

Abstract
Idiosyncratic drug-induced liver injury (IDILI) is an infrequent but potentially serious disease that develops the main reason for post-marketing safety warnings and withdrawals of drugs. Epimedii Folium (EF), the widely used herbal medicine, has shown to cause idiosyncratic liver injury, but the underlying mechanisms are poorly understood. Increasing evidence has indicated that most cases of IDILI are immune mediated. Here, we report that icariside Ⅱ (ICS Ⅱ), the major active and metabolic constituent of EF, causes idiosyncratic liver injury by promoting NLRP3 inflammasome activation. ICS Ⅱ exacerbates NLRP3 inflammasome activation triggered by adenosine triphosphate (ATP) and nigericin, but not silicon dioxide (SiO2), monosodium urate (MSU) crystal or cytosolic lipopolysaccharide (LPS). Additionally, the activation of NLRC4 and AIM2 inflammasomes is not affected by ICS Ⅱ. Mechanistically, synergistic induction of mitochondrial reactive oxygen species (mtROS) is a crucial contributor to the enhancing effect of ICS Ⅱ on ATP- or nigericin-induced NLRP3 inflammasome activation. Importantly, in vivo data show that a combination of non-hepatotoxic doses of LPS and ICS Ⅱ causes the increase of aminotransferase activity, hepatic inflammation and pyroptosis, which is attenuated by Nlrp3 deficiency or pretreatment with MCC950 (a specific NLRP3 inflammasome inhibitor). In conclusion, these findings demonstrate that ICS Ⅱ causes idiosyncratic liver injury through enhancing NLRP3 inflammasome activation and suggest that ICS Ⅱ may be a risk factor and responsible for EF-induced liver injury.
Key words: Epimedii Folium, Icariside Ⅱ, Idiosyncratic drug-induced liver injury, NLRP3 inflammasome, Reactive oxygen species, Mitochondria
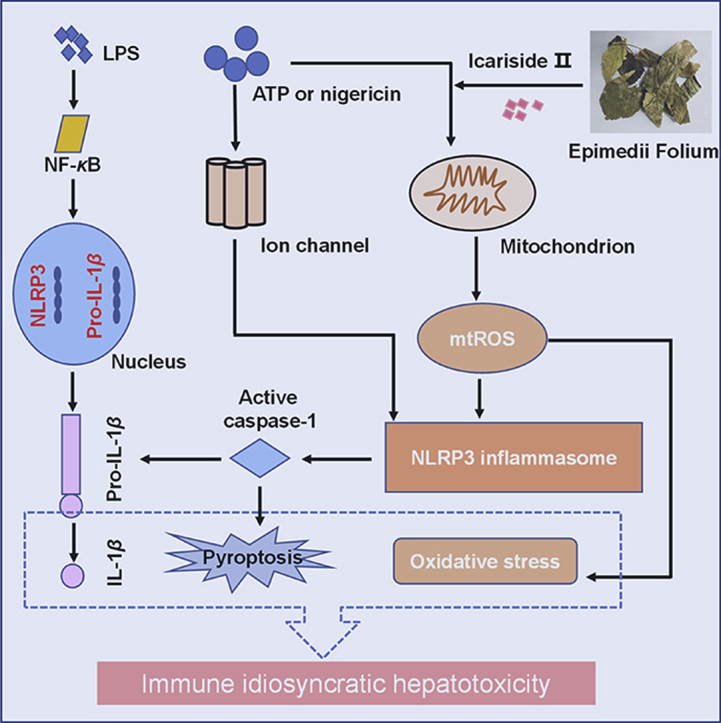
Graphical abstract
The activated NLRP3 inflammasome-mediated IL-1β secretion and subsequently the production of TNF-α, pyroptosis and oxidative stress synergistically induced liver injury after the combination of non-hepatotoxic doses of LPS and ICS Ⅱ. ICS Ⅱ may be a risk factor and responsible for Epimedii Folium-induced idiosyncratic hepatotoxicity.
1. Introduction
Idiosyncratic drug-induced liver injury (IDILI) is a severe adverse reaction that significantly increases the expenditure of drug development on account of a variable latency to onset and a lack of clear dose dependency1,2. Recently, several studies have pointed out that traditional Chinese medicines (TCMs) and anti-tuberculosis drugs are the primary causes of IDILI in China3,4. Specifically, liver injury caused by TCMs, such as Polygonum multiflorum, Gynura segetum, and Ageratina adenophora, has occurred frequently in recent years5, 6, 7. Although emerging evidence has demonstrated that some TCMs can lead to IDILI, the precise pathogenesis underlying this role is still undefined.
NLRP3 inflammasome comprises a sensor molecule NLRP3, an adaptor protein ASC, and an effector molecule pro-caspase-18. NLRP3 inflammasome senses pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), which leads to pro-caspase-1 cleavage, and subsequently induces pyroptosis and triggers inflammatory responses9, 10, 11. Recent researches have demonstrated that excessive activation of NLRP3 inflammasome could lead to various liver diseases, such as IDILI, nonalcoholic/alcoholic steatohepatitis and liver fibrosis12, 13, 14, and plays a critical role in determining the progression, severity and pathology via amplifying the inflammatory response.
Epimedii Folium (EF) is a well-known herbal medicine that has been employed extensively as a tonic, aphrodisiac, and antirheumatic15. In recent years, the possibility that EF and its preparations cause IDILI has been reported frequently16,17, and clinical safety issues associated with EF have drawn increasing attention. In our previous research18, we demonstrated that EF could induce idiosyncratic liver injury in a non-hepatotoxic dose of lipopolysaccharide (LPS)-mediated susceptibility mouse model of IDILI. In addition, interleukins-1β (IL-1β) is a special inflammatory cytokine in EF-induced liver injury. Therefore, we speculated that EF likely induces liver injury by promoting the activation of NLRP3 inflammasome. In the current study, we showed that icariside Ⅱ (ICS Ⅱ), the major active and metabolic constituent of EF, could specifically enhance NLRP3 inflammasome activation induced by adenosine triphosphate (ATP) or nigericin, thereby leading to an uncontrolled immune response and the induction of idiosyncratic liver injury.
2. Materials and methods
2.1. Mice
Female wild-type (WT) C57BL/6 mice (6–8-week-old) were obtained from SPF Biotechnology Co., Ltd (Beijing, China). Nlrp3 knock-out (Nlrp3−/−) mice were from National Center of Biomedical Analysis (NCBA, Beijing, China) supplied by Dr. Tao Li. All mice were permitted water and food freely and held under a 12-h dark/light cycle at 22 ± 2 °C. All of the experimental procedures in our experiments were carried out on the basis of the Guidelines Of Laboratory Animals Care And Use and authorized by the Animal Ethics Committee Of the Fifth Medical Centre, Chinese People's Liberation Army (PLA) General Hospital (Beijing, China).
2.2. Cell culture
Using WT or Nlrp3−/− female C57BL/6 mice (10-week-old), bone marrow cells were extracted into Dulbecco's modified Eagle's medium (DMEM, Macgene, Beijing, China) containing 1% penicillin/streptomycin (Macgene) and 10% fetal bovine serum (Gibco, Rockford, IL, USA). Moreover, 50 ng/mL murine macrophage colony-stimulating factor (MedChemExpress, Monmouth, NJ, USA) was added to DMEM to differentiate cells into bone marrow-derived macrophages (BMDMs). Human THP-1 cells, which were supplied by Dr. Tao Li from NCBA, were grown in Roswell Park Memorial Institute (RPMI) 1640 medium (Macgene) containing 1% penicillin/streptomycin (Macgene) and 10% fetal bovine serum (FBS), and they were stimulated using 100 nmol/L phorbol-12-myristate-13-acetate (PMA, MedChemExpress) overnight to differentiate into macrophages. All of the cultured cells were placed in a humidified 5% CO2 atmosphere at 37 °C.
2.3. Inflammasomes activation
For inducing NLRP3, NLRC4 and AIM2 inflammasomes activation, BMDMs at 1 × 106 cells/mL were seeded 0.5 mL in 24-well plates overnight. Then, 50 ng/mL LPS-PG Ultrapure (InvivoGen, San Diego, CA, USA) or 1 μg/mL Pam3CSK4 (InvivoGen) was used to stimulate BMDMs for 4 h, after which the medium was changed to Opti-MEM (Gibco) containing the main components from EF (epmedin A, epmedin A1, epmedin B, epmedin C, icariin, icaritin, icariside Ⅰ, ICS Ⅱ, anhydroicaritin; TargetMol, Boston, MA, USA), hydrogen peroxide (H2O2, Sigma, Darmstadt, Germany) or N-acetyl-l-cysteine (NAC, MedChemExpress). One hour later, the cells were stimulated for 1 h with ATP (5 mmol/L; Sigma) and nigericin (7.5 μmol/L; InvivoGen) or for 6 h with monosodium urate (MSU, 200 μg/mL; InvivoGen), silicon dioxide (SiO2, 250 μg/mL; InvivoGen) or Lfn-Flic (200 ng/mL, supplied by Dr. Tao Li). Cells were transfected with poly (I:C) (2 μg/mL; InvivoGen), poly(dA:dT) (2 μg/mL; InvivoGen) or LPS (1 μg/mL; InvivoGen) for 6 h through the use of Lipofectamine 2000 (Thermo Fisher Scientific, Rockford, MI, USA) according to the manufacturer's protocols. PMA-primed THP-1 cells were seeded at 1 × 106 cells/mL in 24-well plates overnight. Then, the cells were pretreated with ICS Ⅱ. One hour later, the cells were stimulated with nigericin (7.5 μmol/L) for 1 h. Cell extracts and precipitated supernatants were analyzed by immunoblot.
2.4. Western blot analysis
Cell extracts and precipitated supernatants were lysed with 1 × loading buffer (Dingguo Changsheng Biotechnology, Beijing, China) containing radio immunoprecipitation assay buffer (RIPA, Thermo Fisher Scientific). The samples were denatured in 105 °C for 15 min, and equal amounts of the protein samples were separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto 0.2 μm polyvinylidene fluoride membranes. Then, the membrane was blocked by 5% non-fat milk for 1 h at room temperature. The indicated primary antibodies were incubated at 4 °C overnight and then treated with anti-rat IgG (1:3000; Cell Signaling Technology, Danvers, MA, USA), anti-mouse IgG (1:5000; Jackson ImmunoResearch, West Grove, PA, USA) or anti-rabbit IgG (1:5000; Jackson ImmunoResearch) for 1 h at room temperature. The signals were analyzed using the enhanced chemiluminescent reagent (Promega, Beijing, China). The primary antibodies anti-NLRP3 (1:1000) and anti-mouse caspase-1 (1:1000) were bought from Adipogen (San Diego, CA, USA). Anti-mouse cleaved IL-1β (1:2000) was from R&D systems (Minneapolis, MN, USA). Anti-human caspase-1 (1:2000), anti-mouse caspase-11 (1:1000), anti-mouse cleaved GSDMD (1:500), and anti-human cleaved IL-1β (1:1000) were obtained from Cell Signaling Technology (Boston, MA, USA). Anti-ASC (1:1000) was purchased from ABclonal (Wuhan, China). Anti-GAPDH (1:2000; Proteintech, Chicago, IL, USA) was used as an internal control.
2.5. Caspase-1 activity assay
The activity of caspase-1 in cell culture supernatants was evaluated by Caspase-Glo® one Inflammasome Assay on the basis of the manufacturer's direction (Promega).
2.6. Lactate dehydrogenase (LDH) assay
BMDMs or THP-1 cells were primed with LPS or PMA, and the cells were treated with ICS Ⅱ before stimulated with the inflammasomes stimuli. LDH was measured by an LDH cytotoxicity assay kit (Promega).
2.7. ASC oligomerization
BMDMs at 1 × 106 cells/mL were seeded with 1.0 mL medium in 12-well plates overnight. The following day, the medium was replaced, and cells were primed with 50 ng/mL LPS or 1 μg/mL Pam3CSK4 for 4 h. Next, the medium was changed to Opti-MEM containing ICS Ⅱ. The method to induce inflammasomes activation has been described in Section 2.3. Next, the cells were lysed by Triton Buffer [50 mmol/L Tris–HCl (pH 7.5), 150 mmol/L NaCl, 0.5% Triton X-100, and 1% EDTA-free protease inhibitor cocktail (TargetMol)]). A small amount (2.5%) of the lysates were collected as whole cell lysates. The rest of the lysates were centrifuged at 6000×g for 10 min at 4 °C. The pellets were washed with one mL of ice-cold phosphate buffered saline (PBS) and resuspended in 200 μL PBS. Two millimolar disuccinimidyl suberate (DSS, Abcam, Cambridge, UK) was added to the resuspended pellets, which were incubated at 37 °C for 30 min with rotation. Samples were then centrifuged at 6000×g for 10 min at 4 °C. The cross-linked pellets were resuspended in 60 μL sample buffer and then were boiled and analyzed by immunoblot.
2.8. Intracellular potassium detection
To measure the intracellular potassium, BMDMs at 1 × 106 cells/mL were seeded with 1.0 mL medium overnight in 12-well plates. After being primed with 50 ng/mL LPS for 4 h, cells were treated with ICS Ⅱ for 1 h and then stimulated with different NLRP3 stimuli. Culture medium was thoroughly aspirated and lysed with 3% ultrapure HNO3. Intracellular K+ measurements were performed by inductively coupled plasma optical emission spectrometry.
2.9. Immunoprecipitation assay
For the exogenous NLRP3–NLRP3 interaction experiment, HEK-293T cells (3 × 105 cells/mL) were transfected with Flag-NLRP3 and Myc-NLRP3 in 6-well plates via Lipofectamine 2000. Twelve hours later, ICS Ⅱ was given to assess its impact on NLRP3–NLRP3 interaction. Then, cells were gathered with lysis buffer containing protease inhibitor after 24 h. Next, the lysate was immunoprecipitated with anti-Flag antibody and beads, and then Western blot was used to evaluate the NLRP3–NLRP3 interaction.
2.10. Confocal microscopy
Confocal microscopy analysis, which was carried out to test mitochondrial damage, has been described previously19. BMDMs were plated at 5 × 105 cells/mL on coverslips in 24-well plates overnight. Then, the cells were stimulated with 50 ng/mL LPS for 4 h. After that, ICS Ⅱ was added as described for another 1 h. BMDMs were stimulated by ATP for 1 h and stained with MitoTracker Red (50 nmol/L, Thermo Fisher Scientific), then cells were washed three times by PBS and fixed with 4% paraformaldehyde in PBS for 15 min. After that, cells were washed with PBST three times. Confocal microscopy analysis was carried out by using a Zeiss LSM 700.
2.11. Cell viability assay
The cell counting kit-8 (CCK-8; Dojindo, Shanghai, China) assay was applied to detect the viability of cells. BMDMs were seeded in 96-well growth-medium plate overnight at 1 × 105 cells/well. Then, LPS-primed BMDMs were incubated at 37 °C followed by treatment with H2O2, then these cells were cultured with CCK-8 for 30 min. The optical density (O.D.) values at the wavelength of 450 nm were determined.
2.12. Mitochondrial reactive oxygen species assay
BMDMs were put onto 100 mm diameter culture dish tubes and primed with LPS (50 ng/mL) for 4 h. Then, the LPS-primed BMDMs were transferred into 1.5 mL tubes and treated with ICS Ⅱ for 1 h. Next, the cells were stimulated with ATP, nigericin or SiO2, after which the cells were washed with PBS. For mitochondrial ROS (mtROS) measurement, BMDMs were loaded with 4 μmol/L MitoSOX red mitochondrial superoxide indicator (Invitrogen, Ex/Em: 510/580 nm) for 20 min and washed with PBS. After staining and washing, cells were resuspended in PBS and flow cytometry was conducted to test mtROS production.
2.13. Alanine aminotransferase (ALT) and aspartate transaminase (AST)
Serum ALT and AST levels were measured according to the GPT (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and GOT (Nanjing Jiancheng Bioengineering Institute) assay kit directions.
2.14. Enzyme-linked immunosorbent assay (ELISA)
ELISA measurements of mouse IL-1β (Dakewe, Beijing, China), human IL-1β (Dakewe), mouse tumor necrosis-α (TNF-α, Dakewe), and human TNF-α (Dakewe) in cell culture supernatant and mouse serum IL-1β (R&D systems) were executed on the basis of the manufacturer's directions.
2.15. Levels of oxidative stress parameter
Mouse liver tissue oxidative stress parameter malondialdehyde (MDA) assay (Beyotime, Shanghai, China) was detected according to the manufacturer's directions. Liver samples were homogenized and treated in accordance with the manufacturer's recommendations.
2.16. Assessment of the effects of LPS/ICS Ⅱ cotreatment-induced DILI in vivo
Six to eight-week-old female C57BL/6 mice fasted for 24 h were given 2 mg/kg LPS (from Escherichia coli O55:B5, Sigma) or its saline vehicle via a tail vein (i.v.). Two hours later, ICS Ⅱ (25 and 50 mg/kg, TargetMol) or its vehicle (10% tween 80 + 90% PBS) was administered through intraperitoneal injection. Mice serum was collected 6 h after ICS Ⅱ treatment. The serum IL-1β, TNF-α, ALT and AST levels were tested.
In the second experiment, 6–8-week-old female C57BL/6 WT and Nlrp3−/− mice fasted for 24 h were given 2 mg/kg LPS (Sigma) or its saline vehicle via a tail vein (i.v.). Two hours later, ICS Ⅱ (50 mg/kg) or its vehicle (10% tween 80 + 90% PBS) was administered through intraperitoneal injection. Mice serum and a fraction of liver samples fixed in 10% formalin neutral buffer solution were collected 6 h after ICS Ⅱ treatment. Hematoxylin and eosin (H&E), TUNEL staining and the serum IL-1β, TNF-α, ALT and AST levels were tested. Moreover, liver homogenate was used to detect the activity of caspase-1 after normalization processing of bicinchoninic acid (BCA) protein quantification kit (Solarbio, Beijing, China).
In the third experiment, 6–8-week-old female C57BL/6 mice were fasted for 24 h. Then, MCC950 (50 mg/kg, TargetMol) or its vehicle (10% tween 80 + 90% PBS) was administered through intraperitoneal injection. One hour later, the mice were given 2 mg/kg LPS (Sigma) or its saline vehicle via a tail vein (i.v.). Two hour later, ICS Ⅱ (50 mg/kg, TargetMol) or its vehicle (10% tween 80 + 90% PBS) was administered through intraperitoneal injection. Mice serum and a fraction of liver samples were collected after 6 h. The serum IL-1β, TNF-α, ALT and AST levels, and level of oxidative stress parameter MDA was tested. Moreover, liver homogenate was used to detect the expression of caspase-1, IL-1β and cleaved GSDMD by Western blot after normalization processing using a BCA protein quantification kit (Solarbio).
2.17. Statistical analyses
The software Prism six and SPSS statistics 21.0 were used for statistics and analysis. The testing data are presented as mean ± standard error of mean (SEM). A two-tailed unpaired Student's t-test for two groups or one-way ANOVA for multi groups was conducted to evaluate the significant differences. A P value less than 0.05 was considered to be significant.
3. Results
3.1. Numerous ingredients derived from EF enhance ATP-induced caspase-1 activation and IL-1β production
Nine compounds (epmedin A, epmedin A1, epmedin B, epmedin C, icariin, icaritin, icariside Ⅰ, ICS Ⅱ and anhydroicaritin) derived from EF were selected to test whether they initiated an immune response by activating NLRP3 inflammasomes. First, LPS-primed BMDMs were tested with these compounds (10 μmol/L) for 24 h, but none of them induced caspase-1 maturation and IL-1β secretion (Supporting Information Fig. S1A and S1B), which suggested that they did not serve as direct agonists to induce inflammasome activation. Next, LPS-primed BMDMs were pretreated with these components (10 μmol/L) before ATP stimulation, which is a NLRP3 inflammasome stimulator. The results show that epmedin B, icariside Ⅰ and ICS Ⅱ significantly promoted caspase-1 maturation and IL-1β production that was triggered by ATP in LPS-primed BMDMs (Fig. 1A–C). Besides, TNF-α, an inflammasome-independent cytokine, was not influenced by these compounds under this condition (Fig. 1D). Among these EF constituents, ICS Ⅱ (Supporting Information Fig. S2A), which exhibits the most potent effect on NLRP3 inflammasome activation, may be the main contributor to EF-induced liver injury.
Figure 1.

Effect of the main compositions from Epimedii Folium on NLRP3 inflammasome activation. (A) Caspase-1 activity in supernatants (SN) from LPS-primed BMDMs treated with the main components from EF (10 μmol/L) and then stimulated with ATP (1 h). (B) Western blot analysis of IL-1β (p17), caspase-1 (p20) in culture supernatants (SN) and pro-IL-1β, caspase-1 (p45), NLRP3, ASC in whole cell lysates (WCL) of LPS-primed BMDMs treated with the main components from EF (10 μmol/L) and then stimulated with ATP (1 h). ELISA of IL-1β (C) and TNF-α (D) in SN described in (B). RLU, relative light units. Data are means ± SEM (n = 3); ###P < 0.001 vs. the control group; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. the LPS plus ATP group.
3.2. ICS Ⅱ specifically accelerates NLRP3 inflammasome activation triggered by ATP and nigericin
Next, we evaluated the impact of ICS Ⅱ on NLRP3 inflammasome activation. In the following experiment, we first tested the synergistic effects of ICS Ⅱ, ATP, and LPS on NLRP3 inflammasome activation. The expression of NLRP3 and pro-IL-1β induced by LPS treatment, but not ATP or ICS Ⅱ alone (Fig. S2B and S2C). We subsequently treated LPS-primed BMDMs with ICS Ⅱ alone. The result show that NLRP3 inflammasome was not activated in the absence of ATP (Fig. S2B and S2C). Caspase-1 maturation and IL-1β secretion were induced when BMDMs were cotreat with LPS and ATP. Interestingly, ICS Ⅱ significantly promoted ATP-induced caspase-1 activation and IL-1β production in LPS-primed BMDMs (Fig. S2B, S2C, Supporting Information Fig. S5A and S5B), which was blocked by Nlrp3 deficiency (Fig. S2D–S2H). Then, we assessed the dose–effect relationship of ICS Ⅱ on NLRP3 inflammasome activation triggered by ATP. The results indicated that ICS Ⅱ dose-dependently promoted caspase-1 cleavage and IL-1β secretion that was induced by LPS and ATP in BMDMs, but it had no influence on TNF-α production (Fig. 2A–C and Supporting Information Fig. S3A).
Figure 2.

ICS Ⅱ promotes NLRP3 inflammasome activation stimulated by ATP and nigericin. (A)–(C) Western blot analysis of IL-1β (p17), caspase-1 (p20) in SN and pro-IL-1β, caspase-1 (p45), NLRP3, ASC in WCL (A), caspase-1 activity (B) in SN and ELISA of IL-1β (C) in SN from LPS-primed BMDMs treated with various doses of ICS Ⅱ and then stimulated with ATP (1 h). (D)–(F) Western blot analysis of IL-1β (p17), caspase-1 (p20) in SN and pro-IL-1β, caspase-1 (p45), NLRP3, ASC in WCL (D), caspase-1 activity (E) in SN and ELISA of IL-1β (F) in SN from LPS-primed BMDMs treated with various doses of ICS Ⅱ before nigericin (1 h) stimulation. (G)–(I) Western blot analysis of IL-1β (p17), caspase-1 (p20) in SN and pro-IL-1β, caspase-1 (p45), NLRP3, ASC in WCL (G), caspase-1 activity (H) in SN and ELISA of IL-1β (I) in SN from PMA-primed THP-1 cells treated with various doses of ICS Ⅱ before nigericin (1 h) stimulation. Data are means ± SEM (n = 3); ###P < 0.001 vs. the control group; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. the LPS/PMA plus ATP/nigericin group.
Next, we assessed the impact of ICS Ⅱ on NLRP3 inflammasome activation in BMDMs and THP-1 cells evoked by nigericin. The results show that ICS Ⅱ also dose-dependently enhanced caspase-1 maturation, IL-1β production, GSDMD cleavage and LDH release but had no impact on TNF-α production in response to nigericin in LPS-primed BMDMs (Figs. 2D–F, S3B, S3C, S5A, and S5B) or PMA-primed THP-1 cells (Figs. 2G–I, S3D, and S3E). In addition, NLRP3 inflammasome can be activated by MSU, SiO2, and cytosolic LPS. To determine whether ICS Ⅱ was a common accelerant for NLRP3 inflammasome, we detected the effect of ICS Ⅱ on NLRP3 inflammasome activation induced by those stimuli. Unexpectedly, pretreatment with ICS Ⅱ had no influence on caspase-1 maturation, IL-1β secretion and TNF-α production triggered by MSU (Fig. 3A–C, and Supporting Information Fig. S4A), SiO2 (Fig, S4B–D), or cytosolic LPS (Figs. 3D–F, and S4E). Moreover, ICS Ⅱ had no impact on AIM2 and NLRC4 inflammasomes activation induced by poly(dA:dT) and Lfn-Flic, respectively (Figs. 4G–I, S4F, and S4G). These results indicate that ICS Ⅱ acts as a specific promoter to increase ATP- and nigericin-induced NLRP3 inflammasome activation.
Figure 3.

ICS Ⅱ has no effect on the activation of NLRP3 inflammasome induced by MSU and cytosolic LPS (cLPS), as well as AIM2 and NLRC4 inflammasomes. (A)–(C) Western blot analysis of IL-1β (p17), caspase-1 (p20) in SN and pro-IL-1β, caspase-1 (p45), NLRP3, ASC in WCL (A), caspase-1 activity (B) in SN and ELISA of IL-1β (C) in SN from LPS-primed BMDMs treated with various doses of ICS Ⅱ and then stimulated with MSU (6 h) or LPS-primed BMDMs stimulated with ATP (1 h) in the presence or absence of ICS Ⅱ (5 μmol/L). (D)–(F) Western blot analysis of IL-1β (p17), caspase-1 (p20) in SN and pro-IL-1β, caspase-1 (p45), NLRP3, ASC in WCL (D), caspase-1 activity (E) in SN and ELISA of IL-1β (F) in SN from Pam3CSK4-primed BMDMs treated with various doses of ICS Ⅱ and then transfected with LPS (6 h) or LPS-primed BMDMs stimulated with ATP (1 h) in the presence or absence of ICS Ⅱ (5 μmol/L). (G)–(I) Western blot analysis of IL-1β (p17), caspase-1 (p20) in SN and pro-IL-1β, caspase-1 (p45), NLRP3, ASC in WCL (G), caspase-1 activity (H) in SN and ELISA of IL-1β (I) in SN from LPS-primed BMDMs treated with ICS Ⅱ (5 μmol/L) before ATP (1 h), poly(dA:dT) (6 h) or Lfn-Flic (6 h) stimulation. Data are means ± SEM (n = 3); ###P < 0.001 vs. the control group; ∗∗P < 0.01, ∗∗∗P < 0.001.
Figure 4.

ICS Ⅱ facilitates NLRP3-dependent ASC oligomerization, but has no impact on NLRP3–NLRP3 interaction and intracellular potassium. (A) Western blot analysis of ASC oligomerization in lysates from BMDMs stimulated with LPS and ATP (1 h) and treated with various doses of ICS Ⅱ. (B) Western blot analysis of ASC oligomerization in lysates from LPS-primed BMDMs treated with ICS Ⅱ (5 μmol/L) before ATP (1 h), MSU (6 h), poly(dA:dT) (6 h), Lfn-Flic (6 h) stimulation or Pam3CSK4-primed BMDMs treated with ICS Ⅱ (5 μmol/L) and then transfected with LPS (6 h). (C) Western blots of cell lysates and flag-immunoprecipitation samples from HEK-293T cells transfected with Flag-NLRP3 and Myc-NLRP3. (D) Qualification of potassium efflux in LPS-primed BMDMs treated with various doses of ICS Ⅱ and then stimulated with ATP (1 h). Data are means ± SEM (n = 3).
3.3. ICS Ⅱ promotes ATP- or nigericin-induced ASC oligomerization, but has no effect on the NLRP3–NLRP3 interaction and potassium efflux
Next, we further investigated how ICS Ⅱ affected NLRP3 inflammasome activation induced by ATP and nigericin. ASC oligomerization is the pivotal step for NLRP3, NLRC4 and AIM2 inflammasome activation. LPS- or Pam3CSK4-primed BMDMs were handled with ICS Ⅱ before various activators stimulation. Then, ASC was crosslinked by disuccinimidyl suberate (DSS), and ASC oligomerization was detected through Western blot. The results show that ICS Ⅱ dose-dependently promoted ASC oligomerization that was induced by ATP (Fig. 4A) and nigericin (Supporting Information Fig. S6). However, ICS Ⅱ had no impact on ASC oligomerization induced by MSU, poly(dA:dT), Lfn-Flic or cytosolic LPS (Fig. 4B), which indicated that ICS Ⅱ acted upstream event of ASC oligomerization to exacerbate ATP- or nigericin-induced NLRP3 inflammasome activation.
We then investigated whether ICS Ⅱ affected the direct NLRP3–-NLRP3 interaction. HEK-293T cells were handled with ICS Ⅱ before being transfected with Flag-NLRP3 and Myc-NLRP3, and a co-immunoprecipitation assay was carried out to assess NLRP3–NLRP3 interaction. The results show that ICS Ⅱ did not alter the NLRP3–NLRP3 interaction in HEK-293T cells (Fig. 4C). Potassium efflux is an important upstream event for the activation of NLRP3 inflammasome. We therefore examined whether ICS Ⅱ affect potassium efflux triggered by these stimuli, and the results show that ICS Ⅱ promoted potassium efflux induced by nigericin (Supporting Information Fig. S7A), but not ATP (Fig. 4D) or SiO2 (Fig. S7B). Therefore, potassium efflux may be not responsible for the enhancement effect of ICS Ⅱ on ATP- and nigericin-induced NLRP3 inflammasome activation.
3.4. ICS Ⅱ facilitates ATP- or nigericin-induced NLRP3 inflammasome activation by increasing mitochondrial ROS (mtROS) production
Mitochondrial damage and mtROS are thought to be important upstream events for NLRP3 inflammasome activation20. Next, the impact of ICS Ⅱ on mitochondrial damage was evaluated in the following experiment. The results show that mitochondrial damage was not observed in BMDMs that were treated with ICS Ⅱ (5 μmol/L) (Supporting Information Fig. S8A). The MitoSOX Red Mitochondrial Superoxide Indicator assay was used to record the amount of mtROS production during the course of ATP, nigericin or SiO2 treatment in the presence or absence of ICS Ⅱ. The results show that ICS Ⅱ treatment alone did not induce the mtROS production (Fig. 5A). However, ICS Ⅱ obviously potentiated mtROS production that was triggered by ATP and nigericin rather than SiO2 in LPS-primed BMDMs (Fig. 5A and B). This suggested that synergistic induction of ROS production is a crucial contributor to the enhancement effect of ICS Ⅱ on NLRP3 inflammasomes triggered by ATP and nigericin. To evaluate the role of ROS production on the activation of NLRP3 inflammasomes triggered by ATP or nigericin, LPS-primed BMDMs were treated with oxidizing agent H2O2 or NAC (a scavenger of ROS) before ATP, nigericin or SiO2 stimulation. Western blot analysis show that H2O2 enhanced caspase-1 maturation and IL-1β production in a certain dose range triggered by ATP or nigericin, but not SiO2 (Supporting Information Fig. S9A–S9E). Similarly, NAC treatment suppressed mtROS production significantly (Supporting Information Fig. S10C) and inhibited nigericin-induced caspase-1 activation and IL-1β secretion in a dose-dependent manner (Fig. S10A and S10B). Most importantly, NAC pretreatment eliminated the increase of caspase-1 maturation and IL-1β production when LPS-primed BMDMs were treated with ICS Ⅱ and then stimulated with ATP or nigericin (Fig. 5C and D). These results confirmed that ICS Ⅱ facilitates ATP- or nigericin-induced NLRP3 inflammasome activation by increasing mtROS production.
Figure 5.

ICS Ⅱ facilitates ATP/nigericin-induced NLRP3 inflammasome activation dependent on mitochondrial ROS production. LPS-primed BMDMs were untreated or treated with ICS Ⅱ (2.5 and 5 μmol/L) before no stimulation (A) or stimulation with ATP (B), nigericin or SiO2. BMDMs were loaded with MitoSOX red mitochondrial superoxide indicator (Ex/Em: 510/580 nm). After staining and washing, flow cytometry was conducted to test mtROS production. Caspase-1 activity (C) and Western blot analysis of IL-1β (p17), caspase-1 (p20) in SN and pro-IL-1β, caspase-1 (p45), NLRP3, ASC in WCL (D) from LPS-primed BMDMs treated with ICS Ⅱ (5 μmol/L), NAC (2.5 mmol/L) or ICS Ⅱ (5 μmol/L) plus NAC (2.5 mmol/L), and then stimulated with ATP, nigericin or SiO2. Data are means ± SEM (n = 3); ###P < 0.001 vs. the control group; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
3.5. Combination of LPS and ICS Ⅱ induces liver injury in WT mice but not in Nlrp3−/−mice
Coexisting inflammatory mediators such as LPS should be considered to be determinants of susceptibility to IDILI21. Non-hepatotoxic doses of LPS decrease the threshold for toxicity and/or increase the magnitude of response22. Numerous studies have demonstrated that IDILI can be mimicked in animals through co-exposure to non-hepatotoxic doses of LPS and drugs with the ability to induce IDILI23, 24, 25, 26. A previous study demonstrated that EF alone does not cause liver injury in vivo, but EF combined with non-hepatotoxic doses of LPS could induce hepatotoxicity18. We therefore evaluated whether ICS Ⅱ, an active constituent that promotes NLRP3 inflammasome activation, could result in hepatoxicity in an LPS-mediated susceptibility mouse model of IDILI. The results show that mice treated with ICS Ⅱ alone did not have altered plasma ALT and AST levels compared with those of control mice. However, co-treatment with LPS and ICS Ⅱ resulted in significant increases in the plasma levels of ALT and AST, and this was accompanied by increases in the level of inflammatory cytokines IL-1β and TNF-α compared to those of LPS group (Supporting Information Fig. S11A–S11D). These results suggested that ICS Ⅱ can induce liver injury in an LPS-mediated susceptibility mouse model of IDILI.
To further confirm the role of the NLRP3 inflammasomes on ICS Ⅱ-induced liver injury, WT and Nlrp3−/− mice were used to explore the relationship between NLRP3 inflammasome and the liver injury caused by ICS Ⅱ. The results show that co-treatment with LPS and ICS Ⅱ led to elevation of the serum ALT, AST, IL-1β and TNF-α production in WT mice but not in Nlrp3−/− mice (Fig. 6A–D). Histological analysis of the mouse liver tissues was performed using H&E staining assay. As illustrated in Fig. 6G, co-exposure to LPS and ICS Ⅱ resulted in pathological changes including inflammatory infiltration and hepatocyte focal necrosis, which was not occurred in control group, ICS Ⅱ group, LPS group or any of the Nlrp3−/− groups. Moreover, caspase-1 activity was increased in the livers of WT mice but not in the livers of Nlrp3−/− mice after LPS/ICS Ⅱ treatment (E). Similarly, TUNEL fluorescence staining showed that the TUNEL positive signal was significantly increased in WT mice that were co-exposed to LPS and ICS Ⅱ compared with the other groups (Fig. 6F and G). Taken together, these results suggest that ICS Ⅱ could induce liver injury by promoting NLRP3 inflammasome activation.
Figure 6.

Early liver injury and inflammatory mediator production after LPS/ICS Ⅱ cotreatment. (A)–(D) WT and Nlrp3−/− female C57BL/6 mice were pretreated with LPS (2 mg/kg) through the tail vein. Two hours later, intraperitoneal ICS Ⅱ (50 mg/kg, n = 6) injection was conducted. Six hours after ICS Ⅱ injection, serum levels of ALT (A), AST (B), IL-1β (C), TNF-α (D) were measured by assay kit. (E) Caspase-1 activity in the livers was detected after BCA protein quantification and normalization processing. H&E staining (G) and TUNEL staining (F) were conducted to observe liver injury and TUNEL positive. Data are means ± EM; ##P < 0.01, ###P < 0.001 vs. the control; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001. Scale bar: 100 μm.
3.6. MCC950 pretreatment reverses LPS/ICS Ⅱ-induced liver injury
To further prove the involvement of NLRP3 inflammasome in ICS Ⅱ/LPS-induced liver injury, a specific NLRP3 inflammasome inhibitor (MCC950) was administered before ICS Ⅱ and LPS treatment to suppress the activation of NLRP3 inflammasomes in vivo. In vivo data demonstrate that MCC950 pretreatment before LPS/ICS Ⅱ suppressed serum ALT, AST, IL-1β and TNF-α production (Fig. 7A–D). Because mtROS plays an important role in ICS Ⅱ/LPS-induced IDILI, we next evaluated the level of oxidative stress in the liver. MDA, the end-product of lipid peroxidation, is used widely as a sensitive marker of oxidative stress and contributes to the hepatocyte injury27,28. Similar to ALT and AST activity, the levels of MDA in the ICS Ⅱ/LPS group were higher than that of the other groups (Fig. 7E). In addition, LPS/ICS Ⅱ enhanced the production of IL-1β p17 and caspase-1 p20, and cleaved GSDMD (a downstream effector of pyroptosis) in the liver tissue (Fig. 7F). On the contrary, the above indicators in the liver decreased substantially when MCC950 was administered before LPS/ICS Ⅱ treatment (Fig. 7F). These results suggest that NLRP3 inflammasomes play a critical role in ICS Ⅱ/LPS-induced liver injury.
Figure 7.

MCC950 pretreatment reverses LPS/ICS Ⅱ-induced liver injury. (A)–(D) Female C57BL/6 mice were pretreated with MCC950 (50 mg/kg), then treated with LPS (2 mg/kg) through the tail vein. Two hours later, intraperitoneal ICS Ⅱ (50 mg/kg, n = 6) injection was conducted. Six hours after ICS Ⅱ injection, serum levels of ALT (A), AST (B), IL-1β (C), TNF-α (D) were measured by assay kit. (E) Mouse liver tissue oxidative stress parameter malondialdehyde (MDA) was detected from female C57BL/6 mice pretreated with MCC950 (50 mg/kg) before LPS (2 mg/kg) and ICS Ⅱ (50 mg/kg, n = 6) injection. (F) Western blot analysis of IL-1β (p17), caspase-1 (p20) and cleaved GSDMD in the liver tissue. Data are means ± SEM; ##P < 0.01, ###P < 0.001 vs. the control; ∗P < 0.05, ∗∗∗P < 0.001.
4. Discussion
IDILI caused by the wide use of conventional drugs is recognized broadly; however, there is deficient understanding of the possible liver injury of herbal preparations all over the world29, 30, 31. In recent years, the problem of IDILI caused by TCMs, especially the traditional non-toxic Chinese medicines, has become increasingly serious because of increasing awareness of risk recognition and prevention. Several studies have reported that hepatotoxicity can be induced by two Chinese materia medica preparations, namely Zhuang-gu-guan-jie pills and Xian-ling-gu-bao capsules, both of which contain EF32, 33, 34. Our previous studies demonstrated that EF may cause hepatotoxicity in an LPS-mediated susceptibility mouse model of IDILI. ICS Ⅱ is the main component of EF, and it is also the major pharmacological metabolite of EF35,36. Previous studies have shown that some drugs with the ability to induce DILI, such as amodiaquine and nevirapine, may induce the release of DAMPs from hepatocytes, which leads to the activation of NLRP3 inflammasomes in macrophages37. Although ICS Ⅱ inhibits LPS-induced NF-κB signaling pathway in vitro38,39, our research demonstrated that ICS Ⅱ had no impact on pro-IL-1β expression and TNF-α production in our experiment conditions (stimulated with LPS for 4 h before ICS Ⅱ treatment). We further found that none of the components of EF could directly activate NLRP3 inflammasome, but when LPS-primed BMDMs were pretreated with ICS Ⅱ before stimulation with various NLRP3 inflammasome agonists, ICS Ⅱ specifically enhanced ATP- or nigericin-induced NLRP3 inflammasome activation and pyroptosis.
NLRP3 inflammasome can sense PAMPs and DAMPs, such as extracellular ATP, nigericin, SiO2, and MSU. Our research indicates that ICS Ⅱ can specifically reinforce NLRP3 inflammasome activation and pyroptosis induced by ATP or nigericin, but not MSU, SiO2, or cytosolic LPS. Furthermore, ICS Ⅱ had no impact on AIM2 and NLRC4 inflammasomes activation. Evidence from well-designed studies has indicated that drugs cause IDILI synergistically with other risk factors that include drug properties, host factors, and drug–host interaction, which contribute to influence the susceptibility to hepatotoxicity40,41. For example, methotrexate-related fatty liver disease and its severity levels have been connected with alcohol abuse, type two diabetes and obesity42, 43, 44. The inheritable variant of the HLA-B gene, HLA-B∗1502, increases sensibility to Stevens–Johnson syndrome and toxic epidermal necrolysis induced by carbamazepine45. ICS Ⅱ specifically amplified ATP- or nigericin-induced NLRP3 inflammasome activation, which suggested that ICS Ⅱ should be avoided in treating diseases related to ATP- or nigericin-induced NLRP3 inflammasome activation. However, underlying the other agonist-induced NLRP3 inflammasome activation-related diseases, such as MSU-induced gout, SiO2-induced silicosis, may not be the risk signal of ICS Ⅱ-induced idiosyncratic liver injury.
To evaluate the mechanism by which ICS Ⅱ enhanced NLRP3 inflammasome activation triggered by ATP or nigericin, we assessed the effects of ICS Ⅱ on the upstream and downstream signaling that is associated with NLRP3 inflammasome activation. ICS Ⅱ could promote ASC oligomerization triggered by ATP rather than SiO2. However, ASC oligomerization is essential for all stimuli induction of NLRP3 inflammasome, which suggests that ICS Ⅱ acts on upstream signaling of ASC oligomerization to exacerbate ATP- or nigericin-induced NLRP3 inflammasome activation. MtROS is thought to be important upstream events for NLRP3 inflammasome activation. Interestingly, ATP, nigericin or SiO2 could induce the production of mtROS, but ICS Ⅱ treatment intensified mtROS generation triggered by ATP or nigericin but not SiO2, suggesting that ICS Ⅱ may enhance ATP- or nigericin-induced NLRP3 inflammasome activation by promoting mtROS production. Further, oxidizing agent H2O2 enhanced caspase-1 maturation and IL-1β production triggered by ATP or nigericin, but not SiO2. Similarly, ROS scavenger NAC and ICS Ⅱ co-treatment suppressed caspase-1 maturation and IL-1β production when stimulated with ATP or nigericin. Therefore, induction of mtROS production contributes to the enhancement effect of ICS Ⅱ on NLRP3 inflammasome activation triggered by ATP and nigericin that results in a continuous inflammatory response and ultimately induces liver injury.
NLRP3 inflammasome activation leads to caspase-1-dependent release of IL-1β and pyroptosis. IL-1β serves mainly as a proinflammatory mediator to activate and recruit neutrophils into the tissues46 and subsequently trigger neutrophil tissue infiltration and liver damage47,48. In addition, IL-1β can induce the production of TNF-α49,50, another key cytokine involved in amplifying and perpetuating the liver damage by triggering liver inflammation, neutrophils, and proinflammatory macrophage recruitment51, 52, 53, 54. Moreover, NLRP3 inflammasome activation also leads to caspase-1-dependent pyroptosis8, this distinct form of programmed cell death could mediate liver injury47,55,56. In addition, one of important upstream signaling of NLRP3 inflammasome activation is ROS production57,58, which can lead to mitochondrial dysfunction through an intracellular oxidant stress in hepatocytes leading mainly to oncotic necrosis and less apoptosis52,59. Our in vivo data demonstrated that LPS/ICS Ⅱ enhanced the production of IL-1β p17, TNF-α, cleaved GSDMD (the downstream effector of pyroptosis), and the level of oxidative stress in the liver tissue, accompanied by the rise of ALT and AST, which is reversed by MCC950 pretreatment or Nlrp3 deficiency. These results demonstrated that NLRP3 inflammasome plays a critical role in ICS Ⅱ/LPS-induced liver injury.
5. Conclusions
The study demonstrated that ICS Ⅱ exacerbates NLRP3 inflammasome activation and pyroptosis triggered by ATP and nigericin, but not SiO2, MSU or cytosolic LPS. Synergistic induction of mtROS is a crucial contributor to the enhancing effect of ICS Ⅱ on ATP- or nigericin-induced NLRP3 inflammasome activation. Our in vivo data show that a combination of non-hepatotoxic doses of LPS and ICS Ⅱ causes the increase of ALT and AST, hepatocyte necrosis and hepatic inflammation in WT mice, which is blocked by MCC950 or Nlrp3 deficiency. The data suggest that ICS Ⅱ causes idiosyncratic liver injury through enhancing NLRP3 inflammasome activation and may be a risk factor and responsible for EF-induced liver injury.
Acknowledgments
This work has been supported by National Natural Science Foundation of China (81874368, 81630100, and 81903891), Beijing Nova Program (Z181100006218001, China), National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” (2017ZX09301022 and 2018ZX09101002-001-002, China), the Innovation Groups of the National Natural Science Foundation of China (81721002). Thanks to pathologists Yanhong Tai and Lina Jiang from the Fifth Medical Centre, Chinese PLA General Hospital (Beijing, China) for the analysis of H&E staining.
Author contributions
Zhilei Wang, Guang Xu and Hongbo Wang: investigation, validation, visualization, software, and original draft. Xiaoyan Zhan and Yuan Gao: investigation, formal analysis, and data curation. Nian Chen: visualization and software. Ruisheng Li: methodology and formal analysis. Xueai Song and Yuming Guo: investigation and software. Ruichuang Yang, Ming Niu and Jiabo Wang: visualization and formal analysis. Youping Liu, Xiaohe Xiao and Zhaofang Bai: conceptualization, methodology, validation, resources, data curation, supervision, project administration, review & editing, and funding acquisition.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.03.006.
Contributor Information
Youping Liu, Email: liuyouping@163.com.
Xiaohe Xiao, Email: pharmacy_302@126.com.
Zhaofang Bai, Email: baizf2008@hotmail.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Fontana R.J. Pathogenesis of idiosyncratic drug-induced liver injury and clinical perspectives. Gastroenterology. 2014;146:914–928. doi: 10.1053/j.gastro.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kullak-Ublick G.A., Andrade R.J., Merz M., End P., Benesic A., Gerbes A.L. Drug-induced liver injury: recent advances in diagnosis and risk assessment. Gut. 2017;66:1154–1164. doi: 10.1136/gutjnl-2016-313369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen T., Liu Y., Shang J., Xie Q., Li J., Yan M. Incidence and etiology of drug-induced liver injury in mainland China. Gastroenterology. 2019;156:2230–2241. doi: 10.1053/j.gastro.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Bjornsson E.S. Drug-induced liver injury: an overview over the most critical compounds. Arch Toxicol. 2015;89:327–334. doi: 10.1007/s00204-015-1456-2. [DOI] [PubMed] [Google Scholar]
- 5.Lin L., Ni B., Lin H., Zhang M., Li X., Yin X. Traditional usages, botany, phytochemistry, pharmacology and toxicology of Polygonum multiflorum Thunb.: a review. J Ethnopharmacol. 2015;159:158–183. doi: 10.1016/j.jep.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin G., Wang J.Y., Li N., Li M., Gao H., Ji Y. Hepatic sinusoidal obstruction syndrome associated with consumption of Gynura segetum. J Hepatol. 2011;54:666–673. doi: 10.1016/j.jhep.2010.07.031. [DOI] [PubMed] [Google Scholar]
- 7.Sun W., Zeng C., Liu S., Fu J., Hu L., Shi Z. Ageratina adenophora induces mice hepatotoxicity via ROS-NLRP3-mediated pyroptosis. Sci Rep. 2018;8:16032. doi: 10.1038/s41598-018-34492-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He Y., Hara H., Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elliott E.I., Sutterwala F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015;265:35–52. doi: 10.1111/imr.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou X., Wu Y., Ye L., Wang Y., Zhang K., Wang L. Aspirin alleviates endothelial gap junction dysfunction through inhibition of NLRP3 inflammasome activation in LPS-induced vascular injury. Acta Pharm Sin B. 2019;9:711–723. doi: 10.1016/j.apsb.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z., Xu G., Gao Y., Zhan X., Qin N., Fu S. Cardamonin from a medicinal herb protects against LPS-induced septic shock by suppressing NLRP3 inflammasome. Acta Pharm Sin B. 2019;9:734–744. doi: 10.1016/j.apsb.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu X., Dong L., Lin X., Li J. Relevance of the NLRP3 Inflammasome in the pathogenesis of chronic liver disease. Front Immunol. 2017;8:1728. doi: 10.3389/fimmu.2017.01728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szabo G., Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57:642–654. doi: 10.1016/j.jhep.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 14.Wree A., Eguchi A., McGeough M.D., Pena C.A., Johnson C.D., Canbay A. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59:898–910. doi: 10.1002/hep.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma H., He X., Yang Y., Li M., Hao D., Jia Z. The genus Epimedium: an ethnopharmacological and phytochemical review. J Ethnopharmacol. 2011;134:519–541. doi: 10.1016/j.jep.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L., Wang T., Zhao B.S., Zhang J.X., Yang S., Fan C.L. Effect of 2′′-O-rhamnosyl icariside II, baohuoside I and baohuoside II in herba Epimedii on cytotoxicity indices in HL-7702 and HepG2 cells. Molecules. 2019;24:1263. doi: 10.3390/molecules24071263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong R., Chen Y., Ling J., Xia Z., Zhan Y., Sun E. The toxicity and metabolism properties of Herba Epimedii flavonoids on laval and adult zebrafish. Evid Based Complement Alternat Med. 2019;2019:3745051. doi: 10.1155/2019/3745051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao Y., Wang Z., Tang J., Liu X., Shi W., Qin N. New incompatible pair of TCM: Epimedii Folium combined with Psoraleae Fructus induces idiosyncratic hepatotoxicity under immunological stress conditions. Front Med. 2020;14:68–80. doi: 10.1007/s11684-019-0690-z. [DOI] [PubMed] [Google Scholar]
- 19.He H., Jiang H., Chen Y., Ye J., Wang A., Wang C. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. 2018;9:2550. doi: 10.1038/s41467-018-04947-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dan Dunn J., Alvarez L.A., Zhang X., Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–485. doi: 10.1016/j.redox.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganey P.E., Luyendyk J.P., Maddox J.F., Roth R.A. Adverse hepatic drug reactions: inflammatory episodes as consequence and contributor. Chem Biol Interact. 2004;150:35–51. doi: 10.1016/j.cbi.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Roth R.A., Ganey P.E. Animal models of idiosyncratic drug-induced liver injury—current status. Crit Rev Toxicol. 2011;41:723–739. doi: 10.3109/10408444.2011.575765. [DOI] [PubMed] [Google Scholar]
- 23.Yee S.B., Kinser S., Hill D.A., Barton C.C., Hotchkiss J.A., Harkema J.R. Synergistic hepatotoxicity from coexposure to bacterial endotoxin and the pyrrolizidine alkaloid monocrotaline. Toxicol Appl Pharmacol. 2000;166:173–185. doi: 10.1006/taap.2000.8968. [DOI] [PubMed] [Google Scholar]
- 24.Shaw P.J., Ditewig A.C., Waring J.F., Liguori M.J., Blomme E.A., Ganey P.E. Coexposure of mice to trovafloxacin and lipopolysaccharide, a model of idiosyncratic hepatotoxicity, results in a unique gene expression profile and interferon gamma-dependent liver injury. Toxicol Sci. 2009;107:270–280. doi: 10.1093/toxsci/kfn205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hassan H.M., Guo H., Yousef B.A., Ping-Ping D., Zhang L., Jiang Z. Dexamethasone pretreatment alleviates isoniazid/lipopolysaccharide hepatotoxicity: inhibition of inflammatory and oxidative stress. Front Pharmacol. 2017;8:133. doi: 10.3389/fphar.2017.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luyendyk J.P., Maddox J.F., Green C.D., Ganey P.E., Roth R.A. Role of hepatic fibrin in idiosyncrasy-like liver injury from lipopolysaccharide-ranitidine coexposure in rats. Hepatology. 2004;40:1342–1351. doi: 10.1002/hep.20492. [DOI] [PubMed] [Google Scholar]
- 27.Bao W., Li K., Rong S., Yao P., Hao L., Ying C. Curcumin alleviates ethanol-induced hepatocytes oxidative damage involving heme oxygenase-1 induction. J Ethnopharmacol. 2010;128:549–553. doi: 10.1016/j.jep.2010.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Xu L., Yu Y., Sang R., Li J., Ge B., Zhang X. Protective effects of taraxasterol against ethanol-induced liver injury by regulating CYP2E1/Nrf2/HO-1 and NF-κB signaling pathways in mice. Oxid Med Cell Longev. 2018;2018:8284107. doi: 10.1155/2018/8284107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Navarro V.J., Khan I., Bjornsson E., Seeff L.B., Serrano J., Hoofnagle J.H. Liver injury from herbal and dietary supplements. Hepatology. 2017;65:363–373. doi: 10.1002/hep.28813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teschke R., Frenzel C., Glass X., Schulze J., Eickhoff A. Herbal hepatotoxicity: a critical review. Br J Clin Pharmacol. 2013;75:630–636. doi: 10.1111/j.1365-2125.2012.04395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho J.H., Oh D.S., Hong S.H., Ko H., Lee N.H., Park S.E. A nationwide study of the incidence rate of herb-induced liver injury in Korea. Arch Toxicol. 2017;91:4009–4015. doi: 10.1007/s00204-017-2007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng Y., Liu Y., Wang H., Li J., Ren J., Zhu L. A 26-week repeated dose toxicity study of Xian-ling-gu-bao in Sprague–Dawley rats. J Ethnopharmacol. 2013;145:85–93. doi: 10.1016/j.jep.2012.09.055. [DOI] [PubMed] [Google Scholar]
- 33.Wu H., Zhong Q., Wang J., Wang M., Fang F., Xia Z. Beneficial effects and toxicity studies of Xian-ling-gu-bao on bone metabolism in ovariectomized rats. Front Pharmacol. 2017;8:273. doi: 10.3389/fphar.2017.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu W., Wang T., Sun B., Liu D., Lin Z., Miao Y. Xian-Ling-Gu-Bao induced inflammatory stress rat liver injury: inflammatory and oxidative stress playing important roles. J Ethnopharmacol. 2019;239:111910. doi: 10.1016/j.jep.2019.111910. [DOI] [PubMed] [Google Scholar]
- 35.Xu W., Zhang Y., Yang M., Shen Z., Zhang X., Zhang W. LC–MS/MS method for the simultaneous determination of icariin and its major metabolites in rat plasma. J Pharmaceut Biomed Anal. 2007;45:667–672. doi: 10.1016/j.jpba.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Teo Y.L., Cheong W.F., Cazenave-Gassiot A., Ji S., Logan S., Lee Z.X.K. Pharmacokinetics of prenylflavonoids following oral ingestion of standardized epimedium extract in humans. Planta Med. 2019;85:347–355. doi: 10.1055/a-0806-7673. [DOI] [PubMed] [Google Scholar]
- 37.Kato R., Uetrecht J. Supernatant from hepatocyte cultures with drugs that cause idiosyncratic liver injury activates macrophage inflammasomes. Chem Res Toxicol. 2017;30:1327–1332. doi: 10.1021/acs.chemrestox.7b00065. [DOI] [PubMed] [Google Scholar]
- 38.Song J., Feng L., Zhong R., Xia Z., Zhang L., Cui L. Icariside II inhibits the EMT of NSCLC cells in inflammatory microenvironment via down-regulation of Akt/NF-κB signaling pathway. Mol Carcinog. 2017;56:36–48. doi: 10.1002/mc.22471. [DOI] [PubMed] [Google Scholar]
- 39.Yan N., Wen D.S., Zhao Y.R., Xu S.J. Epimedium sagittatum inhibits TLR4/MD-2 mediated NF-κB signaling pathway with anti-inflammatory activity. BMC Compl Alternative Med. 2018;18:303. doi: 10.1186/s12906-018-2363-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bjornsson E.S. Epidemiology and risk factors for idiosyncratic drug-induced liver injury. Semin Liver Dis. 2014;34:115–122. doi: 10.1055/s-0034-1375953. [DOI] [PubMed] [Google Scholar]
- 41.Chalasani N., Reddy K.R.K., Fontana R.J., Barnhart H., Gu J., Hayashi P.H. Idiosyncratic drug induced liver injury in African–Americans is associated with greater morbidity and mortality compared to caucasians. Am J Gastroenterol. 2017;112:1382–1388. doi: 10.1038/ajg.2017.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenberg P., Urwitz H., Johannesson A., Ros A.M., Lindholm J., Kinnman N. Psoriasis patients with diabetes type 2 are at high risk of developing liver fibrosis during methotrexate treatment. J Hepatol. 2007;46:1111–1118. doi: 10.1016/j.jhep.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 43.Laharie D., Seneschal J., Schaeverbeke T., Doutre M.S., Longy-Boursier M., Pellegrin J.L. Assessment of liver fibrosis with transient elastography and FibroTest in patients treated with methotrexate for chronic inflammatory diseases: a case-control study. J Hepatol. 2010;53:1035–1040. doi: 10.1016/j.jhep.2010.04.043. [DOI] [PubMed] [Google Scholar]
- 44.Aithal G.P. Dangerous liaisons: drug, host and the environment. J Hepatol. 2007;46:995–998. doi: 10.1016/j.jhep.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 45.Usui T., Naisbitt D.J. Human leukocyte antigen and idiosyncratic adverse drug reactions. Drug Metabol Pharmacokinet. 2017;32:21–30. doi: 10.1016/j.dmpk.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 46.Bajt M.L., Farhood A., Jaeschke H. Effects of CXC chemokines on neutrophil activation and sequestration in hepatic vasculature. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1188–G1195. doi: 10.1152/ajpgi.2001.281.5.G1188. [DOI] [PubMed] [Google Scholar]
- 47.Geng Y., Ma Q., Liu Y.N., Peng N., Yuan F.F., Li X.G. Heatstroke induces liver injury via IL-1β and HMGB1-induced pyroptosis. J Hepatol. 2015;63:622–633. doi: 10.1016/j.jhep.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 48.Huebener P., Pradere J.P., Hernandez C., Gwak G.Y., Caviglia J.M., Mu X. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2015;125:539–550. doi: 10.1172/JCI76887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Granowitz E.V., Vannier E., Poutsiaka D.D., Dinarello C.A. Effect of interleukin-1 (IL-1) blockade on cytokine synthesis: II. IL-1 receptor antagonist inhibits lipopolysaccharide-induced cytokine synthesis by human monocytes. Blood. 1992;79:2364–2369. [PubMed] [Google Scholar]
- 50.Dinarello C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 51.Bieghs V., Trautwein C. The innate immune response during liver inflammation and metabolic disease. Trends Immunol. 2013;34:446–452. doi: 10.1016/j.it.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 52.Schwabe R.F., Brenner D.A. Mechanisms of Liver Injury. I. TNF-α-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 53.Wree A., McGeough M.D., Inzaugarat M.E., Eguchi A., Schuster S., Johnson C.D. NLRP3 inflammasome driven liver injury and fibrosis: roles of IL-17 and TNF in mice. Hepatology. 2018;67:736–749. doi: 10.1002/hep.29523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z., Xu G., Zhan X., Liu Y., Gao Y., Chen N. Carbamazepine promotes specific stimuli-induced NLRP3 inflammasome activation and causes idiosyncratic liver injury in mice. Arch Toxicol. 2019;93:3585–3599. doi: 10.1007/s00204-019-02606-3. [DOI] [PubMed] [Google Scholar]
- 55.Zhang L.Y., Zhan D.L., Chen Y.Y., Wang W.H., He C.Y., Lin Y. Aflatoxin B1 enhances pyroptosis of hepatocytes and activation of Kupffer cells to promote liver inflammatory injury via dephosphorylation of cyclooxygenase-2: an in vitro, ex vivo and in vivo study. Arch Toxicol. 2019;93:3305–3320. doi: 10.1007/s00204-019-02572-w. [DOI] [PubMed] [Google Scholar]
- 56.Chen Y.L., Xu G., Liang X., Wei J., Luo J., Chen G.N. Inhibition of hepatic cells pyroptosis attenuates CLP-induced acute liver injury. Am J Transl Res. 2016;8:5685–5695. [PMC free article] [PubMed] [Google Scholar]
- 57.Minutoli L., Puzzolo D., Rinaldi M., Irrera N., Marini H., Arcoraci V. ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev. 2016;2016:2183026. doi: 10.1155/2016/2183026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 59.Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: present concepts. J Gastroenterol Hepatol. 2011;26(Suppl 1):173–179. doi: 10.1111/j.1440-1746.2010.06592.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.