

Abstract
Prevalent resistance to inhibitors that target the influenza A M2 proton channel has necessitated a continued drug design effort, supported by a sustained study of the mechanism of channel function and inhibition. Recent high-resolution X-ray crystal structures present the first opportunity to see how the adamantyl amine class of inhibitors bind to M2 and disrupt and interact with the channel’s water network, providing insight into the critical properties that enable their effective inhibition in wild-type M2. In this work, we examine the hypothesis that these drugs act primarily as mechanism-based inhibitors by comparing hydrated excess proton stabilization during proton transport in M2 with the interactions revealed in the crystal structures, using the Multiscale Reactive Molecular Dynamics (MS-RMD) methodology. MS-RMD, unlike classical molecular dynamics, models the hydrated proton (hydronium-like cation) as a dynamic excess charge defect and allows bonds to break and form, capturing the intricate interactions between the hydrated excess proton, protein atoms, and water. Through this, we show that the ammonium group of the inhibitors is effectively positioned to take advantage of the channel’s natural ability to stabilize an excess protonic charge and act as a hydronium mimic. Additionally, we show that the channel is especially stable in the drug binding region, highlighting the importance of this property for binding the adamantane group. Finally, we characterize an additional hinge point near Val27, which dynamically responds to charge and inhibitor binding. Altogether, this work further illuminates a dynamic understanding of the mechanism of drug inhibition in M2, grounded in the fundamental properties that enable the channel to transport and stabilize excess protons, with critical implications for future drug design efforts.
Introduction
Proton transport (PT) across cellular membranes is a critical component of many biomolecular systems, necessary, for example, to maintain pH gradients,1,2 to drive ATP synthesis,3 and to facilitate the co- or antitransport of other small molecules.4−6 Because of their essential role in such systems, channels and transporters with PT functionality are often targets for drug design to inhibit or control PT: in the case of viruses and bacteria, to slow or prevent infection, but there are myriad other disease applications.7−9 Drug design is notoriously challenging, as both thermodynamic and kinetic factors must be considered but are difficult to predict and control, and its success depends on high quality structures, an understanding of structural dynamics, and a knowledge of the protein’s function and its mechanism. Thus, beyond elucidating mechanisms of PT in order to understand how a specific channel or transporter works, studying the detailed interactions that facilitate PT can provide valuable insights to help guide drug design efforts.
The influenza virus kills up to 650 000 people each year,10 and the impact of the recent global coronavirus pandemic11 emphasizes how critical it is to maintain our focus on understanding and treating viral infections. The influenza A virus matrix 2 (M2) proton channel is a homotetrameric protein responsible for the acidification of the viral interior, a critical step in the influenza infection process.12−14 It is the target of two of the three currently available oral antivirals, amantadine and rimantadine.15,16 While these are effective at blocking PT in wild-type M2, drug-resistant mutants have become the predominant strains, the majority of which contain an S31N mutation.17,18 This widespread resistance requires a continued drug design effort19 informed by a deeper understanding of the PT and drug inhibition mechanisms. Additionally, M2 is considered an archetype for the viroporin family, a class of viral channels considered ideal drug targets.20 The SARS-CoV-2 virus responsible for the COVID-19 pandemic contains two viroporins: protein E and 3.21−23 Thus, viroporins are a critical class of proteins to study as potential therapeutic targets.
M2 is located in the viral capsid and is acid-activated: as the pH of the endosome encapsulating the virus is lowered, the M2 channel becomes activated and facilitates unidirectional proton flow to the viral interior, allowing the virus to escape the endosome and infect the cell. The key residue that controls activation is His37,24−26 which can bind one additional proton and take on a +1 charge. One histidine from each helix forms the His37 tetrad, which can collectively hold a +0 to +4 excess charge, dependent on pH. The channel becomes activated and the C-terminal portion opens (adopting the Inwardopen conformation) upon reaching the +3 state, and PT occurs as the channel cycles through a transporter-like mechanism.27−34
Amantadine and rimantadine belong to the adamantyl amine class of inhibitors, binding in the upper-middle portion of the channel. These drugs were the predecessors of many related adamantane-based compounds featuring a relatively rigid, apolar group and an attached charged group.35−42 Recently, Thomaston et al. published several high-resolution X-ray crystal structures of M2 with amantadine, rimantadine, and a novel spiro-adamantyl amine bound.43 These structures provided the first opportunity to see the specific interactions that facilitate stable inhibitor binding and the disruption of the hydrogen bonded water network otherwise present. Along with an earlier qualitative MD simulation study that guided the design of the spiro-adamantyl amine inhibitors,44 the crystallographic analysis provided potential insights into the mechanism of inhibition, suggesting that the backbone carbonyls of pore-lining residues act as “physiochemical chameleons”, able to engage in both hydrophobic and hydrophilic interactions, and that the drug is tilted off the channel’s axis and interacts with waters in the Ala30 layer. Taken together, it is hypothesized that amantadine acts as a mechanism-based inhibitor, with the ammonium group functioning as a hydronium mimic. Computational studies to date have primarily focused on the means of entry into the channel and location of binding,45−50 but have not deduced specific interactions between the drug, channel, and channel water involved in binding as they relate specifically to similar interactions seen in the PT mechanism.
Proton transport is an inherently quantum mechanical process, as the hydrated proton structure (hydronium-like) exists in a complex hydrogen bonded network that rearranges dynamically as bonds break and form according to the Grotthuss shuttling mechanism.51−53 Thus, classical molecular dynamics (MD) with fixed bonding topology cannot be used to study PT; moreover, ab initio methods are not efficient enough to reach the many nanosecond time scales necessary to obtain sufficient sampling in biomolecular systems that may have important degrees of freedom several orders of magnitude slower than proton shuttling. Multiscale Reactive Molecular Dynamics (MS-RMD)54−57 (and Multistate Empirical Valence Bond, MS-EVB, before it) was developed to efficiently and accurately capture the solvation and delocalization of an excess proton in water, such that the quantum-chemical nature of the hydrated proton can be studied in the context of membrane proteins over the long time scales needed for accurate simulation of such systems. MS-RMD has been successfully applied in several protein systems to predict and explain mechanisms of PT.33,34,58−65
In previous work,33,34 quantum mechanics/molecular mechanics (QM/MM) and MS-RMD were used to calculate potentials of mean force (PMFs; i.e., free energy profiles) of PT through the M2 channel in the +0 to +3 states, providing critical insight into the pH-dependent activation behavior and the role of the His37 tetrad in PT. Most recently, we further analyzed the MS-RMD simulations to explore the detailed interactions between the hydrated excess proton and the channel and found that the proton dynamically, as a function of its position, alters several properties of the protein and pore waters, including the hydrogen bonding network and the protein structure.65 This latter work illustrates how MS-RMD can be used successfully to investigate explicit, dynamic interactions between a hydrated proton and its immediate environment, as well as its indirect effects on other parts of the system. Here, we employ an approach similar to that in this previous work to focus specifically on properties related to drug binding and how the position of the bound drug relates to the overall PT mechanism. Through this analysis, we examine the hypothesis that the adamantyl amine drugs act as mechanism-based inhibitors—by identifying stabilizing interactions between the excess proton and the channel, we show how the drug may similarly be stabilized in support of this hypothesis. Additionally, by examining conformational fluctuations, we show that the drug binding pocket is an especially stable and symmetrical portion of the channel, conducive to binding a roughly spherical drug, and we reveal an additional minor hinge point toward the top of the channel which may be a relevant feature for future drug design efforts.
Methods
Simulations for calculating properties as the proton moves through the top of the channel were run as follows. Starting structures were taken from previous simulations, which were initiated from a crystal structure of the transmembrane portion of the M2 channel (this construct is referred to as M2TM) resolved at room temperature and high pH (PDB: 4QKL(66)) embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer solvated with water. M2TM is the minimum construct necessary to retain proton conduction similar to full-length M2,67 and it has been shown that the presence of amphipathic helices, included in the full-length M2 protein, do not significantly influence the PT mechanism.34 The water and excess proton in the system were modeled using the MS-RMD method, which allows bonds to break and form by taking a linear combination of different bonding topology states at each time step. The MS-EVB version 3.2 parameters68 were used to describe the hydrated excess proton. We refer the reader to previous work for a full description of the method.54−57 The excess proton center of excess charge (CEC) is defined as69
| 1 |
where 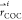 is the
center of charge of the ith diabatic MS-RMD state
and ci2 is the amplitude of that state. The
sum is over all N states. The interactions in the
remainder of the system were defined
by the CHARMM36 force field. Simulations were run in the NVT ensemble at 308.0 K using LAMMPS70 (http://lammps.sandia.gov) with
the MS-RMD package. The collective variable (CV) defined for umbrella
sampling (US) is the z-coordinate of the vector between
the excess proton CEC and the center of mass of the four Gly34 α-carbons,
as in our previous work, such that the CV has negative values at the
top (N-terminal end) of the protein and progresses to positive values
at the bottom (C-terminal end). Simulations were run with the excess
proton at every 0.5 Å along the CV coordinate between −18.0
and 1.0, generating 39 windows. To ensure that the proton remained
in the channel, a cylindrical restraint was added at 8 Å with
a force constant of 10 kcal/mol·Å2 using the
open-source, community-developed PLUMED library.71,72 After a 250 ps MS-RMD equilibration, the replica exchange umbrella
sampling73 technique was used to facilitate
convergence. Production simulations were run for ∼2–4
ns with frames saved every 1 ps.
is the
center of charge of the ith diabatic MS-RMD state
and ci2 is the amplitude of that state. The
sum is over all N states. The interactions in the
remainder of the system were defined
by the CHARMM36 force field. Simulations were run in the NVT ensemble at 308.0 K using LAMMPS70 (http://lammps.sandia.gov) with
the MS-RMD package. The collective variable (CV) defined for umbrella
sampling (US) is the z-coordinate of the vector between
the excess proton CEC and the center of mass of the four Gly34 α-carbons,
as in our previous work, such that the CV has negative values at the
top (N-terminal end) of the protein and progresses to positive values
at the bottom (C-terminal end). Simulations were run with the excess
proton at every 0.5 Å along the CV coordinate between −18.0
and 1.0, generating 39 windows. To ensure that the proton remained
in the channel, a cylindrical restraint was added at 8 Å with
a force constant of 10 kcal/mol·Å2 using the
open-source, community-developed PLUMED library.71,72 After a 250 ps MS-RMD equilibration, the replica exchange umbrella
sampling73 technique was used to facilitate
convergence. Production simulations were run for ∼2–4
ns with frames saved every 1 ps.
For calculating hydrogen bond residence times, longer independent trajectories were run with the CEC restrained in five different positions using US as described above. Each trajectory was run for 1.75–2 ps with frames saved every 10 fs.
Simulation frames were binned by excess proton CEC value for subsequent analyses, which were performed in Python74 using the SciPy,75 NumPy,76 and pandas77 libraries. For hydrogen bond analysis, values were averaged over the four helices. Hydrogen bonds were defined by the following criteria: the donor–acceptor distance must be less than 3.5 Å and the donor–hydrogen–acceptor angle must be greater than 150°. Several hydrogen bond definitions were tested and did not affect the conclusions (not shown). For calculating residence times, a hydrogen bond was considered in place as long as the particular water molecule remained the closest water to the protein atom and the hydrogen bond criteria were met.
Images of molecular structures were rendered in Visual Molecular Dynamics (VMD),78 while other figures were generated using Matplotlib.79
Results and Discussion
If the adamantyl amine drugs are acting as mechanism-based inhibitors as hypothesized, we would expect to see specific aspects of the PT mechanism taken advantage of or replicated by the drug upon drug binding. To test this, we performed MS-RMD simulations of M2 in the +0 His37 charge state with an explicit excess proton to evaluate the hydrogen bonding networks, pore shape, and protein fluctuations throughout PT that relate to drug binding. By focusing on PT in the +0 state, we are studying the process of proton entry and diffusion to His37 in the first key step of channel activation, paralleling inhibitor entry into the channel. We additionally expect this to be a prevalent charge state in drug-bound structures due to the lowered His37 pKa’s.80 Replica exchange umbrella sampling was used to obtain sufficient sampling of proton positions throughout the top portion of the channel, with windows from CECz = −18.0 to 1.0 Å where the coordinate origin is defined as the center of mass of the Gly34 α-carbons. The channel is aligned along the z-axis for all subsequent analyses.
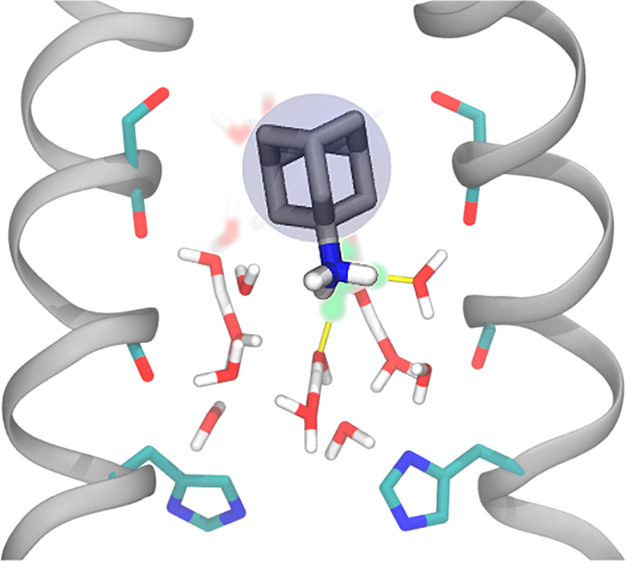
To understand how properties of PT may provide insight into drug binding, we primarily examine variations dependent on proton position. We compared the values of each property when the proton is at the drugs’ ammonium group positions versus other parts of the channel to determine if the drugs could be taking advantage of the channel’s natural ability to stabilize a proton. This idea is highlighted in Figure 1, which shows both the drug-bound crystal structure and a snapshot of a hydrated excess proton in the channel from our simulations. We refer to the drugs’ ammonium nitrogen position along the z-axis in the crystal structure as AmmNz. This value is −1.7 and −1.5 Å for the Inwardclosed amantadine and rimantadine bound structures, respectively (averaged over the two tetramers in each crystal structure).
Figure 1.

Comparison of M2 with amantadine and the hydrated excess proton. (left) X-ray crystal structure of amantadine-bound M2, PDB 6BKK. Water oxygens are shown in red. A blue circle around the adamantane group is depicted to reflect the spherical nature of this group. Hydrogens on the ammonium group of amantadine were added. (right) Snapshot from an MS-RMD trajectory with the most hydronium-like water indicated in green. In both, two opposing chains of M2 are shown in silver. The Ser31 and His37 side chains and the Gly34 and Ala30 backbone carbonyls are shown. The z-coordinate for the system is included on the left, where z = 0 Å is defined as the center of mass of the Gly34 α-carbons.
Flexible Hydrogen Bonds Stabilize the Excess Proton near AmmNz
It has been shown in our previous work that hydrogen bonds within the channel, including those between water and protein atoms, help facilitate proton transport by altering their direction and frequency of interaction as the proton moves through the channel. Here, we focus specifically on water interactions that may help account for excess charge stabilization near AmmNz. In Figure 2, we calculate the occupancy of three different hydrogen bonds between protein atoms and water as a function of the excess proton position in the channel. While the Ala30 hydrogen bond occupancy is constant as the excess proton enters and moves through the top of the channel, as it approaches the Ala30 carbonyls, the occupancy decreases ∼20%. This dip indicates the Ala30 hydrogen bonded waters can flexibly reduce their interaction with the protein as a result of an excess charge in their vicinity. Additionally, this dip is centered at −2.5 Å, near AmmNz. At this point, the role of the waters near Ala30 carbonyls in hydrating the proton is maximized. In a previously published PMF of the +0 His37 charge state,34 there is notably a local minimum near this point, further indicating that an excess charge is relatively stable here. This supports the hypothesis that amantadine and rimantadine are mechanism-based inhibitors and take advantage of the channel’s natural ability to stabilize a hydrated excess proton in order to stabilize the drug’s ammonium group.
Figure 2.
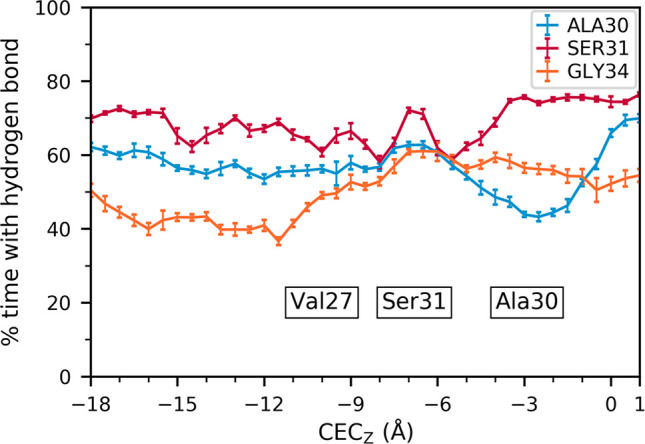
Hydrogen bond occupancy averages and error as a function of hydrated excess proton position between water and the backbone carbonyls of Ala30 and Gly34, and the side chain hydroxyl group of Ser31. Approximate average positions of Val27 side chains, Ser31 side chains, and Ala30 carbonyls are indicated.
The hydrogen bond occupancy of waters with the Gly34 carbonyls increases once the excess proton passes through the Val27 gate and remains fairly consistent across proton positions thereafter, exhibiting little dependence on the hydrated proton position once it is in the channel. The Ser31 side chain water occupancies are shown for comparison, which do not show a noticeable trend based on proton position. Thus, this change in interactions is not a universal effect throughout the channel, but the Ala30 waters seem to be uniquely flexible in this manner. These differences are consistent with drug design studies: while compounds such as spiro-adamantyl amine have been able to displace the water in the Ala30 layer, no designed inhibitors have displaced the water around Gly34.
To further understand how the dynamics of the hydrogen bond network may show how these drugs benefit from the channel’s inherent excess-charge stabilization used in proton transport, we examined the average residence times of hydrogen bonds between water and several important protein atoms. To do this, independent trajectories were run for five different excess proton positions, including two trajectories with the proton completely outside the channel (CECz = −24.0, 24.0 Å) and three when the proton is near AmmNz. These results are shown in Figure 3. The Ala30 water residence times slightly increase when the excess proton is near AmmNz, the Ser31 water residence times do not show any significant difference between the proton outside the channel and at AmmNz, and those of Gly34 waters decrease at AmmNz. The waters hydrogen bonded to His37 imidazole nitrogens show the greatest change in residence times and are shown to highlight the ability of this method to describe such differences.
Figure 3.
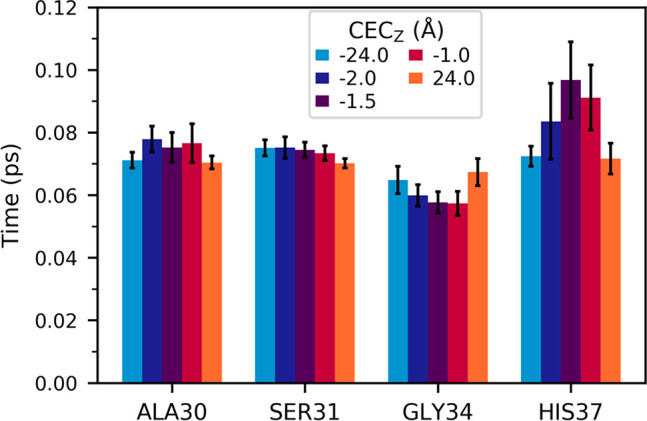
Average hydrogen bond residence times, calculated for the backbone carbonyls of Ala30, Ser31, and Gly34, and the unprotonated nitrogen of the His37 imidazole side chain. The quantities are averaged over the interactions of all four helices, and error bars were calculated using block averaging. Each bar represents the value calculated from one trajectory with the proton restrained at the labeled z-coordinate.
With the above results for Ala30, this may indicate that several waters remain tightly hydrogen bonded to the Ala30 backbone carbonyls, while one or more are bonded less frequently. While the Gly34 hydrogen bonds do not form less frequently (as indicated in Figure 2) with an excess charge in this region, they do exhibit greater dynamics and flexibility. This change indicates an increase in water dynamics when the excess proton is near, which could help stabilize and solvate the excess charge in the Ala30 water layer.
Taken together, these results further support the hypothesis that amantadine and rimantadine act as mechanism-based inhibitors: the channel acts as a scaffold to facilitate PT by harboring flexible protein–water interactions that can adapt and respond to a positive excess charge, with the specific ability to stabilize an excess proton near AmmNz.
Drug Tilt Positions Ammonium Group in Highest CEC Density
One prominent characteristic of the amantadine and rimantadine bound structures is the drug’s tilt within the pore. This tilted conformation is also seen in solid-state NMR studies.81 Given the drug’s threefold symmetry in a fourfold symmetric channel, the ammonium group cannot form hydrogen bonds with all four waters hydrogen bonded to Ala30, leading in part to this tilt. Based on our previous work examining the proton’s path through the channel, we used a similar analysis to examine the density of CEC positions when the excess proton is near the ammonium position in drug-bound structures. Figure 4A shows the difference in CEC density when the proton is near AmmNz compared with the average over all proton positions through the top portion of the channel. This two-dimensional histogram of CEC positions in the xy-plane is calculated for CECz = −1.7 ± 0.2 Å, minus the average over all normalized histograms for CECz positions [−18.0, 1.0] Å binned every 0.2 Å. Interestingly, in this portion of the channel the excess proton prefers to be near the edge of the pore, unlike the predominant preference for the center of the pore throughout the rest of the channel, as indicated by the positive values around the edge and negative values in the center. Figure 4B shows the radial density of the CEC in this same region of the channel. Possible positions of amantadine’s ammonium group nitrogens were calculated based on the drug’s position and tilt in the crystal structure, and their radii are included as dashed lines (these positions were also used to generate the image in Figure 1). These hydrogens can extend to a radius of ∼1.5 Å in this static crystal structure, which means that the slightly off-centered ammonium group directly positions one to two of its hydrogens in the region of the CEC’s highest density. The CEC’s propensity for the edge of the pore indicates that the drug’s tilt in the channel may not only be a necessary component of its binding, but also a thermodynamic advantage. This tilt further allows the drug to act as a hydronium mimic, as the hydrogens of the ammonium group are in the favorable positions of the solvated excess proton.
Figure 4.

(A) Difference in hydrated excess CEC density in the xy-plane when the CEC is located within CECz = −1.7 ± 0.2 Å compared with the average CEC density over all CEC positions in the range z = [−18.0, 1.0]. (B) Average radial density of the proton when CECz = −1.7 ± 0.2 Å. One possible set of positions of amantadine’s ammonium group hydrogens calculated from the drug-bound crystal structure are indicated by blue dashed lines.
The analysis of hydrogen bonding changes and proton densities indicates how the ammonium group is a functional addition to the adamantane scaffold, as the charged group is positioned in a region where the channel is especially adept to stabilize an excess charge. This stabilization relies on flexible water structures and hydrogen bond interactions that can undergo minor changes to accommodate the proton, suggesting that the adamantyl amine inhibitors are acting as hydronium mimics: they take advantage of these inherent features to help solvate the charged ammonium group. The identification of other regions of the channel with increased ability to stabilize an excess charge, such as areas of increased proton density or significantly flexible water interactions, could help provide new targets for drugs to act as hydronium mimics.
Pore Shape and Stability Near Ser31 Are Ideal for Adamantane Binding
Another hypothesis about the adamantyl amine class of inhibitors is that adamantane is effectively spherical and can freely rotate within the channel, but has no rotatable bonds, which minimizes the entropy lost upon binding. This rapid rotation can be seen on the NMR time scale81 and is consistent with the recent Thomaston et al. crystallographic studies, in which the motion was indirectly inferred. Nevertheless, its significance depends on the dynamic nature of the channel: if the protein exhibits great structural fluctuations in the region where the drug binds, then drug binding may induce changes that greatly decrease the entropy and this hypothesis would not fully explain the drugs’ efficacy. To better understand how the channel’s natural dynamics may lend itself to favorable drug binding, we examined the pore shape throughout our trajectories. As an estimate of the asymmetry of the channel, we calculated the eccentricity, which is essentially a measure of how “circular” a given oval is. The eccentricity is defined as
| 2 |
where a and b are the semimajor and semiminor axes, respectively, which we approximate by the distance between α-carbons on opposing helices. A schematic of this is shown in SI Figure 1. Eccentricity can have values between 0 and 1, with 0 indicating a circle and 1 indicating a parabola.
These results are shown in Figure 5. Eccentricity maximum, minimum, and root-mean-square-deviation (RMSD) values are calculated in SI Table 1. The pore-lining residues in the bottom half of the channel, Gly34, His37, and Trp41, all show a greater degree of asymmetry and a wider range of eccentricity values, dependent on the excess proton position, than the pore-lining residues in the top part of the channel. Interestingly, the proton entry at CECz = −17 Å has a pronounced effect on the channel near Trp41, greater than that when the proton nears the center of the channel. Ser31, however, has overall the smallest average eccentricity and the lowest minimum value than the other pore-lining residues during PT in this portion of the channel. Additionally, Ser31 and Val27 have smaller proton position dependent changes in eccentricity than the pore-lining residues in the bottom half of the channel. This result indicates that the Ser31 region is the most symmetrical and stable in the channel.
Figure 5.

Average and standard deviation of the pore eccentricity estimated by α-carbon distances of pore-lining residues as a function of the hydrated excess proton CEC position.
While analyzing the α-carbon distances and eccentricity, we also examined the correlation between these α-carbon distances on opposing helices, shown in Figure 6, to gain further insight into protein motion and conformational fluctuations on the nanosecond time scale. These motions captured here are equilibrium fluctuations in the +0, Inwardclosed state, not necessarily motions driving the transition between Inwardopen and Inwardclosed. The calculated correlations indicate that the channel’s equilibrium structural fluctuations are dominated by alternating inward–outward motions of opposing helices. At each pore-lining residue, the distances are negatively correlated: that is, when helices A and C move farther apart, helices B and D move closer together, and vice versa.
Figure 6.
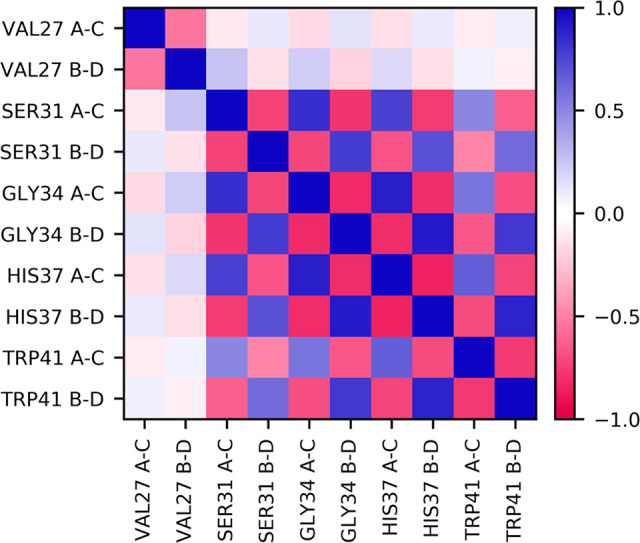
Pearson correlation coefficients of the distances between α-carbons on opposing helices, for all pore-lining residues, when excess proton CECz = −18.0 Å. Each row and column correspond to a specific residue and distance, as labeled, where “A–C” is the distance between the α-carbons of helices 1 and 3 and “B–D” is that of helices 2 and 4. Only those values with a p-value of <0.05 are shown; any other values are set to 0.0.
Gly34 is known to be the hinge point whose kinking controls the large structural change between inward-open and inward-closed conformations, which may falsely lead to the conclusion that the conformational fluctuations at equilibrium above and below Gly34 are decorrelated, with a stable core centered at Gly34. Interestingly, however, the motions at Gly34 are strongly and similarly correlated with the motions at both Ser31 and His37. This correlation indicates that, in this fixed charge state, the Gly34 kink is relatively rigid. Instead, there is a noticeable lack of correlation between Val27 and the other pore-lining residues, suggesting that there is a secondary, minor “hinge” between Val27 and Ser31 that decorrelates the inward–outward motions between the helices above and below this point. This natural hinge observed near Val27 furthers our understanding of Val27 acting as a secondary gate that opens to allow proton and water entry into the channel.82,83 In our simulations, this valve can readily hydrate, particularly in the presence of a nearby excess proton. Moreover, it is frequently closed, which may make passage of a hydrated sodium or chloride ion more difficult. This aspect of the Val27 gate and its relevance for PT and proton selectivity is likely an important feature of the M2 channel and could be further explored in future work.
Given that the adamantane group of the drug is centered in the Ser31 tetrad plane, we hypothesize that these facets of the channel’s dynamics are critical for fully explaining the drugs’ favorable binding. Because of the more circular shape of the pore at the Ser31 tetrad, the spherical adamantane group can fit snugly under the hydrophobic Val27 cleft and block PT. Additionally, the relative stability of the pore in the region of drug binding helps explain why drug binding is thermodynamically favorable. Because the channel exhibits smaller structural fluctuations here than in other regions of the channel, the adamantane-based drugs are able to bind with minimal loss of entropy as the channel does not need to lose flexibility to create a stable drug-binding interface. This has important implications for designing drugs that use scaffolds different from the adamantane group39,84−89 or that interact with drug-resistant mutants such as S31N. While drug binding to more flexible regions of the channel is possible, only modest changes in potency are often observed despite large changes in the size of the drugs. This is likely because of the need to counter the greater loss of entropy resultant from structural changes and reduced fluctuations.
One limitation of this study, however, is the homogeneous POPC bilayer, which is commonly used in experiments and is standard in computational studies but does not capture the complexity of the viral membrane and may influence channel dynamics.90 Thus, these features need to be examined in the more complex membrane to fully understand their physiological relevance.
Conclusions
Altogether, we have shown how the adamantyl amine inhibitors of M2 are suited to exploit various inherent features of the M2 channel that naturally facilitate proton transport, further supporting the claim that they function as mechanism-based inhibitors. The flexible hydrogen bond interactions, measured in both hydrogen bond occupancy and residence times, indicate how the channel is suited to stabilizing an excess charge near AmmNz. Thus, the ammonium group of these inhibitors can act as a hydronium mimic by binding in this region. We also analyzed the pore shape throughout the channel by calculating the eccentricity of the pore based on α-carbon distances. The results from these calculations indicate that the drug binding pocket is an especially stable and symmetrical portion of the channel, conducive to binding a roughly spherical drug. Finally, by examining the correlations between these distances, we found an additional minor hinge point toward the top of the channel which may be a relevant feature for future drug design efforts.
Understanding these features as they relate to drug binding gives further insight into the specific interactions that stabilize the adamantyl amine inhibitors in wild-type M2, and these results suggests that this approach could be used to aid future drug-design efforts to methodically create new inhibitors for S31N mutants. With the recent publication of high resolution influenza B M2 (BM2) structures,91 it is possible to conduct similar studies to elucidate the detailed PT mechanism in BM2 to guide drug design in this functionally similar protein.
This work also shows how similar analyses to understand the details of explicit proton transport mechanisms (not those inferred by water structures alone) could be used in other systems and extended to ion transporters such as the SARS-CoV-2 viroporins, to help inform mechanism-based inhibitor design. Elucidating the inherent features of drug-targetable proton transporters, such as flexible water and hydrogen bonding interactions, preferred proton positions, dynamic pore shapes, and structural fluctuations, can help guide the design of drug scaffolds and added substituents. The MS-RMD simulation methodology utilized in this work has made these studies possible both for M2 and for other important drug targets.
Acknowledgments
The personnel in this research were supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH Grant R01 GM053148). The authors acknowledge The University of Chicago Research Computing Center and the U.S. Department of Defense High Performance Computing Modernization Program for providing computing resources. L.C.W. received funding from a Department of Energy (DOE) Computational Science Graduate Fellowship under Grant DE-FG02-97ER25308.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c06419.
Schematic of eccentricity calculations; average, maximum, minimum, and RMSD values for eccentricity (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Decoursey T. E. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 2003, 83 (2), 475–579. 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- Deamer D. W. Proton permeation of lipid bilayers. J. Bioenerg. Biomembr. 1987, 19 (5), 457–479. 10.1007/BF00770030. [DOI] [PubMed] [Google Scholar]
- Maloney P. C.; Kashket E. R.; Wilson T. H. A protonmotive force drives ATP synthesis in bacteria. Proc. Natl. Acad. Sci. U. S. A. 1974, 71 (10), 3896–900. 10.1073/pnas.71.10.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–8. 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- Stryer L. In Biochemistry; W. H. Freeman & Co.: New York, 1988; Chapters 17 and 22. [Google Scholar]
- Wraight C. A. Chance and design--proton transfer in water, channels and bioenergetic proteins. Biochim. Biophys. Acta, Bioenerg. 2006, 1757 (8), 886–912. 10.1016/j.bbabio.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Aoi W.; Marunaka Y. Importance of pH homeostasis in metabolic health and diseases: crucial role of membrane proton transport. BioMed Res. Int. 2014, 2014, 598986. 10.1155/2014/598986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs G. Proton pump inhibitors and acid-related diseases. Pharmacotherapy 1997, 17 (1), 22–37. 10.1002/j.1875-9114.1997.tb03675.x. [DOI] [PubMed] [Google Scholar]
- Poet M.; Kornak U.; Schweizer M.; Zdebik A. A.; Scheel O.; Hoelter S.; Wurst W.; Schmitt A.; Fuhrmann J. C.; Planells-Cases R.; Mole S. E.; Hubner C. A.; Jentsch T. J. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (37), 13854–9. 10.1073/pnas.0606137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . Influenza (Seasonal). https://www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal) (accessed 2020-03-16).
- World Health Organization . Coronavirus disease (COVID-2019) Situation Report - 143. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200611-covid-19-sitrep-143.pdf (accessed 2020-06-11).
- Pinto L. H.; Lamb R. A. The M2 proton channels of influenza A and B viruses. J. Biol. Chem. 2006, 281 (14), 8997–9000. 10.1074/jbc.R500020200. [DOI] [PubMed] [Google Scholar]
- Pinto L. H.; Holsinger L. J.; Lamb R. A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69 (3), 517–28. 10.1016/0092-8674(92)90452-I. [DOI] [PubMed] [Google Scholar]
- Chizhmakov I. V.; Geraghty F. M.; Ogden D. C.; Hayhurst A.; Antoniou M.; Hay A. J. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J. Physiol. 1996, 494 (2), 329–336. 10.1113/jphysiol.1996.sp021495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Takeuchi K.; Pinto L. H.; Lamb R. A. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J. Virol. 1993, 67 (9), 5585–94. 10.1128/JVI.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M. L. The Next Wave of Influenza Drugs. ACS Infect. Dis. 2017, 3 (10), 691–694. 10.1021/acsinfecdis.7b00142. [DOI] [PubMed] [Google Scholar]
- Dong G.; Peng C.; Luo J.; Wang C.; Han L.; Wu B.; Ji G.; He H. Adamantane-resistant influenza a viruses in the world (1902–2013): frequency and distribution of M2 gene mutations. PLoS One 2015, 10 (3), e0119115. 10.1371/journal.pone.0119115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musharrafieh R.; Ma C.; Wang J. Discovery of M2 channel blockers targeting the drug-resistant double mutants M2-S31N/L26I and M2-S31N/V27A from the influenza A viruses. Eur. J. Pharm. Sci. 2020, 141, 105124. 10.1016/j.ejps.2019.105124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odnovorov A.; Garaev T.; Grebennikova T.; Pleteneva T. Prospects for Specific Influenza Treatment. Syst. Rev. Pharm. 2020, 11 (2), 242–248. 10.5530/srp.2020.2.37. [DOI] [Google Scholar]
- Scott C.; Griffin S. Viroporins: structure, function and potential as antiviral targets. J. Gen. Virol. 2015, 96 (8), 2000–2027. 10.1099/vir.0.000201. [DOI] [PubMed] [Google Scholar]
- Wilson L.; McKinlay C.; Gage P.; Ewart G. SARS coronavirus E protein forms cation-selective ion channels. Virology 2004, 330 (1), 322–31. 10.1016/j.virol.2004.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W.; Zheng B. J.; Xu K.; Schwarz W.; Du L.; Wong C. K.; Chen J.; Duan S.; Deubel V.; Sun B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (33), 12540–5. 10.1073/pnas.0605402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa E.; Merhi G.; Panossian B.; Salloum T.; Tokajian S. SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 2020, 5 (3), e00266. 10.1128/mSystems.00266-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Lamb R. A.; Pinto L. H. Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue. Biophys. J. 1995, 69 (4), 1363–71. 10.1016/S0006-3495(95)80003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Fu R.; Nishimura K.; Zhang L.; Zhou H. X.; Busath D. D.; Vijayvergiya V.; Cross T. A. Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (18), 6865–70. 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman P.; Lamb R. A.; Pinto L. H. Chemical rescue of histidine selectivity filter mutants of the M2 ion channel of influenza A virus. J. Biol. Chem. 2005, 280 (22), 21463–72. 10.1074/jbc.M412406200. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Zaitseva F.; Lamb R. A.; Pinto L. H. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J. Biol. Chem. 2002, 277 (42), 39880–6. 10.1074/jbc.M206582200. [DOI] [PubMed] [Google Scholar]
- Li C.; Qin H.; Gao F. P.; Cross T. A. Solid-state NMR characterization of conformational plasticity within the transmembrane domain of the influenza A M2 proton channel. Biochim. Biophys. Acta, Biomembr. 2007, 1768 (12), 3162–70. 10.1016/j.bbamem.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell J. R.; Chou J. J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451 (7178), 591–5. 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F.; Luo W.; Hong M. Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR. Science 2010, 330 (6003), 505–8. 10.1126/science.1191714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F.; Luo W.; Cady S. D.; Hong M. Conformational plasticity of the influenza A M2 transmembrane helix in lipid bilayers under varying pH, drug binding, and membrane thickness. Biochim. Biophys. Acta, Biomembr. 2011, 1808 (1), 415–23. 10.1016/j.bbamem.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco M. L.; Hansen U. P.; Thiel G.; Moroni A.; Schroeder I. Effect of cytosolic pH on inward currents reveals structural characteristics of the proton transport cycle in the influenza A protein M2 in cell-free membrane patches of Xenopus oocytes. PLoS One 2014, 9 (9), e107406. 10.1371/journal.pone.0107406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R.; Li H.; Swanson J. M.; Voth G. A. Multiscale simulation reveals a multifaceted mechanism of proton permeation through the influenza A M2 proton channel. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (26), 9396–401. 10.1073/pnas.1401997111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R.; Swanson J. M. J.; Madsen J. J.; Hong M.; DeGrado W. F.; Voth G. A. Acid activation mechanism of the influenza A M2 proton channel. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (45), E6955–E6964. 10.1073/pnas.1615471113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalily P. H.; Duncan M. C.; Fedida D.; Wang J.; Tietjen I. Put a cork in it: Plugging the M2 viral ion channel to sink influenza. Antiviral Res. 2020, 178, 104780. 10.1016/j.antiviral.2020.104780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Hu Y.; Xu S.; Zhang Y.; Musharrafieh R.; Hau R. K.; Ma C.; Wang J. In Vitro Pharmacokinetic Optimizations of AM2-S31N Channel Blockers Led to the Discovery of Slow-Binding Inhibitors with Potent Antiviral Activity against Drug-Resistant Influenza A Viruses. J. Med. Chem. 2018, 61 (3), 1074–1085. 10.1021/acs.jmedchem.7b01536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barniol-Xicota M.; Gazzarrini S.; Torres E.; Hu Y.; Wang J.; Naesens L.; Moroni A.; Vazquez S. Slow but Steady Wins the Race: Dissimilarities among New Dual Inhibitors of the Wild-Type and the V27A Mutant M2 Channels of Influenza A Virus. J. Med. Chem. 2017, 60 (9), 3727–3738. 10.1021/acs.jmedchem.6b01758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon N. A.; McGuire K. L.; Wallentine S. K.; Mohl G. A.; Lynch J. D.; Harrison R. G.; Busath D. D. Divalent copper complexes as influenza A M2 inhibitors. Antiviral Res. 2017, 147, 100–106. 10.1016/j.antiviral.2017.10.009. [DOI] [PubMed] [Google Scholar]
- Duque M. D.; Torres E.; Valverde Murillo E.; Barniol-Xicota M.; Guardiola S.; Rey-Carrizo M.; Vazquez S.. Inhibitors of the M2 channel of influenza A virus. In Recent Advances in Pharmaceutical Sciences; Muñoz-Torrero D., Ed.; Transworld Research Network: 2011; p 35. [Google Scholar]
- Wang J.; Ma C.; Balannik V.; Pinto L. H.; Lamb R. A.; Degrado W. F. Exploring the Requirements for the Hydrophobic Scaffold and Polar Amine in inhibitors of M2 from Influenza A Virus. ACS Med. Chem. Lett. 2011, 2 (4), 307–312. 10.1021/ml100297w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.; Zeng S.; Li C.; Jie Y.; Li Z.; Chen L. Identification of hits as matrix-2 protein inhibitors through the focused screening of a small primary amine library. J. Med. Chem. 2010, 53 (9), 3831–4. 10.1021/jm901664a. [DOI] [PubMed] [Google Scholar]
- Drakopoulos A.; Tzitzoglaki C.; McGuire K.; Hoffmann A.; Konstantinidi A.; Kolokouris D.; Ma C.; Freudenberger K.; Hutterer J.; Gauglitz G.; Wang J.; Schmidtke M.; Busath D. D.; Kolocouris A. Unraveling the Binding, Proton Blockage, and Inhibition of Influenza M2 WT and S31N by Rimantadine Variants. ACS Med. Chem. Lett. 2018, 9 (3), 198–203. 10.1021/acsmedchemlett.7b00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomaston J. L.; Polizzi N. F.; Konstantinidi A.; Wang J.; Kolocouris A.; DeGrado W. F. Inhibitors of the M2 Proton Channel Engage and Disrupt Transmembrane Networks of Hydrogen-Bonded Waters. J. Am. Chem. Soc. 2018, 140 (45), 15219–15226. 10.1021/jacs.8b06741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Ma C.; Fiorin G.; Carnevale V.; Wang T.; Hu F.; Lamb R. A.; Pinto L. H.; Hong M.; Klein M. L.; DeGrado W. F. Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. J. Am. Chem. Soc. 2011, 133 (32), 12834–41. 10.1021/ja204969m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nguyen H.; Nguyen H. T.; Le L. T. Investigation of the free energy profiles of amantadine and rimantadine in the AM2 binding pocket. Eur. Biophys. J. 2016, 45 (1), 63–70. 10.1007/s00249-015-1077-y. [DOI] [PubMed] [Google Scholar]
- Llabres S.; Juarez-Jimenez J.; Masetti M.; Leiva R.; Vazquez S.; Gazzarrini S.; Moroni A.; Cavalli A.; Luque F. J. Mechanism of the Pseudoirreversible Binding of Amantadine to the M2 Proton Channel. J. Am. Chem. Soc. 2016, 138 (47), 15345–15358. 10.1021/jacs.6b07096. [DOI] [PubMed] [Google Scholar]
- Homeyer N.; Ioannidis H.; Kolarov F.; Gauglitz G.; Zikos C.; Kolocouris A.; Gohlke H. Interpreting Thermodynamic Profiles of Aminoadamantane Compounds Inhibiting the M2 Proton Channel of Influenza A by Free Energy Calculations. J. Chem. Inf. Model. 2016, 56 (1), 110–26. 10.1021/acs.jcim.5b00467. [DOI] [PubMed] [Google Scholar]
- Gianti E.; Carnevale V.; DeGrado W. F.; Klein M. L.; Fiorin G. Hydrogen-bonded water molecules in the M2 channel of the influenza A virus guide the binding preferences of ammonium-based inhibitors. J. Phys. Chem. B 2015, 119 (3), 1173–83. 10.1021/jp506807y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonov H.; Astrahan P.; Krugliak M.; Arkin I. T. How do aminoadamantanes block the influenza M2 channel, and how does resistance develop?. J. Am. Chem. Soc. 2011, 133 (25), 9903–11. 10.1021/ja202288m. [DOI] [PubMed] [Google Scholar]
- Ioannidis H.; Drakopoulos A.; Tzitzoglaki C.; Homeyer N.; Kolarov F.; Gkeka P.; Freudenberger K.; Liolios C.; Gauglitz G.; Cournia Z.; Gohlke H.; Kolocouris A. Alchemical Free Energy Calculations and Isothermal Titration Calorimetry Measurements of Aminoadamantanes Bound to the Closed State of Influenza A/M2TM. J. Chem. Inf. Model. 2016, 56 (5), 862–76. 10.1021/acs.jcim.6b00079. [DOI] [PubMed] [Google Scholar]
- Knight C.; Voth G. A. The curious case of the hydrated proton. Acc. Chem. Res. 2012, 45 (1), 101–9. 10.1021/ar200140h. [DOI] [PubMed] [Google Scholar]
- de Grotthuss C. J. T. Sur la décomposition de l’eau et des corps qu’elle tient en dissolution á l’aide de l’électricité galvanique. Ann. Chim. 1806, LVIII, 54–74. [Google Scholar]
- Agmon N. The Grotthuss Mechanism. Chem. Phys. Lett. 1995, 244, 456–462. 10.1016/0009-2614(95)00905-J. [DOI] [Google Scholar]
- Knight C.; Lindberg G. E.; Voth G. A. Multiscale reactive molecular dynamics. J. Chem. Phys. 2012, 137 (22), 22A525. 10.1063/1.4743958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T.; Peng Y.; Knight C.; Voth G. A. Computationally Efficient Multiconfigurational Reactive Molecular Dynamics. J. Chem. Theory Comput. 2012, 8 (12), 4863–4875. 10.1021/ct3006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J. G.; Peng Y.; Silverstein D. W.; Swanson J. M. Multiscale Reactive Molecular Dynamics for Absolute pKa Predictions and Amino Acid Deprotonation. J. Chem. Theory Comput. 2014, 10 (7), 2729–2737. 10.1021/ct500250f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Liang R.; Voth G. A.; Swanson J. M. Computationally Efficient Multiscale Reactive Molecular Dynamics to Describe Amino Acid Deprotonation in Proteins. J. Chem. Theory Comput. 2016, 12 (2), 879–91. 10.1021/acs.jctc.5b01109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Swanson J. M.; Voth G. A. Multiscale Simulations Reveal Key Aspects of the Proton Transport Mechanism in the ClC-ec1 Antiporter. Biophys. J. 2016, 110 (6), 1334–45. 10.1016/j.bpj.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R.; Swanson J. M.; Peng Y.; Wikstrom M.; Voth G. A. Multiscale simulations reveal key features of the proton-pumping mechanism in cytochrome c oxidase. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (27), 7420–5. 10.1073/pnas.1601982113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Mayes H. B.; Swanson J. M.; Voth G. A. The Origin of Coupled Chloride and Proton Transport in a Cl(−)/H(+) Antiporter. J. Am. Chem. Soc. 2016, 138 (45), 14923–14930. 10.1021/jacs.6b06683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R.; Swanson J. M. J.; Wikstrom M.; Voth G. A. Understanding the essential proton-pumping kinetic gates and decoupling mutations in cytochrome c oxidase. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (23), 5924–5929. 10.1073/pnas.1703654114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J. L.; Li C.; Brinth A.; Wang Z.; Vogeley L.; Solcan N.; Ledderboge-Vucinic G.; Swanson J. M. J.; Caffrey M.; Voth G. A.; Newstead S. Proton movement and coupling in the POT family of peptide transporters. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (50), 13182–13187. 10.1073/pnas.1710727114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Swanson J. M. J.; Voth G. A. Modulating the Chemical Transport Properties of a Transmembrane Antiporter via Alternative Anion Flux. J. Am. Chem. Soc. 2018, 140 (48), 16535–16543. 10.1021/jacs.8b07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayes H. B.; Lee S.; White A. D.; Voth G. A.; Swanson J. M. J. Multiscale Kinetic Modeling Reveals an Ensemble of Cl(−)/H(+) Exchange Pathways in ClC-ec1 Antiporter. J. Am. Chem. Soc. 2018, 140 (5), 1793–1804. 10.1021/jacs.7b11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins L. C.; Liang R.; Swanson J. M. J.; DeGrado W. F.; Voth G. A. Proton-Induced Conformational and Hydration Dynamics in the Influenza A M2 Channel. J. Am. Chem. Soc. 2019, 141 (29), 11667–11676. 10.1021/jacs.9b05136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomaston J. L.; Alfonso-Prieto M.; Woldeyes R. A.; Fraser J. S.; Klein M. L.; Fiorin G.; DeGrado W. F. High-resolution structures of the M2 channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (46), 14260–5. 10.1073/pnas.1518493112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Polishchuk A. L.; Ohigashi Y.; Stouffer A. L.; Schon A.; Magavern E.; Jing X.; Lear J. D.; Freire E.; Lamb R. A.; DeGrado W. F.; Pinto L. H. Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (30), 12283–8. 10.1073/pnas.0905726106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas R.; Tse Y. L.; Tokmakoff A.; Voth G. A. Role of Presolvation and Anharmonicity in Aqueous Phase Hydrated Proton Solvation and Transport. J. Phys. Chem. B 2016, 120 (8), 1793–804. 10.1021/acs.jpcb.5b09466. [DOI] [PubMed] [Google Scholar]
- Day T. J. F.; Soudackov A. V.; Cuma M.; Schmitt U. W.; Voth G. A. A second generation multistate empirical valence bond model for proton transport in aqueous systems. J. Chem. Phys. 2002, 117 (12), 5839–5849. 10.1063/1.1497157. [DOI] [Google Scholar]
- Plimpton S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117 (1), 1–19. 10.1006/jcph.1995.1039. [DOI] [Google Scholar]
- Bonomi M.; Bussi G.; Camilloni C.; et al. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16 (8), 670–673. 10.1038/s41592-019-0506-8. [DOI] [PubMed] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185 (2), 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Sugita Y.; Kitao A.; Okamoto Y. Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys. 2000, 113 (15), 6042–6051. 10.1063/1.1308516. [DOI] [Google Scholar]
- Rossum G.Python Reference Manual; CWI (Centre for Mathematics and Computer Science): 1995. [Google Scholar]
- Virtanen P.; Gommers R.; Oliphant T. E.; Haberland M.; Reddy T.; Cournapeau D.; Burovski E.; Peterson P.; Weckesser W.; Bright J.; van der Walt S. J.; Brett M.; Wilson J.; Millman K. J.; Mayorov N.; Nelson A. R. J.; Jones E.; Kern R.; Larson E.; Carey C. J.; Polat I.; Feng Y.; Moore E. W.; VanderPlas J.; Laxalde D.; Perktold J.; Cimrman R.; Henriksen I.; Quintero E. A.; Harris C. R.; Archibald A. M.; Ribeiro A. H.; Pedregosa F.; van Mulbregt P.; et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17 (3), 261–272. 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant T. E.A Guide to NumPy; Trelgol Publishing: 2006. [Google Scholar]
- McKinney W.Data Structures for Statistical Computing in Python. Proceedings of the Ninth Python in Science Conference (SciPy 2010); SciPy: 2010; p 56. 10.25080/Majora-92bf1922-00a. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. {VMD} -- {V}isual {M}olecular {D}ynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Hunter J. D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9 (3), 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- Hu J.; Fu R.; Cross T. A. The chemical and dynamical influence of the anti-viral drug amantadine on the M2 proton channel transmembrane domain. Biophys. J. 2007, 93 (1), 276–83. 10.1529/biophysj.106.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady S. D.; Schmidt-Rohr K.; Wang J.; Soto C. S.; Degrado W. F.; Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010, 463 (7281), 689–92. 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana E.; Devane R. H.; Dal Peraro M.; Klein M. L. Computational study of drug binding to the membrane-bound tetrameric M2 peptide bundle from influenza A virus. Biochim. Biophys. Acta, Biomembr. 2011, 1808 (2), 530–7. 10.1016/j.bbamem.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M.; Cross T. A.; Zhou H. X. A secondary gate as a mechanism for inhibition of the M2 proton channel by amantadine. J. Phys. Chem. B 2008, 112 (27), 7977–9. 10.1021/jp800171m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy N. S.; Poongavanam V.; Pratheepa V. Viral M2 ion channel protein: a promising target for anti-influenza drug discovery. Mini Rev. Med. Chem. 2014, 14 (10), 819–830. [PubMed] [Google Scholar]
- Duque M. D.; Ma C.; Torres E.; Wang J.; Naesens L.; Juarez-Jimenez J.; Camps P.; Luque F. J.; DeGrado W. F.; Lamb R. A.; Pinto L. H.; Vazquez S. Exploring the size limit of templates for inhibitors of the M2 ion channel of influenza A virus. J. Med. Chem. 2011, 54 (8), 2646–57. 10.1021/jm101334y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Zhang Z. W.; Cui W.; Chen S. W.; Zhou Y.; Dong J. H.; Jie Y. L.; Wan J. T.; Xu Y.; Hu W. H. Identification of camphor derivatives as novel M2 ion channel inhibitors of influenza A virus. MedChemComm 2015, 6 (4), 727–731. 10.1039/C4MD00515E. [DOI] [Google Scholar]
- Zhao X.; Jie Y.; Rosenberg M. R.; Wan J.; Zeng S.; Cui W.; Xiao Y.; Li Z.; Tu Z.; Casarotto M. G.; Hu W. Design and synthesis of pinanamine derivatives as anti-influenza A M2 ion channel inhibitors. Antiviral Res. 2012, 96 (2), 91–9. 10.1016/j.antiviral.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Rey-Carrizo M.; Torres E.; Ma C.; Barniol-Xicota M.; Wang J.; Wu Y.; Naesens L.; DeGrado W. F.; Lamb R. A.; Pinto L. H.; Vazquez S. 3-Azatetracyclo[5.2.1.1(5,8).0(1,5)]undecane derivatives: from wild-type inhibitors of the M2 ion channel of influenza A virus to derivatives with potent activity against the V27A mutant. J. Med. Chem. 2013, 56 (22), 9265–74. 10.1021/jm401340p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Hu Y.; Wang Y.; Ma C.; Wang J. Expeditious Lead Optimization of Isoxazole-Containing Influenza A Virus M2-S31N Inhibitors Using the Suzuki-Miyaura Cross-Coupling Reaction. J. Med. Chem. 2017, 60 (4), 1580–1590. 10.1021/acs.jmedchem.6b01852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong-Ly K. C.; Nanda V.; Degrado W. F.; Howard K. P. The conformation of the pore region of the M2 proton channel depends on lipid bilayer environment. Protein Sci. 2005, 14 (4), 856–61. 10.1110/ps.041185805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandala V. S.; Loftis A. R.; Shcherbakov A. A.; Pentelute B. L.; Hong M. Atomic structures of closed and open influenza B M2 proton channel reveal the conduction mechanism. Nat. Struct. Mol. Biol. 2020, 27 (2), 160–167. 10.1038/s41594-019-0371-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.