

Abstract

Chiral auxiliaries and asymmetric catalysis are the workhorses of enantioselective transformations, but they still remain limited in terms of either efficiency or generality. Herein, we present an alternative strategy for controlling the stereoselectivity of chemical reactions. Asymmetric catalysis is used to install a transient chiral auxiliary starting from achiral precursors, which then directs diastereoselective reactions. We apply this strategy to a palladium-catalyzed carboetherification/hydrogenation sequence on propargylic amines, providing fast access to enantioenriched chiral amino alcohols, important building blocks for medicinal chemistry and drug discovery. All stereoisomers of the product could be accessed by the choice of ligand and substituent on the propargylic amine, leading to a stereodivergent process.
Currently, most enantioselective transformations rely on two strategies: (i) the use of chiral auxiliaries1 and (ii) asymmetric catalysis.2 The former allows the development of general and robust processes, but requires stoichiometric amounts of enantiopure precursors and multistep procedures. By contrast, asymmetric catalysis relies only on substoichiometric amounts of enantiopure molecules, but it generally requires an intensive optimization at the expense of robustness and generality. To overcome these limitations, we envisioned a catalytic enantioselective method, which would introduce a chiral auxiliary on the substrate from a cheap nonchiral tether in a synthetic useful step (Scheme 1A). This process would require only a catalytic amount of enantiopure species while providing a robust platform for further diastereoselective functionalizations, benefiting from the best aspects of the two traditional strategies. To the best of our knowledge, such an approach has not yet been realized, although different methods for improving asymmetric synthesis have been developed. A seminal work based on the formation of chiral aminals is the “self-reproduction of chirality” reported by Seebach for the stereoselective synthesis of amino acids. In this work, the existing stereocenter on the amino acid first controls the diastereoselective formation of the aminal by condensation with an aldehyde. The latter then shields one face of the enolate.3,4 As another example based on an internal chirality transfer, Maulide and co-workers recently reported a redox-neutral coupling of alkenes and aldehydes via a “catch–release” tethering approach (Scheme 1B).5 However, the resulting functional group (a ketone) remains in the product. Other researchers have worked on the concept of “transient chiral auxiliaries/tethers”, which are easy to install and remove.6−10 For example, Beauchemin and co-workers have used chiral aldehydes in substoichiometric amounts for the Cope-type hydroamination of allyl amines (Scheme 1C).11 However, the scope of these transformations remains limited, and auxiliaries available from the chiral pool are generally required.
Scheme 1. (A) Our Concept: Catalytically Formed Chiral Auxiliaries; (B) Chirality Transfer via Tethering; (C) Transient Chiral Auxiliaries Introduced from Chiral Pool; (D) Implementation for the Stereodivergent Synthesis of Amino Alcohols.

To implement our concept, we considered the palladium-catalyzed carboetherification of propargylic amines,12 based on the use of trifluoroacetaldehyde-derived tethers (Scheme 1D).13−15 The stereocenter formed in this step could direct a subsequent functionalization of the double bond, acting de facto as a chiral auxiliary. The rigid nature of the oxazolidine scaffold containing the stereocenter should secure a high level of diasteroselectivity to the following transformations.
Concerning the following diastereoselective functionalization, we found the hydrogenation of the formed double bond particularly attractive. By comparison, the enantioselective hydrogenation of alkyl- or heteroatom-tetrasubstituted olefins is highly challenging, with only few limited catalytic enantioselective systems reported.16−18 After removal of the tether molecule, this process would provide amino alcohols, key building blocks in synthetic and medicinal chemistry, which have been the focus of intensive methodology development recently.19−26 In particular, the diaryl-substituted amino alcohols obtained using this strategy can be found in antidepressants27,28 and have served as intermediates for the synthesis of antimycotic, antibacterials29 and antiviral molecules.30,31 However, the selective synthesis of one of the four possible stereoisomers of the amino alcohols generally requires multistep processes.
To make this process successful, an enantioselective carboetherification step had to be developed. The reversible formation of the hemiaminal I from the propargylic amine prevents asymmetric induction at this stage (see Supporting Information (SI), section E for more details). Therefore, a dynamic kinetic asymmetric transformation (DYKAT) needs to take place: in the presence of a chiral catalyst, one enantiomer of I should react preferentially to give oxazolidine II enantioselectively. Although palladium-catalyzed DYKATs have been reported,32 the envisaged process is highly challenging, due to the large distance between the chiral metal complex and the stereocenter. To the best of our knowledge, such a DYKAT process has never been realized in the palladium-catalyzed functionalization of alkynes. If successful, the selection of the substitution pattern on the alkyne and on the aryl electrophile, together with the choice of the suitable enantiomer of the chiral ligand on the palladium catalyst would provide a simple enantio- and diastereodivergent access to all four stereoisomers of the amino alcohol. This is especially attractive for medicinal chemistry, as each stereoisomer may have different bioactivity, and the development of stereodivergent methods has been the topic of intensive research in asymmetric catalysis recently.33−35
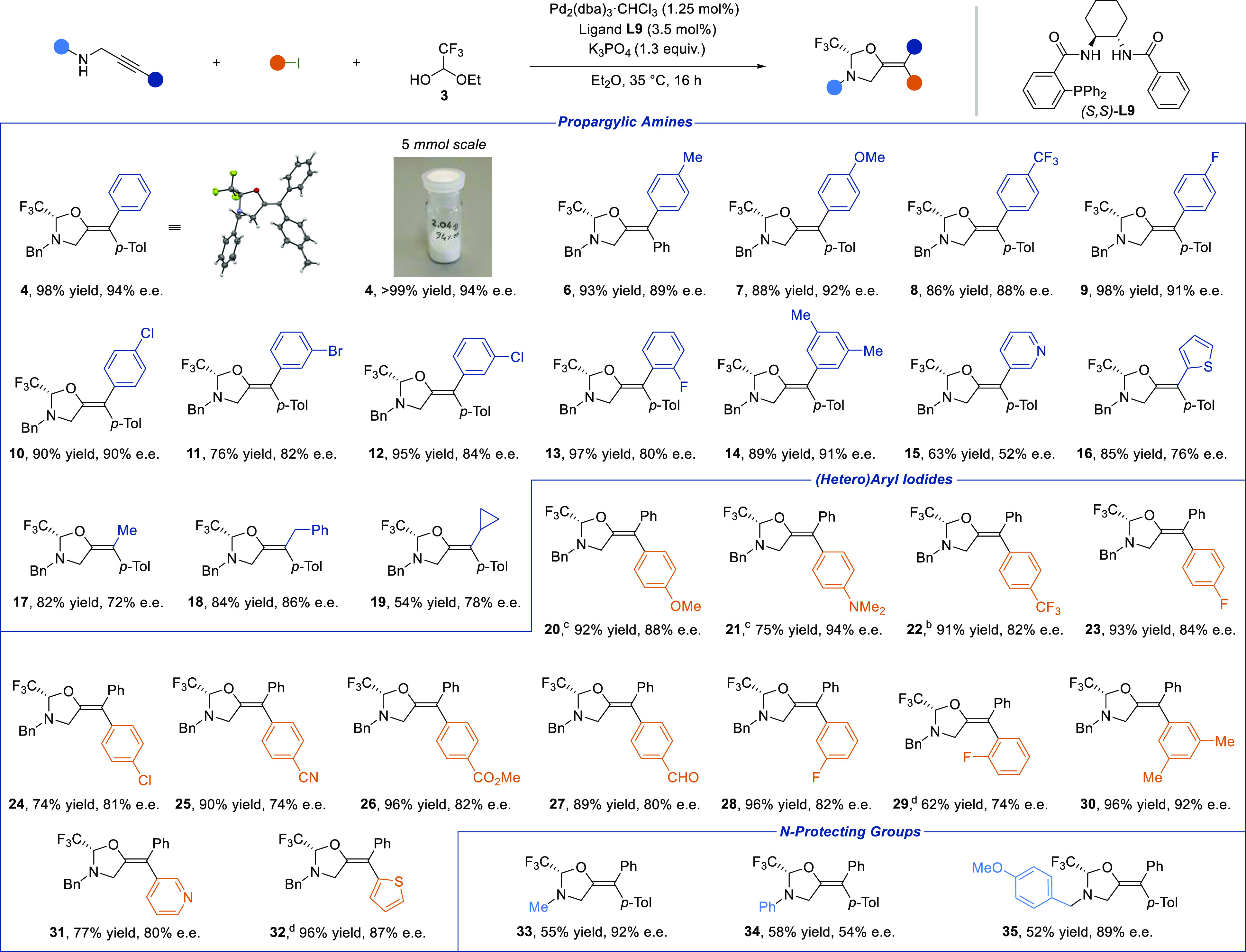
We tested the feasibility of our plan by examining the palladium-catalyzed tethered carboetherification of the readily available propargylic amine 1 with iodotoluene 2 to access tetrasubstituted olefin 4 bearing a chiral oxazolidine fragment (Table 1). 1-Ethoxy trifluoroethanol 3, a commercially available ethyl hemiacetal of trifluoroacetaldehyde, was chosen as the electrophilic molecular tether, and Pd2(dba)3·CHCl3, as the palladium source.12 We first focused on the identification of a suitable ligand that could secure a high level of stereoinduction in the process (for details, see SI, section C). Commonly used bidentate BINAP L1 and Josiphos ligands L2 and L3 were not competent for this reaction (entry 1). The P,N ligands L4 and L5, derived from the corresponding Ugi’s amines,36 delivered 4 in moderate yield and enantiomeric excess (entries 2 and 3), nevertheless demonstrating that a DYKAT was possible. However, higher asymmetric induction could not be achieved with this class of ligands. The P,N ligand S-iPrPhox L6 yielded the desired product in 72% yield and 40% e.e. (entry 4). Promising results were obtained evaluating the Trost type ligands, commonly used for palladium catalyzed asymmetric allylation reactions.37,38 In particular, the commercially available DACH-phenyl Trost ligand L7 delivered product 4 in 49% yield and 64% e.e. (entry 5). Having in mind the previous positive results obtained with P,N ligands, we substituted the 2-(PPh2)-aryl fragment with a 2-pyridine.39 This change increased the e.e. to 74% (entry 6). Surprisingly, the best results were finally obtained employing the benzamide derived L9 lacking a second strongly coordinating site, which delivered quantitatively 4 with 90% e.e. (entry 7). To the best of our knowledge, ligand L9 has been reported only twice in the literature,40,41 and it was not suitable for imparting high stereocontrol, as two strong coordinating sites were required for asymmetric induction. We developed a robust and operationally simple route for accessing both enantiomers of L9 on multigram scale (SI, section B4). Demonstrating the process’s robustness, the reaction could be performed in more “industrially preferred” solvents42 (ethyl acetate, methyl tert-butyl ether and toluene, entries 8, 9 and 10), without loss of yield and enantioselectivity, except for ethyl acetate (entry 8). Finally, the reaction could be scaled up to a 0.40 mmol scale, reducing the catalyst and ligand loading to 1.25 and 3.5 mol %, resulting in an improved stereoselectivity of 94% e.e. (entry 11).
Table 1. Optimization Studies.

| entry | Conditions | 4 yield (%)a | e.e. (%) |
|---|---|---|---|
| 1 | Ligand L1, L2 or L3 | <5 | – |
| 2 | Ligand L4 | 44 | 20 |
| 3 | Ligand L5 | 33 | 30 |
| 4 | Ligand L6 | 72 | 40 |
| 5 | Ligand L7 | 49 | 64 |
| 6 | Ligand L8 | 45 | 74 |
| 7 | Ligand L9 | >95 | 90 |
| 8 | L9, EtOAc instead of Et2O | 80 | 84 |
| 9 | L9, MTBE instead of Et2O | 89 | 89 |
| 10 | L9, Toluene instead of Et2O | 91 | 91 |
| 11b | L9, 0.40 mmol of 1 | >95 | 94 |
NMR yields.
Reaction performed using 1.25 mol % of Pd2(dba)3·CHCl3 and 3.5 mol % of (S,S)-ligand.
The structure of 4, obtained by X-ray single-crystal analysis (Scheme 2A), shows that the trifluoromethyl group is efficiently shielding one of the two enantiotopic faces of the olefin, setting the stage for the stereospecific hydrogenation. Indeed, when we submitted 4 to classical conditions for heterogeneous hydrogenation using Pearlman’s catalyst,43 the desired hydrogenated product 5 was obtained as a single diastereoisomer in 79% yield and 94% e.e. (Scheme 2B). The use of Pearlman’s catalyst also allowed simultaneous removal of the benzyl protecting group.
Scheme 2. (A) X-ray Crystal Structure of the Product 4; (B) Optimized Conditions for the Diastereoselective Hydrogenation.

Various aryl propargylic amines were well tolerated in the reaction, regardless of the position of the substituents on the phenyl ring, as well as their electronic and steric properties (Scheme 3, 4, 6–14). The geometry of the olefin can be switched by just exchanging the aryl group on the alkyne and the aryl iodide (4 vs 6). The reaction tolerates heterocycles such as pyridine and thiophene on the alkyne, although an erosion of the enantioselectivity was observed (15 and 16). Alkyl propargylic amines delivered products 17–19 bearing a methyl, a benzyl, and a cyclopropyl group. The reaction could be performed on a 5 mmol scale providing 2.0 g of 4 (quantitative yield) without erosion of the optical purity. The absolute configuration of the products were assigned by X-ray analysis of 4, confirming in addition that the aryl group coming from the iodide is incorporated in trans position to the oxygen.
Scheme 3. Scope of the Enantioselective Carboetherification.
Reactions performed on a 0.40 mmol scale using 1.3 equiv of aryl iodide and 1.4 equiv of 1-ethoxy trifluoroethanol (3). Isolated yields and HPLC enantiomeric excess are given.
Dichloroethane (DCE) instead of Et2O.
Using 2.5 mol % of Pd2(dba)3·CHCl3 and 7 mol % of ligand.
DCE at 60 °C.
The investigation of the scope of the iodoarene showed that numerous synthetically useful functional groups, including ethers, amines, halogens, esters, nitriles or aldehydes, are well tolerated independently from their electronic and steric properties or position on the benzene ring (20–30). 2-Iodothiophene and 3-iodopyridine delivered products 31 and 32 in good yields. Finally, N-methyl, N-phenyl and N-para-methoxybenzyl (PMB) propargyl amines delivered products 33–35 in 52–58% yield and 54–92% e.e.
The obtained enantioenriched tetrasubstituted olefins were then submitted to the optimized conditions for the diastereoselective hydrogenation (Scheme 4). Products 36–55 were all obtained as single diastereoisomers, confirming the robustness of our approach. Scale-up was straightforward and compound 5 could be obtained in 72% yield on 1.2 mmol scale without erosion of stereoselectivity. Heterocycles and functional groups containing coordinating N or S atoms and chlorides were not tolerated in the hydrogenation step (for details, see SI, Section D5). The nitrile and the carbonyl group within 25 and 27 were reduced to the corresponding amine 48 and alcohol 50.44 Interestingly, 5, 36, 45, 46, 47 and 54 are precursors of bioactive compounds with antidepressive activity,27,28 while the amino alcohols derived from 36 and 47 are intermediates for the synthesis of patented antiviral drugs candidates.30 Remarkably, our method provides a high level of asymmetric induction even in the presence of sterically and electronically similar aryl substituents on the olefin, thus overcoming a common obstacle in the development of catalytic asymmetric reactions. Finally, to confirm the traceless nature of our strategy, we performed a mild acidic hydrolysis of the hemiaminal in 5. The enantioenriched amino alcohol 56 was obtained in 76% yield without loss in optical purity.
Scheme 4. Scope of the Stereoselective Hydrogenation.

Reactions performed on a 0.2 mmol scale using Pd(OH)2/C (∼20 wt %). Isolated yields and HPLC enantiomeric excess are given. Product 56 was obtained after treating 5 with TsOH·H2O (7 equiv) in a 2:1 THF/H2O mixture at room temperature for 16 h, the trifluoroacetate salt was obtained after purification by reversed phase preparative HPLC.
Pd/C (∼5 wt %) was used instead of Pd(OH)2/C.
We then demonstrated that this strategy provides a simple stereodivergent access to the four possible stereoisomers of chiral diaryl aminoalcohols by a judicious selection of the substrates and the ligands (Scheme 5). Starting from the benzyl propargyl amine 57, a sequence of (i) Sonogashira coupling, (ii) enantioselective carboetherification and (iii) diastereoselective hydrogenation leads to all the stereoisomers of the desired products 5 and 36. Permuting the iodoarenes in the cross-coupling and in the carboetherification steps allows the tuning of the E,Z geometry of the double bond. This selective process, combined with the choice of the enantiomer of the ligand, and the diasteroselectivity of the hydrogenation provide a selective access to the four stereoisomers of the diaryl amino alcohol precursors.
Scheme 5. Diastereo- and Enantiodivergent Access to Chiral Aminoalcohol Precursors.

See SI for detailed reaction conditions.
In summary, we have developed an innovative strategy to control the stereoselectivity of asymmetric transformations.45 Our approach first capitalizes on the tools of asymmetric catalysis to forge a chiral oxazolidine from broadly available propargylic amines. This stereogenic element is then used to control the selectivity of the asymmetric hydrogenation of the tetrasubstituted double bond, giving access to valuable chiral amino alcohol precursors. The key for success was the first use of a “truncated” monophosphine Trost-type ligand to induce high enantioselectivity in an unprecedented DYKAT process. Combined with a Sonogashira cross-coupling, our approach gives a stereodivergent access to the four stereoisomers of protected diaryl amino alcohols in high yield and enantioselectivity. New opportunities for the design and development of asymmetric functionalizations of olefins can be expected based on the combination of the enantioselective introduction of a transient chiral auxiliary followed by a diastereoselective transformation. Such processes are currently under investigation in our laboratory.
Acknowledgments
This work is supported by the European Research Council (ERC Consolidator Grant SeleCHEM, No. 771170 and EPFL. We thank Dr. R. Scopelliti from ISIC at EPFL for X-ray analysis. This publication was created as part of NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss National Science Foundation.
Glossary
Abbreviations
- Me
Methyl
- Bu
Butyl
- Pr
Propyl
- Cy
Cyclohexyl
- Ph
Phenyl
- Bn
Benzyl
- p-Tol
p-Tolyl
- EtOAc
Ethyl acetate
- MTBE
Methyl tertbutyl ether
- Et2O
Diethyl ether
- MeOH
Methanol
- AcOH
Acetic acid
- dba
Dibenzylideneacetone.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c09177.
Author Contributions
‡ L.B., M.P., and P.D.G.G. contributed equally.
The authors declare no competing financial interest.
Notes
Crystallographic data for the product 4 and 5 have been deposited at the Cambridge Crystallographic Data Centre, accession numbers CCDC 2020478 and 2020479, respectively. Raw HPLC, NMR, MS and IR data is available at https://doi.org/ 10.5281/zenodo.4046256.
Supplementary Material
References
- Gnas Y.; Glorius F. Chiral Auxiliaries - Principles and Recent Applications. Synthesis 2006, 2006, 1899–1930. 10.1055/s-2006-942399. [DOI] [Google Scholar]
- Noyori R. Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angew. Chem., Int. Ed. 2002, 41, 2008–2022. . [DOI] [PubMed] [Google Scholar]
- Seebach D.; Boes M.; Naef R.; Schweizer W. B. Alkylation of Amino Acids without Loss of the Optical Activity: Preparation of α-Substituted Proline Derivatives. A case of Self-Reproduction of Chirality. J. Am. Chem. Soc. 1983, 105, 5390–5398. 10.1021/ja00354a034. [DOI] [Google Scholar]
- Polt R.; Seebach D. Stereoselective Alkylation of Glycine Units in Dipeptide Derivatives: “Chirality Transfer” via a Pivalaldehyde N,N-Acetal Center. J. Am. Chem. Soc. 1989, 111, 2622–2632. 10.1021/ja00189a042. [DOI] [Google Scholar]
- Li J.; Preinfalk A.; Maulide N. Enantioselective Redox-Neutral Coupling of Aldehydes and Alkenes by an Iron-Catalyzed “Catch–Release” Tethering Approach. J. Am. Chem. Soc. 2019, 141, 143–147. 10.1021/jacs.8b12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F. L.; Hong K.; Li T. J.; Park H.; Yu J. Q. Functionalization Of C(Sp(3))-H Bonds Using a Transient Directing Group. Science 2016, 351, 252–256. 10.1126/science.aad7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L. J.; Hong K.; Luo F.; Hu L.; Ewing W. R.; Yeung K. S.; Yu J. Q. Pd-II-Catalyzed Enantioselective C(sp(3))-H Arylation of Cyclobutyl Ketones Using a Chiral Transient Directing Group. Angew. Chem., Int. Ed. 2020, 59, 9594–9600. 10.1002/anie.202000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q. J.; Zhang S.; Zhan B. B.; Shi B. F. Atroposelective Synthesis of Axially Chiral Biaryls by Palladium-Catalyzed Asymmetric C-H Olefination Enabled by a Transient Chiral Auxiliary. Angew. Chem., Int. Ed. 2017, 56, 6617–6621. 10.1002/anie.201701849. [DOI] [PubMed] [Google Scholar]
- Dhawa U.; Tian C.; Wdowik T.; Oliveira J. C. A.; Hao J. P.; Ackermann L. Enantioselective Pallada-Electrocatalyzed C-H Activation by Transient Directing Groups: Expedient Access to Helicenes. Angew. Chem., Int. Ed. 2020, 59, 13451–13457. 10.1002/anie.202003826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxtoby L. J.; Li Z. Q.; Tran V.; Erbay T. G.; Deng R. H.; Liu P.; Engle K. M. A Transient-Directing-Group Strategy Enables Enantioselective Reductive Heck Hydroarylation of Alkenes. Angew. Chem., Int. Ed. 2020, 59, 8885–8890. 10.1002/anie.202001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald M. J.; Schipper D. J.; Ng P. J.; Moran J.; Beauchemin A. M. A. Catalytic Tethering Strategy: Simple Aldehydes Catalyze Intermolecular Alkene Hydroaminations. J. Am. Chem. Soc. 2011, 133, 20100–20103. 10.1021/ja208867g. [DOI] [PubMed] [Google Scholar]
- Greenwood P. D. G.; Grenet E.; Waser J. Palladium-Catalyzed Carbo-Oxygenation of Propargylic Amines using in Situ Tether Formation. Chem. - Eur. J. 2019, 25, 3010–3013. 10.1002/chem.201900020. [DOI] [PubMed] [Google Scholar]
- Orcel U.; Waser J. Palladium-Catalyzed Vicinal Amino Alcohols Synthesis from Allyl Amines by In Situ Tether Formation and Carboetherification. Angew. Chem., Int. Ed. 2015, 54, 5250–5254. 10.1002/anie.201500636. [DOI] [PubMed] [Google Scholar]
- Orcel U.; Waser J. One-Pot Three-Component Synthesis of Vicinal Diamines via In Situ Aminal Formation and Carboamination. Angew. Chem., Int. Ed. 2016, 55, 12881–12885. 10.1002/anie.201607318. [DOI] [PubMed] [Google Scholar]
- Muriel B.; Orcel U.; Waser J. Palladium-Catalyzed Carboamination of Allylic Alcohols Using a Trifluoroacetaldehyde-Derived Tether. Org. Lett. 2017, 19, 3548–3551. 10.1021/acs.orglett.7b01524. [DOI] [PubMed] [Google Scholar]
- Schrems M. G.; Neumann E.; Pfaltz A. Iridium-Catalyzed Asymmetric Hydrogenation of Unfunctionalized Tetrasubstituted Olefins. Angew. Chem., Int. Ed. 2007, 46, 8274–8276. 10.1002/anie.200702555. [DOI] [PubMed] [Google Scholar]
- Bigler R.; Mack K. A.; Shen J.; Tosatti P.; Han C.; Bachmann S.; Zhang H. M.; Scalone M.; Pfaltz A.; Denmark S. E.; Hildbrand S.; Gosselin F. Asymmetric Hydrogenation of Unfunctionalized Tetrasubstituted Acyclic Olefins. Angew. Chem., Int. Ed. 2020, 59, 2844–2849. 10.1002/anie.201912640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biosca M.; Magre M.; Pàmies O.; Diéguez M. Asymmetric Hydrogenation of Disubstituted, Trisubstituted, and Tetrasubstituted Minimally Functionalized Olefins and Cyclic β-Enamides with Easily Accessible Ir–P,Oxazoline Catalysts. ACS Catal. 2018, 8, 10316–10320. 10.1021/acscatal.8b03170. [DOI] [Google Scholar]
- Bergmeier S. C. The synthesis of vicinal amino alcohols. Tetrahedron 2000, 56, 2561–2576. 10.1016/S0040-4020(00)00149-6. [DOI] [Google Scholar]
- Donohoe T. J.; Callens C. K. A.; Flores A.; Lacy A. R.; Rathi A. H. Recent Developments in Methodology for the Direct Oxyamination of Olefins. Chem. - Eur. J. 2011, 17, 58–76. 10.1002/chem.201002323. [DOI] [PubMed] [Google Scholar]
- Kurandina D.; Yadagiri D.; Rivas M.; Kavun A.; Chuentragool P.; Hayama K.; Gevorgyan V. Transition-Metal- and Light-Free Directed Amination of Remote Unactivated C(sp3)–H Bonds of Alcohols. J. Am. Chem. Soc. 2019, 141, 8104–8109. 10.1021/jacs.9b04189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F.; Alom N.-E.; Ariyarathna J. P.; Naß J.; Li W. Regioselective Formal [3 + 2] Cycloadditions of Urea Substrates with Activated and Unactivated Olefins for Intermolecular Olefin Aminooxygenation. Angew. Chem., Int. Ed. 2019, 58, 11676–11680. 10.1002/anie.201904662. [DOI] [PubMed] [Google Scholar]
- Yang H.; Zheng W.-H. Chiral-Organotin-Catalyzed Kinetic Resolution of Vicinal Amino Alcohols. Angew. Chem., Int. Ed. 2019, 58, 16177–16180. 10.1002/anie.201909700. [DOI] [PubMed] [Google Scholar]
- Li X.; Hall D. G. Stereodivergent Asymmetric Synthesis of α,β-Disubstituted β-Aminoalkylboronic Acid Derivatives via Group-Selective Protodeboronation Enabling Access to the Elusive Anti Isomer. J. Am. Chem. Soc. 2020, 142, 9063–9069. 10.1021/jacs.0c03207. [DOI] [PubMed] [Google Scholar]
- Schwarz J. L.; Kleinmans R.; Paulisch T. O.; Glorius F. 1,2-Amino Alcohols via Cr/Photoredox Dual-Catalyzed Addition of α-Amino Carbanion Equivalents to Carbonyls. J. Am. Chem. Soc. 2020, 142, 2168–2174. 10.1021/jacs.9b12053. [DOI] [PubMed] [Google Scholar]
- Nakafuku K. M.; Zhang Z.; Wappes E. A.; Stateman L. M.; Chen A. D.; Nagib D. A. Enantioselective Radical C–H Amination for the Synthesis Of β-Amino Alcohols. Nat. Chem. 2020, 12, 697–704. 10.1038/s41557-020-0482-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J. A.; Hadley M. S.. Diphenylpropylamines. G.B. Patent No. 1,448,437, 1973.
- Clark J. A.; Clark M. S. G.; Gardner D. V.; Gaster L. M.; Hadley M. S.; Miller D.; Shah A. Substituted 3-Amino-1,1-Diaryl-2-Propanols as Potential Antidepressant Agents. J. Med. Chem. 1979, 22, 1373–1379. 10.1021/jm00197a018. [DOI] [PubMed] [Google Scholar]
- Prucher H.; Uhl J.; Kurmeier H. A.; Rudolph V.; Wahlig H., Basic Thioethers. U.S. Patent No 4,285,961, 1981.
- Jain R.; Koester D. C.; Manning J. R.; Sutton J. C.; Taft B. R.; Wan L.; Zhao Q.. Tricyclic Compounds Useful to Treat Orthomyxovirus Infections. U.S. Patent Application No 16/083,089, 2019.
- Hilpert H.; Kreis L.; Lerner C.; Humm R.; Muser T.; Kuhn B., Pyrimidone Derivatives and Their Use in the Treatment, Amelioration or Prevention of a Viral Disease. U.S. Patent No 9,988,390, 2018.
- Trost B. M.; Bunt R. C.; Lemoine R. C.; Calkins T. L. Dynamic kinetic asymmetric transformation of diene monoepoxides: A practical asymmetric synthesis of vinylglycinol, vigabatrin, and ethambutol. J. Am. Chem. Soc. 2000, 122, 5968–5976. 10.1021/ja000547d. [DOI] [Google Scholar]
- Krautwald S.; Sarlah D.; Schafroth M. A.; Carreira E. M. Enantio- and Diastereodivergent Dual Catalysis: α-Allylation of Branched Aldehydes. Science 2013, 340, 1065–1068. 10.1126/science.1237068. [DOI] [PubMed] [Google Scholar]
- Lin L.; Feng X. Catalytic Strategies for Diastereodivergent Synthesis. Chem. - Eur. J. 2017, 23, 6464–6482. 10.1002/chem.201604617. [DOI] [PubMed] [Google Scholar]
- Kaldre D.; Klose I.; Maulide N. Stereodivergent Synthesis of 1,4-Dicarbonyls by Traceless Charge–Accelerated Sulfonium Rearrangement. Science 2018, 361, 664–667. 10.1126/science.aat5883. [DOI] [PubMed] [Google Scholar]
- Oost R.; Rong J.; Minnaard A. J.; Harutyunyan S. R. Synthesis of New Derivatives of Copper Complexes of Josiphos Family Ligands for Applications in Asymmetric Catalysis. Catal. Sci. Technol. 2014, 4, 1997–2005. 10.1039/C4CY00180J. [DOI] [Google Scholar]
- Trost B. M.; Crawley M. L. Asymmetric transition-metal-catalyzed allylic alkylations: Applications in total synthesis. Chem. Rev. 2003, 103, 2921–2943. 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Machacek M. R.; Aponick A. Predicting the Stereochemistry of Diphenylphosphino Benzoic Acid (DPPBA)-Based Palladium-Catalyzed Asymmetric Allylic Alkylation Reactions: A Working Model. Acc. Chem. Res. 2006, 39, 747–760. 10.1021/ar040063c. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Oslob J. D. Asymmetric Synthesis of (−)-Anatoxin-a via an Asymmetric Cyclization Using a New Ligand for Pd-Catalyzed Alkylations. J. Am. Chem. Soc. 1999, 121, 3057–3064. 10.1021/ja983617d. [DOI] [Google Scholar]
- Trost B. M.; Breit B.; Organ M. G. On the Nature of the Asymmetric Induction in a Palladium Catalyzed Allylic Alkylation. Tetrahedron Lett. 1994, 35, 5817–5820. 10.1016/S0040-4039(00)78192-5. [DOI] [Google Scholar]
- Huang D. S.; Liu X. Q.; Li L. J.; Cai Y. D.; Liu W. G.; Shi Y. Enantioselective Bromoaminocyclization of Allyl N-Tosylcarbamates Catalyzed by a Chiral Phosphine-Sc(OTf)3 Complex. J. Am. Chem. Soc. 2013, 135, 8101–8104. 10.1021/ja4010877. [DOI] [PubMed] [Google Scholar]
- Prat D.; Wells A.; Hayler J.; Sneddon H.; McElroy C. R.; Abou-Shehada S.; Dunn P. J. CHEM21 selection guide of classical- and less classical-solvents. Green Chem. 2016, 18, 288–296. 10.1039/C5GC01008J. [DOI] [Google Scholar]
- Pearlman’s Catalyst. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Inc.: 2010. 10.1002/9780470638859.conrr483. [DOI] [Google Scholar]
- Product 55 was obtained with higher e.e. compared to its precursor 34. The reasons for this surprising result are unclear at this stage. As 34 was not fully soluble under these conditions, a plausible explanation would be a lower solubility of the racemate salt, leading to a chiral resolution during hydrogenation.
- A previous version of this work appeared in a preprint: Buzzetti L.; Purins M.; Greenwood P. D. G.; Waser J.. Enantioselective Carboetherification/Hydrogenation for the Synthesis of Amino Alcohols via a Catalytically-Formed Chiral Auxiliary ChemRxiv, August 25, 2020, ver. 1, 10.26434/chemrxiv.12855218.v1. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.