

The mechanisms that enable integral proteins to be targeted to the inner nuclear membrane are poorly understood. Herpes simplex virus 1 (HSV-1) glycoprotein M (gM) is an interesting candidate, as it is dynamically relocalized from nuclear envelopes to the trans-Golgi network (TGN) in a virus- and time-dependent fashion. However, it was, until now, unclear how gM was directed to the nucleus or evaded that compartment later on. Through a proteomic study relying on a proximity-ligation assay, we identified several novel gM interacting partners, many of which are involved in vesicular transport. Analysis of select proteins revealed that XPO6 is required for gM to leave the nuclear membranes late in the infection. This was unexpected, as XPO6 is an exportin specifically associated with actin/profilin nuclear export. This raises some very interesting questions about the interaction of HSV-1 with the exportin machinery and the cargo specificity of XPO6.
KEYWORDS: herpes, herpes simplex, herpesvirus, intracellular transport, host-pathogen interactions, UL10, BioID, XPO6, exportin, RANBP20, Ran-binding protein 20, Ran, inner nuclear membrane, INM, nuclear egress
ABSTRACT
The glycoprotein M of herpes simplex virus 1 (HSV-1) is dynamically relocated from nuclear membranes to the trans-Golgi network (TGN) during infection, but molecular partners that promote this relocalization are unknown. Furthermore, while the presence of the virus is essential for this phenomenon, it is not clear if this is facilitated by viral or host proteins. Past attempts to characterize glycoprotein M (gM) interacting partners identified the viral protein gN by coimmunoprecipitation and the host protein E-Syt1 through a proteomics approach. Interestingly, both proteins modulate the activity of gM on the viral fusion machinery. However, neither protein is targeted to the nuclear membrane and consequently unlikely explains the dynamic regulation of gM nuclear localization. We thus reasoned that gM may transiently interact with other molecules. To resolve this issue, we opted for a proximity-dependent biotin identification (BioID) proteomics approach by tagging gM with a BirA* biotinylation enzyme and purifying BirA substrates on a streptavidin column followed by mass spectrometry analysis. The data identified gM and 170 other proteins that specifically and reproducibly were labeled by tagged gM at 4 or 12 h postinfection. Surprisingly, 35% of these cellular proteins are implicated in protein transport. Upon testing select candidate proteins, we discovered that XPO6, an exportin, is required for gM to be released from the nucleus toward the TGN. This is the first indication of a host or viral protein that modulates the presence of HSV-1 gM on nuclear membranes.
IMPORTANCE The mechanisms that enable integral proteins to be targeted to the inner nuclear membrane are poorly understood. Herpes simplex virus 1 (HSV-1) glycoprotein M (gM) is an interesting candidate, as it is dynamically relocalized from nuclear envelopes to the trans-Golgi network (TGN) in a virus- and time-dependent fashion. However, it was, until now, unclear how gM was directed to the nucleus or evaded that compartment later on. Through a proteomic study relying on a proximity-ligation assay, we identified several novel gM interacting partners, many of which are involved in vesicular transport. Analysis of select proteins revealed that XPO6 is required for gM to leave the nuclear membranes late in the infection. This was unexpected, as XPO6 is an exportin specifically associated with actin/profilin nuclear export. This raises some very interesting questions about the interaction of HSV-1 with the exportin machinery and the cargo specificity of XPO6.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a DNA virus that replicates its genome and incorporates them in newly assembled capsids in the nucleus. These capsids then leave the nucleus by budding through the inner nuclear membrane, in the process, forming transient perinuclear virions that next fuse with the outer nuclear membrane to release naked nucleocapsids in the cytoplasm (1). The maturation of these viral particles depends in part on the recruitment of so-called tegument proteins and the acquisition of a final envelope embedding several viral glycoproteins. Though the identity of the compartment where the virus acquires its final envelope is disputed (2, 3), many viral glycoproteins, tegument proteins, and capsids accumulate at the trans-Golgi network (TGN), suggesting the virus acquires its mature envelope from that compartment (4–8). However, these glycoproteins are not static and are detectable in other cellular compartments during the infection. One interesting case is glycoprotein M (gM), an integral protein found at the TGN but also the cell surface and both the inner and outer nuclear membranes (9, 10). The presence of gM in these compartments is dynamic and is mostly on nuclear membranes at 4 h postinfection (hpi). By 12 hpi, gM is much more abundant and primarily accumulates at the TGN (11). From there, it actively moves back and forth with the plasma membrane (12). In contrast, gM never accumulates at the nucleus in transfected cells, indicating that gM is actively directed to that compartment during the infection (11–14). The mechanism underlying this nuclear localization is not known nor are the viral or host proteins implicated.
The HSV-1 gM protein is conserved throughout herpesviruses (15), suggesting an important role. Oddly, it is nonessential for HSV-1 and only reduces viral infectivity in tissue culture by 1 log when absent (16). In contrast, its absence in animal models attenuates viral yields by 100-fold in the related pseudorabies virus (PRV) (17). Nonetheless, gM does harbor several interesting functions. First, it modulates the viral fusion machinery, both positively and negatively. For instance, in concert with gN, gM promotes the endocytosis of gD and gH/gL (components of the viral fusion machinery) in transfected cells, which hampers cell-cell fusion (12). In contrast, gM stimulates virally induced fusion in cells infected with viral syncytial mutants (18). Similarly, gM interacts with gN, which stimulates HSV-1-induced syncytia in infected cells (12, 19), while binding of gM to the host protein E-Syt1 suppresses cell-cell fusion (20). In parallel, gM is also required for the efficient entry of the virus at the cell surface, which is particularly evident when syncytial strains are used (18). Though unproven, it is tempting to speculate that gM also regulates viral nuclear egress, since core components of the viral fusion machinery are present on the nuclear envelope and are required for the fusion of perinuclear virions with the outer nuclear membrane, at least for HSV-1 though not the related PRV (21–24). Second, gM acts as a molecular carrier by piggy-backing gN to the TGN from the endoplasmic reticulum (ER) (13, 14, 19). Finally, gM is also required for the incorporation of VP22 into viral particles and, along with gE and gI, capsid re-envelopment (25). While nonessential, gM nevertheless plays important roles during an infection.
It is not clear if gM is actively targeted and retained at the nuclear membranes early during the infection or if an active mechanism is needed to release it from that compartment. What is clear is this undoubtedly requires the participation of the virus. Given that past attempts to identify gM interacting partners did not shed any light on gM’s presence at the nuclear membranes, we reasoned that such partners may fall off during purification or only transiently bind gM. Consequently, we opted for a proximity-dependent biotin identification (BioID) approach, which consisted of adding a BirA enzymatic moiety to gM. More specifically, a mutant form of this biotinylation enzyme was used (R118G or BirA*), which is more promiscuous and efficient than its wild-type version yet selective, as it only acts on molecules within a 10-nm range (26–29). The main advantage of this proximity-dependent biotinylation assay is that transient or weak interactions can be monitored by affinity chromatography over streptavidin columns. It is also amenable to the study of integral proteins. This approach, coupled to mass spectrometry at both early (4 hpi) and later times (12 hpi), led to the identification of gM as well as 170 cellular proteins reproducibly and specifically interacting with gM. Interestingly, proteins implicated in vesicular transport were strongly represented and constituted the largest functional group (35% of all hits). Upon screening of select candidate proteins, we discovered that XPO6, an exportin known to promote actin and profilin nuclear exit, is required for gM release from the nucleus at late times. This unexpected and exciting finding opens up new research avenues.
RESULTS
Generation of cell lines.
To define novel gM interacting partners, particularly, transient interactors, we generated a gM chimera that includes at its carboxyl terminus the proximity ligation BirA* protein. We further added a hemagglutinin (HA) tag to monitor the presence of BirA* by fluorescence microscopy or Western blotting. It should be noted that gM remains functional when tagged at one end or the other (11, 14, 19, 30). One key question for the success of this approach is the orientation of gM in membranes. Based on its pseudovirus homolog, gM is predicted to contain 8 transmembrane domains, with both its termini in the cytosol (9, 12). However, this was never validated experimentally. This is critical, since we needed to know where the BirA* would end up in the cells to design appropriate controls. To resolve this, we opted to probe the orientation of HSV-1 gM BirA*HA by flow cytometry. Hence, cells overexpressing gM BirA*HA were either mock treated or permeabilized and then incubated with HA primary antibodies and fluorescently labeled secondary antibodies. As indicated in Fig. 1, a HA signal could only be seen when cells were permeabilized, indicating the presence of the HA tag (i.e., gM carboxyl tail) within the cells rather than on their external surface (Fig. 1B and D). As control, cells were also monitored for CD81, a plasma membrane protein expressed on a variety of cells, using a monoclonal antibody known to recognize a portion exposed to the extracellular milieu. As expected, CD81 was readily detected in both permeabilized and nonpermeabilized cells (Fig. 1A and C). This indicated that the carboxyl terminus of gM is on the cytoplasmic side of the plasma membrane (Fig. 1E). By extrapolation, this implies that the carboxyl tail of gM would also be cytoplasmic when gM is at the ER (where gM is synthesized), the outer nuclear membrane, or the TGN. Thus, a proper control for these conditions would be to design a BirA*HA construct expressed in the cytosol. However, gM is also found in the inner nuclear membrane (10), where its carboxyl tail should be in the nuclear matrix, where our first control would not be effective. To take into consideration these situations, two gM negative controls were engineered, namely, BirA*HA to monitor the biotinylation of irrelevant cytosolic proteins and nuclear localization signal (NLS) BirA*HA to rule out nonspecific signals within the nucleus by adding an SV40 nuclear localization signal to BirA* (see Materials and Methods).
FIG 1.

The C-terminal tail (Ct) of gM is cytoplasmic. Control untransfected cells or expressing gM BirA*HA were monitored by flow cytometry to define the localization of gM’s Ct. The cells were mock treated (A and B) or permeabilized (C and D) to determine the presence of the antigenic epitopes within the cells or at their surface, respectively. (A and C) Control untransfected cells were unstained (red) or stained with an anti-CD81 antibody coupled to APC-Cy7-A (gray). (B and D) Both cell types were unstained (red) or stained with an anti-HA antibody (green), no first antibody (orange), and anti-γ-tubulin antibody (blue). CD81 is a surface marker in HEK293 cells, and γ-tubulin is a cytoplasmic marker. (E) Schematic representation of gM-BirA*HA based on the above results. HA, hemagglutinin tag; NFA, no first antibody. Results are representative of two independent experiments.
Two other key questions needed to be addressed before the BioID approach could be launched. It was critical to show first the correct localization of gM when tagged and, second, the functionality of BirA* when appended to gM. Though gM tagging does not typically impede its normal localization and function (11, 14, 19, 30), we nonetheless verified that it is the case for the BirA* and HA dually tagged gM. First, gM localization was monitored by immunofluorescence using transiently expressing 143B cells, which we routinely use to follow up HSV-1 (6, 7, 11, 19). As shown by Western blotting, the constructs displayed the expected molecular weights (35 kDa for BirA*HA and 86 kDa for gM BirA*HA) (Fig. 2C). Moreover, immunofluorescence studies indicated that gM BirA*HA primarily accumulated at the TGN in mock-infected cells at 12 h postinfection (hpi) but at the nuclear envelope at 4 hpi when probed with HA (Fig. 2A) or gM (Fig. 2B) antibodies. This is in agreement with past reports that indicate that gM accumulates at the TGN in the absence of other viral proteins and is actively relocalized during an infection (11, 13, 19). BirA* was also functional, as total cell lysates from cells incubated with biotin exhibited several biotinylated proteins, as revealed with streptavidin-horseradish peroxidase (HRPO) (Fig. 2D). Hence, both gM and BirA* worked as expected in transient transfections, and so we could move forward.
FIG 2.

gM BirA*HA relocates from the nuclear membrane at 4 hpi toward the TGN at 12 hpi in transiently expressing cells. (A and B) 143B cells were seeded on coverslips and transiently transfected with pcDNA5FRT gM BirA*HA for 24 h. Cells were then infected with HSV-1ΔgM2 (to only detect exogenous gM) at an MOI of 5 for 4 h or 12 h to exclusively monitor the tagged gM. Cells were stained with anti-HA and anti-TGN antibodies (A) or with anti-NPC and anti-gM antibodies (B). In all cases, Hoechst was used to label the nuclei, and images were captured by confocal microscopy. (C) The expression level of each construct was determined by Western blotting using anti-HA or anti-gM antibodies. (D) Cells were incubated for 24 h with d-biotin (50 μM), total cell lysates were harvested, and BirA* activity was monitored by Western blotting using streptavidin-HRPO (Strep-HRPO). In panels C and D, 25 μg of materials was loaded on the gels. An immunoblot against γ-tubulin was used as a loading control. Results are representative of three independent experiments. NT, not transfected. The molecular masses of the markers are indicated to the left of the gels.
For maximal efficacy of our approach, we opted to generate stable cell lines expressing gM BirA*HA or the controls BirA*HA and NLS BirA*HA. To this end, a recombinase system to rapidly introduce the BirA* constructs into HEK293 Flp-In cells was used (31). As shown in Fig. 3A, Western blot analyses of lysates from these cells indicated that HA antibodies once again detected bands at the expected molecular weights. As expected, only the gM BirA*HA cell line was gM positive when probed with gM-specific antibodies. To evaluate if the NLS signal added to BirA* indeed targeted the protein to the nucleus, we separately analyzed cytoplasmic (Cyt) or nuclear (Nu) extracts by Western blotting (see Materials and Methods). Analysis of those fractions by Western blotting indicated that NLS BirA*HA was indeed strongly enriched in the nuclear fraction (Fig. 3A and B). Note that some nuclear pore proteins were detected in the cytoplasmic fraction, likely due to residual nuclear membranes when preparing the extracts (Fig. 3B). Once again, these cell lines expressed functional BirA*, since their incubation with biotin yielded protein bands detectable by streptavidin-HRPO (Fig. 3C). To ensure that the BirA*HA tag did not interfere with the proper localization of gM in these cells, the cell lines were once again monitored by immunofluorescence using antibodies directed against HA, TGN, or nuclear pores (Fig. 4). These experiments showed that BirA*HA was essentially cytoplasmic, while NLS BirA*HA was nuclear. In uninfected cells, gM BirA*HA was at the TGN, in agreement with past reports that gM accumulates in that compartment in the absence of other viral proteins (see the introduction). Upon infection with a viral strain lacking gM (HSV-1ΔgM2) (19, 32), the endogenous gM relocated to nuclear membranes at 4 hpi but returned to the TGN at 12 hpi, as expected. The BirA*HA dual tag on gM was therefore fully functional and properly located.
FIG 3.

The BirA*HA and gM BirA*HA cell lines harbor a functional biotinylation enzyme. Total cell lysates were prepared from the HEK293 FLP-In, HEK293 BirA*HA, and HEK293 gM BirA*HA cell lines. For HEK293 NLS BirA*HA, the cell lysates were additionally subfractionated into cytoplasmic (Cyt) and nuclear (Nu) fractions to monitor the functionality of the NLS tag. (A) The expression of each construct was determined by Western blotting using anti-HA or anti-gM antibodies as indicated to the right of the blots. As control, GAPDH, which is cytoplasmic, was also monitored. (B) To confirm the enrichment of the cytoplasmic and nuclear fractions, they were probed for the presence of nuclear pore complexes (NPC) with a mixture of antibodies detecting p152, p110, and p62. (C) To evaluate if the BirA* constructs were enzymatically functional, the cell lines were incubated for 24 h with d-Biotin (50 μM), and BirA* activity was probed by Western blotting using streptavidin-HRPO (Strep-HRPO) to detect biotinylated proteins. In panels A and C, GAPDH was used as a loading control. Thirty micrograms was loaded in all lanes. NT, not transfected. Results are representative of three independent experiments. The molecular masses of the markers are indicated to the left of the gels.
FIG 4.

gM BirA*HA shuttles as expected between the nuclear membrane and the TGN. HEK293 BirA*HA (A), HEK293 NLS-BirA*HA (B), and HEK293 gM BirA*HA (C) cell lines were mock treated or infected for 4 or 12 h with HSV-1ΔgM2. The TGN and nuclei were then labeled (with antibodies and Hoechst, respectively), and expression of the constructs was monitored with anti-HA antibodies. Scale bars, 10 μm. Results are representative of three independent experiments.
Proteomics.
To identify novel gM interaction partners, the cell lines expressing gM BirA*HA or controls BirA*HA and NLS BirA*HA were incubated with biotin for 24 h. The cells were further infected with HSV-1ΔgM2 for the last 4 or 12 h (Fig. 5A). Total cell lysates were subsequently prepared, biotinylated proteins were enriched over streptavidin columns, and bead bound proteins were analyzed by Western blotting and mass spectrometry (Fig. 5B and C). Only hits with 95% thresholds (proteins and peptides), a minimum of two peptides, absence from control samples (i.e., BirA*HA or NLS BirA*HA), and presence in the three independent replicates were considered. A total of 2,455 different proteins were identified in the independent experiments, of which, 171 proteins were deemed specific according to the above-described criteria (Fig. 5D). Interestingly, the only viral protein reproducibly detected in this study was gM (at both 4 and 12 hpi). In contrast, 9 host proteins were unique to the 4 hpi condition, 126 were unique to the 12 hpi condition, and 35 host proteins were common to both times. Given the long incubation times with biotin and the constitutive expression of gM in the cell lines, the 12 hpi condition likely included proteins that interacted with gM at various times and intracellular compartments. This means that some of the hits identified as common to the 4 and 12 h conditions may in fact be unique to the shorter 4-hpi time. Most interestingly, proteins implicated in vesicular transport constituted a large proportion of the hits (56% at 4 hpi, 32% at 12 hpi, and 43% at both 4 and 12 hpi, with an overall rate of 35% for all proteins) (Fig. 5D and 6; see also Table S1 in the supplemental material). Evidence for interactions between gM and the core viral fusion machinery or E-Syt1 was not conclusive, as gB, gH, gL, and E-Syt1 were also detected in control samples (Table 1), while gD was not detected in any of the triplicates.
FIG 5.

Biotinylated protein enrichment and MS analysis. (A) Schematic presentation of the strategy adopted for the d-biotin incubation and infection of the cell lines. Cells were incubated with d-biotin (50 μM, 24 h) and infected with HSV-1ΔgM2 for the last 4 or 12 h. (B and C) HEK293 BirA*HA, HEK293 NLS BirA*HA, and HEK293 gM BirA*HA preincubated with biotin were infected for 4 or 12 hpi, and total cell lysates were harvested. A 10% fraction of these total lysates (TL) was set aside, and remaining samples were captured on streptavidin columns. BB, bead bound. All were analyzed by Western blotting (2 μg loaded) using streptavidin-HRPO. (D) A Venn diagram shows the number of proteins identified at 4 hpi, 12 hpi, or at both times that were identified in all three independent experiments but absent from the BirA*HA and NLS BirA*HA controls.
FIG 6.

Diversity of protein classes identified by BioID-MS that interact with HSV-1 gM. The 170 host proteins identified in our proteomics approach (Fig. 5D) were analyzed by the software PANTHER to delineate their protein classes and then plotted in Excel. PC numbers refer to the protein class as defined by PANTHER.
TABLE 1.
Numbers of peptides detected in triplicate experiments
| Peptide | Time of infection (hpi) | No. of peptides |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NLS BirA*HA |
Bir*HA |
gM BirA*HA |
||||||||
| Exp 1 | Exp 2 | Exp 3 | Exp 1 | Exp 2 | Exp 3 | Exp 1 | Exp 2 | Exp 3 | ||
| ESyt-1 | 4 | 6 | 14 | 18 | 13 | 43 | 58 | 49 | 41 | 27 |
| 12 | 7 | 8 | 15 | 15 | 30 | 44 | 38 | 40 | 44 | |
| gB (UL27) | 4 | 2 | 8 | |||||||
| 12 | 45 | 15 | 16 | 21 | 4 | 2 | 34 | 25 | 20 | |
| gH (UL22) | 4 | |||||||||
| 12 | 20 | 2 | 9 | 18 | 6 | 3 | 30 | 12 | 11 | |
| gD (US6) | 4 | 3 | 4 | |||||||
| 12 | 3 | 6 | 3 | |||||||
| gL (UL1) | 4 | |||||||||
| 12 | 4 | 6 | 8 | 9 | 5 | |||||
XPO6 is required for gM nuclear exit at late times.
As mentioned above, vesicular transport protein interacting with gM constituted a significant portion of the hits identified by BioID and mass spectrometry (MS). While a screen of all hits may be warranted, we chose to test select candidates based on their function and presence at relevant membranes. This included MTMR6, SCAMP3, SNX1, SNX2, and VPS33B, which all modulate protein transport at the ER, the Golgi compartment, or TGN or act in the cytoplasm and which interacted specifically with gM at 4 hpi (see Table S1). Given its role in maintaining the nuclear envelope structure, we also included XPO6. Finally, we also included TMEM43B, since it mediates nuclear membrane integrity. To functionally determine if any of these molecules impact gM localization, we depleted them by RNA interference using Dicer-substrate short interfering RNAs (dsiRNAs), which are deemed more efficient than conventional siRNAs (33). Fortuitously, these reagents did not have any major impact on cell viability, with 80% or more of the cells being metabolically active even after 48 h of dsiRNA treatment (Fig. 7A). Furthermore, the dsiRNAs were efficient and reduced their respective mRNA targets by 65% to 85% (Fig. 7B). Interestingly, the virus had no impact on endogenous levels of the targets, as measured by reverse transcription-quantitative PCR (RT-qPCR) (Fig. 7C), and the reagents were equally efficient in either mock-treated or infected cells at 4 (Fig. 7D) or 12 hpi (data not shown). When dsiRNA-treated cells were subsequently infected with wild-type HSV-1, gM localization was, however, unperturbed in any of those conditions at 4 hpi, with a primarily nuclear punctate staining (Fig. 8). Since XPO6 and TMEM43 specifically interacted with gM at 12 hpi, we next probed their impact at both time points. Again, gM localized to the nucleus at 4 hpi and was chased to the TGN at 12 hpi in untreated but also dsiTMEM43-treated cells (Fig. 9A). To our surprise, dsiXPO6 strongly prohibited the relocalization of gM from the nuclear envelope to the TGN (Fig. 9B). The depletion of XPO6 did not have pleiotropic effects, since it did not alter the localization of lamin B receptor (LBR), another protein found on the inner nuclear membrane. It did also not noticeably modulate viral yields, despite a significant reduction of XPO6 at the protein level (average reduction of 63% in two independent experiments) (Fig. 10). Thus, XPO6 appears to specifically interact with gM and modulate its transport.
FIG 7.

Inhibition by RNA interference of select targets implicated in vesicular transport. (A) 143B cells were transfected for 48 h with LipoJet alone or with dsiRNAs targeting SNX1, SNX2, SCAMP3, VPS33B, MTMR6, TMEM43, or XPO6. Their viability was measured with alamarBlue. (B) In parallel, the effect of the dsiRNAs on the relative gene expression (%) was measured by RT-qPCR. The mean values and SEMs from three independent experiments are shown. The data are normalized on the LipoJet alone (A) or nonspecific RNAi (dsiCTL) (B). (C) HSV-1 had minimal impact on the endogenous levels of the targets, as assessed by RT-qPCR. Cells were mock treated or infected with wild-type HSV-1 at an MOI of 5 for 4 or 12 hpi. (D) The dsiRNAs were equally efficient in mock and infected scenarios. The relative expression of the hits in mock-infected cells or cells infected with HSV-1 (MOI of 5, 4 hpi) were analyzed by RT-qPCR. The means values and SEMs from three independent experiments are shown. The data are normalized to the dsiCTL. Bilateral Student’s t tests were performed to detect significant hits compared to the reagent alone or dsiCTL. *, P < 0.05; **, P < 0,01; ***, P < 0,001; ****, P < 0,0001.
FIG 8.

Inhibition of SNX1, SNX2, SCAMP3, VPS33B, MTMR6, and TMEM43 does not affect the localization of gM at 4 hpi. 143B cells seeded on coverslips were transfected for 48 h with LipoJet reagent alone or dsiRNA targeting the above-listed proteins and then infected for 4 hpi with wild-type HSV-1. Immunofluorescence was performed using antibodies recognizing gM or lamin B receptor (LBR), an inner nuclear membrane marker. Scale bars, 20 μm. Note that we previously showed that the punctate gM is associated with nuclear invaginations (11).
FIG 9.
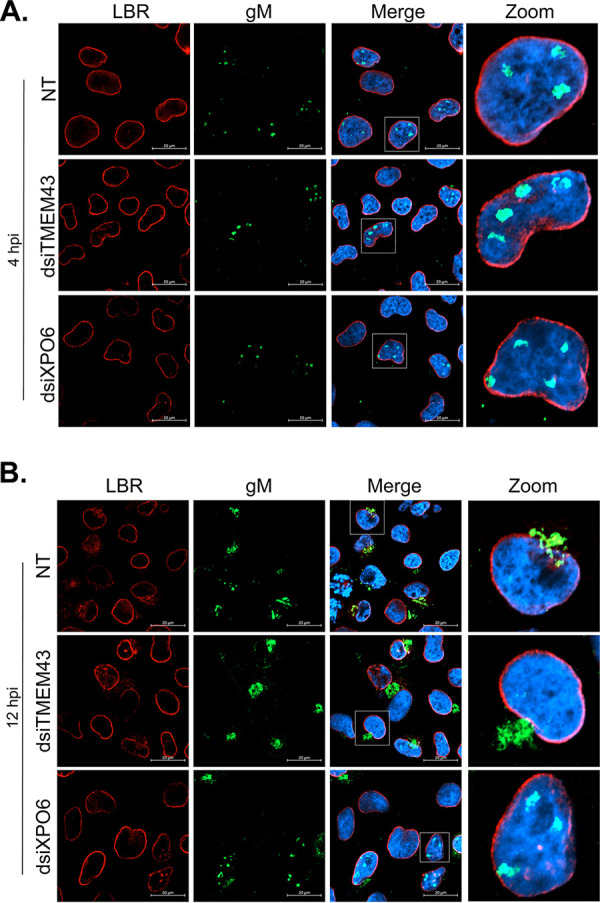
XPO6 is required for the nuclear export of gM at 12 hpi. 143B cells seeded on coverslips were transfected for 48 h with LipoJet reagent or dsiRNA targeting XPO6 and TMEM43 and then infected for 4 or 12 h with wild-type HSV-1. Immunofluorescence was performed as for Fig. 8 at either 4 (A) or 12 (B) hpi. Scale bars, 20 μm. NT, nontransfected cells.
FIG 10.

XPO6 depletion does not impact viral yields. (A) Equal numbers of 143B cells were transfected for 48 h with nonspecific RNAi (dsiCTL) or dsiXPO6. Since none of the tested antibodies detected endogenous XPO6 proteins, we immunoprecipitated the protein then probed RNAi efficacy by Western blotting (top). As control, we probed γ-tubulin levels in the original total lysates used for the IP (bottom). (B) 143B cells were not treated (NT) or transfected with control RNAi or those targeting XPO6 for 48 h and then infected with HSV-1 at an MOI of 2. The supernatants were harvested at 18 hpi and viral titers were determined on Vero cells.
DISCUSSION
To identify novel HSV-1 gM interacting partners that may have been missed in previous studies, we opted for a BioID method. Hence, BirA*HA was tagged to gM, and proximal proteins were identified by mass spectrometry. Importantly, neither the tag nor gM overexpression altered the normal localization of gM in either transient transfections or in established cell lines (Fig. 2 and 4), and BirA* was functional under both conditions (Fig. 2 and 3). We also determined the orientation of gM on membranes and found that its tagged carboxyl terminus is cytoplasmic when gM is at the cell surface (Fig. 1). By extension, this implies the carboxyl tail of gM is also cytoplasmic when the protein is anchored at the TGN or outer nuclear membrane or during its synthesis at the ER. This also means the carboxyl tail of gM is within the nuclear matrix when the protein is on the inner nuclear membrane. These experiments were essential to determine proper controls to limit false positives. We therefore analyzed two parallel constructs lacking gM, namely, BirA*HA, which is cytosolic, and NLS BirA*HA, which is nuclear (Fig. 3 and 4). This analysis revealed 170 potential new targets. This was a bit surprising at first, but not unusual of protein interactomes. Moreover, the present study does not tell us if these interactions are direct or indirect, and false positives may indeed have slipped by.
The proteomics analysis led to the identification of 9 distinct host proteins labeled at 4 hpi, 126 host proteins at 12 hpi, and 35 host proteins plus the HSV-1 gM protein at both 4 and 12 hpi. It should be noted that given the long incubation periods with biotin, the 12-h time point is cumulative and thus includes proteins that may interact with gM at earlier times. The absence of gN and pUL20 may at first seem perplexing given that gM directly binds gN and that pUL20 coimmunoprecipitates (co-IPs) with gM in infected cell lysates (18, 19). It remains unclear why these proteins were undetected, but this is not unique to the present study, as both proteins were also undetected in a previous study in our lab (34) and in an independent study (35). We can only say that this is not due to the general loss of small proteins, since pUS9 (6 kDa) and pUL45 (18 kDa) were indeed detected in some of our samples, albeit nonreproducibly. It is also not a general lack of detection of integral proteins, as most of the viral glycoproteins were sporadically identified. Similarly, the absence of E-Syt1, another known gM binding partner (20), was puzzling. Upon a closer look at the data, E-Syt1 was indeed detected in all three independent replicates at 4 and 12 hpi but was excluded since it was also present in the controls NLS BirA*HA and BirA*HA (Table 1). Similarly, the identification of gB, gD, and gH/gL, the core components of the viral fusion machinery, was anticipated, since gM modulates its activity. Interestingly, gB, gH, and gL were indeed detected in the 12 hpi triplicates but absent at 4 hpi, consistent with a late requirement for viral fusion (11). Unfortunately, they were also found in the negative controls, and so we cannot conclude that they bind gM based upon the present proximity-dependent assay (Table 1). It remains to be seen if the controls used were perhaps too stringent and if gM does interact with the core viral fusion machinery late in infection.
All biotinylated proteins specific to the gM BirA*HA condition, aside from gM itself, were of cellular origin and belonged to a limited number of protein classes, with a large contingent regulating vesicular protein transport (Fig. 6). These included 5 of the 9 proteins uniquely interacting with gM at 4 hpi, namely, MTMR6, SCAMP3, SNX1, SNX2, and VPS33B (see Table S1 in the supplemental material for detailed functions). However, testing their impact on gM localization by RNA interference and immunofluorescence failed to reveal any role (Fig. 7 and 8). Two other proteins were also noted. The first one is TMEM43, which maintains nuclear envelope integrity and retains emerin at the inner nuclear membrane (36). The second one is XPO6, an exportin that mediates nuclear protein export (37) and which was also identified under the 12 hpi condition. RNA interference experiments revealed that XPO6, but not TMEM43, was required for gM to leave the nuclei (Fig. 9). It is worth mentioning that while XPO6 was only detected with an average of 2.3 peptides per experiment (with 95% confidence), it is an unlikely contaminant given its absolute absence in our control samples. Moreover, an analysis with Crapome, which is a repository of common contaminants (see Materials and Methods), indicated that XPO6 is absent among 716 distinct proteomics experiments using BirA*, the same buffers, and the same line used as the present study. We conclude that XPO6 is a de facto novel gM interacting protein.
The implication of XPO6 on gM nuclear localization was a rather surprising finding considering XPO6 specifically promotes the nuclear export of actin and profilin in a Ran-dependent fashion (37). Interesting, such an interaction between a karyopherin and a viral protein destined for the inner nuclear membrane is not new. For instance, importin-α-16, the drosophila homolog of the human KPNA4 importin, drives the envelope protein of the occlusion-derived virus, a baculovirus, to the inner nuclear membrane (38). In that scenario, the importin remains bound to the viral protein on the nuclear envelope, suggesting a targeting/retention role of the importin. In contrast, XPO6 rather favors the release of gM from the nucleus, hinting that other molecules target or retain gM in that compartment. Potential candidates include, of course, the many other hits found in this study and which await further characterization. In the case of XPO6, the data suggest that it is highjacked by the virus, presumably through direct or indirect gM binding. This raises several important questions. Does XPO6 have a larger cargo specificity than previously reported? Does gM nuclear export also depend on the Ran GTPase (note that XPO6 is also referred to as Ran-binding protein 20)? Conversely, could it be that gM nuclear export instead depends on the level of nuclear actin or profilin, since XPO6 limits their presence in the nucleus? Given the debate on the possible transport of herpesviruses on nuclear actin (39–44), it would be interesting to see if the virus perturbs actin/profilin nuclear transport or alters the function of XPO6. Our RT-qPCR experiments suggest such an impact is not merely affecting XPO6 transcription (Fig. 7C), though some transcriptomic studies report that XPO6 is downregulated by HSV-1 in some cell types but not others (45–47). Perhaps, and most intriguingly for the present study, is why XPO6 only promotes gM nuclear exit late in infection.
The main advantages of proximity-dependent biotinylation over conventional cofractionation assays are the possible identification of weak or transient interactions, compatibility with membrane-bound baits, and efficient recovery of labeled proteins. Conversely, the main caveats of BirA* are its cumulative effect owing to the long incubations with biotin to efficiently label the proteins, yielding less-dynamic views of the reality, and the loss of relevant targets through the degradation of short-lived proteins. Novel tags (BioID2, miniTurbo, and TurboID) have since emerged to label proteins within minutes rather than hours, which should be interesting approaches in the future (48, 49). Despite its limitation, BirA* has nonetheless been successfully used in a variety of settings (50), including the present study. It remains to be seen what protein hits, aside from XPO6, also modulate HSV-1 gM nuclear localization. Moreover, the molecular mechanism of gM targeting to the inner nuclear membrane remains to be clarified.
MATERIALS AND METHODS
Cells and viruses.
The cells 143B TK− (ATCC) and the HEK293 Flp-In (kindly provided by Jason Young) were grown at 37°C, 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Medicorp), 2 mM l-glutamine (G7513; Sigma-Aldrich), and 100 U/ml penicillin-streptomycin (P4333; Sigma-Aldrich). The DMEM was supplemented with 100 μg/ml Zeocin (ant-zn-05; InvivoGen) for the HEK293 Flp-In cells or 15 μg/ml bromodeoxyuridine (BUdR) in the case of 143B TK− cells. However, prior to all transfections and infections, cells were grown 24 h without BUdR or Zeocin. The cells were regularly tested for the absence of mycoplasma. The wild-type strain HSV-1 17+ provided by Beate Sodeik and the virus HSV-1ΔgM2 supplied by Gus Kousoulas (32) were produced in BHK cells and titrated with Vero cells.
Antibodies.
The polyclonal anti-gM PAS980 was provided by Lynn Enquist (immunofluorescence [IF], 1:150; immunoblotting, 1:2,000). Other antibodies were purchased commercially: monoclonal anti-HA (IF, 1:500; immunoblot, 1:2,500; flow cytometry, 1:50) (SC-7392; Santa Cruz Biotechnology), polyclonal anti-TGN46 (IF, 5 μg/ml) (Ab-50595; Abcam), monoclonal anti-LBR (IF, 1:1,000) (Ab-232731; Abcam), monoclonal anti-nuclear pore complex (NPC) MAb 414 (IF, 1:5,000) (Covance Research Products), monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (immunoblot, 1:10,000) (MAB374; Millipore), monoclonal anti-γ-tubulin (flow cytometry, 1:100) (T6557; Sigma), monoclonal anti-CD81-allophycocyanin (APC) (TAPA-1; flow cytometry, 1:50) (number 349509; BioLegend), and polyclonal anti-XPO6 (immunoprecipitation [IP], 4 μg; immunoblot, 1:1,000) (11408-1-AP; Proteintech). Streptavidin-HRPO was used (1:10,000) (Ab-7403; Abcam) to detect biotinylated proteins by immunoblotting. All secondary antibodies used for immunofluorescence were ordered from Molecular Probes, and those for immunoblots were obtained from Jackson ImmunoResearch.
Constructs.
Vector pcDNA5FRT was sent to Mutagenex to insert BirA*(R118G)-HA (Addgene number 36047) or UL10 BirA*HA, which codes for HSV-1 gM coupled to the BirA*HA tags, and then sequenced. Plasmid pcDNA5FRT NLS BirA*HA was made using the QuikChange Lightning site-directed mutagenesis kit (210518; Agilent) to insert the SV40 NLS sequence in the pcDNA5FRT BirA*HA plasmid according to the manufacturer’s instructions. The primers used were CTTACGATGTACCGGATTACGCAGCCGAAAAAAAAACGCAAAGTTCGAGTCTAGAG (SV40 NLS underlined, forward primer) and CTCTAGACTCGAACTTTGCGTTTTTTTTTCGGCTGCGTAATCCGGTACATCGTAAG (SV40 NLS underlined, reverse primer) and were acquired from IDT. Ultracompetent X10-Gold bacteria provided in the mutagenesis kit were transformed by adding 1 μl of PCR product and placing on ice for 10 min. Heat shock was performed for 45 s in a water bath at 42°C without shaking. Bacteria were then grown at 37°C, 300 rpm for 1 h in Luria Bertani medium (LB) without antibiotics. The preparation was then spun at 1,000 × g for 1 min, and the pellet was kept in 100 μl of LB prior to plating the transformed bacteria on LB agar with ampicillin (50 μg/ml) and incubating at 37°C overnight.
Generations of stable cell lines.
Plasmids pcDN5FRT BirA*HA, pcDNA5FRT NLS BirA*HA, and pcDNA5FRT UL10 BirA*HA were cotransfected with pOG44 in HEK293 Flp-In cells using LipoD293 (SL100688; SignaGen) with a ratio of 3:1 LipoD (μg):DNA (μg) according to the manufacturer’s instructions. The transformed cells were selected in the presence of 100 μg/ml hygromycin B (450-141-xl; Multicell). Colonies were obtained after 3 to 4 weeks, and a single-colony selection was performed with cloning cylinder and grown separately. Cells were then cultured in complete DMEM in the presence of 100 μg/ml hygromycin B to maintain the selection.
FACS analysis.
HEK293 gM BirA*HA and HEK293 Flp-In cells were harvested from 10-cm dishes with collect buffer (5 mM EDTA, 1% phosphate-buffered saline [1× PBS]) and incubated for 10 min at 37°C. Complete DMEM was added to the cells, which were then scraped and collected in a 15-ml flask. Cells were concentrated by centrifugation at 250 × g for 5 min at 4°C. The pellet was washed twice with 1× PBS at 4°C. After counting the cells, another centrifugation at 250 × g for 5 min at 4°C was made to resuspend the cells in an appropriate volume to seed the cells (2 × 106 cells) into a 96-well plate. For the nonpermeabilized condition, the cells were blocked with blocking buffer (0.5% bovine serum albumin [BSA], 2% normal FBS in 1× PBS) for 30 min at 4°C with agitation, washed with fluorescence-activated cell sorter (FACS) buffer (0.5% BSA, 0.05% sodium azide in 1× PBS), and centrifuged at 250 × g for 2 min at 4°C. First, antibodies against HA or γ-tubulin were diluted in FACS buffer and added to the cells for 30 min at 4°C. To wash the samples, FACS buffer was added followed by centrifugation at 250 × g at 4°C for 2 min and removal of the supernatant. Secondary antibodies, anti-mouse Alexa Fluor 647 (1:1,000) (A-31571; Molecular Probes) or anti-CD81-APC, were diluted in FACS buffer and added to the cells for 1 h at 4°C in the dark. Cells were washed again as described above and then washed twice with 1× PBS. For the permeabilized condition, the same protocol as describe above was used, except that BD Cytofix/Cytoperm (51-2090KZ; BD Biosciences) was used for 20 min at 4°C prior the use of blocking buffer. In addition, the FACS Buffer and the 1× PBS were replaced by the PermWash (51-2091KZ; BD biosciences) for both the washes and for the antibody dilutions. For both conditions, the cells were resuspended in 1× PBS in FACS tubes prior to data acquisition with FACSDiva software 8.0.2 (BD Biosciences). Final analyses were conducted with FlowJo version 10.4.1 (TreeStar).
Cytosol and nuclear isolation.
Nuclei were isolated from HEK293 NLS BirA*HA cells using lysis buffer (225 mM mannitol, 75 mM sucrose, 5 mM HEPES [pH 7.5], 1 mM EGTA, complete with fresh protease inhibitors). Cells were kept in the lysis buffer on ice for 30 min, vortexed every 10 min for 30 s to break the cells, and then centrifuged at 1,000 × g for 5 min at 4°C to pellet the nuclei. The supernatant, i.e., the cytoplasmic fraction, was kept aside, while the nuclei were washed twice with the lysis buffer to remove any cytoplasmic contaminants. Three cycles of freeze-thaw were performed to break the nuclei. Then, the samples were passed through a 18G1/2 needle 5 times to eject the sample with high pressure to be sure that all the nuclei were disrupted prior to using the 27G1/2 needle to shear any residual DNA. Samples were analyzed as described below.
Western blotting.
HEK293 BirA*HA and HEK293 gM BirA*HA cells were harvested to collect the whole-cell lysate using radioimmunoprecipitation assay (RIPA) buffer (20% SDS, 1% NP-40, 1% deoxycholic acid, 150 mM NaCl, 10 mM Tris-HCl [pH 7.4], with a fresh cocktail of protease inhibitors) and incubated for 30 min at 4°C. Samples were centrifuged at 14,000 × g for 15 min at 4°C. Protein levels in the supernatants were quantified by using the Bradford assay (Bio-Rad). Samples were loaded into SDS-PAGE gels (10% gels or 5% to 20% gradient gels). Proteins were transferred at a constant current of 300 mA for 90 min to a polyvinylidene difluoride (PVDF) membrane (number 1620177; Bio-Rad). The membranes were blocked with 5% BSA (A7906; Sigma-Aldrich) in Tris-buffered saline–Tween 20 (TBST; 13.7 mM NaCl, 0.27 mM KCl, 0.2 mM KH2PO4, 1 mM Na2HPO4, 0.1% Tween 20) for 1 h at room temperature (RT). Primary antibodies were diluted in 5% BSA-TBST and incubated with the membranes for 1 h or overnight at 4°C. Membranes were washed 3 times for 5 min with TBST and incubated for 1 h at RT with the appropriate secondary antibodies conjugated to horseradish peroxidase diluted in 5% BSA-TBST. The membranes were incubated 5 min with Clarity Western ECL substrate (Bio-Rad), and detection was performed on a ChemiDoc MP system (Bio-Rad). The results were analyzed using Image Lab software version 6.0 (Bio-Rad).
XPO6 immunoprecipitation.
143B TK− cells were grown on 10-cm dishes and transfected with either nonspecific dsiRNA or dsiRNA targeting XPO6. Forty-eight hours posttransfection, the cells were lysed for 30 min at 4°C with rotation in IP lysis buffer (1% Triton, 1 mM EDTA, 150 mM NaCl, 50 mM Tris-HCl [pH 7.5], with a fresh cocktail of protease inhibitors). Samples were centrifuged at 14,000 × g for 20 min at 4°C, and the supernatants were incubated with rabbit preimmune serum for 1 h. These lysates were then incubated with protein A agarose (Roche) for 1 h at 4°C with rotation. After centrifugation at 2,000 × g for 5 min to remove the beads, the precleared lysates were incubated overnight at 4°C with rabbit anti-XPO6 antibody. Fresh protein A agarose beads were added for 2 h, and the immune complexes were washed three times with IP lysis buffer to remove unbound proteins. The bead-bound material and total lysates were analyzed by Western blotting.
Infections.
Prior each infection, cells were grown in pretreated dishes or 6-well plates with poly-lysine (P8920; Sigma-Aldrich) to avoid the detachment of the cells during the infection. Cells were mock treated or infected with HSV-1ΔgM2 or wild-type HSV-1 at a multiplicity of infection (MOI) of 2 or 5 for 1 h at 37°C for the adsorption and then grown in appropriate DMEM at 37°C as specified in the figure legends. For plaque assays, cells were infected for 18 h, and the supernatants were titrated on Vero cells.
Immunofluorescence microscopy.
All coverslips were pretreated with poly-lysine to ensure the tight binding of the cells to the glass. Cells were fixed with 3% paraformaldehyde in PBS for 30 min at 4°C. Two washing steps were performed with PBS prior to the permeabilization with filtered 0.1% Triton X-100 (T9284; Sigma) in 1× PBS for 4 min at room temperature (RT). Samples were then washed twice with 1× PBS, and nonspecific protein binding sites were blocked with 10% fetal calf serum (FCS) for 30 min at RT. Samples were labeled overnight at RT with primary antibodies diluted in 10% FCS and washed three times with 1× PBS for 5 min. Secondary antibodies diluted in PBS were applied for 45 min at RT and then washed three times. Coverslips were placed on slides previously prepared with droplets of Dako solution (53023; Dako) containing 10 μg/ml Hoechst 33342 (Sigma-Aldrich) overnight. The cells were examined with an Axiophot epifluorescence microscope (Zeiss Axio-imager Z2).
BioID protocol and purification of biotinylated proteins.
The protocol used for the BioID purification has been adapted to our needs from Anne-Claude Gingras’s lab protocol (51), itself adapted from the protocol of K. J. Roux (52). Cell lines HEK293 BirA*HA, HE293 NLS BirA*HA, and HEK293 gM BirA*HA were each grown in one 24.5-cm dish per condition to reach 75% confluence and incubated with d-biotin (50 μM) for 24 h. Before the end of the incubation with d-biotin, cells were infected for 4 h or 12 h with HSV-1ΔgM2 at an MOI of 5 so that the d-biotin incubation and the infection ended at the same time. Cells were washed three times with 1× PBS and then scraped and collected in 15-ml tubes. Samples were centrifuged at 500 × g for 5 min at 4°C, and supernatants were removed and RIPA buffer was added to the cell pellets. Two cycles of sonication were applied with 30 pulses at an amplification of 30% and at power 3 on ice with a Branson Sonifier 450. Samples were then cleared of debris by centrifugation at 16,000 × g for 10 min at 4°C. To check sample enrichment, 10% of the total lysates were put aside for subsequent analysis by Western blotting. Meanwhile, streptavidin-Sepharose beads (170969-01; GE Healthcare) were resuspended and washed three times in 1 ml of RIPA buffer by centrifugation at 300 × g for 1 min at 4°C to equilibrate the beads in the buffer. The remaining 90% of the cell lysates were added to 70 μl of beads and incubated for 3 h at 4°C followed by centrifugation at 300 × g for 1 min at 4°C. The beads were washed twice with RIPA buffer with centrifugation at 300 × g for 1 min at 4°C between each wash. Finally, the beads were resuspended in 100 μl of ammonium bicarbonate (ABC; 50 mM) and quantified with a Pierce bicinchoninic acid (BCA) protein assay kit using naked beads as a baseline.
Biotinylated protein digestion and LC-MS/MS.
Bead-bound samples were sent to the proteomic platform of Montreal Clinical Research Institute (IRCM), and the digestion of the samples was performed as follows. Proteins on beads were digested with 0.5 μg of trypsin (sequencing-grade modified trypsin; Promega) overnight at 37°C with agitation. The supernatants were collected, and the beads washed twice with 100 μl of water. The supernatants of each wash were pooled and then reduced and alkylated. The reduction step was performed with 9 mM dithiothreitol at 37°C for 30 min, and after a cool down of 10 min, the alkylation step was made with 17 mM iodoacetamide at RT for 20 min in the dark. The supernatants were acidified with trifluoroacetic acid to remove salts, and all detergent residues were removed by MCX (Waters Oasis MCX96-well elution plate) according to the manufacturer’s instructions. After elution in 10% of ammonium hydroxide-90% methanol (vol/vol), samples were dried with a SpeedVac, reconstituted under agitation for 15 min in 15 μl of 5% formic acid, and charged in a C18 self-pack column (75-μm inside diameter [i.d.], 150 mm) installed in the Easy-nLC II system (Proxeon Biosystems). The buffers used for the chromatography were 0.2% formic acid (A) and 90% acetonitrile-0.2% formic acid (B). Peptides were eluted on a gradient with a flow of 250 nl/min. The high-performance liquid chromatography (HPLC) system was coupled to a mass spectrometry Q Exactive (Thermo Scientific) via a Nanospray Flex ion source. The voltages of Nanospray and S-lens were fixed to 1.3 to 1.8 kV and 50 V, respectively. The capillary temperature was 225°C. The sounding spectra of the scanning mass spectrometry (m/z 36 to 2,000) were acquired with a resolution of 70,000. The 16 most intense peptide ions were fragmented in the collision cell and tandem mass spectrometry (MS/MS) spectra were analyzed with an Orbitrap with the dynamical exclusion set to 7 s.
Protein identification.
The peak list files were generated with Proteome Discoverer (version 2.1) using the following parameters: minimum mass set to 500 Da, maximum mass set to 6,000 Da, no grouping of MS/MS spectra, precursor charge set to auto, and minimum number of fragment ions set to 5. The identification of proteins was performed with Mascot (version 2.6; Matrix Science) with the human RefSeq coupled to the HSV-1 database. The data were then transferred to Scaffold (version 4.8) for further analysis. Only hits that respected the following criteria were considered: protein and peptide threshold (>95%), a minimum number of 2 peptides, and presence of the proteins in the three independent triplicates. Protein functions were probed with PANTHER software (53), while Venn diagrams were created using Venny 2.0 (54). All hits were also tested against the Crapome V2.0 (https://reprint-apms.org/).
dsiRNA treatments.
143B TK− cells grown in 6-well plates or 96-well plates were transfected for 48 h with the transfection agent LipoJet (SL100468; SignaGen Laboratories) according to the instructions of the manufacturer. Cells were transfected with the dsiRNA targeting the proteins NC1 (50 nM), SNX1 (25 nM), SNX2 (50 nM), SCAMP3 (25 nM), VPS33B (25 nM), MTMR6 (50 nM), TMEM43 (50 nM), and XPO6 (50 nM) (IDT).
Cell viability.
Cell viability was assessed by using the alamarBlue (BuF012A; Bio-Rad). Ten percent alamarBlue was added to 143B cells grown in 96-well plates and treated with dsiRNAs for 48 h. The cells were incubated for 3 h at 37°C with the alamarBlue before the cell viability was measured using the fluorescence Spectromax (Gemini EM; excitation at 560 nm and emission at 590 nm). The percentages of cell viability were normalized with the transfection agent LipoJet condition.
RT-qPCR.
Total RNA was extracted from cells transfected with dsiRNA as indicated above using an SV Total RNA isolation system kit (Z3100; Promega). The RNA was converted to cDNA with a reverse transcriptase kit according to the manufacturer’s instructions (Applied Biosystems). The cDNAs were next analyzed by qPCR with a LightCycler 96 (Roche) and appropriate primers (see Table 2). Genes were quantified using a standard curve and normalized to the endogenous control (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]). All the PCRs were performed by using SYBR green (Molecular Probes).
TABLE 2.
PCR primers used in this study
| Target | Primer sequence (5′→3′) |
|
|---|---|---|
| Forward | Reverse | |
| SNX1 | CAACAAAGCCACAGATGCC | TTGTCCTCAGAGCTCCCAAG |
| SNX2 | AACTCCAATGGCCCAAAACC | AAAGATCACGGGAGCAGACA |
| VPS33B | CGTCTTCGGTGGTGCTTATG | CCTTTCTCTCTGCCCAGGAA |
| SCAMP3 | TCGTCCAGGATGTGCTCTTT | CGGCGGTATAAGGAGTGGAT |
| MTMR6 | GGTGCATTGTTCCGATGGTT | ACTGTTCGGTCAAATGCCAC |
| TMEM43 | GCTAGTCCCATTCTCCACCA | ACAGAAGGCAAAGGCTTTCA |
| XPO6 | CAGTTCCCTGTGGTGGAGTT | GCTTCCTTGTCTCCAAGACG |
| GAPDH | GAGTCAACGGATTTGGTCGT | TTGATTTTGGAGGGATCTCG |
Statistical analysis.
All data are expressed as the means values ± standard deviations of the means (SEMs). Statistical analyses were performed using bilateral Student’s t tests using GraphPad Prism, version 8 (GraphPad Software).
Supplementary Material
ACKNOWLEDGMENTS
We thank Denis Faubert of the IRCM proteomics platform for his support and suggestions.
The research was supported by grants from the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 2.Henaff D, Radtke K, Lippé R. 2012. Herpesviruses exploit several host compartments for envelopment. Traffic 13:1443–1449. doi: 10.1111/j.1600-0854.2012.01399.x. [DOI] [PubMed] [Google Scholar]
- 3.Lippé R. Transport intracellulaire des Alphaherpesvirinae. 2020. Virologie 24:210–230. doi: 10.1684/vir.2020.0851. [DOI] [PubMed] [Google Scholar]
- 4.Mettenleiter TC. 2006. Intriguing interplay between viral proteins during herpesvirus assembly or: the herpesvirus assembly puzzle. Vet Microbiol 113:163–169. doi: 10.1016/j.vetmic.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 5.Harley CA, Dasgupta A, Wilson DW. 2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J Virol 75:1236–1251. doi: 10.1128/JVI.75.3.1236-1251.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turcotte S, Letellier J, Lippé R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J Virol 79:8847–8860. doi: 10.1128/JVI.79.14.8847-8860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Remillard-Labrosse G, Mihai C, Duron J, Guay G, Lippé R. 2009. Protein kinase D-dependent trafficking of the large Herpes simplex virus type 1 capsids from the TGN to plasma membrane. Traffic 10:1074–1083. doi: 10.1111/j.1600-0854.2009.00939.x. [DOI] [PubMed] [Google Scholar]
- 8.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 82:5198–5211. doi: 10.1128/JVI.02681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baines JD, Roizman B. 1993. The UL10 gene of herpes simplex virus 1 encodes a novel viral glycoprotein, gM, which is present in the virion and in the plasma membrane of infected cells. J Virol 67:1441–1452. doi: 10.1128/JVI.67.3.1441-1452.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baines JD, Wills E, Jacob RJ, Pennington J, Roizman B. 2007. Glycoprotein M of herpes simplex virus 1 is incorporated into virions during budding at the inner nuclear membrane. J Virol 81:800–812. doi: 10.1128/JVI.01756-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Nagel CH, Sodeik B, Lippé R. 2009. Early, active, and specific localization of herpes simplex virus type 1 gM to nuclear membranes. J Virol 83:12984–12997. doi: 10.1128/JVI.01180-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crump CM, Bruun B, Bell S, Pomeranz LE, Minson T, Browne HM. 2004. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J Gen Virol 85:3517–3527. doi: 10.1099/vir.0.80361-0. [DOI] [PubMed] [Google Scholar]
- 13.Striebinger H, Zhang J, Ott M, Funk C, Radtke K, Duron J, Ruzsics Z, Haas J, Lippé R, Bailer SM. 2015. Subcellular trafficking and functional importance of herpes simplex virus type 1 glycoprotein M domains. J Gen Virol 96:3313–3325. doi: 10.1099/jgv.0.000262. [DOI] [PubMed] [Google Scholar]
- 14.Striebinger H, Funk C, Raschbichler V, Bailer SM. 2016. Subcellular trafficking and functional relationship of the HSV-1 glycoproteins N and M. Viruses 8:83. doi: 10.3390/v8030083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montague MG, Hutchison CA III. 2000. Gene content phylogeny of herpesviruses. Proc Natl Acad Sci U S A 97:5334–5339. doi: 10.1073/pnas.97.10.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baines JD, Roizman B. 1991. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J Virol 65:938–944. doi: 10.1128/JVI.65.2.938-944.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dijkstra JM, Gerdts V, Klupp BG, Mettenleiter TC. 1997. Deletion of glycoprotein gM of pseudorabies virus results in attenuation for the natural host. J Gen Virol 78:2147–2151. doi: 10.1099/0022-1317-78-9-2147. [DOI] [PubMed] [Google Scholar]
- 18.Kim IJ, Chouljenko VN, Walker JD, Kousoulas KG. 2013. Herpes simplex virus 1 glycoprotein M and the membrane-associated protein UL11 are required for virus-induced cell fusion and efficient virus entry. J Virol 87:8029–8037. doi: 10.1128/JVI.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Kasmi I, Lippé R. 2015. Herpes simplex virus 1 gN partners with gM to modulate the viral fusion machinery. J Virol 89:2313–2323. doi: 10.1128/JVI.03041-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Kasmi I, Khadivjam B, Lackman M, Duron J, Bonneil E, Thibault P, Lippé R. 2017. Extended synaptotagmin 1 interacts with herpes simplex virus 1 glycoprotein m and negatively modulates virus-induced membrane fusion. J Virol 92:e01281-17. doi: 10.1128/JVI.01281-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci U S A 104:10187–10192. doi: 10.1073/pnas.0703790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J Virol 83:3115–3126. doi: 10.1128/JVI.01462-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright CC, Wisner TW, Hannah BP, Eisenberg RJ, Cohen GH, Johnson DC. 2009. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J Virol 83:11847–11856. doi: 10.1128/JVI.01397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klupp B, Altenschmidt J, Granzow H, Fuchs W, Mettenleiter TC. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J Virol 82:6299–6309. doi: 10.1128/JVI.00386-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuchs W, Klupp BG, Granzow H, Hengartner C, Brack A, Mundt A, Enquist LW, Mettenleiter TC. 2002. Physical interaction between envelope glycoproteins E and M of pseudorabies virus and the major tegument protein UL49. J Virol 76:8208–8217. doi: 10.1128/jvi.76.16.8208-8217.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi-Rhee E, Schulman H, Cronan JE. 2004. Promiscuous protein biotinylation by Escherichia coli biotin protein ligase. Protein Sci 13:3043–3050. doi: 10.1110/ps.04911804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cronan JE. 2005. Targeted and proximity-dependent promiscuous protein biotinylation by a mutant Escherichia coli biotin protein ligase. J Nutr Biochem 16:416–418. doi: 10.1016/j.jnutbio.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 28.Roux KJ, Kim DI, Raida M, Burke B. 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim DI, Birendra KC, Zhu W, Motamedchaboki K, Doye V, Roux KJ. 2014. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci U S A 111:E2453–E2461. doi: 10.1073/pnas.1406459111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lau SY, Crump CM. 2015. HSV-1 gM and the gK/pUL20 complex are important for the localization of gD and gH/L to viral assembly sites. Viruses 7:915–938. doi: 10.3390/v7030915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Gorman S, Fox DT, Wahl GM. 1991. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science 251:1351–1355. doi: 10.1126/science.1900642. [DOI] [PubMed] [Google Scholar]
- 32.Chouljenko DV, Kim IJ, Chouljenko VN, Subramanian R, Walker JD, Kousoulas KG. 2012. Functional hierarchy of herpes simplex virus 1 viral glycoproteins in cytoplasmic virion envelopment and egress. J Virol 86:4262–4270. doi: 10.1128/JVI.06766-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kubo T, Zhelev Z, Ohba H, Bakalova R. 2007. Modified 27-nt dsRNAs with dramatically enhanced stability in serum and long-term RNAi activity. Oligonucleotides 17:445–464. doi: 10.1089/oli.2007.0096. [DOI] [PubMed] [Google Scholar]
- 34.Loret S, Guay G, Lippé R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bell C, Desjardins M, Thibault P, Radtke K. 2013. Proteomics analysis of herpes simplex virus type 1-infected cells reveals dynamic changes of viral protein expression, ubiquitylation, and phosphorylation. J Proteome Res 12:1820–1829. doi: 10.1021/pr301157j. [DOI] [PubMed] [Google Scholar]
- 36.Liang WC, Mitsuhashi H, Keduka E, Nonaka I, Noguchi S, Nishino I, Hayashi YK. 2011. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol 69:1005–1013. doi: 10.1002/ana.22338. [DOI] [PubMed] [Google Scholar]
- 37.Stuven T, Hartmann E, Gorlich D. 2003. Exportin 6: a novel nuclear export receptor that is specific for profilin.actin complexes. EMBO J 22:5928–5940. doi: 10.1093/emboj/cdg565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braunagel SC, Cox V, Summers MD. 2009. Baculovirus data suggest a common but multifaceted pathway for sorting proteins to the inner nuclear membrane. J Virol 83:1280–1288. doi: 10.1128/JVI.01661-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forest T, Barnard S, Baines JD. 2005. Active intranuclear movement of herpesvirus capsids. Nat Cell Biol 7:429–431. doi: 10.1038/ncb1243. [DOI] [PubMed] [Google Scholar]
- 40.Feierbach B, Piccinotti S, Bisher M, Denk W, Enquist LW. 2006. Alpha-herpesvirus infection induces the formation of nuclear actin filaments. PLoS Pathog 2:e85. doi: 10.1371/journal.ppat.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bosse JB, Virding S, Thiberge SY, Scherer J, Wodrich H, Ruzsics Z, Koszinowski UH, Enquist LW. 2014. Nuclear herpesvirus capsid motility is not dependent on F-actin. mBio 5:e01909-14. doi: 10.1128/mBio.01909-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bosse JB, Hogue IB, Feric M, Thiberge SY, Sodeik B, Brangwynne CP, Enquist LW. 2015. Remodeling nuclear architecture allows efficient transport of herpesvirus capsids by diffusion. Proc Natl Acad Sci U S A 112:E5725–E5733. doi: 10.1073/pnas.1513876112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilkie AR, Lawler JL, Coen DM. 2016. A role for nuclear F-actin induction in human cytomegalovirus nuclear egress. mBio 7:e01254-16. doi: 10.1128/mBio.01254-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bosse JB, Enquist LW. 2016. The diffusive way out: herpesviruses remodel the host nucleus, enabling capsids to access the inner nuclear membrane. Nucleus 7:13–19. doi: 10.1080/19491034.2016.1149665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu B, Li X, Huo Y, Yu Y, Zhang Q, Chen G, Zhang Y, Fraser NW, Wu D, Zhou J. 2016. Cellular responses to HSV-1 infection are linked to specific types of alterations in the host transcriptome. Sci Rep 6:28075. doi: 10.1038/srep28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pheasant K, Moller-Levet CS, Jones J, Depledge D, Breuer J, Elliott G. 2018. Nuclear-cytoplasmic compartmentalization of the herpes simplex virus 1 infected cell transcriptome is co-ordinated by the viral endoribonuclease vhs and cofactors to facilitate the translation of late proteins. PLoS Pathog 14:e1007331. doi: 10.1371/journal.ppat.1007331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wyler E, Franke V, Menegatti J, Kocks C, Boltengagen A, Praktiknjo S, Walch-Ruckheim B, Bosse J, Rajewsky N, Grasser F, Akalin A, Landthaler M. 2019. Single-cell RNA-sequencing of herpes simplex virus 1-infected cells connects NRF2 activation to an antiviral program. Nat Commun 10:4878. doi: 10.1038/s41467-019-12894-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varnaitė R, MacNeill SA. 2016. Meet the neighbors: mapping local protein interactomes by proximity-dependent labeling with BioID. Proteomics 16:2503–2518. doi: 10.1002/pmic.201600123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mair A, Xu SL, Branon TC, Ting AY, Bergmann DC. 2019. Proximity labeling of protein complexes and cell-type-specific organellar proteomes in Arabidopsis enabled by TurboID. Elife 8:e47864. doi: 10.7554/eLife.47864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trinkle-Mulcahy L. 2019. Recent advances in proximity-based labeling methods for interactome mapping. F1000Res 8:135. doi: 10.12688/f1000research.16903.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kean MJ, Couzens AL, Gingras AC. 2012. Mass spectrometry approaches to study mammalian kinase and phosphatase associated proteins. Methods 57:400–408. doi: 10.1016/j.ymeth.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 52.Roux KJ. 2013. Marked by association: techniques for proximity-dependent labeling of proteins in eukaryotic cells. Cell Mol Life Sci 70:3657–3664. doi: 10.1007/s00018-013-1287-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, Thomas PD. 2019. Protocol update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 14:703–721. doi: 10.1038/s41596-019-0128-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oliveros JC. 2007. Venny. An interactive tool for comparing lists with Venn's diagrams. https://bioinfogp.cnb.csic.es/tools/venny/index.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.