same patient at different points, may yield misleading results. However, given the remarkable symmetry of the VF area in right and left eyes that we observed in this cohort and the fact that VF area is a measure of visual function rather than an anatomical end point, we judge VF testing as a suitable marker and end point for a phase I or II safety and efficacy trial.
Macular OCT findings may well limit a patient’s eligibility to participate in a gene therapy trial, since the preferred injection site for the therapeutic vector to date is the central retina. Generally, macular findings in patients with RP include cystoid macular edema in 6% to 25%, ERM in 1% to 27%, and macular holes in 1% to 5% of patients with RP.30,31,32 In our cohort, CME was present in 25%, ERM in 7%, and macular holes in 4%. Thus, the frequency of macular OCT findings in PDE6A-associated RP was similar to that in the general population of patients with RP. Mild CME may not be as much of a limitation for a patient to participate in a gene therapy trial, and the same applies to mild ERMs without traction. Yet, extensive CME, as well as marked ERM with traction and any type of macular hole, would be expected to increase the risk of complications attributable to the surgical procedure, irrespective of the investigational new drug, thus having a potential effect on safety and efficacy observations.
Among the most frequent variants found in our cohort, the variant c.998 + 1G>A;p.? led to a more severe phenotype with respect to VF and BCVA when present in a homozygous state and when compared with individuals homozygous for the c.304C>A;p.(R102S) variant. The third most frequent variant c.2053G>A;p.(V685M) was mostly present in a (compound) heterozygous state, making it difficult to compare this genotype with the 2 other most frequent variants mentioned with respect to the severity of the phenotype. Our clinical findings are thus in line with the fact that c.998 + 1G>A;p.? is considered a null allele, in contrast with c.304C>A;p.(R102S).
Genetic Findings of Pathogenicity of Identified Variants
Because we were unable to validate biallelism in all of the patients, the pathogenicity of single variants rather than genotypes will be discussed. Of the 33 putatively pathogenic variants identified in this study, 7 are nonsense variants and 5 are frameshift variants. These are considered to constitute loss-of-function alleles. Furthermore, we identified 8 canonical splice variants, 7 of which affect invariable donor sites and 1 of which affects an invariable acceptor site.
Because nonsense, canonical splice site, and frameshift variants have a strong weight in the ACMG scoring system, these classes of variants are consequently classified either as pathogenic or likely pathogenic (Table 2). On the other hand, missense variants that lack segregation data and functional analyses to support a damaging outcome are always classified as variants of uncertain significance per the ACMG guidelines. The potential pathogenicity of the missense variants affecting codon 102 and 389 of the PDE6A gene (c.304C>A;p.R102S, c.305G>A;p.R102H, and c.1166C>T;p.P389L) is supported by the fact that corresponding sequence variations in PDE6C have been shown to have a causal association with PDE6C-associated autosomal recessive achromatopsia.33 Their effect on PDE6 function has further been substantiated by functional in vitro studies performed on the catalytic subunit of cone photoreceptor phosphodiesterase PDE6C, the PDE6 paralog in cone photoreceptors.33 Grau and coworkers33 demonstrated that the corresponding PDE6C missense variants c.310C>T;p.R104W and c.1172C>T;p.P391L led to a highly significant reduction in phosphodiesterase activity to almost baseline levels. Since codons 104 and 391 of PDE6C are homologous to codons 102 and 389 of PDE6A, the results obtained from functional studies performed on PDE6C can be transferred to PDE6A. Evidence for the pathogenicity of the missense variants c.1684C>T;p.R562W and c.2053G>A;p.V685M is derived from the functional analysis of Pde6a mouse models: Sothilingam and coworkers11 showed that homozygous (R562W/R562W and V685M/V685M) and compound heterozygous (R562W/V685M) Pde6a mutant animals expressed only residual amounts of Pde6a protein, resulting in photoreceptor cell death. Functional studies or animal models are not available for the remaining 7 missense variants identified in this study. Yet, the respective amino acid residues are located either between cGMP-specific phosphodiesterases, adenylyl cyclases and FhlA (GAF) domains (c.743T>A;p.V248D), within the second GAF domain (c.959A>G;p.D320G, c.1235T>C;p.F412S), or in the catalytic domain of PDE6A (c.1610T>C;p.I537T, c.1689C>A;p.H563Q, c.1705C>A;p.Q569K, c.1862T>G;p.L621R). Specifically, the variants c.1684C>T;p.R562W and c.1689C>A;p.H563Q affect the Zn2+-binding motif, which is invariant among phosphodiesterases. All affected amino acid residues are highly conserved among vertebrates, not only for PDE6A, but also within PDE6B and PDE6C (eFigure 2 in the Supplement). Consequently, in silico assessment using 5 different prediction tools predicted damaging outcomes on protein function for all 7 variants (Table 2). However, without functional data supportive of damaging outcomes, their pathogenicity cannot be unequivocally proven.
Last, we have identified a novel in frame deletion/insertion variant, c.63_68del;p.(K21_Y23delinsN). It localizes near the N-terminus of PDE6A, upstream of the first GAF domain, and affects 3 amino acid residues that are conserved among vertebrate PDE6A and partially conserved among human PDE6B and PDE6C polypeptides. It has been shown that dimerization of PDE6 is mediated by multiple regions in the N-terminal domain.34 We therefore hypothesize that the c.63_68del;p.(K21_Y23delinsN) variant may impair dimerization, thereby leading to loss of function of PDE6A.
Limitations
True homozygosity and the absence of pathogenic variants in other genes that are known to cause inherited retinal dystrophies are key eligibility criteria for inclusion in a gene therapy trial. One limiting factor with respect to the clinical aspect (and course) of PDE6A-associated RP and subretinal gene therapy in our cohort may be macular pathology, which would increase the risk of complications attributable to the surgical procedure if the injection site is the central retina. However, given that PDE6 is rod specific, from a mechanistical standpoint, it would be more promising to treat regions outside the macula with high rod density early in life.
Conclusions
In summary, we observed and described the genetic and ophthalmologic characteristics in 57 patients with RP associated with pathogenic biallelic variants in the PDE6A gene. Regarding the genetic findings, 17 of the PDE6A variants found in these patients appeared to be novel. Regarding the clinical findings, disease was highly symmetrical between right and left eyes and visual impairment was mild or moderate in 90% of patients, providing a window of opportunity for gene therapy. These data suggest that individuals with PDE6A-associated RP may be eligible for gene therapy.
eFigure 1. Visual field maps, optical coherence tomography, and fundus autofluorescence images of patients homozygous for c.304C>A;p.(R102S) variant and c.998+1G>A;p.? variant.
eFigure 2. Comparative amino acid sequence analysis of photoreceptor specific PDE6A in different vertebrate species.
References
- 1.Retinal Information Network Table of contents. https://sph.uth.edu/retnet. Updated 2020. Accessed September 14, 2020.
- 2.Huang SH, Pittler SJ, Huang X, Oliveira L, Berson EL, Dryja TP. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat Genet. 1995;11(4):468-471. doi: 10.1038/ng1295-468 [DOI] [PubMed] [Google Scholar]
- 3.Dryja TP, Finn JT, Peng YW, McGee TL, Berson EL, Yau KW. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci U S A. 1995;92(22):10177-10181. doi: 10.1073/pnas.92.22.10177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Cote RH. cGMP signaling in vertebrate retinal photoreceptor cells. Front Biosci. 2005;10(1-3):1191-1204. doi: 10.2741/1612 [DOI] [PubMed] [Google Scholar]
- 5.Dryja TP, Rucinski DE, Chen SH, Berson EL. Frequency of mutations in the gene encoding the alpha subunit of rod cGMP-phosphodiesterase in autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1999;40(8):1859-1865. [PubMed] [Google Scholar]
- 6.Khateb S, Nassisi M, Bujakowska KM, et al. Longitudinal clinical follow-up and genetic spectrum of patients with rod-cone dystrophy associated with mutations in PDE6A and PDE6B. JAMA Ophthalmol. 2019;137(6):669-679. doi: 10.1001/jamaophthalmol.2018.6367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan SY, Ali S, Naeem MA, et al. Splice-site mutations identified in PDE6A responsible for retinitis pigmentosa in consanguineous Pakistani families. Mol Vis. 2015;21:871-882. [PMC free article] [PubMed] [Google Scholar]
- 8.Sharon D, Ben-Yosef T, Goldenberg-Cohen N, et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum Mutat. 2020;41(1):140-149. doi: 10.1002/humu.23903 [DOI] [PubMed] [Google Scholar]
- 9.Petersen-Jones SM, Entz DD, Sargan DR. cGMP phosphodiesterase-alpha mutation causes progressive retinal atrophy in the Cardigan Welsh corgi dog. Invest Ophthalmol Vis Sci. 1999;40(8):1637-1644. [PubMed] [Google Scholar]
- 10.Sakamoto K, McCluskey M, Wensel TG, Naggert JK, Nishina PM. New mouse models for recessive retinitis pigmentosa caused by mutations in the Pde6a gene. Hum Mol Genet. 2009;18(1):178-192. doi: 10.1093/hmg/ddn327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sothilingam V, Garcia Garrido M, Jiao K, et al. Retinitis pigmentosa: impact of different Pde6a point mutations on the disease phenotype. Hum Mol Genet. 2015;24(19):5486-5499. doi: 10.1093/hmg/ddv275 [DOI] [PubMed] [Google Scholar]
- 12.Wert KJ, Davis RJ, Sancho-Pelluz J, Nishina PM, Tsang SH. Gene therapy provides long-term visual function in a pre-clinical model of retinitis pigmentosa. Hum Mol Genet. 2013;22(3):558-567. doi: 10.1093/hmg/dds466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wert KJ, Sancho-Pelluz J, Tsang SH. Mid-stage intervention achieves similar efficacy as conventional early-stage treatment using gene therapy in a pre-clinical model of retinitis pigmentosa. Hum Mol Genet. 2014;23(2):514-523. doi: 10.1093/hmg/ddt452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis RJ, Hsu C-W, Tsai Y-T, et al. Therapeutic margins in a novel preclinical model of retinitis pigmentosa. J Neurosci. 2013;33(33):13475-13483. doi: 10.1523/JNEUROSCI.0419-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulze-Bonsel K, Feltgen N, Burau H, Hansen L, Bach M. Visual acuities “hand motion” and “counting fingers” can be quantified with the Freiburg visual acuity test. Invest Ophthalmol Vis Sci. 2006;47(3):1236-1240. doi: 10.1167/iovs.05-0981 [DOI] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073-1081. doi: 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- 18.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248-249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361-362. doi: 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- 20.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. doi: 10.1093/nar/gkr407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745-2747. doi: 10.1093/bioinformatics/btv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jespersgaard C, Fang M, Bertelsen M, et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci Rep. 2019;9(1):1219. doi: 10.1038/s41598-018-38007-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glöckle N, Kohl S, Mohr J, et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet. 2014;22(1):99-104. doi: 10.1038/ejhg.2013.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riazuddin SA, Zulfiqar F, Zhang Q, et al. Mutations in the gene encoding the alpha-subunit of rod phosphodiesterase in consanguineous Pakistani families. Mol Vis. 2006;12:1283-1291. [PubMed] [Google Scholar]
- 25.Perez-Carro R, Corton M, Sánchez-Navarro I, et al. Panel-based NGS reveals novel pathogenic mutations in autosomal recessive retinitis pigmentosa. Sci Rep. 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soens ZT, Branch J, Wu S, et al. Leveraging splice-affecting variant predictors and a minigene validation system to identify mendelian disease-causing variants among exon-captured variants of uncertain significance. Hum Mutat. 2017;38(11):1521-1533. doi: 10.1002/humu.23294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collin RWJ, van den Born LI, Klevering BJ, et al. High-resolution homozygosity mapping is a powerful tool to detect novel mutations causative of autosomal recessive RP in the Dutch population. Invest Ophthalmol Vis Sci. 2011;52(5):2227-2239. doi: 10.1167/iovs.10-6185 [DOI] [PubMed] [Google Scholar]
- 28.Birtel J, Gliem M, Mangold E, et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS One. 2018;13(12):e0207958. doi: 10.1371/journal.pone.0207958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corton M, Blanco MJ, Torres M, Sanchez-Salorio M, Carracedo A, Brion M. Identification of a novel mutation in the human PDE6A gene in autosomal recessive retinitis pigmentosa. Clin Genet. 2010;78(5):495-498. doi: 10.1111/j.1399-0004.2010.01487.x [DOI] [PubMed] [Google Scholar]
- 30.Hagiwara A, Yamamoto S, Ogata K, et al. Macular abnormalities in patients with retinitis pigmentosa. Acta Ophthalmol. 2011;89(2):e122-e125. doi: 10.1111/j.1755-3768.2010.01866.x [DOI] [PubMed] [Google Scholar]
- 31.Gorovoy IR, Gallagher DS, Eller AW, Mayercik VA, Friberg TR, Schuman JS. Cystoid macular edema in retinitis pigmentosa patients without associated macular thickening. Semin Ophthalmol. 2013;28(2):79-83. doi: 10.3109/08820538.2012.760614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giusti C, Forte R, Vingolo EM. Clinical pathogenesis of macular holes in patients affected by retinitis pigmentosa. Eur Rev Med Pharmacol Sci. 2002;6(2-3):45-48. [PubMed] [Google Scholar]
- 33.Grau T, Artemyev NO, Rosenberg T, et al. Decreased catalytic activity and altered activation properties of PDE6C mutants associated with autosomal recessive achromatopsia. Hum Mol Genet. 2011;20(4):719-730. doi: 10.1093/hmg/ddq517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muradov KG, Boyd KK, Martinez SE, Beavo JA, Artemyev NO. The GAFa domains of rod cGMP-phosphodiesterase 6 determine the selectivity of the enzyme dimerization. J Biol Chem. 2003;278(12):10594-10601. doi: 10.1074/jbc.M208456200 [DOI] [PubMed] [Google Scholar]
- 35.Madeira F, Madhusoodanan N, Lee J, Tivey ARN, Lopez R. Using EMBL-EBI services via web interface and programmatically via web services. Curr Protoc Bioinformatics. 2019;66(1):e74. doi: 10.1002/cpbi.74 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eFigure 1. Visual field maps, optical coherence tomography, and fundus autofluorescence images of patients homozygous for c.304C>A;p.(R102S) variant and c.998+1G>A;p.? variant.
eFigure 2. Comparative amino acid sequence analysis of photoreceptor specific PDE6A in different vertebrate species.