

Abstract
A light-switchable transgene system called LightOn gene expression system could regulate gene expression with a high on/off ratio under blue light, and have great potential for spatiotemporally controllable gene expression. We developed a nanoparticle drug delivery system (NDDS) to achieve tumor microenvironment-responsive and targeted delivery of diphtheria toxin A (DTA) fragment-encoded plasmids to tumor sites. The expression of DTA was induced by exposure to blue light. Nanoparticles composed of polyethylenimine and vitamin E succinate linked by a disulfide bond, and PEGylated hyaluronic acid modified with RGD peptide, accumulated in tumor tissues and were actively internalized into 4T1 cells via dual targeting to CD44 and αvβ3 receptors. The LightOn gene expression system was able to control target protein expression through regulation of the intensity or duration of blue light exposure. In vitro studies showed that light-induced DTA expression reduced 4T1 cell viability and induced apoptosis. Furthermore, the LightOn gene expression system enabled spatiotemporal control of the expression of DTA in a mouse 4T1 tumor xenograft model, which resulted in excellent antitumor effects, reduced tumor angiogenesis, and no systemic toxicity. The combination of the LightOn gene expression system and NDDS may be an effective strategy for treatment of breast cancer.
KEY WORDS: Light-switchable gene expression system, Nanoparticle drug delivery system, Microenvironment-responsive, Diphtheria toxin, Breast cancer
Graphical abstract
The pGDTA@PVHRs NPs actively targeted 4T1 cells and dissociated in the tumor microenvironment to release gene. Subsequently, the LightOn gene expression system could spatiotemporally control the expression of DTA via blue light irradiation to inhibit tumor growth, which may represent an effective strategy for breast cancer treatment.
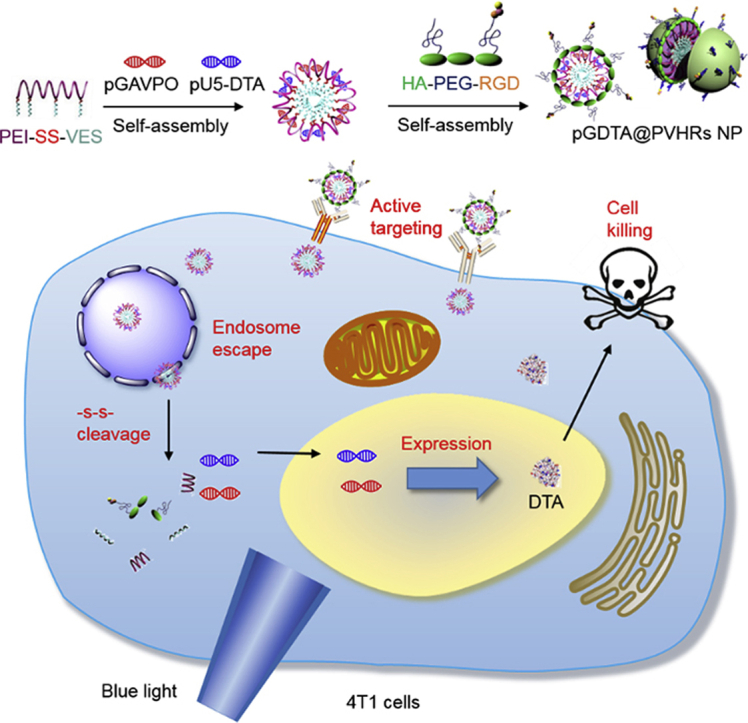
1. Introduction
Gene therapy, which modifies genetic information in tumor cells, is becoming an increasingly popular form of cancer treatment1, 2, 3. More than 1000 gene therapy clinical trials have been conducted in the last decade4. However, poor spatiotemporal resolution5 and off-target expression6,7 have limited clinical application of gene therapy for cancer treatment. To overcome these obstacles, different endogenous or exogenous promoters were designed and utilized to regulate gene expression in specific cell populations8, 9, 10. Light is an ideal inducer of gene expression because it is easy to obtain, highly tunable, non-toxic, and can provide high spatiotemporal resolution11. Recently, the field of optogenetics, in which use of light and genetically encoded light-sensitive proteins are used to control the behavior of living cells and organisms, has grown dramatically. Our previous study reported on a simple and robust light-switchable transgene system (LightOn gene expression system)12,13, which consisted of a single, synthetic light-sensitive transcription factor, GAVPO, and a reporter gene. GAVPO can homodimerize and bind to its specific promoter upon exposure to blue light, resulting in transcription of target genes. Use of the LightOn gene expression system with a single component can allow for precise control of gene expression with high spatiotemporal resolution and high induction efficiency. The LightOn gene expression system encoded with a suicide gene may be able to control the expression of toxin proteins such as diphtheria toxin (DT)14 and Pseudomonas exotoxin15 in tumor cells, which may be a promising anti-cancer strategy.
Diphtheria toxin, a 62,000 Dalton protein excreted by the bacterium Corynebacterium diphtheria, consists of fragment A and B linked by a disulfide bond16. Fragment A of DT (DTA) inhibits protein synthesis through NAD+-dependent ADP ribosylation of elongation factor 2, which may be an effective strategy to treat chemotherapy-resistant tumors17. First-generation18 and second-generation19 DT therapies utilized full-length DT protein and diphtheria toxin A, which resulted in severe toxicity and purification difficulties20. Third-generation DT, known as recombinant immunotoxin14, was composed of targeting antibody fragments and toxin domains to enhance tumor targeting and permeability. Ontak®, the first immunotoxin approved by the US Food and Drug Administration (FDA) in 1999, was comprised of DTA and target fragment IL-2 for treatment of cutaneous T-cell lymphoma21. Although Ontak® was effective, and improved patient quality of life, vascular leak toxicity limited its clinical use20. Use of a special promoter to drive DTA expression has shown great potential for treatment of tumors20,22. The LightOn gene expression system encoding DTA may be a new generation of DT application. Blue light could penetrate through the skin to the tumor site, which could trigger gene expression to inhibit tumor growth without damaging normal tissues. Furthermore, the expression level and site of application could be regulated entirely by irradiation with blue light, which may provide a simple and focused treatment strategy12. This strategy could incorporate a suicide gene to treat breast cancer without the development of multidrug resistance or killing of normal body cells23.
Effective delivery of genes in vivo is a major obstacle to gene therapy. Nonspecific distribution, rapid clearance, and low transfection efficiency limit the clinical application of gene therapy24,25. Recently, nano-formulations such as micelles, liposomes, hybrid nanoparticles (NP), and exosomes have attracted increasing attention due to their biocompatibility, tumor specificity, and high transfection efficiency26, 27, 28. Vectors less than 200 nm in diameter can be engineered with favorable properties to prolong blood circulation and allow for passive accumulation in tumor tissues through the enhanced permeability and retention effect (EPR)29. Targeting moieties modified on nano-vectors can actively target tumor specific receptors, resulting in reduced toxicity and immunogenic issues in normal tissues30. Cationic polymers such as polyethyleneimine (PEI) can condense gene and promote endosomal escape ability through the ‘proton-sponge’ effect31, resulting in efficient gene transfection. Moreover, tumor specific environments such as lower pH32, higher glutathione (GSH)33 levels, and higher reactive oxygen species (ROS)34 levels can be used to trigger gene release in the cytoplasm. Use of a light-switchable system incorporated into a nanoparticle drug delivery system (NDDS) may be an ideal strategy for treatment of tumors.
To exploit the potency of diphtheria toxin and minimize its side effects, a new strategy combining the LightOn gene expression system with an NDDS was designed to allow for spatiotemporal control of DTA expression. As shown in Fig. 1, a polymer comprised of PEI and vitamin E succinate (VES) linked by a disulfide bond self-assembled in water to form cationic PEI-SS-VES micelles, which were able to compress light-switchable plasmid DNA (pDNA) through electrostatic interactions. Arginine-glycine-aspartic acid (RGD) peptide, a targeting motif to αvβ3 integrin on the surface of tumor cells and neovascular endothelial cells35, was conjugated to PEGylated hyaluronic acid (HA) to form the anionic polymer HA-PEG-RGD. Then, HA-PEG-RGD was coated on the surface of pDNA-loaded micelles to form the final nanoparticulate formulation, pDNA@PVHRs nanoparticles. The PEG coating provided excellent stability and improved blood circulation. Hyaluronic acid and RGD promoted accumulation in tumors by actively targeting to the CD44 receptor36 and the αvβ3 integrin receptor37, respectively. In addition, this intelligent nanoparticles were able to escape from endosomes due to the ‘proton-sponge’ effect gifted by PEI, and were able to disintegrate rapidly through the stimuli of hyaluronidase and GSH overexpressed in tumor microenvironment. The LightOn gene expression system allowed for spatiotemporal transfection with DTA in response to exposure to blue light to kill 4T1 tumor cells in vitro and in vivo. The combination of the LightOn gene expression system and nanoparticle gene carrier exhibited effective 4T1 tumor inhibition with negligible toxicity, which resulted in a promising nano-platform for targeted gene delivery and breast cancer therapy.
Figure 1.

Scheme of DTA-based light dynamic therapy combining nanoparticle drug delivery system with LightOn gene expression system for breast cancer.
2. Materials and methods
2.1. Materials
All restriction enzymes, T4 ligase, and T4 polynucleotide kinase were purchased from Thermo Scientific (Waltham, USA). Branched polyethyleneimine (PEI, MW = 25 kDa), hyaluronic acid (HA, MW = 10 kDa), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 2-hydroxyethyl disulfide, dichloromethane, 4-dimethylaminopyridine (DMAP), coumarin-6 (C6), YOYO-1, LysoTracker Red and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DIR) were purchased from Sigma‒Aldrich Corporation (St. Louis, USA). NHS-PEG2000-MAL was purchased from Beijing JenKem Technology Co., Ltd. (Beijing, China). The c (RGDfK) peptide was purchased from GL Biochem Co., Ltd. (Shanghai, China). Vitamin E succinate (VES) was purchased from TCI Development Co., Ltd. (Shanghai, China). Dialysis bags (molecular weight cut-off: 8 kDa) were purchased from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), phosphate-buffered saline (PBS), trypsin, and penicillin–streptomycin (5000 U/mL) were purchased from Gibco Co., Ltd. (New York, USA). Cell-counting-kit 8 (CCK-8), Hoechst 33342, hyaluronidase (HAase) and annexin V-FITC/propidium iodide (PI) apoptosis detection kits were purchased from Shanghai Maokang Biotech Co., Ltd. (Shanghai, China). Anti-CD31 (GB13063), Cy3-conjugated donkey anti-goat IgG (GB21404), and DAPI (G1012) were purchased from Wuhan Servicebio Technology Co., Ltd. (Wuhan, China). Diphtheria toxin A ELISA kit was purchased from Shanghai Enzyme-linked Biotechnology Corporation (Shanghai, China). Alanine transaminase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and serum creatinine (CRE) ELISA kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). All other chemicals were purchased from Titan Scientific Co., Ltd. (Shanghai, China).
2.2. Cell culture and animals
The mouse breast cancer cell line 4T1 was purchased from the Institute of Biochemistry & Cell Biology, Chinese Academy of Sciences (China). Cells were cultured in DMEM supplemented with 10% FBS and streptomycin-penicillin (1%, v/v). Cells were incubated at 37 °C in a humidified 5% CO2 atmosphere. Cells were passaged using 0.25% trypsin/EDTA solution when nearly confluent.
Female Balb/c mice (6‒8 weeks of age, 20 ± 0.5 g) were purchased from Slaccas Experimental Animal Co., Ltd. (Shanghai, China). All animal experiments were performed in accordance with the guidelines, and with the approval, of the Ethics Committee of East China University of Science and Technology.
2.3. LightOn gene expression system construction
The pGAVPO regulator plasmid containing the light-inducible transcription factor GAVPO and the reporter plasmids pU5-DTA and pU5-mCherry, which use DTA and mCherry as reporter genes, were constructed as previously described12. The light-inducible trans-activators consisted of the Gal4 DNA binding domain, the VVD light sensor, and a transactivation domain (Supporting Information Fig. S1). The synthetic proteins were constitutively expressed using the CMV promoter. The expression of DTA (Supporting Information Fig. S2) and mCherry (Supporting Information Fig. S3) was controlled by a Gal4-responsive promoter assembled by placing a 5X UASG element adjacent to a TATA box.
2.4. Preparation of pDNA loaded HA-PEG-RGD/PEI-SS-VES nanoparticles
The synthesis of PEI-SS-VES and HA-PEG-RGD was stated in Supporting Information. An ethanolic solution containing PEI-SS-VES was added to a flask and tried under nitrogen atmosphere. The dried film was rehydrated with water and sonicated at 100 W for 5 min to obtain PEI-SS-VES (PVs) micelles. Polyethyleneimine-VES (PV) micelles were also prepared. For C6- or DIR-loaded micelles, 0.5% (wt%) C6 or DIR was added to the ethanolic PEIVES solution, as previously described. Plasmid DNA and PVs micelles were vortex-mixed at different N/P ratios, then held at room temperature for 15 min to prepare pDNA@PVs micelles. Hyaluronic acid, HA-PEG, or HA-PEG-RGD were vortex-mixed with pDNA@PVs micelles at weight ratio of 4:1 according to our previous study38, then held at room temperature for 15 min to prepare pDNA-loaded PEI-SS-VES/HA (PVHs) NP, PEI-SS-VES/HA-PEG (PVHPs) NP, or PEI-SS-VES/HA-PEG-RGD (PVHRs) NP. Plasmid DNA loading efficiency was measured using 2% agarose gel electrophoresis assay.
To assess the DNA-binding ability of the nanoparticles, pDNA@PVHRs NP at various N/P ratios were mixed with 4 mL of 6X loading buffer, and analyzed using 0.6% agarose gel electrophoresis assay. The particle sizes and zeta potentials of various nanoparticles were measured using dynamic light scattering (Nanosizer ZS 90, Malvern, UK). The morphology of pDNA@PVHRs NP was visualized using JEM-1400transmission electron microscopy (TEM, JEOL, Tokyo, Japan). To determine the stability of pDNA@PVHRs NP, the nanoparticles were dispersed in 10 mmol/L PBS (pH 7.4 or pH 5.5), incubated at 4 °C. At predetermined time points, size distribution and zeta potential were measured using dynamic light scattering. To assess the degradation of PVHRs NP, nanoparticles incubated with 1 mg/mL HAase and/or 10 mmol/L GSH for 30 min and size distribution and zeta potential were measured using dynamic light scattering. To assess the degradation of PVHRs NP, nanoparticles incubated with 1 mg/mL HAase and/or 10 mmol/L GSH for 30 min and then visualized using TEM.
2.5. In vitro pDNA release
PEI-SS-VES micelles and PVHRs NP containing 5 μg of YOYO-1-labeled pDNA were dispersed in 2 mL of deionized water with or without 10 mmol/L GSH, then shaken at 37 °C. At predetermined time points, 50 μL of supernatant was withdrawn after centrifugation at 14,000 × g for 30 min (Jinzhong, GL-18 B, Shanghai, China), and an equal volume of deionized water was added. The amount of YOYO-1 labeled pDNA in the supernatant was monitored using a microplate reader (Synergy 2, BioTek, USA) set to 490/535 nm excitation/emission. For tumor-microenvironment responsive release, PVHRs NP containing 5 μg of YOYO-1-labeled pDNA was incubated with 1 mg/mL HAase and/or 10 mmol/L GSH and the amount of YOYO-1 labeled pDNA was monitored in the same way.
2.6. In vitro cellular uptake
4T1 cells were seeded into 6-well plates at 1 × 105 cells/well. After culturing for 24 h, the cells were treated with C6-loaded PV micelles, PVs micelles, PVHPs NP, or PVHRs NP for 2 h39. For competition assay, excess HA or RGD was added 30 min prior to C6@PVHRs NP. Cellular uptake was measured using fluorescence microscopy. To further quantify uptake efficiency, the cells were dissociated following the incubation procedure, then washed twice with PBS at 1200×g. The cells were resuspended in PBS at 1 × 106 cells/mL and analyzed using flow cytometry (FACSJazz, BD Biosciences, USA).
To evaluate the subcellular localization of PVHRs nanoparticles, 4T1 cells were seeded into 6-well plates at 1 × 105 cells/well. After culturing for 24 h, the cells were treated with PVHRs NP containing 1 μg of YOYO-1-labeled pDNA for 2 h. The cells were stained with LysoTracker Red for 30 min, then washed, fixed, and stained with Hoechst 33258 solution. The cells were visualized using A1R confocal laser scanning microscopy (Nikon, Tokyo, Japan).
2.7. Cytotoxicity analysis of PVHRs NP
Cell Counting Kit-8 (CCK-8) was used for cytotoxicity studies. 4T1 cells were seeded into a 96-well plate at 1 × 104 cells/well and incubated with RPMI 1640 medium for 24 h at 37 °C. The medium was replaced with serum-free RPMI 1640 medium containing 5 to 100 μg/mL of PVs micelles, PVHs NP, PVHPs NP, or PVHRs NP. After 24 h of incubation, 100 μL of CCK-8 was added, and the cells were incubated for an additional 2 h. Absorbance was measured at 450 nm using a microplate reader.
2.8. Light-induced gene expression
To evaluate light-induced gene expression of the LightOn gene expression system, LED lamps were controlled with a timer to adjust the overall dose of blue light illumination during the specified period (Supporting Information Figs S4A and S4B). The red fluorescent protein mCherry was used as a reporter gene, and gene expression was detected using a fluorescent microscope. 4T1 cells were seeded into 6-well plates at 1 × 105 cells/well. After culturing for 24 h. Then cells were treated with PVHRs NP containing 2 μg of pGCherry (1 μg of pGAVPO and 1 μg of pU5-mCherry), and incubated overnight. The cells were incubated in the dark or exposed to blue light irradiation (2 W/m2) over a predetermined time course. The expression of mCherry was determined using a fluorescence microscope.
2.9. Inhibition of proliferation of 4T1 cells in vitro
2.9.1. Cytotoxicity
To evaluate DTA-induced cell death, 4T1 cells were seeded into a 96-well plate at 1 × 104 cells/well and transfected with PVHRs NP containing 2 μg of pGDTA (1 μg of pGAVPO and 1 μg of pU5-DTA). The cells were cultured under blue light at different powers (0 to 2 W/m2) for 24 h. The cells treated with PVHRs NP without pGDTA were also cultured under blue light to investigate light cytotoxicity. Quantitative analysis of cell viability was conducted using CCK-8 assay as described in section 2.7.
2.9.2. Cell apoptosis
4T1 cells (5 × 104 cells/well) were seeded into 6-well plates. After 24 h, the cells were treated with 4 μg of pGDTA (2 μg of pGAVPO and 2 μg of pU5-DTA). After incubation with or without exposure to blue light (2 W/m2) for 24 h, the cells were collected, washed, and stained with annexin V-FITC and propidium iodide (PI). The cells were analyzed for apoptosis using flow cytometry.
2.9.3. Wound-healing assay
4T1 cells were seeded into 24-well plates at 5 × 104 cells/well and cultured for 24 h. When the cells reached confluence, a sterile pipette tip was used to scratch the monolayer. The cells were then incubated with different formulations containing 4 μg of pGDTA (2 μg of pGAVPO and 2 μg of pU5-DTA) with or without exposure to blue light (2 W/m2) for 24 h. Wound healing was observed using a microscope at 0 and 24 h, respectively. The relative recover area ratios of wound healing were calculated using ImageJ.
2.9.4. Tumor spheroid growth inhibition
Tumor spheroids were prepared by seeding 5 × 103 4T1 cells into 96-well plates pre-coated with 2% (w/v) low gelling temperature agarose gel (70 μL per well)40. After 4 days of activation, homogeneous and compact tumor spheroids were sorted and treated with pGDTA@PVs micelles, pGDTA@PVHPs NP, or pGDTA@PVHRs NP (1 μg of pGAVPO and 1 μg of pU5-DTA) with exposure to blue light (2 W/m2). Cells treated with an equal volume of PBS with or without irradiation were used as negative controls. The tumor inhibition efficacy of the formulations was evaluated using microscopy.
2.9.5. Enzyme-linked immunoassay (ELISA) analysis of DTA expression in vitro
4T1 cells (5 × 104 cells/well) were seeded into 6-well plates. After culturing for 24 h, the cells were incubated with different formulations containing 4 μg of pGDTA (2 μg of pGAVPO and 2 μg of pU5-DTA) with blue light irradiation (2 W/m2) for 24 h. The expression levels of DTA in 4T1 cells and in the supernatants were analyzed using ELISA.
2.10. In vivo biodistribution
4T1 cells (2 × 107 cells) were injected into the mammary glands of BALB/c mice to establish a xenograft breast cancer model. When the tumor size reached 200 mm3, DIR-labeled PVs micelles, PVHs NP, PVHPs NP, or PVHRs NP were intravenous injected at a dose of 100 μg/kg. Mice were anesthetized at different time points (4, 8, 24, and 48 h), and real-time images were captured using an In-Vivo Multispectral System FX (Kodak, USA) with 750 nm excitation and 780 nm emission filters.
2.11. In vivo antitumor efficiency
A 4T1 xenograft breast cancer model was established in BALB/c mice as described in section 2.10. When tumor size reached approximately 100 mm3, tumor-bearing mice were randomly divided into 6 groups (n = 5). Mice were intravenously injected with saline, pGDTA@PVs micelles, pGDTA@PVHPs NP, or pGDTA@PVHRs NP once every three days for a total of three injections. Each dose contained 50 μg of pGDTA/mouse (10 μg of pGAVPO and 40 μg of pU5-DTA). Twelve hours after each dose, mice in the LightOn groups were illuminated from below using a blue LED lamp (90 mW/cm2) for 8 h (Fig. S4C). The abdominal fur of the mice was removed by shaving the mice to allow for better illumination with 8% sodium sulfide. Mice treated with equal volumes of saline or pGDTA@PVHRs NP without illumination were used as negative controls. Body weights and tumor volumes ([major axis] × [minor axis]2/2) were measured and recorded every other day. The survival rate of mice was calculated based on the time of death from the beginning of treatment and a tumor volume over 2000 mm3 was also regarded as a criteria of death. Nine days after the final dose of blue light, mice were sacrificed, and tumors were dissected and photographed, then fixed with paraformaldehyde. The tumors were sliced and subjected to hematoxylin and eosin (H&E), TUNEL, and CD31 staining. The sections were examined using a DM IL microscope (Leica, Wetzlar, Germany).
2.12. Biosafety
To evaluate the side effects of pGDTA@PVHRs NP, major organs (heart, liver, spleen, lung and kidney) were deparaffinized, sliced, and stained using H&E as described in section 2.11. To further evaluate the side effects of pGDTA-loaded nanoparticles, blood samples were collected at the end of the treatment regimen, and serum levels of ALT, AST, BUN and CRE were analyzed using ELISA kit according to the manufacturer's instructions.
2.13. Statistical analysis
Multiple group comparisons were conducted using one-way analysis of variance (ANOVA). All data were analyzed using IBM SPSS Statistics 17.0. All data are presented as the mean ± SD. P-values less than 0.05 were considered statistically significant.
3. Results and discussion
3.1. Synthesis of PEIVES and HA-PEG-RGD
The chemical structure of PEIVES and HA-PEG-RGD were shown in Fig. 2A. The synthetic steps for formation of PEIVES are shown in Supporting Information Fig. S5. Vitamin E succinate was modified with a disulfide bond to obtain VES-SS-COOH. The structure of PEI-SS-VES and VES-SS-COOH was determined using 1H NMR (Supporting Information Figs S6A and S6B). The proton peak at 2.10 ppm (-ph-CH3) and 2.81 ppm (-CH3-COOH) demonstrated successful synthesis of VES-SS-COOH, which was further confirmed using ESI-TOF-MS (Fig. S6C). VES-SS-COOH was reacted with PEI to obtain PEIVES. The appearance of a proton peak at 4.01 ppm demonstrated successful synthesis of PEIVES. In addition, absorption peaks at 3440 and 1640 cm−1 in the FT-IR spectrum were assigned to (-NH-) and amide groups, which further demonstrated successful synthesis (Fig. S6D). The degree of substitution (DS) of the amine groups of PEI with carboxyl groups of VESCOOH was 5.8% and the molecular weight of PEIVES was 48,620 g/mol.
Figure 2.

(A) Molecular structures of cationic core PEI-SS-VES and multifunctional outer layer HA-PEG-RGD. (B) Formation of pGDTA@PVHRs NP, which were constituted of pGDTA, PEI-SS-VES and HA-PEG-RGD (schematic). (C) Gel electrophoresis assay of pGDTA@PVHRs NP at different N/P ratio. (D) TEM image of pGDTA@PVHRs NP. (E) Particle size and zeta potential changes of pDNA@PVHRs NP at pH 7.4. Data were presented as mean ± SD (n = 3). (F) Particle size change of PVHRs NP incubated in 1 mg/mL HAase and/or 10 mmol/L GSH solution (pH 7.4) for 30 min. (G) Particle size and zeta potential changes of PVHRs NP incubated in 1 mg/mL HAase at pH 7.4 or pH 5.5. Data were presented as mean ± SD (n = 3). (H) In vitro gene release of PVs micelles and PVHRs NP loading YOYO-1 labeled pGDTA in the PBS (pH 7.4) with or without 10 mmol/L GSH. Data were presented as mean ± SD (n = 3). HAase- and redox-responsive release profile of pGDTA from PVHRs NP incubating with 1 mg/mL HAase and/or 10 mmol/L GSH at (I) pH 7.4 and (J) pH 5.5. Data were presented as mean ± SD (n = 3).
The synthetic route for HA-PEG-RGD is shown in Supporting Information Fig. S7. Hyaluronic acid was first thiolated41 to promote click reaction with the maleimides of MAL-PEG-NHS. Then, HA-PEG-NHS was modified with RGD to obtain HA-PEG-RGD. The 1H NMR spectrum of HA-PEG-RGD (Supporting Information Fig. S8) had proton peaks at 7.22 ppm, 3.62 ppm, and 1.91 ppm, which were attributed to the benzene protons of RGD, the methyl group of PEG, and the acetyl group of HA, respectively, which indicated successful synthesis of HA-PEG-RGD. The DS of the carboxyl groups of HA-SH with maleoyl groups of NHS-PEG2000-MAL was 5.1% and the molecular weight of PEIVES was 13,490 g/mol.
3.2. Preparation of pDNA-loaded nanoparticles
The LightOn gene expression system consists of a light-inducible transactivator plasmid and a gene expression plasmid. Following light activation, GAVPO homodimerizes, interacts with UASG elements (5X UASG), and initiates the expression of DTA13. To prepare nanostructures via self-assembly with amphiphilic polymers, PVs micelles were prepared using dialysis. Plasmid DNA was condensed on the surface of PVs micelles through electrostatic interactions, and then the micelles were coated onto the anionic polymer HA-PEG-RGD to form nanoparticles (Fig. 2B). To investigate gene loading capacity, the N/P ratio of gene-loaded nanoparticles was evaluated using agarose gel electrophoresis. As shown in Supporting Information Fig. S9A and Fig. 2C, pDNA migration was completely inhibited in gel wells when the N/P ratio was greater than 6. This N/P ratio was selected for further study. The average size and zeta potential of pDNA@PVs micelles were 65.2 ± 1.3 nm and 32.5 ± 2.5 mV, respectively, with a narrow PDI of 0.10. An ideal nanocarrier should maintain structural integrity in the bloodstream, and should have active tumor targeting ability42,43. The negative charge of HA prevented unwanted protein absorption, and PEG modification extended stability in the blood circulation44,45. Supporting Information Table S1 showed the physicochemical properties of pDNA@PVHRs NP in different weight ratios of PEIVES/HA-PEG-RGD. A slight increase of average size (from 65.2 to 74.3 nm) and a reversion of zeta potential (from 32.5 to −20.3 mV) were observed as the weight ratio of PEIVES/HA-PEG-RGD increased from 1/0 to 1/4. Considering the narrow nanosize distribution and the preferred negative charge, the ratio of 1/4 (PEI-SS-VES/HA-PEG-RGD) was selected for further study. Transmission electron microscopy images showed that pDNA@PVHRs NP exhibited a spherical and core‒shell structure, with the size of approximately 70 nm (Fig. 2D), which met the size criterion of less than 200 nm for EPR effects. Moreover, pDNA@PVHRs NP exhibited stability for 28 days, with only slight changes in size and zeta potential due to the negative surface charge and the shielding effects of PEG (Fig. 2E and B). To evaluate the protective effects of nanoparticles on pDNA stability, pDNA@PVHRs nanoparticles were incubated with DNase I for different time intervals46, then analyzed using agarose gel electrophoresis. As shown in Fig. S9C, the nanoparticles protected pDNA against DNase I for at least 6 h.
To confirm the tumor microenvironment-responsive degradation of PVHRs NP, nanoparticles were incubated with HAase and/or GSH. As shown in Fig. 2F, the size of PVHRs NP dramatically increased with the presence of GSH, suggesting the cleavage of disulfide bonds lead to the disintegration of PVHRs nanoparticles. HAase in tumor microenvironment can promote HA degradation47. To further investigate the effect of hyaluronidase on PVHRs NP, nanoparticles were incubated with 1 mg/mL HAase for 8 h. As shown in Fig. 2G, the zeta potential of PVHRs NP increased from −20.3 to −8.2 mV at pH 7.4 and from −19.5 to −0.5 mV at pH 5.5, respectively. HAase induced higher zeta potential increase of PVHRs NP at pH 5.5, which might be due to the higher hyaluronidase activity at acidic pH, resulting the more detachment of HA layer.
3.3. In vitro pDNA release
Nanotechnology can prevent rapid clearance of free pDNA. To evaluate the reduction-responsive profile of the nanoparticles produced in this study, YOYO-1-labeled pDNA was loaded in PVs micelles and PVHRs NP. As shown in Fig. 2H, the 72 h-cumulative release percentages of pDNA from pDNA@PVs micelles and pDNA@PVHRs NP were 28.1% and 10.3%, respectively. The slow release profile of pDNA@PVHRs NP may have been due to the shielding effects of PEG. Concentration differences in GSH in the bloodstream (<5 μmol/L) and tumor cells (2–10 mmol/L) may be used to promote intracellular drug release in tumor cells48. Glutathione-cleavable disulfide bonds have been used as efficient intracellular redox-responsive components for drug release. After incubation with 10 mmol/L GSH for 72 h, the cumulative release of pDNA@PVs micelles and pDNA@PVHRs NP sharply increased to 57.1% and 56.3%, respectively. Cleavage of the disulfide bond led to rapid dissociation of the nanoparticles, resulting in the enhanced release of entrapped pDNA. The total release of pDNA was still relatively low due to electrostatic interactions between pDNA and cationic PEI. Consistent with the release profiles observed in PBS, PVHRs nanoparticles were excellent nanocarriers that exhibited excellent stability in the blood circulation, and selectively released pDNA in the tumor site. For the tumor microenvironment-responsive gene release study, PVHRs NP showed a faster pDNA release at pH 7.4 (Fig. 2I) and pH 5.5 (Fig. 2J) after incubating with HAase, proving the detachment of HA could improve gene release. PVHRs NP exhibited highest in vitro gene release after co-incubating with HAase and GSH, suggesting the complete disassociation of nanoparticles could enhance gene release. These results demonstrated PVHRs nanoparticles could rapidly degrade in tumor microenvironment and trigger gene release in a timely fashion.
3.4. Cellular uptake
To investigate cellular uptake, 4T1 cells were treated with different nanoparticles loaded with the hydrophobic dye coumarin-6. As shown in Fig. 3A and B, fluorescent signal was higher in C6@PVs micelles than that in C6@PV micelles, which suggested that cleavage of disulfide bonds triggered dissociation of the micelles. The fluorescence intensity of C6@PVHPs NP was significantly higher than that of C6@PVs micelles, which may have resulted from selective targeting to CD44, which was overexpressed in 4T1 cells, through endocytosis. C6@PVHRs NP induced 1.7-fold greater fluorescence than C6@PVHPs NP due to overexpression of αvβ3 integrin receptors in 4T1 cells, which highlighted the dual targeting ability of HA and RGD. To further confirm receptor-mediated endocytosis, 4T1 cells were pretreated with excess free HA or RGD to competitively block the corresponding receptors49. The green fluorescence in the HA or RGD pretreated groups was not significantly different than that observed in response to treatment with C6@PVHPs NP, which demonstrated receptor-mediated internalization. These results demonstrated the effective active targeting ability of PVHRs nanoparticles due to HA and RGD surface modifications.
Figure 3.

In vitro cellular uptake studies qualitatively by (A) fluorescence microscopy, and quantitatively by (B) flow cytometry in 4T1 cells. 4T1 cells were treated with C6-loaded nanoparticles 2 h before image. To further study the mechanism of cellular uptake, excess amount of HA or RGD was added and incubation for 30 min before uptake. ∗∗P < 0.01 compared with C6@PVs micelle, ##P < 0.01 compared with C6@PVHRs NP. Data were presented as mean ± SD (n = 3). (C) Confocal images of 4T1 cells treated with pDNA@PVHRs NP for 2 h pDNA was labeled with YOYO-1, the lysosomes were stained with LysoTracker Red, while the nuclei were stained with Hoechst 33342. (D) Cell viability of 4T1 cells after 24 h incubation with nanocarriers of different concentrations. Data were presented as mean ± SD (n = 3). (E) Time-dependent gene expression of mCherry after different irradiation time. Cells were incubated with pGCherry@PVHRs NP for 6 h in dark, fresh medium was replaced and cells were cultured under blue light (2 W/m2) for 4, 6 or 8 h. (F) Mean fluorescence intensity of mCherry was quantitatively analyzed by ImageJ. Data were presented as mean ± SD (n = 3).
The intracellular localization of pDNA@PVHRs nanoparticles were monitored using confocal scanning laser microscopy. The plasmids and lysosomes were labeled with YOYO-1 and LysoTracker Red, respectively. As shown in Fig. 3C, green fluorescence of the pDNA was not co-localized with red fluorescence of the lysosomes, which indicated successful lysosomal escape of PVHRs nanoparticles through the “proton sponge” effect of PEI31.
3.5. Nanocarrier cytotoxicity
The CCK-8 assay was used to evaluate the cytotoxicity of different nanoparticles. As shown in Fig. 3D, all blank nanoparticles showed negligible cytotoxicity from 5 to 100 μg/mL. Treatment with 0.05–0.5 mg/mL PVHRs NP resulted in a low hemolysis ratio (Fig. S9D). These results suggested that PVHRs nanoparticles may exhibit superior biocompatibility and hemocompatibility.
3.6. Light-induced gene expression
To investigate the spatial controllability of the LightOn gene expression system in vitro, 4T1 cells were transfected with the mCherry reporter and pGAVPO, then irradiated with blue light. As shown in Fig. 3E and F, mCherry fluorescence increased rapidly after exposure of cells to blue light, while very little fluorescence signal was observed in cells kept in the dark, which confirmed that gene expression was strictly triggered by exposure to blue light with fast kinetics. Moreover, fluorescence increased with irradiation time, which indicated time-dependent gene expression in response to blue light. These results showed that the LightOn gene expression system allowed for spatiotemporal control of gene expression with high sensitivity and low non-specific expression.
3.7. In vitro tumor inhibition
3.7.1. In vitro cytotoxicity
We investigated blue light-induced toxicity. As shown in Fig. S9E, blue light did not induce cell death from 0.3 to 2 W/cm2. Cytotoxicity caused by light-induced DTA expression was evaluated in 4T1 cells across a range of blue light intensities. All groups showed dose-dependent decreases in cell viability across the range of 0.3 to 2 W/cm2 (Fig. 4A). The pU5-DTA reporter gene and pGAVPO gene (pGDTA) were used to inhibit tumor cell growth through light-induced expression of DTA. After 24 h of incubation, intensity-dependent decreases in cell viability were observed. Furthermore, pGDTA@PVHRs NP significantly inhibited 4T1 cell growth, as evidenced by 28.5% cell viability after 24 h of blue light exposure, which suggested effective tumor inhibition in vitro. To maximize in vitro tumor inhibition, 2 W/cm2 was selected for further experiments.
Figure 4.

(A) Cell viability of 4T1 cells after 24 h incubation with pGDTA loaded nanoparticles under blue light radiation with different intensity. ∗∗P < 0.01. Data were presented as mean ± SD (n = 3). (B) Apoptosis study of 4T1 cells induced by pGDTA loaded nanoparticles. (C) Total apoptosis was quantified by flow cytometry. Cells were incubated with pGDTA loaded nanoparticles for 12 h in dark, fresh medium was replaced and cells were incubated with or without blue light irradiation (2 W/m2). ∗∗P < 0.01, ∗∗∗P < 0.001. Data were presented as mean ± SD (n = 3). (D) Wound healing ability of 4T1 cells. Cells were incubated with pGDTA loaded nanoparticles for 12 h in dark, fresh medium was replaced and cells were incubated with or without blue light irradiation for 24 h (2 W/m2), scale bar: 500 μm. (E) Quantitative analysis of relative recover area ratio in different groups. ∗∗∗P < 0.001. Data were presented as mean ± SD (n = 3). (F) In vitro 3D tumor spheroid growth inhibition. pGDTA-loaded nanoparticles were incubated with 4T1 tumor spheroids for 12 h in dark. Fresh medium was replaced and tumor spheroids were incubated under blue light for 5 days under blue light irradiation (2 W/m2). (G) Real DTA level of intracellular and extracellular samples. Cells were incubated with pGDTA loaded nanoparticle for 12 h in dark, fresh medium was replaced and cells were incubated under blue light radiation for 24 h ∗∗∗P < 0.001. Data were presented as mean ± SD (n = 3).
3.7.2. Apoptosis assay
Apoptosis of 4T1 cells following incubation with different pGDTA-loaded nanoparticles with or without exposure to blue light was evaluated using flow cytometry (Fig. 4B and C). All groups exhibited negligible apoptosis, which demonstrated that DTA was not expressed in the absence of light stimulation. After irradiation, all pGDTA-loaded nanoparticles induced apoptosis, and pGDTA@PVHRs NP induced the greatest level of apoptosis of 4T1 cells (65.1%), which indicated that this formulation had the best targeting ability of PVHRs NP evaluated in this study. These results indicated that pGDTA@PVHRs nanoparticles could allow for spatiotemporal control of DTA expression, leading to effective tumor apoptosis in vitro.
3.7.3. Wound-healing assay
Migration of 4T1 cells was evaluated by wound healing assay50. Migration was significantly inhibited in response to pGDTA@PVs micelles and pGDTA@PVHRs NP following exposure to blue light (Fig. 4D). Decreased migration may have been due to high transfection efficiency and light-induced DTA expression of the nanoparticles. Furthermore, the scratch area in cells treated with pGDTA-loaded nanoparticles widened following exposure to blue light, which demonstrated excellent antitumor effects in vitro (Fig. 4E). These results suggested that pGDTA@PVHRs nanoparticles exhibited light-controllable inhibition of cell migration.
3.7.4. Tumor spheroid inhibition
4T1 spheroids were employed to mimic 3D solid tumors in vitro40. 4T1 cells were cultured in 96-well plates pre-coated with 1% (w/v) low gelling temperature agarose gel to form tumor spheroids. After one week of incubation, 4T1 spheroids were incubated with different nanoparticles and exposed to blue light for five days. Anti-proliferation effects were evaluated using microscopy (Fig. 4F). Significant tumor growth inhibition was observed in all pGDTA-treated groups, as evidenced by gradual collapse and loss of integrity following blue light irradiation. Moreover, pGDTA@PVHRs nanoparticles exhibited the best anti-proliferation ability, which agreed with the results of the apoptosis assay.
3.7.5. Detection of DTA
The intracellular and extracellular DTA concentrations after 24 h of exposure to blue light were evaluated using a DTA ELISA kit. As shown in Fig. 4G, treatment with pGDTA@PVHRs NP resulted in the highest intracellular and extracellular DTA concentrations, with a total concentration of 7.67 ng/mL, which may have been due to excellent cellular uptake efficiency of this nanocarrier. The relatively high extracellular concentration of DTA likely resulted from release from apoptotic cells.
3.8. Pharmacokinetics and biodistribution study
Supporting Information Fig. S10A showed relative profiles of mean plasma DIR concentration over time following the intravenous injection of different nanoparticle systems loaded with DIR; Supporting Information Table S2 showed the corresponding pharmacokinetic parameters. The plasma concentration of DIR decreased rapidly following DIR treatment. Furthermore, the mean area under the curves (AUC) for the plasma concentration of DIR-loaded nanoparticles was significantly higher than that of free DIR (∗P < 0.05), thus demonstrating that the nanocarriers were stable. The AUC0–24h of the DIR@PVHRs NP treatment increased from 226.9 to 529.5 μg·h/L compared with that of PVs micelle treatment. The corresponding mean residence time (MRT) of the DIR@PVHRs NP was 1.9-fold longer than that of the PVs micelle treatment, thus indicating a longer duration of circulation in the blood; we believe that this characteristic was attributable to the PEG modification on the surface of the nanoparticles.
To investigate the in vivo targeting ability of PVHRs nanoparticles, the near-infrared dye DIR was loaded into different nanoparticles, and the nanoparticles were intravenously injected into 4T1 tumor-bearing mice. At pre-determined timepoints, mice were anesthetized and imaged using an In-Vivo Multispectral System. As shown in Fig. 5A, DIR@PVHs NP exhibited better accumulation in tumors than DIR@PVs micelles because HA actively targets CD44, which is overexpressed on tumor cells. However, the nanoparticles showed significant liver accumulation due to non-specific uptake by the reticuloendothelial system (RES)51 and high expression of CD44 in liver cells. Treatment with DIR@PVHPs NP and DIR@PVHRs NP showed greater tumor accumulation at 24 and 48 h due to the ability of PEG to increase circulation time. The excellent targeting ability of DIR@PVHRs NP may have been due to the dual-targeting ability of HA and RGD, which resulted in active targeting to CD44 and αvβ3 receptors on 4T1 cells, respectively. The quantitative analysis of DIR uptake in both tumors and main organs at 4, 8, and 24 h post-treatment is shown in Fig. S10B; the corresponding AUC0–24h data are summarized in Supporting Information Table S3. The concentrations of DIR in tumors following treatment with PVHs NP, PVHPs NP, and PVHRs NP were 1.3-, 2.3-, and 2.8-folds higher compared with those following treatment with PVs micelles, respectively; these data were consistent with in vivo observations. Ex vivo imaging of tissue distribution showed the strongest DIR fluorescence 24 h after the injection of PVHRs NP, thus confirming the enhanced accumulation of PVHRs NP in tumors (Fig. S10C).
Figure 5.

Evaluation of in vivo 4T1 tumor inhibition. (A) Biodistribution of DIR loaded nanoparticles, among which the dashed circles indicate the tumor foci of mice. In vivo fluorescence images of 4T1-bearing mice after intravenous injection with different DIR loaded nanoparticles for 4, 8, 24 and 48 h. (B) Schematic diagram of 4T1 bearing Balb/c mice treated with blue light radiation after intravenous injection of samples. (C) The tumor growth profiles after different treatments with or without blue light irradiation Data were presented as mean ± SD (n = 5). ∗∗P < 0.01, ∗∗∗P < 0.001. (D) Tumor morphology of different groups on day 16. (E) Body weight and (F) survival rate of mice after different treatments with or without blue light irradiation. Data were presented as mean ± SD (n = 5). (G) Immunofluorescence staining of H&E, TUNEL and CD31 for tumor tissue sections, scale bar: 50 μm. Quantitative analysis of (H) TUNEL and (I) CD31 positive ratio in different groups were calculated by ImageJ. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001. Data were presented as mean ± SD (n = 3).
3.9. In vivo antitumor efficiency
In vivo antitumor efficacy was evaluated in 4T1 tumor-bearing mice. When tumor size increased to approximately 100 mm3, 200 μL of different formulations (0.5 mg/mL pGAVPO and 2 mg/mL pU5-DTA) were intravenously injected a total of three times each. Twelve hours after administration, the mice were exposed to blue light or placed in the dark for 8 h (Fig. 5B). As shown in Fig. 5C and D, all pGDTA-loaded nanoparticles exposed to blue light demonstrated remarkable suppression of tumor growth, but exposure to blue light alone did not inhibit tumor growth. Administration of pGDTA@PVHRs NP resulted in the strongest antitumor effect, with a tumor inhibition rate of 71.7%, which may have been due to superior tumor targeting ability. Notably, the tumor volumes in the pGDTA@PVHRs NP group were reduced following exposure to blue light, which demonstrated light-dependent tumor cell death. The body weights of the mice were monitored and recorded every other day. As shown in Fig. 5E, there were no significant differences in weight among the mice exposed to the LightOn gene expression system, which suggested that light therapy did not induce systemic toxicity. The median survive time of pGDTA@PVHRs NP prolonged from 25 to 40 days compared with saline group, exhibiting the best survival rate (Fig. 5F). These results demonstrated light-induced antitumor effects in response to treatment with the LightOn gene expression system, which precisely expressed the suicide gene DTA in the illuminated area but did not express DTA in the absence of blue light.
To further evaluate antitumor activity, tumor specimens from mice were collected, sliced, and subjected to H&E, TUNEL, and CD31 staining. As shown in Fig. 5G, all pGDTA loaded nanocarrier groups induced extensive tumor necrosis after blue light irradiation, which demonstrated excellent tumor inhibition. In addition, the number of TUNEL-positive cells (green) significantly increased following exposure to blue light, and treatment with pGDTA@PVHRs NP resulted in the highest TUNEL-positive ratio, which indicated the best antitumor efficacy (Fig. 5H). Staining for CD31 was used to evaluate blood vessel content in tumor tissues52. Reduced CD31 fluorescence in pGDTA-loaded nanocarrier groups indicated that light therapy inhibited tumor angiogenesis. Notably, pGDTA@PVHRs NP exhibited the greatest inhibition of tumor angiogenesis, which may have been due to the targeting ability of RGD to αvβ3, which is highly expressed tumor neovascular cells (Fig. 5I)35. These results indicated that pGDTA@PVHRs nanoparticles were able to induce the expression of DTA to kill tumor cells, and also normalized the tumor vasculature to reduce the nutrient supply to the tumor, which resulted in excellent inhibition of tumor growth53.
3.10. Biosafety
Non-specific expression of DTA could result in tissue damage. To investigate the potential for systemic toxicity in response to pGDTA@PVHRs nanoparticles, non-tumorous organs were collected following therapy, sliced, and then stained with H&E (Fig. 6A). No significant necrosis was observed in the heart, liver, spleen, lung, or kidney following therapy, which showed that pGDTA@PVHRs NP with blue light exposure, resulted in neglectable toxicity. The treatment groups did not show any differences in biochemical parameters compared with the treatment groups (Fig. 6B), which indicated that pGDTA@PVHRs nanoparticles were safe. Although some nanoparticles accumulated in the livers of the mice, no obvious tissue damage was observed. Light-induced gene expression was limited to a depth of 1 mm or less in the liver12, so the remaining fur could block normal tissues from blue light penetration, avoiding unwanted DTA expression in other tissues. Consistent with in vivo antitumor efficiency, DTA expression can be triggered in certain tumor sites by blue light irradiation and avoid systemic toxicity, which maximize the antitumor efficiency of DTA treatment and minimize its side effects. In conclusion, these results indicated that the nanoparticles evaluated in this study were biocompatible.
Figure 6.

Safety evaluation of DTA-based treatment in mice. (A) H&E stained major organs from different treatment groups. (scale bar: 100 μm) (B) Serum levels of AST, ALT, CRE and BUN after treatment. Data were presented as mean ± SD (n = 3).
4. Conclusions
In summary, a multifunctional gene delivery system based on the polymers PEIVES and HA-PEG-RGD was successfully constructed to encapsulate the LightOn transgene system. The stability of PVHRs nanoparticles was excellent, and they showed redox-sensitive characteristics, and good tumor accumulation. Importantly, the LightOn gene expression system was effectively delivered to 4T1 cells due to CD44 receptor- and αvβ3 integrin receptor-mediated endocytosis. Encapsulation of the LightOn gene expression system resulted in structure-, dose- and spatiotemporal-dependent gene expression through exposure to blue light. Moreover, in vitro and in vivo studies showed that DTA expression following administration of pDNA@PVHRs nanoparticles with exposure to blue light resulted in excellent tumor growth inhibition. Furthermore, we showed that the LightOn gene expression system formulations developed in this study exhibited excellent biosafety. Therefore, pDNA@PVHRs nanoparticles exhibited excellent antitumor properties by effectively utilizing a light-switchable gene expression system encapsulated in a nanoparticle delivery system, which may be a promising platform for treatment of breast cancer.
Footnotes
Peer review under the responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.04.010.
Contributor Information
Xianjun Chen, Email: xianjunchen@ecust.edu.cn.
Feng Gao, Email: fgao@ecust.edu.cn.
Acknowledgments
This work was supportedby Shanghai Municipal Natural Science Foundation (No. 17ZR1406600, China), Science and Technology Commission of Shanghai Municipality (No.10DZ2220500, China), The Shanghai Committee of Science and Technology (Grant No. 11DZ2260600, China) and National Natural Science Foundation of China (Grant No.81973700).
Author contributions
Feng Gao and Xianjun Chen designed the research. Xinyu Hou and Chenting Shou carried out the experiments and performed data analysis. Jiajun Xu and Muye Heparticipated part of the experiments. Zeting Yuan, Yu Zheng Zhao and Yi Yang provided experimental drugs and quality control. Xinyu Hou wrote the manuscript. Feng Gao and Minbo Lan revised the manuscript. All of the authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351–360. doi: 10.1038/nature15818. [DOI] [PubMed] [Google Scholar]
- 2.Birkeland A.C., Ludwig M.L., Spector M.E., Brenner J.C. The potential for tumor suppressor gene therapy in head and neck cancer. Discov Med. 2016;21:41–47. [PMC free article] [PubMed] [Google Scholar]
- 3.Rupaimoole R., Slack F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 4.Ginn S.L., Amaya A.K., Alexander I.E., Edelstein M., Abedi M.R. Gene therapy clinical trials worldwide to 2017: an update. J Gene Med. 2018;20 doi: 10.1002/jgm.3015. [DOI] [PubMed] [Google Scholar]
- 5.Kim J., Mirando A.C., Popel A.S., Green J.J. Gene delivery nanoparticles to modulate angiogenesis. Adv Drug Deliv Rev. 2017;119:20–43. doi: 10.1016/j.addr.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pahle J., Walther W. Vectors and strategies for nonviral cancer gene therapy. Expet Opin Biol Ther. 2016;16:443–461. doi: 10.1517/14712598.2016.1134480. [DOI] [PubMed] [Google Scholar]
- 7.Li J., Wang Y., Zhu Y., Oupicky D. Recent advances in delivery of drug-nucleic acid combinations for cancer treatment. J Contr Release. 2013;172:589–600. doi: 10.1016/j.jconrel.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim B., Zimmermann M., Barry N.A., Goodman A.L. Engineered regulatory systems modulate gene expression of human commensals in the gut. Cell. 2017;169:547–558. doi: 10.1016/j.cell.2017.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen C., Yue D.X., Lei L.Y., Wang H.R., Lu J., Zhou Y. Promoter-operating targeted expression of gene therapy in cancer: current stage and prospect. Mol Ther Nucleic Acids. 2018;11:508–514. doi: 10.1016/j.omtn.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perri F., Longo F., Giuliano M., Sabbatino F., Favia G., Ionna F. Epigenetic control of gene expression: potential implications for cancer treatment. Crit Rev Oncol Hematol. 2017;111:166–172. doi: 10.1016/j.critrevonc.2017.01.020. [DOI] [PubMed] [Google Scholar]
- 11.Zhang K., Cui B.X. Optogenetic control of intracellular signaling pathways. Trends Biotechnol. 2015;33:92–100. doi: 10.1016/j.tibtech.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X.J., Zhang D.S., Su N., Bao B.K., Xie X., Zuo F.T. Visualizing RNA dynamics in live cells with bright and stable fluorescent RNAs. Nat Biotechnol. 2019;37:1287–1293. doi: 10.1038/s41587-019-0249-1. [DOI] [PubMed] [Google Scholar]
- 13.Wang X., Chen X.J., Yang Y. Spatiotemporal control of gene expression by a light-switchable transgene system. Nat Methods. 2012;9:266–269. doi: 10.1038/nmeth.1892. [DOI] [PubMed] [Google Scholar]
- 14.Allahyari H., Heidari S., Ghamgosha M., Saffarian P., Amani J. Immunotoxin: a new tool for cancer therapy. Tumour Biol. 2017;39:1–11. doi: 10.1177/1010428317692226. [DOI] [PubMed] [Google Scholar]
- 15.Dhillon S. Moxetumomab pasudotox: first global approval. Drugs. 2018;78:1763–1767. doi: 10.1007/s40265-018-1000-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen M.S., Chang P. Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat Chem Biol. 2018;14:236–243. doi: 10.1038/nchembio.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cocco E., Deng Y., Shapiro E.M., Bortolomai I., Lopez S., Lin K. Dual-targeting nanoparticles for in vivo delivery of suicide genes to chemotherapy-resistant ovarian cancer cells. Mol Canc Therapeut. 2017;16:323–333. doi: 10.1158/1535-7163.MCT-16-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buzzi S., Maistrello I. Inhibition of growth of Erlich tumors in Swiss mice by diphtheria toxin. Can Res. 1973;33:2349–2353. [PubMed] [Google Scholar]
- 19.Mizuguchi H., Nakanishi M., Nakanishi T., Nakagawa T., Nakagawa S., Mayumi T. Application of fusogenic liposomes containing fragment A of diphtheria toxin to cancer therapy. Br J Canc. 1996;73:472–476. doi: 10.1038/bjc.1996.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung L.S., Fu J., Kumar A., Urbanowski M.E., Ihms E.A., Parveen S. Second-generation IL-2 receptor-targeted diphtheria fusion toxin exhibits antitumor activity and synergy with anti-PD-1 in melanoma. Proc Natl Acad Sci U S A. 2019;116:3100–3105. doi: 10.1073/pnas.1815087116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Figgitt D.P., Lamb H.M., Goa K.L. Denileukin diftitox. Am J Clin Dermatol. 2000;1:67–72. doi: 10.2165/00128071-200001010-00008. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y., Li Z., Xu Z., Tang H.Y., Guo W.X., Sun X.X. Use of the XRCC2 promoter for in vivo cancer diagnosis and therapy. Cell Death Dis. 2018;9:420. doi: 10.1038/s41419-018-0453-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denkert C., Liedtke C., Tutt A., von Minckwitz G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. 2017;389:2430–2442. doi: 10.1016/S0140-6736(16)32454-0. [DOI] [PubMed] [Google Scholar]
- 24.Dougherty G.J., Dougherty S.T. Exploiting the tumor microenvironment in the development of targeted cancer gene therapy. Canc Gene Ther. 2009;16:279–290. doi: 10.1038/cgt.2008.72. [DOI] [PubMed] [Google Scholar]
- 25.Wang J.P., Meng F.F., Kim B.K., Ke X., Yeo Y. In-vitro and in-vivo difference in gene delivery by lithocholic acid-polyethyleneimine conjugate. Biomaterials. 2019;217:119296. doi: 10.1016/j.biomaterials.2019.119296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Z.X., Liu X.R., Zhu D.C., Wang Y., Zhang Z., Zhou X.F. Nonviral cancer gene therapy: delivery cascade and vector nanoproperty integration. Adv Drug Deliv Rev. 2017;115:115–154. doi: 10.1016/j.addr.2017.07.021. [DOI] [PubMed] [Google Scholar]
- 27.Miller J.B., Zhang S., Kos P., Xiong H., Zhou K., Perelman S.S. Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew Chem Int Ed Engl. 2017;56:1059–1063. doi: 10.1002/anie.201610209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y., Thambi T., Lee D.S. Co-delivery of drugs and genes using polymeric nanoparticles for synergistic cancer therapeutic effects. Adv Healthc Mater. 2018;7:1700886. doi: 10.1002/adhm.201700886. [DOI] [PubMed] [Google Scholar]
- 29.Golombek S.K., May J.N., Theek B., Appold L., Drude N., Kiessling F. Tumor targeting via EPR: strategies to enhance patient responses. Adv Drug Deliv Rev. 2018;130:17–38. doi: 10.1016/j.addr.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye Y.J., Sun Y., Zhao H.L., Lan M.B., Gao F., Song C. A novel lactoferrin-modified β-cyclodextrin nanocarrier for brain-targeting drug delivery. Int J Pharm. 2013;458:110–117. doi: 10.1016/j.ijpharm.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 31.He M.Y., Huang L., Hou X.Y., Zhong C., Bachir Z.A., Lan M.B. Efficient ovalbumin delivery using a novel multifunctional micellar platform for targeted melanoma immunotherapy. Int J Pharm. 2019;560:1–10. doi: 10.1016/j.ijpharm.2019.01.027. [DOI] [PubMed] [Google Scholar]
- 32.Wang H.B., Liu G.Y., Dong S.H., Xiong J.J., Du Z.L., Cheng X. A pH-responsive AIE nanoprobe as a drug delivery system for bioimaging and cancer therapy. J Mater Chem B. 2015;3:7401–7407. doi: 10.1039/c5tb01169h. [DOI] [PubMed] [Google Scholar]
- 33.Li M., Zhao L.W., Zhang T., Shu Y., He Z.G., Ma Y. Redox-sensitive prodrug nanoassemblies based on linoleic acid-modified docetaxel to resist breast cancers. Acta Pharm Sin B. 2019;9:421–432. doi: 10.1016/j.apsb.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang X.Q., Wu J.R., He M.Y., Hou X.Y., Wang Y., Cai X.R. Combined cancer chemo-photodynamic and photothermal therapy based on ICG/PDA/TPZ-loaded nanoparticles. Mol Pharm. 2019;16:2172–2183. doi: 10.1021/acs.molpharmaceut.9b00119. [DOI] [PubMed] [Google Scholar]
- 35.Prokopiou E.M., Ryder S.A., Walsh J.J. Tumour vasculature targeting agents in hybrid/conjugate drugs. Angiogenesis. 2013;16:503–524. doi: 10.1007/s10456-013-9347-8. [DOI] [PubMed] [Google Scholar]
- 36.Luo Z.J., Dai Y., Gao H.L. Development and application of hyaluronic acid in tumor targeting drug delivery. Acta Pharm Sin B. 2019;9:1099–1112. doi: 10.1016/j.apsb.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu J., He M., Hou X., Wang Y., Shou C., Cai X. Safe and efficacious diphtheria toxin-based treatment for melanoma: combination of a Light-On gene-expression system and nanotechnology. Mol Pharm. 2020;17:301–315. doi: 10.1021/acs.molpharmaceut.9b01038. [DOI] [PubMed] [Google Scholar]
- 38.He M., Wang Y., Chen X., Zhao Y., Lou K., Wang Y. Spatiotemporally controllable diphtheria toxin expression using a light-switchable transgene system combining multifunctional nanoparticle delivery system for targeted melanoma therapy. J Contr Release. 2020;319:1–14. doi: 10.1016/j.jconrel.2019.12.015. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y.H., Sun J., Cao W., Yang J.H., Lian H., Li X. Dual targeting folate-conjugated hyaluronic acid polymeric micelles for paclitaxel delivery. Int J Pharm. 2011;421:160–169. doi: 10.1016/j.ijpharm.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 40.Lu Z.Z., Long Y., Cun X.L., Wang X.H., Li J.P., Mei L. A size-shrinkable nanoparticle-based combined anti-tumor and anti-inflammatory strategy for enhanced cancer therapy. Nanoscale. 2018;10:9957–9970. doi: 10.1039/c8nr01184b. [DOI] [PubMed] [Google Scholar]
- 41.Ouasti S., Donno R., Cellesi F., Sherratt M.J., Terenghi G., Tirelli N. Network connectivity, mechanical properties and cell adhesion for hyaluronic acid/PEG hydrogels. Biomaterials. 2011;32:6456–6470. doi: 10.1016/j.biomaterials.2011.05.044. [DOI] [PubMed] [Google Scholar]
- 42.Wen Y.T., Zhang Z.X., Li J. Highly efficient multifunctional supramolecular gene carrier system self-assembled from redox-sensitive and zwitterionic polymer blocks. Adv Funct Mater. 2014;24:3874–3884. [Google Scholar]
- 43.Yin H.R., Wang H.Z., Li Z.F., Shu D., Guo P.X. RNA micelles for the systemic delivery of anti-miRNA for cancer targeting and inhibition without ligand. ACS Nano. 2019;13:706–717. doi: 10.1021/acsnano.8b07948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong L., Xu L., Liu Y.Y., Li Q.S., Zhao D.Y., Li Z.B. Transformative hyaluronic acid-based active targeting supramolecular nanoplatform improves long circulation and enhances cellular uptake in cancer therapy. Acta Pharm Sin B. 2019;9:397–409. doi: 10.1016/j.apsb.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong G., Yang C., Liu S., Zheng Y., Lou W., Teo J.Y. Polymers with distinctive anticancer mechanism that kills MDR cancer cells and inhibits tumor metastasis. Biomaterials. 2019;199:76–87. doi: 10.1016/j.biomaterials.2019.01.036. [DOI] [PubMed] [Google Scholar]
- 46.Kim M.H., Na H.K., Kim Y.K., Ryoo S.R., Cho H.S., Lee K.E. Facile synthesis of monodispersed mesoporous silica nanoparticles with ultralarge pores and their application in gene delivery. ACS Nano. 2011;5:3568–3576. doi: 10.1021/nn103130q. [DOI] [PubMed] [Google Scholar]
- 47.Liu R., Hu C., Yang Y.Y., Zhang J.Q., Gao H.L. Theranostic nanoparticles with tumor-specific enzyme-triggered size reduction and drug release to perform photothermal therapy for breast cancer treatment. Acta Pharm Sin B. 2019;9:410–420. doi: 10.1016/j.apsb.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q.J., Chen M., Chen D.Y., Wu L.M. One-pot synthesis of diphenylalanine-based hybrid nanospheres for controllable pH- and GSH-responsive delivery of drugs. Chem Mater. 2016;28:6584–6590. [Google Scholar]
- 49.Bhatnagar P., Kumari M., Pahuja R., Pant A.B., Shukla Y., Kumar P. Hyaluronic acid-grafted PLGA nanoparticles for the sustained delivery of berberine chloride for an efficient suppression of Ehrlich ascites tumors. Drug Deliv Transl Res. 2018;8:565–579. doi: 10.1007/s13346-018-0485-9. [DOI] [PubMed] [Google Scholar]
- 50.Kang X.J., Zheng Z.N., Liu Z.H., Wang H.Y., Zhao Y.G., Zhang W.Y. Liposomal codelivery of doxorubicin and andrographolide inhibits breast cancer growth and metastasis. Mol Pharm. 2018;15:1618–1626. doi: 10.1021/acs.molpharmaceut.7b01164. [DOI] [PubMed] [Google Scholar]
- 51.Goel S., Ferreira C.A., Dogra P., Yu B., Kutyreff C.J., Siamof C.M. Size-optimized ultrasmall porous silica nanoparticles depict vasculature-based differential targeting in triple negative breast cancer. Small. 2019;15 doi: 10.1002/smll.201903747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang B., Ding Y.P., Zhao X.Z., Han X.X., Yang N., Zhang Y.L. Delivery of small interfering RNA against Nogo-B receptor via tumor-acidity responsive nanoparticles for tumor vessel normalization and metastasis suppression. Biomaterials. 2018;175:110–122. doi: 10.1016/j.biomaterials.2018.05.034. [DOI] [PubMed] [Google Scholar]
- 53.Martin J.D., Panagi M., Wang C., Khan T.T., Martin M.R., Voutouri C. Dexamethasone increases cisplatin-loaded nanocarrier delivery and efficacy in metastatic breast cancer by normalizing the tumor microenvironment. ACS Nano. 2019;13:6396–6408. doi: 10.1021/acsnano.8b07865. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.