

RSV is a highly contagious virus that causes multiple infections in infants within their first year of life. It can also easily cause infection in elderly or immunocompromised individuals, suggesting that individual differences in immunity play an important role in RSV infection. Therefore, exploring the pathogenic mechanisms of RSV and identifying essential genes which inhibit RSV infection are necessary to develop an effective strategy to control RSV infection. Here, we report that the IFN-inducible gene GBP5 potently inhibits RSV replication by reducing the cell-associated levels of the RSV small hydrophobic (SH) protein, which is a viroporin. In contrast, the RSV G protein was shown to upregulate the expression of the DZIP3 protein, an E3 ligase that degrades GBP5 through the proteasomal pathway. Our study provides important information for the understanding of the pathogenic mechanisms of RSV and host immunity as well as the complicated interplay between the virus and host.
KEYWORDS: RSV replication, IFN-γ, GBP5, DZIP3, ubiquitination and degradation
ABSTRACT
Guanylate binding protein 5 (GBP5) belongs to the GTPase subfamily, which is mainly induced by interferon gamma (IFN-γ) and is involved in many important cellular processes, including inflammasome activation and innate immunity against a wide variety of microbial pathogens. However, it is unknown whether GBP5 inhibits respiratory syncytial virus (RSV) infection. In this study, we identified GBP5 as an effector of the anti-RSV activity of IFN-γ and found that in children, the weaker immune response, especially the weaker IFN-γ response and the decreased GBP5 expression, leads to RSV susceptibility. Furthermore, we revealed that GBP5 reduced the cell-associated levels of the RSV small hydrophobic (SH) protein, which was identified as a viroporin. In contrast, overexpression of the SH protein rescued RSV replication in the presence of GBP5. The GBP5-induced decrease in intracellular SH protein levels is because GBP5 promotes the release of the SH protein into the cell culture. Moreover, the GBP5 C583A mutants with changes at the C terminus or the GBP5 ΔC mutant lacking the C-terminal region, which impairs GBP5 localization in the Golgi, could not inhibit RSV infection, whereas the GTPase-defective GBP5 maintained RSV inhibition, suggesting that Golgi localization but not the GTPase activity of GBP5 is required for RSV inhibition. Interestingly, we found that RSV infection or RSV G protein downregulates GBP5 expression by upregulating DZIP3, an E3 ligase, which induces GBP5 degradation through the K48 ubiquitination and proteasomal pathways. Thus, this study reveals a complicated interplay between host restrictive factor GBP5 and RSV infection and provides important information for understanding the pathogenesis of RSV.
IMPORTANCE RSV is a highly contagious virus that causes multiple infections in infants within their first year of life. It can also easily cause infection in elderly or immunocompromised individuals, suggesting that individual differences in immunity play an important role in RSV infection. Therefore, exploring the pathogenic mechanisms of RSV and identifying essential genes which inhibit RSV infection are necessary to develop an effective strategy to control RSV infection. Here, we report that the IFN-inducible gene GBP5 potently inhibits RSV replication by reducing the cell-associated levels of the RSV small hydrophobic (SH) protein, which is a viroporin. In contrast, the RSV G protein was shown to upregulate the expression of the DZIP3 protein, an E3 ligase that degrades GBP5 through the proteasomal pathway. Our study provides important information for the understanding of the pathogenic mechanisms of RSV and host immunity as well as the complicated interplay between the virus and host.
INTRODUCTION
Respiratory syncytial virus (RSV) is one of the major pathogens most commonly responsible for causing severe acute lower respiratory tract infections (ALRTIs) in children younger than 5 years (1). Shi et al. reported that in 2015, nearly 33 million cases of new ALRTIs caused by RSV led to approximately 3.2 million (2.7 to 3.8 million) hospital admissions and 59,600 (48,000 to 74,500) in-hospital deaths in children under 5 years of age (2). Therefore, RSV infection is a burden on both public health and society worldwide (3, 4). As there are currently no approved preventive RSV vaccines or effective drugs on the market, an improved understanding of the pathogenesis of RSV and host innate immunity is urgently needed (5). RSV is a single-stranded RNA enveloped virus belonging to the Pneumoviridae family (6). It contains 10 genes, which produce 11 proteins in the following order: NS1, NS2, N, P, M, SH, F, G, M2-1, M2-2, and L (5). The small hydrophobic (SH) gene encodes a glycoprotein as a surface protein (7). Although several studies have reported that the RSV SH protein is not essential for virus replication in vitro (8–10), other studies have demonstrated that SH-defective RSV exhibits site-specific restriction in the respiratory tracts of mice or was attenuated in chimpanzees (8, 11–13). This research led to the discovery that the RSV SH protein accumulates within the lipid raft structures of the Golgi complex during virus infection (14, 15). Furthermore, the SH protein was demonstrated to form pentameric ion channels by using a random coil and β-sheet structure, which induces membrane permeability and acts as a viroporin (11, 16, 17). Notably, some studies have demonstrated that the RSV SH protein plays an important role in RSV pathogenesis, as SH prevents apoptosis, therefore allowing for continuous virion production (18, 19).
Interferons (IFNs), which are produced in response to RSV infection, play important roles in regulating immune defenses, the rate of virus clearance, and progress to severe disease (20, 21). It is well known that two RSV nonstructural proteins, NS1 and NS2, as well as the envelope G glycoprotein, strongly suppress type I IFN production in RSV-infected cells (20, 22, 23). This is consistent with a clinical study that showed that RSV infection failed to induce a robust type I IFN response (24–27). RSV infection elicits higher levels of the IFN lambda subtype (IFN-λ), which positively correlates with the severity of RSV bronchiolitis (28). IFN-γ has a strong protective role against RSV (21). The large GTPase guanylate binding protein family members (GBPs), including GBP5, are the main target genes of the IFN response and may be responsible for up to 20% of all proteins induced by IFN-γ (29–31). Among them, GBP5 acts as an activator of NLRP3 inflammasome assembly and plays a role in innate immunity and inflammation (32, 33). Due to this, GBP5 has been reported to inhibit many kinds of pathogens, including Brucella abortus (34), Francisella novicida (35), and Toxoplasma gondii (36). In addition, GBP5 was identified as a host restrictor of HIV and was shown to target the envelope glycoproteins. A recent study reported that GBP2/5 exerts broad antiviral activity by suppressing furin-mediated processing of the envelope proteins (29, 37). Moreover, Feng et al. showed that GBP5 also inhibits influenza A virus (IAV) infection by enhancing the expression of IFN and IFN-related effectors (38).
In this study, we identified GBP5 as an inhibitor of RSV replication for the first time and revealed that GBP5 inhibited the expression of the RSV SH protein. Moreover, we showed that localization in the Golgi of GBP5 but not its GTPase activity is required for RSV inhibition. In addition, we found that RSV utilizes its G protein but not IFNs to upregulate the expression of the DZIP3 protein, an E3 ubiquitin ligase (39), which antagonizes the restriction of GBP5 through ubiquitination and degradation of the GBP5 pathway.
RESULTS
IFNs, especially IFN-γ, inhibit RSV replication.
The role of IFNs in RSV infection has been a subject of interest for decades. Since high type III IFN (IFN-λ) expression levels are correlated with the severity of RSV bronchiolitis in children (28), we investigated the effect of type I IFN (IFN-α) and type II IFN (IFN-γ) on RSV replication. A549 cells were treated with IFN-α or IFN-γ and then infected with RSV at a multiplicity of infection (MOI) of 0.1. Then, we measured RSV viral protein levels (Fig. 1A), viral mRNA levels (Fig. 1B), and virus titers (Fig. 1C) at different times and found that IFN-γ is more efficient in restricting RSV infection than IFN-α. On the other hand, RSV infection induces IFN production (20, 27, 40). Thus, we measured IFN-α and IFN-γ mRNA levels after RSV infection in A549 cells and observed that IFN-γ is produced in larger amounts than IFN-α (Fig. 1D), which is consistent with previous studies showing that the RSV NS1 and NS2 proteins downregulate signal transducer and activator of transcription 2 (STAT2) protein expression, thus blocking type I IFN signaling (22, 23). However, we found that NS1 and NS2 had no effect on the expression and phosphorylation of STAT1 (Fig. 1E). Thus, our results show that RSV infection induced a higher production of IFN-γ because IFN-γ/STAT1 is responsible for the transcriptional activation of IFN-responsive genes, which is also consistent with previous studies (22, 23). These results suggest that IFN-γ leads to a stronger inhibition of RSV replication than IFN-α.
FIG 1.

IFNs restrict RSV replication. A549 cells were treated with IFN-α (1,000 U/ml) or IFN-γ (1,000 U/ml) for 24 h, then infected with RSV at an MOI of 0.1, and harvested at different time postinfection. Viral proteins (A) and viral mRNA (B) in the cell lysates were detected by Western blotting and RT-qPCR, respectively. (C) The virus titer in the supernatant was measured using the endpoint dilution titer method. (D) IFN mRNA levels in A549 cells infected with RSV at an MOI of 0.1 were detected at different times via RT-qPCR. For RT-qPCR, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a control. (E) The effect of the RSV NS1 and NS2 proteins on STAT1 expression and phosphorylation. For statistical analysis, n = 3.
GBP5 is the major effector of the anti-RSV activity of IFN-γ.
Due to the fact that a series of host restrictive factors, including APOBEC3 family members, SAMHD1, BST-2, Mx2, and GBP5, were identified as IFN-inducible proteins and present potent antiviral activity (29, 41–43), we examined the mRNA expression levels of these factors after treatment with IFN-α or IFN-γ. As expected, IFN-γ more efficiently induced GBP1, GBP5, Mx2, and BST2 expression than did IFN-α, while IFN-α induced Mx2 expression (Fig. 2). We next investigated the effects of the four upregulated restrictive factors on RSV replication and found that GBP5 causes a decrease in the expression of RSV viral proteins in HEK293T cells, while BST2, Mx2, and GBP1 had no effect on RSV replication (Fig. 3A). We also measured the mRNA levels of the RSV targeting N protein and RSV viral titers in cell lysates and supernatants, respectively, and the results showed a similar trend (Fig. 3B and C). In order to confirm that GBP5 suppresses RSV replication, an A549 cell line stably expressing GBP5 was constructed. After confirming that stable expression of GBP5 had no effect on cell viability using the CCK8 kit (Fig. 3D), the cells were infected with RSV at an MOI of 0.1. Our results showed that RSV replication was suppressed at 24 and 48 h by GBP5, which was measured by detecting the RSV viral proteins, viral mRNA levels, and viral titers (Fig. 3E to G). Furthermore, we constructed cells which stably expressed a control short hairpin RNA (shRNA) or shRNA targeting GBP5 in A549 cells expressing GBP5. After confirmation that GBP5 knockdown had no effect on cell viability (Fig. 3H), GBP5 shRNA cells and control shRNA cells were infected with RSV at an MOI of 0.1 or were grown in Dulbecco’s modified Eagle’s medium (DMEM) as a control in the absence or presence of IFN-γ. The results showed that GBP5 knockdown produced more RSV viral proteins, increased viral mRNA levels, and stronger viral titers than for control shRNA cells with or without IFN-γ treatment (Fig. 3I to K). However, IFN-γ treatment led to the production of fewer viral proteins, lower viral mRNA levels, and weaker viral titers than for cells that did not receive IFN-γ treatment, demonstrating that GBP5 is a substantial but not unique effector in RSV restriction by IFN-γ. Taken together, our results show that as an IFN-γ-inducible protein, GBP5 is a major effector of the anti-RSV activity of IFN-γ.
FIG 2.

Detection of host restrictive factors stimulated by IFN-α and IFN-γ. A549 cells were treated with IFN-α (1,000 U/ml) or IFN-γ (1,000 U/ml) for 24 h and then harvested. Then, the mRNA levels of host restrictive factors were detected by RT-qPCR. GAPDH was used as a control (n = 3).
FIG 3.

GBP5 restricts RSV replication in vitro. (A to C) HEK293T cells seeded in 6-well plates were transfected with 0.5 μg or 1.5 μg of plasmids expressing host factors. At 24 h posttransfection, the cells were infected with RSV at an MOI of 0.1 and then harvested at 48 h postinfection. (D to G) A549 cells stably expressing GBP5-HA and control cells were constructed and infected with RSV at an MOI of 0.1 for 24 or 48 h. (I to K) A549-sh GBP5 cells or control cells were treated or not with IFN-γ for 24 h, as indicated, and then infected with RSV at an MOI of 0.1 for 24 or 48 h. Viral proteins (A, E, and I) and viral mRNA (B, F, and J) levels in the cell lysates were detected by Western blotting and RT-qPCR, respectively. (C, G, and K) Virus titers in the supernatant were measured using the endpoint dilution titer method. For RT-qPCR, GAPDH was used as a control. (D and H) The viability of the stable cell lines constructed was detected using the CCK8 assay. For statistical analysis, n = 3.
RSV sensitivity is negatively correlated with GBP5 expression in PBMCs.
It is well known that children under 5 years of age are more susceptible to RSV infection than adults. In order to investigate the correlation between GBP5 expression and RSV sensitivity in children and adults, peripheral blood mononuclear cells (PBMCs) from healthy donors (10 children and 10 adults) were harvested and treated or not with IFN-γ for 24 h. The cells were then infected with RSV at an MOI of 0.1 as indicated. The mRNA levels of the RSV N protein, IFN-γ, and GBP5 in cell lysates at 48 h postinfection were then measured. IFN-γ efficiently suppressed RSV replication in PBMCs from adults but failed to do so in the PBMCs from children (Fig. 4A). However, the IFN-γ and GBP5 mRNA levels in the PBMCs from adults could be greatly upregulated by IFN-γ treatment or RSV infection alone or by the combination of the IFN-γ treatment and RSV, while in children it could be upregulated only by IFN-γ treatment alone and not by RSV infection (Fig. 4B and C). RSV infection even attenuated the effects of IFN-γ stimulation, such as increased GBP5 expression, in the PBMCs from children (Fig. 4B and C). Moreover, we observed that the PBMCs of one child donor infected with RSV showed higher levels of RSV replication than those of other children, which might be due to the weaker IFN-γ response and lower GBP5 expression (Fig. 4A to C). These results suggest that decreased GBP5 expression caused by a weaker immune response in children is mainly responsible for their susceptibility to RSV infection.
FIG 4.
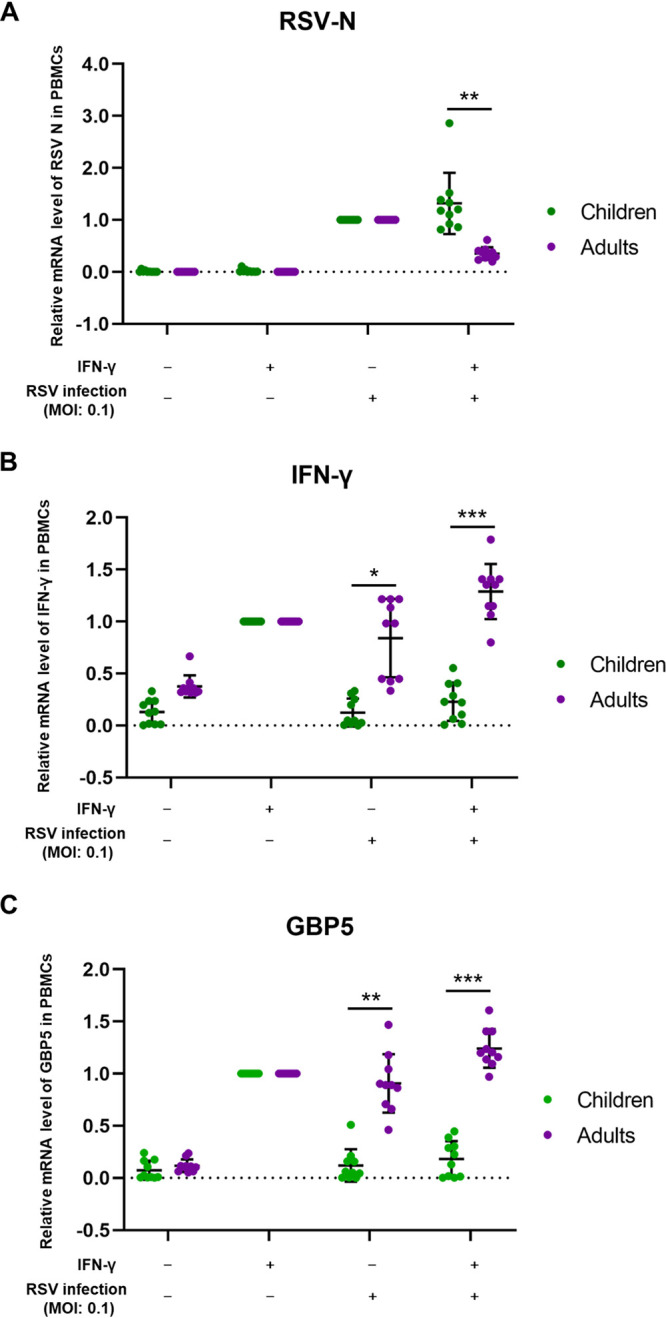
GBP5 expression is a major determinant of RSV sensitivity. PBMCs isolated from 20 healthy donors (10 children and 10 adults) were either treated with IFN-γ or infected with RSV at an MOI of 0.1 for 48 h, respectively, or treated with a combination of the two. RSV-N (A), IFN-γ (B), and GBP5 (C) mRNA levels in the PBMCs of children and adults were detected by RT-qPCR.
GBP5 downregulates the expression of the intracellular RSV SH protein.
In order to explore the mechanism by which GBP5 suppresses RSV replication, we investigated the effects of GBP5 on the expression of various RSV viral proteins and found that only the RSV SH viral protein was downregulated by GBP5 (Fig. 5A). Furthermore, we found by measuring viral protein expression and viral mRNA levels that treatment with an increased dose of the SH protein could rescue RSV replication in the presence of GBP5, whereas the SH protein alone slightly increased RSV replication in the absence of GBP5, indicating that the SH protein neutralized the GBP5-induced inhibition of RSV (Fig. 5B and C). We also observed that GBP5 had no effect on the mRNA levels of SH (Fig. 5D). Interestingly, treatment with MG132 (inhibitor of the proteasome 26s subunit) or Baf-A1 (inhibitor of lysosome acidification) did not affect the GBP5-mediated downregulation of the SH protein, indicating that this process is independent of the proteasome or lysosomal pathway (Fig. 5E). However, the coimmunoprecipitation (co-IP) assay showed that GBP5 could interact with the SH protein (Fig. 5F). The fluorescence resonance energy transfer (FRET) assay further showed that the RSV SH protein directly interacts with GBP5, as shown in Fig. 5G, due to the fact that bleaching of the fluorescence from GBP5-yellow fluorescent protein (YFP) enhanced the intensity of the fluorescence from SH-cyan fluorescent protein (CFP). Moreover, the colocalization assay showed that GBP5 and SH protein mainly colocalized at the cytoplasmic membrane (Fig. 5H), as previous reports had shown that the SH protein is localized mainly at the cytoplasmic membrane (44).
FIG 5.

GBP5 restricts RSV by downregulating SH expression in the cytoplasm. (A) Plasmids containing the viral proteins were cotransfected with GBP5 or the control vector VR1012 into HEK293T cells. At 48 h posttransfection, the cells were harvested and detected by immunoblotting. (B and C) The HEK293T cells were cotransfected with increasing doses of SH (0.1, 0.3, and 0.9 μg) and GBP5 or a negative-control vector for 24 h and then infected with RSV at an MOI of 0.5 for another 48 h. The cells were then harvested and analyzed by immunoblotting (B) and RT-qPCR (C). GAPDH was used as a control (n = 3). (D) The effect of GBP5 on RSV SH mRNA levels. (E) GBP5-mediated downregulation of SH is not associated with the proteasomal or lysosomal pathway. HEK293T cells cotransfected with SH and GBP5 or VR1012 were treated with DMSO, 10 μM MG132, or 10 nM Baf-A1 for 10 h before the cells were harvested. Protein levels in the harvested cells were detected by immunoblotting. (F) The interaction between GBP5 and SH. HEK293T cells transfected with GBP5-Flag and SH-HA were harvested 48 h posttransfection and subjected to an IP assay. The input and elution were detected by immunoblotting. (G) GBP5 directly interacts with the SH protein. HeLa cells transfected with GBP5-YFP and SH-CFP were analyzed using a FRET assay. The fluorescence of YFP and CFP was captured before and after bleaching. The areas within the white dotted line are bleached. Bars correspond to 10 μm. (H) Localization of GBP5 and the SH protein. HeLa cells transfected with GBP5-HA and SH-Flag alone or in combination were analyzed using an immunofluorescence assay. Bars correspond to 10 μm. (H) HEK293T cells were transfected with GBP5-Flag and SH-HA alone or in combination. Cells and supernatants were harvested 48 h posttransfection. Protein levels in the cell lysates and concentrated supernatant were detected by immunoblotting. (J) Schematic diagram of the sucrose density gradient centrifugation process. (K) HEK293T cells transfected with GBP5-Flag and SH-HA alone or in combination. The supernatants were harvested and separated via sucrose density gradient centrifugation. The samples harvested from different sucrose interpositions were analyzed by immunoblotting. (L) Samples between 30% and 45% sucrose in panel G were mixed and concentrated via ultracentrifugation. Then, the concentrated samples were analyzed via electron microscopy. Bars correspond to 100 nm.
Since the downregulation of the SH protein caused by GBP5 is not associated with the proteasome and lysosome pathways, we boldly speculate that GBP5 might promote an increase in the release of the SH protein from cells. To confirm our hypothesis, we compared the expression levels of intracellular SH proteins and those found in the culture media in the presence and absence of GBP5. We found that in the presence of GBP5, intracellular SH protein levels were reduced but SH protein levels in the culture media were increased (Fig. 5I). The different components of the supernatant were further separated by density gradient centrifugation and were then detected by immunoblotting (Fig. 5J). We found that the SH protein was present in the denser sucrose intercalation but not in that of CD9, which is considered a marker of exosomes (Fig. 5K). In order to reveal the mode by which the SH protein is secreted from cells, the separated samples were negatively stained and analyzed by electron microscopy. We found that there were microvesicles with diameters of 100 to 200 nm in the culture media of cells transfected with the SH protein (Fig. 5L). Thus, we speculate that the SH protein could form microvesicles, while GBP5 could promote the secretion of the SH protein through these microvesicles. However, more evidence is needed to confirm this hypothesis in the future.
Localization in the Golgi is needed for RSV inhibition by GBP5.
GTPase family members function as GTP/GDP-regulated switches and exhibit GTPase activity. They are also characterized by having a carboxyl-terminal CaaX motif (where “C” stands for cysteine) for prenylation, which leads to the attachment of a lipid hydrophobic moiety (45). Previous studies have shown that GBP5 is located in the Golgi (45). In order to further explore the essential GBP5 domain required for suppressing RSV replication, GBP5 GTPase-defective or prenylation-defective mutants (outside the Golgi) were constructed and evaluated to determine their ability to restrict RSV infection (Fig. 6A). We found that the GTPase-defective GBP5 (KS51/52AA, T75S, and D182N) mutations had no effect on RSV restriction. However, the prenylation-defective mutants C583A and ΔC (lacking the C-terminal domain), which were also reported to translocate outside the Golgi, were found to affect RSV inhibition (Fig. 6B and C). Moreover, the C583A and ΔC mutants failed to downregulate intracellular SH protein levels (Fig. 6D). These results suggest that the location of GBP5 in the Golgi is essential for RSV inhibition.
FIG 6.

Localization in the Golgi is essential for GBP5 restriction of RSV replication. (A) Schematic diagram of mutants and truncation of GBP5. (B and C) HEK293T cells were transfected with the wild type, mutants, and truncation of GBP5 or VR1012 for 24 h and then infected with RSV (MOI: 0.5) for another 48 h. Cells were then harvested and viral protein levels were detected by immunoblotting (B), while viral mRNA levels were detected by RT-qPCR (C). For RT-qPCR, GAPDH was used as a control (n = 3). (D) The wild type, mutants, and truncation of GBP5-Flag or VR1012 were cotransfected with SH-HA into HEK293T cells. At 48 h posttransfection, cells were harvested and detected by immunoblotting. β-Actin was used as a loading control.
The viral G protein triggers GBP5 degradation through ubiquitination.
Previous studies have demonstrated that viruses always antagonize the restriction of host factors through multiple mechanisms (43, 46). As we observed that GBP5 expression was moderately downregulated at 48 h post-RSV infection (Fig. 3E, lanes 6 and 8), we investigated which RSV viral protein was involved in this process. Thus, we cotransfected GBP5 and the indicated RSV viral protein expression vector or the control vector into HEK293T cells and found that the RSV G protein obviously downregulated GBP5 expression (Fig. 7A and B). Treatment with MG132 or Baf-A1 showed that MG132, but not Baf-1, rescued GBP5 expression (Fig. 7C). Moreover, we found that the G protein could enhance the K48 ubiquitination of GBP5 (Fig. 7D). These results indicated that the G protein downregulates GBP5 through ubiquitination and the proteasome pathway.
FIG 7.

The RSV G protein downregulates GBP5 expression. (A) GBP5-HA was cotransfected with viral proteins coupled with an HA tag or VR1012 into HEK293T cells. At 48 h posttransfection, the cells were harvested and detected by immunoblotting. (B) GBP5-HA was cotransfected with gradient G-Flag into HEK293T cells. At 48 h posttransfection, the cells were harvested and detected by immunoblotting. (C) HEK293T cells cotransfected with GBP5-HA and G-Flag or empty vector (VR1012) for 24 h were treated with DMSO, 10 μM of MG132, or 10 nM Baf-A1 for an additional 10 h. The protein levels in the cells were then detected by immunoblotting. (D) GBP5-HA was cotransfected with G-Myc and Ub-K48-Flag or Ub-K63-Flag into HEK293T cells. At 24 h posttransfection, the cells treated with 10 μM MG132 for 10 h and then harvested and analyzed via an immunoprecipitation assay. The input and elution were detected by immunoblotting.
The E3 ligase DZIP3, which is upregulated by the G protein, directly interacts with and degrades GBP5 through the proteasome pathway.
The RSV G protein is a structural protein of the RSV. However, when analyzing the sequence of the G protein, we did not find a domain which was similar to any E3 ligase. Therefore, we suspected that the G protein might hijack or upregulate an E3 ligase to induce the ubiquitination and degradation of GBP5. Proteins coimmunoprecipitated with GBP5 were harvested via a co-IP assay and identified by mass spectrometry. A total of 9 E3 ligases were selected for further investigation (Table 1). Moreover, the mRNA levels of these 9 E3 ligases were detected after RSV infection or G protein overexpression. Among them, only DZIP3 was found to be upregulated by both RSV infection and G protein overexpression (Fig. 8A and B) but not IFN (Fig. 8C). In order to investigate mechanisms by which RSV infection or G protein overexpression induced DZIP3 upregulation, we examined the epigenetic modifications in the promoter of DZIP3 and histone 3 K4 methylation (H3K4me) according to the predictions obtained via UCSC Genome Browser analysis. The chromatin immunoprecipitation (ChIP) assay showed that RSV G increased H3K4 methylation, which might lead to DZIP3 upregulation (Fig. 8D). We next confirmed that DZIP3 induced the degradation of GBP5 and also found that MG132 treatment could rescue GBP5 expression (Fig. 9A). Moreover, the co-IP and FRET assays showed that GBP5 specifically interacted with DZIP3 (Fig. 9B and C). In addition, we confirmed that DZIP3 induced K48, but not K63, ubiquitination of GBP5 (Fig. 9D).
TABLE 1.
E3 ligases binding to GBP5 detected by mass spectrometrya
| Protein name | Relative abundance (%) |
No. of peptides (confidence > 95%) |
|---|---|---|
| RNF115 | 5.92 | 7 |
| UFD1 | 4.89 | 6 |
| DZIP3 | 4.59 | 6 |
| OTUB1 | 3.69 | 4 |
| XIAP | 2.82 | 4 |
| CBL | 2.65 | 3 |
| MKRN2 | 1.92 | 4 |
| RFWD3 | 1.68 | 3 |
| UBR4 | 0.5 | 1 |
The protein of interest is in bold.
FIG 8.

DZIP3 is upregulated by RSV and the G protein but not IFNs. (A) The mRNA of E3 ligases from HEK293T cells infected with RSV at an MOI of 1.0 for 24 h were extracted and detected by RT-qPCR. (B) The mRNAs of E3 ligases from HEK293T cells which were transfected with the indicated viral proteins for 48 h were extracted and detected by RT-qPCR. (C) The mRNA of DZIP3 and Mx2 from A549 cells which were treated with IFN-α or IFN-γ were extracted and detected by RT-qPCR. (D) The RSV G protein increased the H3K4 methylation mark on the DZIP3 promoter. The anti-H3K4me ChIP assay in HEK293T cells shows the levels of H3K4me on the DZIP3 promoter following G protein expression.
FIG 9.

DZIP3 degrades GBP5 through ubiquitination and the proteasome pathway. (A) HEK293T cells cotransfected with GBP5-HA and DZIP3-Flag or VR1012 for 24 h were treated with DMSO or 10 μM MG132 for 10 h. Cell protein levels were then detected by immunoblotting. (B) HEK293T cells cotransfected with GBP5-HA and DZIP3-Flag or VR1012 were treated with 10 μM MG132 for 10 h at 24 h posttransfection. Then, the cells were harvested and analyzed using an IP assay. The input and elution were detected via immunoblotting. (C) HeLa cells transfected with GBP5-YFP and DZIP3-CFP were used to perform a FRET assay. The fluorescence of YFP and CFP was captured before and after bleaching. The areas within the white dotted line are bleached. Bars correspond to 10 μm. (D) GBP5-HA was cotransfected with DZIP3-Myc and Ub-K48-Flag or Ub-K63-Flag into HEK293T cells. At 24 h posttransfection, the cells were treated with 10 μM MG132 for 10 h and then harvested and used to perform an IP assay. The input and elution were detected by immunoblotting. (E and F) DZIP3-KO A549 cells and control cells were infected with RSV at an MOI of 1.0 and harvested at different time points. Protein levels in cell lysate (E) and viral mRNA levels in the supernatant (F) were detected by immunoblotting and RT-qPCR, respectively. For RT-qPCR, GAPDH was used as a control (n = 3). (G) DZIP3-shutdown HEK293T cells and control cells were cotransfected with GBP5-HA and G-Flag or VR1012. Cells were then harvested and analyzed by immunoblotting at 48 h posttransfection.
To confirm that DZIP3 is essential for RSV-induced inhibition of GBP5 restriction, RSV replication was examined in a DZIP3 knockout (DZIP3-KO) cell line, which was constructed in A549 cells with a lentivirus infection system and confirmed to have no effect on cell viability (data not shown). The results showed that RSV had a weaker replication in DZIP3 knockout A549 cells (Fig. 9E and F), while endogenous GBP5 expression was higher in DZIP3 knockout cells than in shRNA-negative control cells (Fig. 9E). Moreover, we confirmed that the RSV G protein failed to downregulate GBP5 expression in DZIP3 knockdown HEK293T cells, which further supports our hypothesis that the G protein or RSV infection mediates the downregulation of GBP5 in a DZIP3-dependent manner (Fig. 9G). DZIP3 knockdown in HEK293T cells also confirmed that DZIP3 has no effect on cell viability (data not shown). Therefore, our data show that DZIP3 is upregulated by the RSV G protein and interacts with GBP5, thereby inducing GBP5 ubiquitination and degradation through the proteasome pathway.
DISCUSSION
RSV is the most common cause of lower respiratory tract infection and has higher morbidity and mortality in high-risk patients, such as premature infants and severely immunocompromised patients. Thus, the role of IFNs in RSV infection as important components of the innate immunity has been an attractive research topic. However, this role has yet to be uncovered. A large number of clinical studies have shown that the concentration of IFN-γ in RSV infections is inversely correlated with disease severity (21, 47–49). In this study, we found that RSV infection led to the production of more IFN-γ than IFN-α, while IFN-γ showed a stronger ability to inhibit RSV (Fig. 1).
GBP5 is an IFN-γ-inducible gene (29–31), which belongs to the GTPase family, which is involved in a variety of cellular processes, including inflammasome assembly, vesicle trafficking, and innate immunity (50). GBP5 has been reported to be an inhibitor of various bacteria and viruses, including HIV-1 and IAV (29, 38), to activate the inflammasome (32, 34, 36, 38), and to trigger caspase-1-dependent pyroptosis, as well as to promote the release of the inflammatory cytokines interleukin 1β (IL-1β) and IL-18 (35). We demonstrated that among the IFN-γ-inducible host restrictive factors, GBP5 potently inhibits RSV replication and is a major effector of the anti-RSV activity of IFN-γ (Fig. 3). Accumulating evidence has shown that premature or weaker immune responses correlate with disease severity in children with RSV infection (20, 26, 27, 51). We further found that lower IFN-γ production and lower GBP5 expression in the PBMCs of children in response to IFN treatment or RSV infection lead to greater sensitivity to RSV infection than that of adults (Fig. 4). This phenomenon explains why RSV is prevalent in children or immunocompromised patients but not in adults.
A previous study found that GBP5 restricts HIV by interfering with the processing and virion incorporation of the viral envelope glycoprotein (Env) (29). In a recent study, the same group found that GBP2/5 reduced the replication of multiple viruses by inhibiting the activity of furin, which is a cellular protease that mediates the processing of diverse viral glycoproteins (37). The RSV SH protein is one of three glycoproteins comprising the surface proteins of RSV and can form pentamers that construct pores at the plasma membrane that mediate membrane permeability (11). Therefore, the SH protein was identified as a viroporin. However, the exact function of the SH protein remains unknown. In this study, we demonstrated that GBP5 specifically interacts with the RSV SH protein and promotes SH oversecretion from cells, resulting in the downregulation of intracellular SH protein levels and increased SH protein levels in the culture media (Fig. 5). This suggests that GBP5 directly restricts RSV replication by interfering with SH function. Although our results are different from those in previous studies, which found that recombinant RSV lacking the entire SH gene grows efficiently in certain cell lines (8), we observed that SH overexpression rescued RSV replication inhibited by GBP5 (Fig. 5B). Similar to the inhibitory effect of GBP5 on HIV-1 (29), localization in the Golgi but not its GTPase activity is essential for GBP5-mediated RSV inhibition (Fig. 6). RSV also encodes another glycoprotein, namely, F, which is cleaved by furin (47, 52). Thus, it is worth investigating in the future whether GBP5 inhibits furin-mediated RSV F protein processing, which might also contribute to RSV inhibition.
As an inhibitor of RSV infection, GBP5 expression was found to be reduced by virus replication (Fig. 3E). It is well known that viruses usually utilize their encoded proteins or host factors to overcome the restriction of host restrictive factors (42, 43, 46, 53). We observed that the RSV G protein or RSV infection could downregulate GBP5 expression by upregulating the mRNA levels of DZIP3, which is identified as an E3 ligase (Fig. 8). DZIP3 was mainly reported to be an RNA-binding ligase that causes histone H2A monoubiquitination, which represses transcription (54). DZIP3 also regulates the differentiation of stem cells (55, 56). We found via a FRET assay that DZIP3 directly interacts with GBP5 and degrades GBP5 through K48 polyubiquitination and the proteasome pathway (Fig. 9). Therefore, our results revealed a novel function of DZIP3 and identified DZIP3 as a negative regulator of GBP5 inhibition in RSV infection. Interestingly, we found that DZIP3 was not an IFN-α- or IFN-γ-inducible protein (Fig. 8C).
Thus, our study showed for the first time the complicated interplay between RSV infection and the IFN-γ-inducible protein GBP5 (Fig. 10) and further supported the hypothesis that the SH protein is a viroporin required for RSV replication. However, further studies will be needed to precisely reveal the molecular mechanism by which SH protein acts as viroporin. Our study also provides information for the development of innovative drug targets against RSV infection.
FIG 10.

Proposed inhibition mechanism of the IFN-γ-inducible protein GBP5 on RSV infection. RSV infection induces IFN-γ production, which stimulates GBP5 expression. In the presence of GBP5, the RSV SH protein, which is an important viroporin, is oversecreted from the cells, resulting in defective RSV, whereas infectious RSV was produced in the absence of GBP5. Meanwhile, RSV infection upregulates DZIP3 expression, which as an E3 ligase induces GBP5 degradation through the proteasomal pathway.
MATERIALS AND METHODS
Plasmid construction.
The cDNAs of BST2, Mx2, GBP1, GBP5, and DZIP3 were reverse transcribed from the mRNAs which were extracted from HEK293T cells and A549 cells. The cDNAs of viral proteins, including NS1, NS2, SH, M2-1, M2-2, N, P, and G, were reverse transcribed from viral mRNAs which were extracted from RSV. Fragments of these genes were amplified by PCR and constructed into the VR1012 vector, which was generously provided by Vical (San Diego, CA) conjugated with a hemagglutinin (HA) tag, a Flag tag, or a Myc tag. A fragment of GBP5 was amplified from GBP5-HA and constructed into pCDNA3-YFP. Fragments of SH and DZIP3 were digested and collected from SH-HA and DZIP3-HA, respectively, and constructed into pECFP-C1. Mutants and a truncation of GBP5 were constructed by PCR. Ub-K48-flag and Ub-K63-flag were described previously (53). Primers used for plasmids construction are listed in Table S1 in the supplemental material.
Construction of stable cell lines.
Sh-GBP5 and sh-DZIP3 cell lines were constructed from A549 cells through a lentiviral system which was described previously (53). For DZIP3 knockout cell line construction, CRISPR-control (K010) lentiviral plasmids were obtained from Applied Biological Materials (Richmond, BC, Canada). CRISPR-DZIP3 was constructed by PCR with the primers 5′-AAAGGACGAAACACCAGGAAAGGCTTGCTCGTCAAGTTTTAGAGCTAGAAATAG-3′ and 5′-TGGTGTTTCGTCCTTTCCACAAGAT-3′. Viruses containing guide RNA (gRNA) and Cas9 were produced in HEK293T cells and then used to infect A549 cells. After 48 h of positive infection, A549 cells were treated with 1 μg/ml of puromycin. Then monoclonal cells were screened in 96-well plates, and DZIP3 expression in surviving cells was detected with Western blotting.
Cell culture and viruses.
HEK293T (American Type Culture Collection [ATCC], Manassas, VA; catalog no. CRL-11268), HEp-2 (ATCC; catalog no. CCL-23), and A549 (ATCC; catalog no. CCL-185) cells were cultured as monolayers in Dulbecco’s modified Eagle’s medium (DMEM; HyClone, Logan, UT) supplemented with 10% heat-inactivated (56°C, 30 min) fetal calf serum (FCS; GIBCO BRL, Grand Island, NY) and maintained at 37°C with 5% CO2 in a humidified atmosphere. Peripheral blood mononuclear cells (PBMCs) were prepared from healthy donors (after informed consent was obtained) by density gradient centrifugation (Lymphoprep; Axis-Shield, UK). PBMCs were maintained in RPMI 1640 medium (HyClone) with 10% heat-inactivated FCS. The RSV A2 strain was a gift from Jilin University College of Life Science professor Chunlai Jiang.
Virus titration.
For titration, as described previously (57), HEp-2 cells were infected with 1:10 dilutions of the supernatants including viruses in the presence of 2% fetal bovine serum (FBS). After 4 to 5 days of incubation in 7% CO2 at 37°C, the endpoint dilution titer (50% tissue culture infective dose [TCID50]) was determined by crystal violet staining of the monolayers and calculated by the Reed-Muench formula.
Transfection and infection.
Plasmids transfections were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. For RSV infection, A549 or HEK29T cells were seeded in 6-well plates and grown to 70% confluence. After being washed with phosphate-buffered saline (PBS), cells were incubated with RSV in 37°C for 2 h, and then supernatants were replaced with fresh medium containing 10% FBS and maintained in 37°C and 5% CO2. For PBMC infection, 2 × 105 cells were incubated with RSV for 2 h. After being washed with PBS, infected cells were supplemented with fresh medium containing 10% FBS and maintained in 37°C and 5% CO2.
IB and antibodies.
For immunoblotting (IB), cells used in this study were harvested and boiled in 1× loading buffer (0.08 M Tris [pH 6.8] with 2.0% SDS, 10% glycerol, 0.1 M dithiothreitol, and 0.2% bromophenol blue), followed by a separation on a 12% polyacrylamide gel. Proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane for Western blot analysis. The membranes were incubated with primary antibodies, followed by a corresponding horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson ImmunoResearch, Suffolk, UK) diluted 1:10,000. Proteins were visualized using an ultrasensitive ECL chemiluminescence detection kit (Proteintech; catalog no. B500024) and then imaged and scanned by a laser imaging system (Azure Biosystems). Several membranes shown in the figures (Fig. 3I, Fig. 5A and K, and Fig. 7B) have a column defect due to long-term use of the charge-coupled-device (CCD) camera pixels, which need to be corrected periodically.
The following antibodies were used in this study: anti-hemagglutinin (anti-HA) monoclonal antibody (MAb) (Biolegend, catalog no. 901513), anti-β-actin MAb (Genscript; catalog no. A00702), anti-Flag MAb (Sigma; catalog no. F1804), anti-RSV polyclonal antibody (PAb) (Millipore; catalog no. AB1128), anti-GBP5 PAb (BBI; catalog no. D222408), anti-DZIP3 PAb (Genetex; catalog no. GEX124219), anti-STAT1 MAb (Cell Signaling Technology; catalog no. 14994), anti-phospho-STAT1 MAb (Cell Signaling Technology; catalog no. 9167), and anti-CD9 PAb (BBI; catalog no. D264336). For confocal microscopy, anti-HA antibody PAb (Invitrogen; 71-5500), anti-Flag MAb (Sigma; catalog no. F1804), goat anti-mouse IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor Plus 488 (Invitrogen; A32723), and goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 568 (Invitrogen; A11011), were used.
RNA extraction and RT-qPCR.
For reverse transcription-quantitative PCR (RT-qPCR), viral or cell RNA was extracted from cells and supernatants with TRIzol reagent (Invitrogen), diethyl pyrocarbonate (DEPC)-treated water, and RNase inhibitor (New England BioLabs, Ipswich, MA). The cDNA was generated by the Transcriptor first-strand cDNA synthesis kit (Roche; catalog no. 04896866001) and oligo(dT) 18 primers or random primer according to the supplier’s instructions. RT-qPCR was carried out on an Mx3005P instrument (Agilent Technologies, Stratagene, La Jolla, CA) with the FastStart universal SYBR green master mix (Roche; catalog no. 04913914001) and designed primers targeting the conserved region sequences of RSV-N, DZIP3, IFNs, GBP1, GBP5, APOBEC3A-H, BST2, Mx2, SAMHD1, and E3 ligases in this study. The RT-qPCR assay was performed in a 20-μl volume as the introduction of this kit. Amplification of the target fragment was carried out as follows: 95°C for 2 min, followed by 40 cycles of 95°C for 15 s, 55°C for 15 s, and 68°C for 20 s. All primers for qPCR are presented in Table S2.
Co-IP assay.
The coimmunoprecipitation (co-IP) assays were performed as previously reported (53). For SH-HA immunoprecipitation, HEK293T cells transfected with GBP5-Flag and SH-HA or VR1012 were treated with MG132 (Sigma) for 12 h prior to harvest. The cells were then harvested and washed twice with cold PBS, followed by disruption with lysis buffer (PBS containing 1% Triton X-100 and complete protease inhibitor cocktail [Roche]) at 4°C for 1 h. Cell lysates were clarified by centrifugation at 10,000 × g for 30 min at 4°C. Anti-HA agarose beads (Roche) were mixed with the precleared cell lysates and incubated at 4°C for 4 h on an end-over-end rocker. The reaction mixtures were then washed six times with cold wash buffer (20 mM Tris-HCl [pH 7.5], 100 mM NaCl, 0.1 mM EDTA, 0.05% Tween 20) and subsequently analyzed by immunoblotting.
Ultracentrifugation and collection of microvesicles.
For concentration of the vesicles in the supernatants, 10 ml of supernatant after centrifugation at 800 g for 5 min in room temperature was added upon the 1 ml of 20% (w/v) sucrose solution, and ultracentrifugation was performed at 100,000 × g for 2 h in 4°C with an Optima L-100 XP ultracentrifuge (Beckman Coulter, Brea, CA). The sediment at the bottom of the tube was resuspended in 100 μl of PBS. For density gradient centrifugation, 4 ml of supernatant after centrifugation at 800 × g for 5 min in room temperature was add upon the density gradient sucrose solution (0.9 ml/gradient), and ultracentrifugation was performed at 100,000 × g for 18 h in 4°C with an Optima L-100 XP ultracentrifuge (Beckman Coulter). Vesicles in intercalations of sucrose were collected for further tests.
Electron microscopy.
The microvesicles isolated by density gradient centrifugation and concentrated by ultracentrifugation were negative stained and analyzed with an electron microscope (Hitachi; H-7650).
Confocal microscopy.
For localization of GBP5 and SH, HeLa cells seeded in 6-well glass-bottom plates were transfected with SH-Flag and GBP5-HA, respectively, or combined. The cells were treated with 10 μM MG132 to avoid degradation for 12 h prior to fixing, then fixed in 4% paraformaldehyde at room temperature for 15 min, washed with PBS, permeabilized in 0.1% Triton X-100 for 5 min, washed in PBS, blocked in 2% bovine serum albumin (BSA) for 1 h, and then incubated at room temperature for 2 h with mouse anti-Flag antibody at 1:1,000 and anti-HA PAb at 1:250. Following a wash, cells were incubated with goat anti-mouse IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor Plus 488, goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 568, and donkey anti-sheep IgG (H+L) cross-adsorbed secondary antibody at room temperature for 1 h. After being washed with cold PBS and treated with 5 μg/ml of 4′,6-diamidino-2-phenylindole (DAPI; Sigma; catalog no. D8417) for 1 min, cells were analyzed by using a laser scanning confocal microscope (Olympus; FV 3000).
Mass spectrometry.
HEK293T cells transfected with GBP5-HA and treated with MG132 or dimethyl sulfoxide (DMSO) were subjected to co-IP assay with HA beads (Roche), and the elution was analyzed by mass spectrum. Mass spectrum analysis was performed by the National Center for Protein Science (Beijing, China).
Fluorescence resonance energy transfer (FRET) analysis.
HeLa cells seeded in 6-well glass-bottom plates were transfected with GBP5-YFP (1 μg) and enhanced cyan fluorescent protein-SH (ECFP-SH) or ECFP-DZIP3 (1 μg). The cells were treated with 10 μM MG132 to avoid degradation for 12 h prior to fixing, then fixed in 4% paraformaldehyde at room temperature for 15 min, and washed with PBS 3 times. Fluorescent images of samples were then acquired with an Olympus FV 3000 confocal imaging system.
ChIP-qPCR.
Chromatin immunoprecipitation (ChIP) was performed with a chromatin immunoprecipitation kit (Millipore; 17-371). Approximately 4 × 106 cells were used for each immunoprecipitation (IP). Briefly, 293T cells were transfected with RSV G expression vector or negative-control vector for 48 h. For cross-linking of proteins to DNA, the transfected cells were treated with 1% formaldehyde (Sigma-Aldrich) for 10 min at room temperature. Unreacted formaldehyde was quenched by 125 mM glycine for 5 min at room temperature and centrifugation at 700 × g for 5 min at 4°C. The cell pellets were washed with cold PBS three times and resuspended in 1 ml of SDS lysis buffer containing 1× protease inhibitor cocktail II. The cell lysates were sonicated on wet ice with 5 sets of 10-s pulses using a Cole-Parmer instrument to acquire the appropriate DNA fragment size between 150 and 900 bp and were centrifuged at 12,000 × g for 10 min at 4°C. The supernatants were collected and precleared by incubation with 60 μl of protein G agarose for 1 h at 4°C with rotation. The agarose was discarded by centrifugation at 5,000 × g for 1 min at 4°C, and the supernatants were transferred into new tubes for immunoprecipitation. Normal rabbit IgG (CST; 2729) and anti-H3K4me3 antibody (Abcam; 8580) were separately added to the RSV-G and negative vector groups, respectively. Immunoprecipitation was carried out overnight at 4°C with rotation with normal rabbit IgG or rabbit anti-H3K4me3 antibody, and incubation was continued with 60 μl of protein G agarose for another 2 h at 4°C with rotation. Low-salt immune complex wash buffer, high-salt immune complex wash buffer, and LiCl immune complex wash buffer were used sequentially one time each to wash the agarose, and Tris-EDTA (TE) was then used for two washes on a rotating platform, with centrifugation at 5,000 × g for 1 min. Then, 100 μl of elution buffer was added to each antibody/agarose complex for 15 min at room temperature, and the supernatants were collected by centrifugation at 3,000 × g for 1 min. The supernatants were incubated first with 8 μl of 5 M NaCl at 65°C for 4 h to reverse the DNA-protein cross-links and then with RNase A to remove RNA. Five volumes of bind reagent A were added into 1 volume of the supernatants. The supernatant/bind reagent A mixture was transferred to a filter in a collection tube and was then centrifuged at 12,000 × g for 30 s. Wash reagent B was used to wash the spin filter in the collection tube by centrifugation at 12,000 × g two times for 30 s each, and 50 μl of elution buffer C was added directly to the center of the spin filter membrane. The eluate was collected by centrifugation at 12,000 × g for 30 s and was then analyzed immediately by RT-qPCR.
Ethics statement.
PBMCs were from 7 healthy volunteer blood donors (3 children, 2 to 5 years old, and 4 adults, 30 to 36 years old). This study was approved by the Ethics Review Committee of the First Hospital of Jilin University and complied with the guidelines and principles of the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report. Informed consent was signed by all research participants.
Statistical analysis.
The data are representative of those from three experiments. Details of statistical analysis are provided in figure legends. All data are expressed as means ± standard deviations (SDs). Statistical comparisons between two groups were made using a Student t test. Significant differences are indicated in figures as follows: *, P < 0.05, **, P < 0.01 and ***, P < 0.001. P values of less than 0.05 are considered to represent a statistically significant difference. “ns” stands for no significance.
Supplementary Material
ACKNOWLEDGMENTS
We thank C. L. Jiang for RSV. We thank C. Y. Dai for providing essential reagents.
This work was supported in part by funding from the National Natural Science Foundation of China (no. 81672004 and 31270202 to W.Z. and 81701987 to Z.L.), the Jilin University Science and Technology Innovative Research Team (2017TD-05), the Science and Technology Department of Jilin Province (20190101003JH and 20190905008SF), the finance department of Jilin Province (2018SCZWSZX-004 and 2018SCZWSZX-063), the Key Laboratory of Molecular Virology, Jilin Province (20102209), the Youth Foundation of the First Hospital of Jilin University (JDYY82017003 and JDYY102019007), and the Graduate Innovation Fund of Jilin University.
Z.L., W.Z., and X.Q. conceived and designed the experiments, analyzed data, and wrote the manuscript. Z.L., X.Q., X.L., C.H., Z.Z., and X.Y. performed the experiments. S.H. and H.W. contributed reagents, materials, and analysis tools.
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Scheltema NM, Gentile A, Lucion F, Nokes DJ, Munywoki PK, Madhi SA, Groome MJ, Cohen C, Moyes J, Thorburn K, Thamthitiwat S, Oshitani H, Lupisan SP, Gordon A, Sanchez JF, O'Brien KL, Group PS, Gessner BD, Sutanto A, Mejias A, Ramilo O, Khuri-Bulos N, Halasa N, de-Paris F, Pires MR, Spaeder MC, Paes BA, Simoes EAF, Leung TF, da Costa Oliveira MT, de Freitas Lazaro Emediato CC, Bassat Q, Butt W, Chi H, Aamir UB, Ali A, Lucero MG, Fasce RA, Lopez O, Rath BA, Polack FP, Papenburg J, Roglic S, Ito H, Goka EA, Grobbee DE, Nair H, Bont LJ, PERCH Study Group. 2017. Global respiratory syncytial virus-associated mortality in young children (RSV GOLD): a retrospective case series. Lancet Glob Health 5:e984–e991. doi: 10.1016/S2214-109X(17)30344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi T, McAllister DA, O’Brien KL, Simoes EAF, Madhi SA, Gessner BD, Polack FP, Balsells E, Acacio S, Aguayo C, Alassani I, Ali A, Antonio M, Awasthi S, Awori JO, Azziz-Baumgartner E, Baggett HC, Baillie VL, Balmaseda A, Barahona A, Basnet S, Bassat Q, Basualdo W, Bigogo G, Bont L, Breiman RF, Brooks WA, Broor S, Bruce N, Bruden D, Buchy P, Campbell S, Carosone-Link P, Chadha M, Chipeta J, Chou M, Clara W, Cohen C, de Cuellar E, Dang D-A, Dash-Yandag B, Deloria-Knoll M, Dherani M, Eap T, Ebruke BE, Echavarria M, de Freitas Lázaro Emediato CC, Fasce RA, Feikin DR, Feng L, Gentile A, Gordon A, Goswami D, Goyet S, Groome M, Halasa N, Hirve S, Homaira N, Howie SRC, Jara J, Jroundi I, Kartasasmita CB, Khuri-Bulos N, Kotloff KL, Krishnan A, Libster R, Lopez O, Lucero MG, Lucion F, Lupisan SP, Marcone DN, McCracken JP, Mejia M, Moisi JC, Montgomery JM, Moore DP, Moraleda C, Moyes J, Munywoki P, Mutyara K, Nicol MP, Nokes DJ, Nymadawa P, da Costa Oliveira MT, Oshitani H, Pandey N, Paranhos-Baccalà G, Phillips LN, Picot VS, Rahman M, Rakoto-Andrianarivelo M, Rasmussen ZA, Rath BA, Robinson A, Romero C, Russomando G, Salimi V, Sawatwong P, Scheltema N, Schweiger B, Scott JAG, Seidenberg P, Shen K, Singleton R, Sotomayor V, Strand TA, Sutanto A, Sylla M, Tapia MD, Thamthitiwat S, Thomas ED, Tokarz R, Turner C, Venter M, Waicharoen S, Wang J, Watthanaworawit W, Yoshida L-M, Yu H, Zar HJ, Campbell H, Nair H, RSV Global Epidemiology Network. 2017. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 390:946–958. doi: 10.1016/S0140-6736(17)30938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Díez-Domingo J, Pérez-Yarza EG, Melero JA, Sánchez-Luna M, Aguilar MD, Blasco AJ, Alfaro N, Lázaro P. 2014. Social, economic, and health impact of the respiratory syncytial virus: a systematic search. BMC Infect Dis 14:544. doi: 10.1186/s12879-014-0544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amand C, Tong S, Kieffer A, Kyaw MH. 2018. Healthcare resource use and economic burden attributable to respiratory syncytial virus in the United States: a claims database analysis. BMC Health Serv Res 18:294. doi: 10.1186/s12913-018-3066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins PL, Melero JA. 2011. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res 162:80–99. doi: 10.1016/j.virusres.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amarasinghe GK, Aréchiga Ceballos NG, Banyard AC, Basler CF, Bavari S, Bennett AJ, Blasdell KR, Briese T, Bukreyev A, Caì Y, Calisher CH, Campos Lawson C, Chandran K, Chapman CA, Chiu CY, Choi K-S, Collins PL, Dietzgen RG, Dolja VV, Dolnik O, Domier LL, Dürrwald R, Dye JM, Easton AJ, Ebihara H, Echevarría JE, Fooks AR, Formenty PBH, Fouchier RAM, Freuling CM, Ghedin E, Goldberg TL, Hewson R, Horie M, Hyndman TH, Jiāng D, Kityo R, Kobinger GP, Kondō H, Koonin EV, Krupovic M, Kurath G, Lamb RA, Lee B, Leroy EM, Maes P, Maisner A, Marston DA, Mor SK, Müller T, Mühlberger E, Ramírez VMN, Netesov SV, Ng TFF, Nowotny N, Palacios G, Patterson JL, Pawęska JT, Payne SL, Prieto K, Rima BK, Rota P, Rubbenstroth D, Schwemmle M, Siddell S, Smither SJ, Song Q, Song T, Stenglein MD, Stone DM, Takada A, Tesh RB, Thomazelli LM, Tomonaga K, Tordo N, Towner JS, Vasilakis N, Vázquez-Morón S, Verdugo C, Volchkov VE, Wahl V, Walker PJ, Wang D, Wang L-F, Wellehan JFX, Wiley MR, Whitfield AE, Wolf YI, Yè G, Zhāng Y-Z, Kuhn JH. 2018. Taxonomy of the order Mononegavirales: update 2018. Arch Virol 163:2283–2294. doi: 10.1007/s00705-018-3814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson PR, Spriggs MK, Olmsted RA, Collins PL. 1987. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: extensive sequence divergence between antigenically related proteins. Proc Natl Acad Sci U S A 84:5625–5629. doi: 10.1073/pnas.84.16.5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bukreyev A, Whitehead SS, Murphy BR, Collins PL. 1997. Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site-specific attenuation in the respiratory tract of the mouse. J Virol 71:8973–8982. doi: 10.1128/JVI.71.12.8973-8982.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karron RA, Buonagurio DA, Georgiu AF, Whitehead SS, Adamus JE, Clements-Mann ML, Harris DO, Randolph VB, Udem SA, Murphy BR, Sidhu MS. 1997. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc Natl Acad Sci U S A 94:13961–13966. doi: 10.1073/pnas.94.25.13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Techaarpornkul S, Barretto N, Peeples ME. 2001. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J Virol 75:6825–6834. doi: 10.1128/JVI.75.15.6825-6834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carter SD, Dent KC, Atkins E, Foster TL, Verow M, Gorny P, Harris M, Hiscox JA, Ranson NA, Griffin S, Barr JN. 2010. Direct visualization of the small hydrophobic protein of human respiratory syncytial virus reveals the structural basis for membrane permeability. FEBS Lett 584:2786–2790. doi: 10.1016/j.febslet.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin H, Zhou H, Cheng X, Tang R, Munoz M, Nguyen N. 2000. Recombinant respiratory syncytial viruses with deletions in the NS1, NS2, SH, and M2-2 genes are attenuated in vitro and in vivo. Virology 273:210–218. doi: 10.1006/viro.2000.0393. [DOI] [PubMed] [Google Scholar]
- 13.Whitehead SS, Bukreyev A, Teng MN, Firestone CY, St Claire M, Elkins WR, Collins PL, Murphy BR. 1999. Recombinant respiratory syncytial virus bearing a deletion of either the NS2 or SH gene is attenuated in chimpanzees. J Virol 73:3438–3442. doi: 10.1128/JVI.73.4.3438-3442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rixon HW, Brown G, Aitken J, McDonald T, Graham S, Sugrue RJ. 2004. The small hydrophobic (SH) protein accumulates within lipid-raft structures of the Golgi complex during respiratory syncytial virus infection. J Gen Virol 85:1153–1165. doi: 10.1099/vir.0.19769-0. [DOI] [PubMed] [Google Scholar]
- 15.Triantafilou K, Kar S, Vakakis E, Kotecha S, Triantafilou M. 2013. Human respiratory syncytial virus viroporin SH: a viral recognition pathway used by the host to signal inflammasome activation. Thorax 68:66–75. doi: 10.1136/thoraxjnl-2012-202182. [DOI] [PubMed] [Google Scholar]
- 16.Araujo GC, Silva RH, Scott LP, Araujo AS, Souza FP, de Oliveira RJ. 2016. Structure and functional dynamics characterization of the ion channel of the human respiratory syncytial virus (hRSV) small hydrophobic protein (SH) transmembrane domain by combining molecular dynamics with excited normal modes. J Mol Model 22:286. doi: 10.1007/s00894-016-3150-6. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, To J, Verdia-Baguena C, Dossena S, Surya W, Huang M, Paulmichl M, Liu DX, Aguilella VM, Torres J. 2014. Inhibition of the human respiratory syncytial virus small hydrophobic protein and structural variations in a bicelle environment. J Virol 88:11899–11914. doi: 10.1128/JVI.00839-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuentes S, Tran KC, Luthra P, Teng MN, He B. 2007. Function of the respiratory syncytial virus small hydrophobic protein. J Virol 81:8361–8366. doi: 10.1128/JVI.02717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin Y, Bright AC, Rothermel TA, He B. 2003. Induction of apoptosis by paramyxovirus simian virus 5 lacking a small hydrophobic gene. J Virol 77:3371–3383. doi: 10.1128/jvi.77.6.3371-3383.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hijano DR, Vu LD, Kauvar LM, Tripp RA, Polack FP, Cormier SA. 2019. Role of type I interferon (IFN) in the respiratory syncytial virus (RSV) immune response and disease severity. Front Immunol 10:566. doi: 10.3389/fimmu.2019.00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell CD, Unger SA, Walton M, Schwarze J. 2017. The human immune response to respiratory syncytial virus infection. Clin Microbiol Rev 30:481–502. doi: 10.1128/CMR.00090-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, Buick R, Stevenson NJ, Touzelet O, Gadina M, Power UF, Johnston JA. 2007. Respiratory syncytial virus NS1 protein degrades STAT2 by using the elongin-cullin E3 ligase. J Virol 81:3428–3436. doi: 10.1128/JVI.02303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swedan S, Andrews J, Majumdar T, Musiyenko A, Barik S. 2011. Multiple functional domains and complexes of the two nonstructural proteins of human respiratory syncytial virus contribute to interferon suppression and cellular location. J Virol 85:10090–10100. doi: 10.1128/JVI.00413-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Comerlato Scotta M, Greff Machado D, Goecks Oliveira S, de Moura A, Rhoden Estorgato G, de Souza APD, Nery Porto B, de Araújo PD, Sarria EE, Pitrez PM, Jones MH, Araújo Pinto L, Tetelbom Stein R, Polack FP, Mattiello R. 2019. Evaluation of nasal levels of interferon and clinical severity of influenza in children. J Clin Virol 114:37–42. doi: 10.1016/j.jcv.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Ioannidis I, McNally B, Willette M, Peeples ME, Chaussabel D, Durbin JE, Ramilo O, Mejias A, Flano E. 2012. Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J Virol 86:5422–5436. doi: 10.1128/JVI.06757-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krilov LR, Hendry RM, Godfrey E, McIntosh K. 1987. Respiratory virus infection of peripheral blood monocytes: correlation with ageing of cells and interferon production in vitro. J Gen Virol 68:1749–1753. doi: 10.1099/0022-1317-68-6-1749. [DOI] [PubMed] [Google Scholar]
- 27.Marr N, Wang TI, Kam SH, Hu YS, Sharma AA, Lam A, Markowski J, Solimano A, Lavoie PM, Turvey SE. 2014. Attenuation of respiratory syncytial virus-induced and RIG-I-dependent type I IFN responses in human neonates and very young children. J Immunol 192:948–957. doi: 10.4049/jimmunol.1302007. [DOI] [PubMed] [Google Scholar]
- 28.Selvaggi C, Pierangeli A, Fabiani M, Spano L, Nicolai A, Papoff P, Moretti C, Midulla F, Antonelli G, Scagnolari C. 2014. Interferon lambda 1–3 expression in infants hospitalized for RSV or HRV associated bronchiolitis. J Infect 68:467–477. doi: 10.1016/j.jinf.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krapp C, Hotter D, Gawanbacht A, McLaren PJ, Kluge SF, Sturzel CM, Mack K, Reith E, Engelhart S, Ciuffi A, Hornung V, Sauter D, Telenti A, Kirchhoff F. 2016. Guanylate binding protein (GBP) 5 is an interferon-inducible inhibitor of HIV-1 infectivity. Cell Host Microbe 19:504–514. doi: 10.1016/j.chom.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 30.Martens S, Howard J. 2006. The interferon-inducible GTPases. Annu Rev Cell Dev Biol 22:559–589. doi: 10.1146/annurev.cellbio.22.010305.104619. [DOI] [PubMed] [Google Scholar]
- 31.Rupper AC, Cardelli JA. 2008. Induction of guanylate binding protein 5 by gamma interferon increases susceptibility to Salmonella enterica serovar Typhimurium-induced pyroptosis in RAW 264.7 cells. Infect Immun 76:2304–2315. doi: 10.1128/IAI.01437-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, Frase S, Zhu Q, Malireddi RKS, Kuriakose T, Peters JL, Neale G, Brown SA, Yamamoto M, Kanneganti TD. 2016. IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase-11-NLRP3 inflammasomes. Cell 167:382–396.e17. doi: 10.1016/j.cell.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, MacMicking JD. 2012. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336:481–485. doi: 10.1126/science.1217141. [DOI] [PubMed] [Google Scholar]
- 34.Cerqueira DM, Gomes MTR, Silva ALN, Rungue M, Assis NRG, Guimaraes ES, Morais SB, Broz P, Zamboni DS, Oliveira SC. 2018. Guanylate-binding protein 5 licenses caspase-11 for gasdermin-D mediated host resistance to Brucella abortus infection. PLoS Pathog 14:e1007519. doi: 10.1371/journal.ppat.1007519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S, Dussurgey S, Dick MS, Kistner A, Rigard M, Degrandi D, Pfeffer K, Yamamoto M, Henry T, Broz P. 2015. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat Immunol 16:476–484. doi: 10.1038/ni.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matta SK, Patten K, Wang Q, Kim BH, MacMicking JD, Sibley LD. 2018. NADPH oxidase and guanylate binding protein 5 restrict survival of avirulent type III strains of Toxoplasma gondii in naive macrophages. mBio 9:e01393-18. doi: 10.1128/mBio.01393-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braun E, Hotter D, Koepke L, Zech F, Groß R, Sparrer KMJ, Müller JA, Pfaller CK, Heusinger E, Wombacher R, Sutter K, Dittmer U, Winkler M, Simmons G, Jakobsen MR, Conzelmann K-K, Pöhlmann S, Münch J, Fackler OT, Kirchhoff F, Sauter D. 2019. Guanylate-binding proteins 2 and 5 exert broad antiviral activity by inhibiting furin-mediated processing of viral envelope proteins. Cell Rep 27:2092–2104.e10. doi: 10.1016/j.celrep.2019.04.063. [DOI] [PubMed] [Google Scholar]
- 38.Feng J, Cao Z, Wang L, Wan Y, Peng N, Wang Q, Chen X, Zhou Y, Zhu Y. 2017. Inducible GBP5 mediates the antiviral response via interferon-related pathways during influenza A virus infection. J Innate Immun 9:419–435. doi: 10.1159/000460294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kreft SG, Nassal M. 2003. hRUL138, a novel human RNA-binding RING-H2 ubiquitin-protein ligase. J Cell Sci 116:605–616. doi: 10.1242/jcs.00261. [DOI] [PubMed] [Google Scholar]
- 40.Hillyer P, Mane VP, Chen A, Dos Santos MB, Schramm LM, Shepard RE, Luongo C, Le Nouen C, Huang L, Yan L, Buchholz UJ, Jubin RG, Collins PL, Rabin RL. 2017. Respiratory syncytial virus infection induces a subset of types I and III interferons in human dendritic cells. Virology 504:63–72. doi: 10.1016/j.virol.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, Hue S, Barclay WS, Schulz R, Malim MH. 2013. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502:559–562. doi: 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, Bieniasz PD. 2013. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502:563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simon V, Bloch N, Landau NR. 2015. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol 16:546–553. doi: 10.1038/ni.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gan SW, Tan E, Lin X, Yu D, Wang J, Tan GM, Vararattanavech A, Yeo CY, Soon CH, Soong TW, Pervushin K, Torres J. 2012. The small hydrophobic protein of the human respiratory syncytial virus forms pentameric ion channels. J Biol Chem 287:24671–24689. doi: 10.1074/jbc.M111.332791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Britzen-Laurent N, Bauer M, Berton V, Fischer N, Syguda A, Reipschläger S, Naschberger E, Herrmann C, Stürzl M. 2010. Intracellular trafficking of guanylate-binding proteins is regulated by heterodimerization in a hierarchical manner. PLoS One 5:e14246. doi: 10.1371/journal.pone.0014246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z, Ning S, Su X, Liu X, Wang H, Liu Y, Zheng W, Zheng B, Yu XF, Zhang W. 2018. Enterovirus 71 antagonizes the inhibition of the host intrinsic antiviral factor A3G. Nucleic Acids Res 46:11514–11527. doi: 10.1093/nar/gky840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bont L, Heijnen CJ, Kavelaars A, van Aalderen WM, Brus F, Draaisma JM, Pekelharing-Berghuis M, van Diemen-Steenvoorde RA, Kimpen JL. 2001. Local interferon-gamma levels during respiratory syncytial virus lower respiratory tract infection are associated with disease severity. J Infect Dis 184:355–358. doi: 10.1086/322035. [DOI] [PubMed] [Google Scholar]
- 48.Garcia C, Soriano-Fallas A, Lozano J, Leos N, Gomez AM, Ramilo O, Mejias A. 2012. Decreased innate immune cytokine responses correlate with disease severity in children with respiratory syncytial virus and human rhinovirus bronchiolitis. Pediatr Infect Dis J 31:86–89. doi: 10.1097/INF.0b013e31822dc8c1. [DOI] [PubMed] [Google Scholar]
- 49.Semple MG, Dankert HM, Ebrahimi B, Correia JB, Booth JA, Stewart JP, Smyth RL, Hart CA. 2007. Severe respiratory syncytial virus bronchiolitis in infants is associated with reduced airway interferon gamma and substance P PLoS One 2:e1038. doi: 10.1371/journal.pone.0001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pilla-Moffett D, Barber MF, Taylor GA, Coers J. 2016. Interferon-Inducible GTPases in host resistance, inflammation and disease. J Mol Biol 428:3495–3513. doi: 10.1016/j.jmb.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sommer C, Resch B, Simoes EA. 2011. Risk factors for severe respiratory syncytial virus lower respiratory tract infection. Open Microbiol J 5:144–154. doi: 10.2174/1874285801105010144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Konig P, Giesow K, Schuldt K, Buchholz UJ, Keil GM. 2004. A novel protein expression strategy using recombinant bovine respiratory syncytial virus (BRSV): modifications of the peptide sequence between the two furin cleavage sites of the BRSV fusion protein yield secreted proteins, but affect processing and function of the BRSV fusion protein. J Gen Virol 85:1815–1824. doi: 10.1099/vir.0.80010-0. [DOI] [PubMed] [Google Scholar]
- 53.Li Z, Huan C, Wang H, Liu Y, Liu X, Su X, Yu J, Zhao Z, Yu XF, Zheng B, Zhang W. 2019. TRIM21-mediated proteasomal degradation of SAMHD1 regulates its antiviral activity. EMBO Rep doi: 10.15252/embr.201847528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou W, Zhu P, Wang J, Pascual G, Ohgi KA, Lozach J, Glass CK, Rosenfeld MG. 2008. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell 29:69–80. doi: 10.1016/j.molcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inoue D, Aihara H, Sato T, Mizusaki H, Doiguchi M, Higashi M, Imamura Y, Yoneda M, Miyanishi T, Fujii S, Okuda A, Nakagawa T, Ito T. 2015. Dzip3 regulates developmental genes in mouse embryonic stem cells by reorganizing 3D chromatin conformation. Sci Rep 5:16567. doi: 10.1038/srep16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saha S, Murmu KC, Biswas M, Chakraborty S, Basu J, Madhulika S, Kolapalli SP, Chauhan S, Sengupta A, Prasad P. 2019. Transcriptomic analysis identifies RNA binding proteins as putative regulators of myelopoiesis and leukemia. Front Oncol 9:692. doi: 10.3389/fonc.2019.00692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun Y, Jain D, Koziol-White CJ, Genoyer E, Gilbert M, Tapia K, Panettieri RA, Hodinka RL, López CB. 2015. Immunostimulatory defective viral genomes from respiratory syncytial virus promote a strong innate antiviral response during infection in mice and humans. PLoS Pathog 11:e1005122. doi: 10.1371/journal.ppat.1005122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.