

Virus entry into target cells is the first step for virus infection. Understanding the overall entry mechanism, including the binding mechanism of specific virus glycoproteins with cellular receptors, can be useful for the design of small molecule inhibitors and vaccine development. Recently, EphA2 was identified as an important entry receptor for both KSHV and EBV. In the present study, we investigated the required binding sites within EphA2 and EBV gH/gL that mediate the interaction of these two proteins allowing entry into epithelial cells and found that it differed in compared to the interaction of KSHV gH/gL with EphA2. Our discoveries may uncover new potential interventional strategies that block EBV and KSHV infection of target epithelial cells.
KEYWORDS: EPH receptors, Epstein-Barr virus, Kaposi’s sarcoma-associated herpesvirus, herpesviruses, virus entry
ABSTRACT
Both Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) are human gammaherpesviruses and are important in a variety of malignancies. Eph family receptor tyrosine kinase A2 (EphA2) is a cellular receptor for KSHV and EBV. Previous studies identified five conserved residues (ELEFN50-54) in the N-terminal domain of KSHV gH that are critical for Eph binding and KSHV infection. However, the specific domains of EBV gH/gL important for EphA2 binding are not well described. We found that the KSHV gH (ELEFN50-54) motif is important for higher KSHV fusion and that EBV gH/gL does not utilize a similar motif for fusion activity. We previously identified that an EBV gL N-glycosylation mutant (gL-N69L/S71V) was hyperfusogenic in epithelial cells but not in B cells. To determine whether this glycosylation site may be the binding region for EphA2, we compared the EphA2 binding activity of EBV gH/gL and the EBV gH/gL-N69L/S71V mutant. We found that EBV gH/gL-N69L/S71V had higher binding affinity for EphA2, indicating that the EBV gL N-glycosylation site might be responsible for inhibiting the binding of gH/gL to EphA2. Loss of N-glycosylation at this site may remove steric hindrance that reduces EBV gH/gL binding to EphA2. In addition, the mutations located in the large groove of EBV gH/gL (R152A and G49C) also have decreased binding with EphA2. Taken together, our data indicate that the binding site of EphA2 on EBV gH/gL is at least in part proximal to the EBV gL glycosylation site, which in part accounts for differences in EphA2 binding affinity by KSHV.
IMPORTANCE Virus entry into target cells is the first step for virus infection. Understanding the overall entry mechanism, including the binding mechanism of specific virus glycoproteins with cellular receptors, can be useful for the design of small molecule inhibitors and vaccine development. Recently, EphA2 was identified as an important entry receptor for both KSHV and EBV. In the present study, we investigated the required binding sites within EphA2 and EBV gH/gL that mediate the interaction of these two proteins allowing entry into epithelial cells and found that it differed in compared to the interaction of KSHV gH/gL with EphA2. Our discoveries may uncover new potential interventional strategies that block EBV and KSHV infection of target epithelial cells.
INTRODUCTION
Herpesviruses are a large, diverse family of double-stranded DNA enveloped viruses capable of infecting a wide range of hosts and causing a variety of diseases. There are nine human herpesviruses, including herpes simplex virus types 1 and 2, varicella-zoster virus (VZV), human cytomegalovirus, Epstein-Barr virus (EBV), and Kaposi’s sarcoma-associated herpesvirus (KSHV) (1). Among these nine viruses, EBV and KSHV belong to the gammaherpesvirus family and are oncogenic viruses associated with a variety of human malignancies (2, 3).
EBV was the first identified human oncogenic herpesvirus and infection is associated with Burkitt lymphoma and Hodgkin lymphoma both originating from B lymphocytes. EBV is also associated with nasopharyngeal and gastric carcinoma, which are of epithelial cell origin. These two cancer types reflect the two major target cell types for EBV infection in humans (3). KSHV is associated with Kaposi’s sarcoma (KS) and two lymphoproliferative disorders: primary effusion lymphoma and multicentric Castleman’s disease. Compared to EBV, KSHV has a broader cell tropism, including endothelial cells, B cells, monocytes, epithelial cells, dendritic cells, macrophages, and fibroblasts (4), whereas EBV infection is typically only observed in B cells and gastric and oral epithelial cells.
EBV and KSHV entry into target cells is a complex multistep process and is initiated by the binding and interaction of viral envelope glycoproteins with cellular receptors. The core fusion machinery for both EBV and KSHV includes gH/gL and gB. EBV gB is a class III viral fusogen that activates membrane fusion of virus and host cell membranes (5). While gB activates fusion, gH/gL regulates fusion by triggering the conformational change of gB from prefusion to postfusion form following receptor binding, resulting in membrane fusion (6). Multiple receptors for EBV and KSHV infection have been identified. Among these receptors, Ephrin receptor A2 (EphA2) is an entry receptor for both EBV and KSHV (7–9). It has also been shown that EphA4, EphA5, and EphA7 are entry receptors for KSHV infection in HEK293 cells and BJAB cells, respectively (10–12). EphA receptors belong to the ephrin receptor family, a large family of receptor tyrosine kinases that activate multiple diverse signaling pathways. The activation of ephrin receptor involves receptor tyrosine phosphorylation and is induced by cell surface-anchored ephrin ligands (13). It has been shown that KSHV interacts with EphA2 receptor to amplify signaling essential for productive infection (14). Binding of gH/gL to EphA2 triggers EphA2 phosphorylation and endocytosis, a major pathway of KSHV entry (9). There are 14 Eph receptors divided into two classes, A and B, based on sequence and binding affinity with their ligands. Their functions include boundary formation, cell migration, angiogenesis, proliferation, and cell differentiation (13). Interestingly, EphA2 has been identified as a host receptor for many pathogens, including Cryptococcus neoformans, hepatitis C virus, EBV, and KSHV (7, 9, 15, 16).
Recently, the binding site for the KSHV gH/gL complex within EphA2 and the corresponding interaction site on the KSHV gH/gL complex were identified (17, 18). This information has remained elusive for EBV. In the present study, we aimed to identify the binding regions of EphA2 and EBV gH/gL, allowing a better understanding of both EBV- and KSHV-directed membrane fusion for the development of novel interventional strategies.
RESULTS
The binding of KSHV gH/gL and EBV gH/gL to EphA2 differs.
EphA2 is the entry receptor for both KSHV and EBV gH/gL; however, it has a different affinity for KSHV and EBV gH/gL. The affinity of EphA2 with KSHV gH/gL is higher than that with EBV gH/gL (10). EphA2 is a membrane protein with extracellular portions that contain a ligand-binding extracellular domain (LBD), a cysteine-rich region (CYS), and two fibronectin regions (FBN). EphA2 and EphA4 share about 51% similarity at the amino acid level. EphA2, but not EphA4, can mediate EBV fusion. Previously, the LBD domain of EphA2 was found important for EphA2 function in EBV fusion (7–9, 17). However, it is not known whether LBD is the only domain that is important for fusion. To investigate this, we used a series of EphA2/EphA4 chimeras that we previously constructed, as well as additional constructs as illustrated in Fig. 1A (10). We found that substitution of each of the EphA4 extracellular domains with the EphA2 extracellular domains increased fusion activity beginning with the EphA2 LBD domain and ending with the FN domains (Fig. 1B). These results indicate each domain of the EphA2 ectodomain is important in EBV fusion. In contrast, it was shown that only the LBD is important for KSHV fusion (9, 10). This suggests that the binding of KSHV and EBV gH/gL with EphA2 is different.
FIG 1.
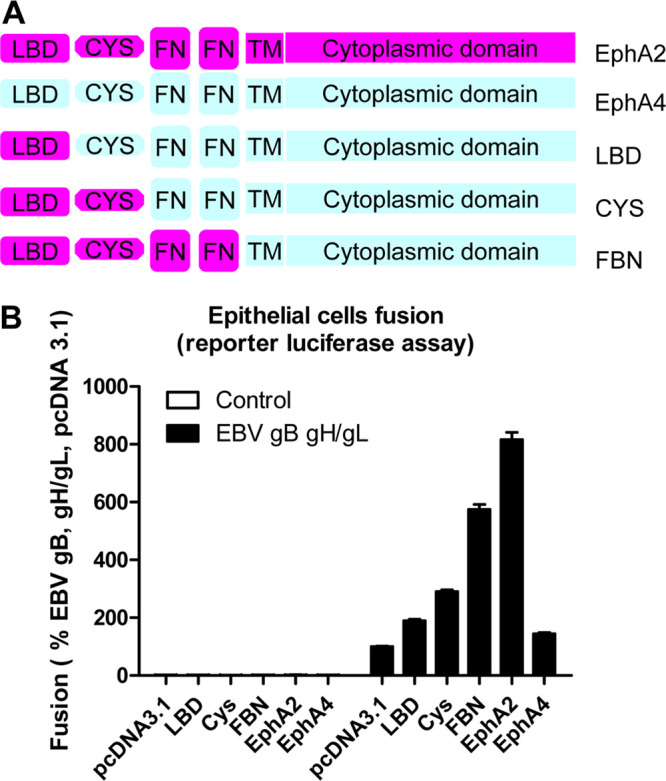
The ectodomain length of EphA2 is important for EBV fusion activity. (A) Schematic drawing of the EphA2, EphA4, and, EphA2/EphA4 chimeras. LBD, ligand-binding domain; CYS, cysteine-rich region; FN, fibronectin region. (B) EBV fusion with HEK293T cells transfected with control plasmid pcDNA3.1, EphA2, EphA4, or EphA2/EphA4 chimeras, as indicated. The fusion activity of HEK293T cells transfected with pcDNA 3.1 was set to 100%.
KSHV gH/gL mediates higher fusion activity compared to EBV gH/gL, and heterologous gH/gL complexes are not functional.
In our previous studies, we found that KSHV gH/gL can mediate fusion in the presence of EBV gB in HEK293T cells (10, 19). The fusion activity is three times higher than wild-type (WT) EBV fusion when KHSV gH/gL is paired with EBV gB (Fig. 2A). As previously shown (10), this may be due to the higher binding affinity of KSHV gH/gL to EphA2 compared to that of EBV gH/gL. To determine whether gH and/or gL play a role in this higher fusion activity, we tested fusion function of different combinations of EBV and KSHV gH/gL in the presence of EBV gB (Fig. 2A). We found that EBV gH and KSHV gL or the reciprocal combination did not mediate fusion in the presence of EBV gB using a luciferase reporter fusion assay (Fig. 2A). This is consistent with our previous studies that heterologous complexes of EBV/KSHV gH/gL are nonfunctional in fusion since they are not expressed at the cell surface (19). We also tested fusion activity using a syncytium formation assay. We found that KSHV gH/gL in combination with EBV gB had larger syncytia compared to EBV gB/gH/gL (Fig. 2B, compare subpanels b and c). Syncytium formation is barely detectable when the gH/gL complex components are not from the same virus or when using KSHV gB (Fig. 2B, subpanels d, e, and f).
FIG 2.
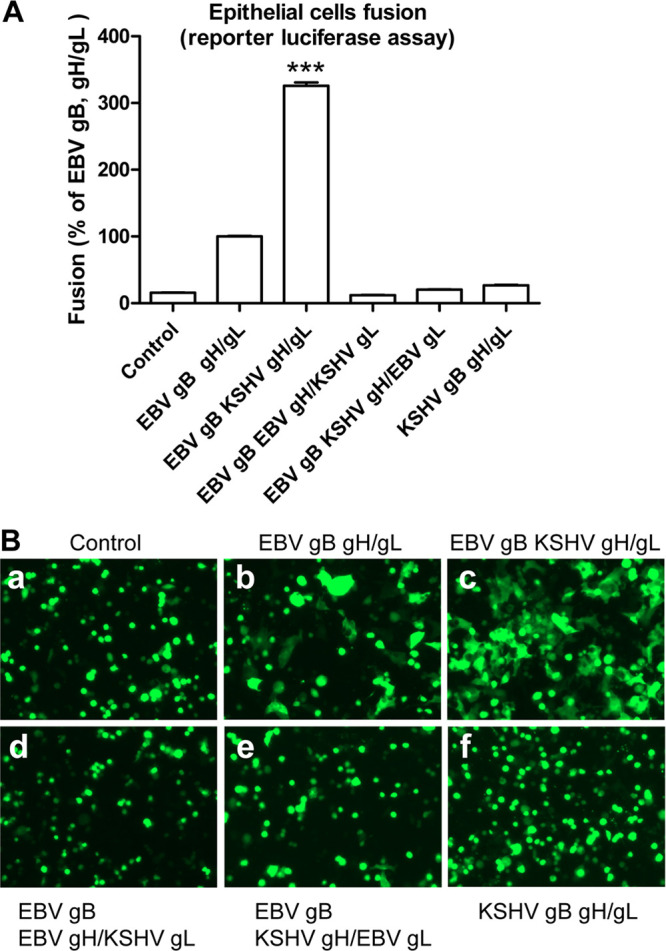
KSHV gH/gL mediate higher fusion activity compared to EBV gH/gL, and heterologous gH/gL is not functional. CHO-K1 cells were transfected with T7 luciferase plasmid (A) or GFP reporter (B) plasmid with either control plasmid, EBV gB/gH/gL, EBV gB with KSHV gH/gL, or other combinations as indicated. Transfected CHO-K1 cells were overlaid with HEK293T cells expressing T7 polymerase. Fusion activity was measured either by luciferase assay (A) standardized to EBV gB/gH/gL fusion activity which was set to 100% or by syncytium formation assay (B) which was visualized and captured with a EVOS fluorescence microscope. ***, P < 0.001 (Student t test), compared to EBV gB/gH/gL fusion activity.
The KSHV gH Eph-binding motif ELEFN50-54 is important for KSHV-mediated fusion but does not increase EBV fusion activity when this motif replaces the native EBV gH DIEGH28-32 motif.
Previous studies from the Hahn laboratory identified a conserved Eph family receptor-binding motif ELEFN50-54 contained within KSHV gH and rhesus monkey rhadinovirus (RRV) (18). The conserved ELEFN50-54 residues are located in the N-terminal domain of KSHV gH and are critical for Eph-binding and infection. Homology-based structural prediction of the KSHV gH/gL complexes based on the EBV gH/gL crystal structure indicates that the ELEFN50-54 motif would be located in a beta-hairpin on gH, which is likely stabilized by gL and is optimally positioned for protein-protein interactions (18). Within the same region of EBV gH, a DIEGH28-32 motif is found that is somewhat similar to the KSHV gH motif with the first three amino acids (DIE) being similar in properties to the ELE found in KSHV gH, whereas the last two amino acids of the ELEFN motif are dissimilar, being GH in EBV gH and FN in KSHV gH (Fig. 3A). The DIEGH28-32 motif is also located in a beta-hairpin on EBV gH, as is the KSHV ELEFN50-54. To investigate whether the KSHV ELEFN50-54 is important for the higher fusion activity of KSHV gH compared to EBV gH, we mutated the KSHV gH ELEFN50-54 to ELAAN and found the fusion activity was decreased (Fig. 3B), as expected from previous results from the Hahn laboratory (18). However, when the EBV gH DIEGH28-32 sequence was changed to ELEFN50-54, there was no change in fusion, suggesting that this region of EBV gH does not play as dominant a role in EphA2 binding and fusion in the context of EBV gH/gL (Fig. 3C).
FIG 3.

The KSHV Eph binding motif ELEFN50-54on gH is important for KSHV fusion but does not increase EBV fusion. (A) Alignment of EBV and KSHV gH showing KSHV Eph binding motif ELEFN50-54 and the corresponding amino acids DIEGH28-32 on EBV gH. (B) CHO-K1 cells transiently transfected with T7 luciferase plasmid, along with vector plasmid (control), EBV gB plus KSHV His gH/gL, and KSHV His ELAAN50-54/gL, were overlaid with HEK293T expressing T7 polymerase 24 h posttransfection. The luciferase activity was monitored 24 h after overlay and normalized to cells, with EBV gB with KSHV His gH/gL set to 100%. The data are means plus the SEM for three independent experiments. (C) CHO-K1 cells were transiently transfected with T7 luciferase plasmid, along with vector plasmid (control), EBV gB plus EBV gH/gL, EBV gH ELEFN28-32/gL, or KSHV gH/gL. At 24 h posttransfection, transfected CHO-K1 cells were overlaid with HEK293T expressing T7 polymerase, and the luciferase activity was monitored 24 h after overlay and normalized to cells with EBV gB/gH/gL, which was set to 100%. The data are means plus the SEM for three independent experiments.
EBV gL lacks 37 carboxy-terminal amino acids compared to the KSHV gL.
Since gH/gL forms a complex and both of them are important for the fusion function, we next examined whether gL plays a role in the increased fusion activity of KSHV. To compare the overall amino acid homology between EBV gL and KSHV gL, we aligned the relevant amino sequence of the two proteins. The alignment indicated that the first 137 amino acids of EBV and KSHV gL have 26% of the amino acids being identical and 46% being similar. Most interestingly in aligning the two gLs, it is readily apparent that KSHV gL has an additional 37 amino acids compared to EBV gL (Fig. 4A). To test whether the length of gL is important for increased KSHV fusion activity, we truncated KSHV gL to be similar in length to EBV gL (131 amino acids). We found that fusion was nearly identical to WT KSHV gL, indicating that the additional 37 amino acids of KSHV gL are not important for KHSV gH/gL mediated fusion activity (Fig. 4B).
FIG 4.

Deletion of the additional 37 amino acids in the KSHV gL C-terminal tail absent in the EBV gL tail has no impact on KSHV fusion activity. (A) Alignment of EBV gL and KSHV gL using EMBOSS Needle, indicating that 26% of the amino acids are identical and 46% are similar. The alignment shows the 37 additional C-terminal amino acids found in KSHV gL. (B) CHO-K1 cells were transiently transfected with T7 luciferase plasmid alone with vector plasmid (control), EBV gB together with EBV gH/gL, KSHV gH/gL, and KSHV gH/gL 131 stop. At 24 h posttransfection, transfected CHO-K1 cells were overlaid with HEK293T expressing T7 polymerase, and the luciferase activity was monitored 24 h after overlay and normalized to cells with EBV gB/gH/gL set to 100%. The data are means plus the SEM for three independent experiments.
The EBV gL N-glycosylation site is important for binding EphA2.
Previously, we found mutation of a EBV gL glycosylation site (gH/gL-N69L/S71V) was hyperfusogenic in epithelial cell fusion compared to B cell fusion (20). Interestingly, the EBV gL-N69L/S71V hyperfusogenic phenotype is not observed when EphA2 is overexpressed in our cell-based fusion assay (Fig. 5A). However, higher fusion levels are observed in control pcDNA 3.1-transfected cells (Fig. 5A). To exclude the possibility that loss of the hyperfusogenic phenotype was due to a saturation of fusion activity in the presence of EphA2, we tested EBV fusion activity in the presence of another hyperfusogenic phenotype mutant, EBV gB843 (Fig. 5B) (21). In contrast to EBV gH/gL-N69L/S71V, we still observed the gB843 hyperfusogenic phenotype in the presence of EphA2. From these data, we hypothesized that the EBV gL glycosylation site mutant altered EphA2 interactions since overexpression of EphA2 has a similar effect as expression of the EBV gH/gL-N69L/S71V mutant in fusion function. To examine whether glycosylation of this site might decrease KSHV fusion, we mutated the corresponding amino acids (TG66-67) in KSHV gL to NGSN66-69 (Fig. 4A) and examined the glycosylation of KSHV gL and its effect on fusion activity. We found that mutation of TG66-67 to NGSN66-69 did not alter fusion activity (Fig. 6A). Western blotting data showed that the molecular weight increased for the KSHV His gL NGSN66-69 mutant (∼26 kDa compared to the WT KSHV His gL of ∼22 kDa) as expected (Fig. 6B), confirming the glycosylation of KSHV gL. Based on the EBV gH/gL crystal structure, the EBV gH DIEGH28-32 sequence that corresponds to the KSHV EphA2 binding motif ELEFN50-54 is located in a beta-hairpin on EBV gH (Fig. 6C). Interestingly, the EBV gL glycosylation site NGSN69-72 is located in another beta-hairpin on EBV gL, which is symmetrical to the beta-hairpin on EBV gH (Fig. 6C). These observations indicate that EBV and KSHV might use similar hairpin structures but distinct sites for EphA2 binding.
FIG 5.
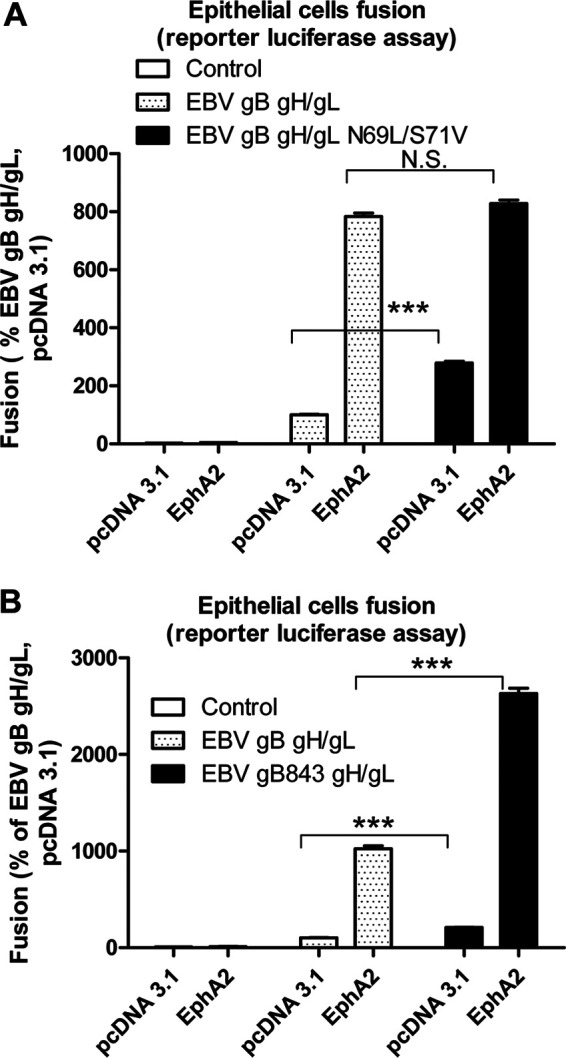
The N-glycosylation site within EBV gL is likely the EphA2 binding site for EBV fusion. (A) CHO-K1 cells transfected with T7 luciferase plasmid, together with either vector plasmid (control), EBV gB with EBV gH/gL, or EBV gH/gL-N69L/S71V, were overlaid with HEK293 cells transfected with control plasmid pcDNA 3.1, EphA2, or EphA4. EBV fusion with HEK293 cells transfected with pcDNA 3.1 was set to 100%. The fusion activity was normalized to EBV fusion with pcDNA 3.1-transfected cells, which was set to 100%. The data are means plus the SEM for three independent experiments. (B) Virus-free EBV or EBV gB843 fusion with WT HEK293T cells that overexpress EphA2 or EphA4. The bars represent the fusion activity, and the data are means plus the SEM for three independent experiments.
FIG 6.
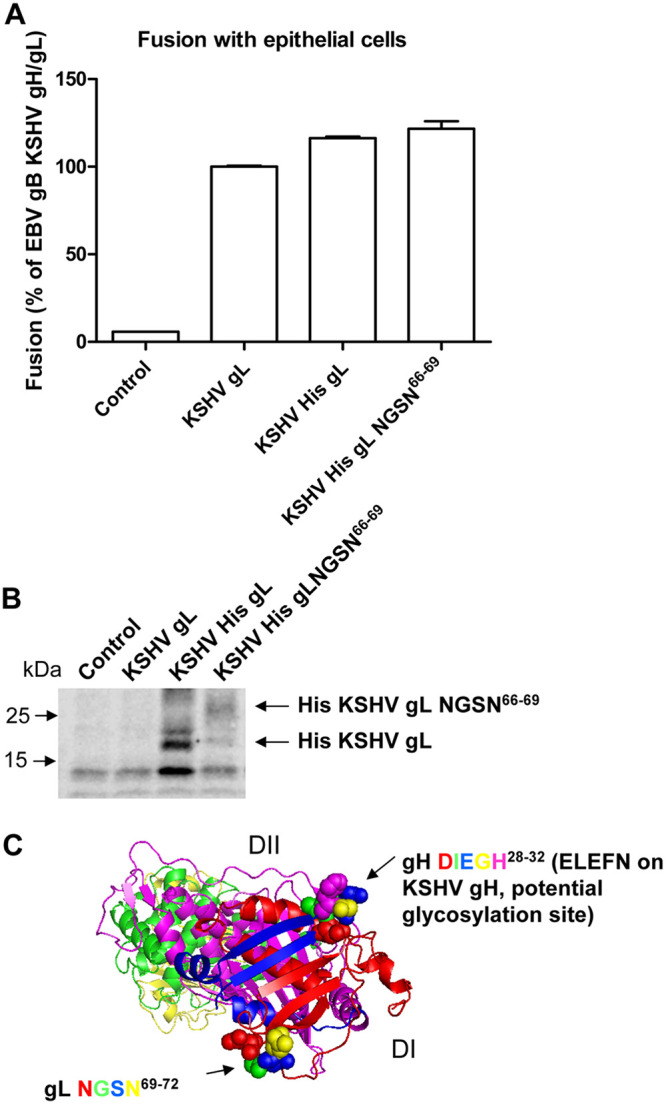
An increase in KSHV gL N-glycosylation does not have effect on KSHV fusion activity. (A) CHO-K1 cells were transiently transfected with T7 luciferase plasmid alone with vector plasmid (control), EBV gB together with KSHV gH/gL, KSHV gH/His gL, and KSHV gH/His gL NGSN66-69. At 24 h posttransfection, transfected CHO-K1 cells were overlaid with HEK293T cells expressing T7 polymerase, and the luciferase activity was monitored 24 h after overlay and normalized to cells with EBV gB with KSHV gH/gL set to 100%. The data are means plus the SEM for three independent experiments. (B) CHO-K1 cells seeded in 6-well plates were transiently transfected with vector plasmid (control), KSHV gH/gL, KSHV gH/His gL, and KSHV gH/His gL NGSN66-69. At 24 h after transfection, the cells were dissociated, washed once with ice-cold PBS, and lysed with 100 μl of 1× SDS lysis buffer. Proteins were separated on Bio-Rad 10% mini-Protean TGX gels. Western blot analyses were performed using a monoclonal anti-His antibody (Calbiochem, OB05) at 1:1,000 to detect the KSHV His gL or KSHV His gL NGSN66-69 (∼26 kDa for KSHV His gL NGSN66-6 compared to the WT KSHV His gL of ∼22 kDa). (C) Structure diagram of gH/gL. gH consists of four domains: DI (blue), DII (magenta), DIII (green), and DIV (yellow). gL is colored red and interacts with gH in DI. The DIEGH28-32 sequence on gH is represented by blue spheres, and the NGSN69-72 sequence on gL is represented by colored spheres.
The EBV gH/gL-N69L/S71V has higher affinity for EphA2 compared to WT EBV gH/gL.
We hypothesized that the glycosylation of EBV gL might block binding of EBV gH/gL to EphA2 due to steric hindrance. To test this hypothesis, we compared the binding affinity of EBV gH/gL and EBV gH/gL-N69L/S71V mutant to EphA2. We first examined binding of soluble EphA-Fc to cells expressing EBV gH/gL or EBV gH/gL-N69L/S71V. Cells were washed to remove unbound EphA2-Fc, and then lysed and bound EphA2-Fc was determined using antibodies to the Fc region of EphA2 by Western blotting. Expression of EBV gH (∼80 kDa) or EBV gL expression was monitored using anti-EBV gH/gL polyclonal antibodies directed against EBV gL (Fig. 7A). As expected, the EBV gL glycosylation mutant (∼21 kDa) migrates more rapidly than the WT gH/gL (∼25 kDa) (Fig. 7A), and there was more EphA2-Fc (∼180 kDa) associated with EBV gH/gL-N69L/S71V. The quantification data from three independent experiments showed that the binding of EphA2-Fc with EBV gH/gL-N69L/S71V is about two times greater than WT EBV gH/gL (Fig. 7B). To further confirm these results, we used a cell enzyme-linked immunosorbent assay (CELISA) assay to investigate the binding of the soluble EphA2-Fc to cells expressing EBV gH/gL or EBV gH/gL-N69L/S71V. CHO-K1 cells seeded in 6-well plates were transfected with soluble EphA2-Fc only or soluble EphA2-Fc with vector control plasmid, EBV gH/gL, and EBV gH/gL-N69L/S71V. Transfected cells were seeded in 96-well plates in triplicate posttransfection. The cells were then washed with ice-cold phosphate-buffered saline (PBS) three times, and the gH/gL-associated EphA2 (Fig. 7C) or surface expression of EBV gH/gL (Fig. 7D) was determined by CELISA using anti-human IgG to detect bound EphA2-Fc or E1D1 anti-gH/gL antibodies.
FIG 7.
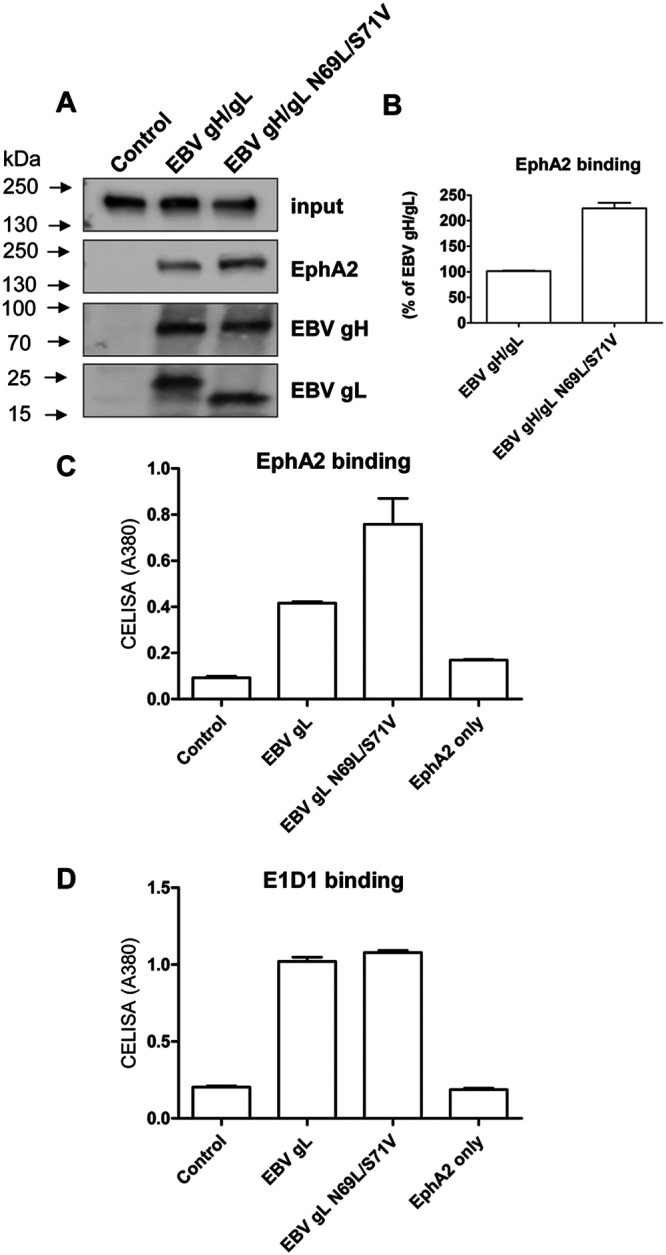
EBV gH/gL-N69L/S71V has a higher affinity with EphA2 compared to WT EBV gH/gL. (A) CHO-K1 cells seeded in 6-well plates were transfected with control plasmid pcDNA3.1, EBV gH/gL, or EBV gH/gL-N69L/S71V. After 24 h, the cells were washed twice with ice-cold PBS and incubated with supernatants from EphA2-Fc-transfected cells isolated 24 h posttransfection for 2 h at 4°C. The cells were then washed with ice-cold PBS three times and lysed with 100 μl of 1× SDS lysis buffer. EphA2-Fc (∼180 kDa) bound to the cells expressing EBV gH/gL or EBV gH/gL-N69L/S71V was then analyzed using antibodies to the Fc region of EphA2 by Western blotting. The expression of the EBV gH/gL or EBV gH/gL-N69L/S71V complex was monitored by analyzing EBV gH (∼80 kDa) or EBV gL (∼21 kDa for gL-N69L/S71V and ∼25 kDa for WT EBV gL) expression using anti-EBV gH/gL polyclonal antibodies directed against EBV gH/gL. (B) Quantification data of associated EphA2-Fc from three independent experiments. (C) CHO-K1 cells seeded in 6-well plates were transfected with soluble EphA2-Fc only or soluble EphA2-Fc with vector control plasmid, EBV gH/gL, and EBV gH/gL-N69L/S71V. Transfected cells were seeded in 96-well plates in triplicate posttransfection. The cells were then washed with ice-cold PBS three times and gH/gL-associated EphA2 (C) or surface expression of EBV gH/gL (D) was determined by CELISA using anti-human IgG or E1D1 anti-gH/gL antibodies.
Overall, we found that EBV gH/gL-N69L/S71V had about two times greater EphA2 binding affinity than WT EBV gH/gL, indicating that the EBV gL glycosylation site plays a role in the binding of the EBV gH/gL complex with EphA2.
Our previous studies indicated that the flexibility between the large groove of EBV gH/gL is important for epithelial cell fusion but not B cell fusion (22). When we introduced disulfide bonds between domain I (DI) and domain II (DII) by mutating G49 to C, which forms a disulfide bond with C153, or by mutating the bulky R152 to A, we observed decreased fusion in epithelial cell fusion but not B cell fusion (22). Thus, we hypothesized that the large groove might also be part of the EphA2 binding region. To test this, we examined the binding of our published gH/gL groove mutants (22) with EphA2 and found that both R152A and G49C have decreased binding with EphA2 (Fig. 8). These data demonstrate the binding of EBV gH/gL with EphA2 is affected by residues in both domain I and domain II, which differs from KSHV gH/gL in which only domain I and especially the conserved KSHV ELEFN50-54 motif appears to be important for binding (18). Based on our presented data, we propose a model of KSHV gH/gL and EBV gH/gL binding to EphA2 shown in Fig. 9C and D. The crystal structure of EBV gH/gL (Fig. 9A) (23) and EphA2 (Fig. 9B) (24) are shown for a better illustration. Our hypothetical binding model of EBV gH/gL shows both domain I and II of gH/gL binding to EphA2 (Fig. 9D) but less tightly compared to that of KSHV gH/gL (Fig. 9C). This weaker binding may result from steric hindrance of the EBV glycosylation site which is not found in KSHV gH/gL. This may also explain why KSHV gH/gL has higher affinity with EphA2 compared to EBV gH/gL.
FIG 8.
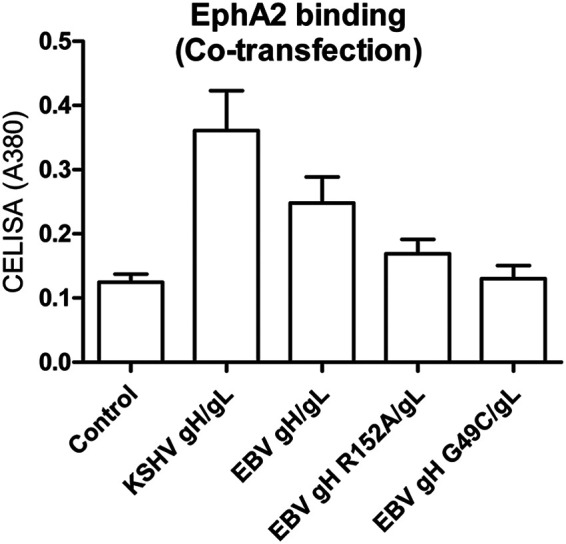
EBV gH/gL R152A and G49C have a lower affinity with EphA2 than WT EBV gH/gL. CHO-K1 cells seeded in 6-well plates were transfected with soluble EphA2-Fc only or soluble EphA2-Fc with vector control plasmid, KSHV gH/gL, EBV gH/gL, and EBV gH G49C/gL. Transfected cells were seeded in 96-well plates in triplicate posttransfections. The cells were then washed with ice-cold PBS three times, and gH/gL-associated EphA2 was determined by CELISA using anti-human IgG.
FIG 9.

Model of EphA2 binding to KSHV gH/gL and EBV gH/gL. (A) EBV gH consists of four domains: DI (blue), DII (magenta), DIII (green), and DIV (yellow) (23). gL is colored red and interacts with gH in DI. Structural data are not available for KSHV gH/gL. The EBV gL glycosylation site (NGSN69-72) is shown as spheres in green. (B) The EphA2 ectodomain structure consists of the ligand-binding domain (LBD) in red, the cysteine-rich region (CYS) in green, and two fibronectin regions (FN) in dark blue (24). Our hypothetical model of KSHV and EBV gH/gL binding to EphA2 is shown (see panels C and D), with domain colors that are the same as in panels A and B. (C) Previous results have shown that an ELEFN50-54 motif found in KSHV gH DI is essential for KSHV fusion and entry, indicating that it may be the primary determinate for KSHV binding to EphA2 (18), with the other domains of EphA2 playing little if any role in binding, as indicated by the dotted line and question mark. (D) For EBV gH/gL binding to EphA2, we propose that EBV gH/gL binds less tightly than KSHV to gH/gL as a result of the EBV gH/gL glycosylation (NGSN69-72 green sphere) that is not present in KSHV gH/gL due to steric hindrance. We propose that the increased fusion seen with the glycosylation-negative mutant may result from increased binding due to removal of steric hindrance preventing DI binding to EphA2 and also other domains and, in particular, the large groove between gH/gL domains DI and DII. This is indicated as dotted line and question mark. Interestingly, this would provide an explanation why gp42 inhibits epithelial fusion since gp42 snakes across the surface of DI through DIV of gH/gL that may prevent access of domains within DI, DII, DIII, and DIV required for EphA2 binding to gH/gL.
In the present study, we identified the region EphA2 binds to on EBV gH/gL, which is the EBV gL N-glycosylation site, and the large groove between DI and DII. The binding site of EphA2 on EBV gH/gL (gL NGSN69-72) is different from the binding site of EphA2 on KSHV gH/gL (gH ELEFN50-54). Unlike KSHV gH/gL, for which the EphA2 LBD is the major binding region, the binding region of EBV gH/gL on EphA2 not only involves the LBD region but also other regions in the EphA2 ectodomain. The different binding mode between KSHV and EBV gH/gL may explain the different affinity between KSHV gH/gL and EBV gH/gL for EphA2 (Fig. 9). Further research, such as protein crystallography and/or cryo-electron microscopy (Cryo-EM), to determine the structure of gH/gL bound to EphA2 and targeted mutational studies will identify commonalities and differences in the binding interactions of EBV gH/gL and KSHV gH/gL with EphA2.
DISCUSSION
Virus entry into target cells is an essential process for virus infection. Understanding the entry process may aid the development of small molecule inhibitors to prevent virus infection and aid in vaccine design. Eph family receptors, especially EphA2, have been reported as host receptors and factors for many pathogens, including KSHV, EBV, Cryptococcus neoformans, hepatitis C virus, Chlamydia trachomatis, and malaria parasites (15, 16, 25). In addition, Ephrin B2 has been identified as a receptor for Nipah virus (26). It has also been shown that KSHV uses EphA4 and EphA7 as host entry receptors in addition to EphA2 (10, 11). EBV does not use EphA4 as a receptor, indicating receptor specificity of these viruses. The use of multiple EphA receptors may be one reason that KSHV has a broader cell tropism compared to EBV. It has been shown that both KSHV and EBV gH/gL bind to the LBD of EphA4, which can compete with the natural ephrin ligands (7–9, 17). A conserved, distinct binding motif (ELEFN50-54) on DI of both KSHV and RRV gH/gL has been identified (18); however, the specific interaction site on EBV gH/gL is not known.
Previously studies showed that both KSHV gH/gL and EBV gH/gL interact with the ephrin ligand binding region of EphA2 (7–9, 17), indicating that the binding mechanism of EphA2 with KSHV or EBV gH/gL or EphA2 with ephrin ligand may be similar. Our recent study identifying EphA4 as a new receptor for KSHV found the ligand-binding regions of EphA2 and EphA4 are interchangeable for KSHV fusion and infection (10), confirming that the ligand binding region is important for KSHV infection. Previous studies found that deletion of the individual EphA2 ectodomains (LBD, CYS, and FBN) all decreased EBV infection (8), indicating that all of the regions play some role in EBV infection. However, it is not known whether the decreased infection is due to structural disruption of the EphA2 ectodomain. To further determine whether the entire ectodomain of EphA2 is important for EBV fusion activity, we took the advantage of the finding that EphA2 but not EphA4 can mediate fusion (7). We utilized previous EphA2/EphA4 chimeras that we constructed that have increasing length of the EphA2 ectodomain (Fig. 1A). We found that the increasing length of the EphA2 ectodomain results in a proportional increase in fusion activity (Fig. 1B). These data confirmed that the full ectodomain plays a role in EBV fusion which differs from KSHV in which only the LBD has been shown to be important for binding and fusion (9, 10).
When using a luciferase fusion assay to measure KSHV fusion, fusion activity is very low when all of the fusion machinery consists of KSHV glycoproteins. In contrast, when KSHV gH/gL is combined with EBV gB, the fusion activity is about three times higher than the WT EBV fusion activity. Previously, our lab demonstrated that KSHV gH/gL has higher binding affinity with EphA2 compared to EBV gH/gL (10). This also suggests that EBV and KSHV may use different mechanisms for binding to EphA2. Although KSHV gH/gL can function with EBV gB for fusion, KSHV and EBV gH/gL complexes cannot function in fusion if gH and gL are from different viruses (Fig. 2). This is likely due to the fact that these proteins evolved together in KSHV and EBV, where the association of gH with gL is required for correct folding, cell surface trafficking, and membrane presentation of the complex (27). This is consistent with the previous findings that heterologous complexes of EBV/KSHV gH/gL are nonfunctional in fusion since they are not expressed at the cell surface (19). Interestingly, the Hutt-Fletcher lab compared EBV gL to VZV gL. These researchers found that VZV gL could substitute for EBV gL and apparently form a functional EBV gH/VZV gL complex although the gL proteins of alphaherpesviruses and gammaherpesviruses do not have a significant percentage of amino acid sequence homology (28). This indicates the importance of gH/gL complex formation for its function.
Previously, the Hahn lab identified that both KSHV and rhesus monkey rhadinovirus gHs contain a conserved Eph family receptor-binding motif ELEFN50-54. The corresponding sequence on EBV gH is DIEGH28-32 by sequence alignment (18). Mutation of KSHV gH ELEFN50-54 to ELAAN50-54 decreased the fusion activity (Fig. 3A). To identify whether EBV also uses a similar region for binding EphA2 as KSHV, we mutated the DIEGH28-32 motif in EBV gH to the corresponding KSHV gH EphA2-binding motif ELEFN50-54 and expected to see higher fusion activity. However, the fusion activity of the EBV gH mutant was the same as that for WT EBV gH (Fig. 3B). These data confirmed that this motif is important for KSHV fusion but does not increase EBV fusion activity. EBV gLs, as well as the gLs of other herpesviruses, function as chaperones for gH processing and transport to the cell surface (28). KSHV gH does not require KSHV gL to function as a chaperone (29). Studies with EBV gL demonstrated that, in addition to functioning as a chaperone protein, gL might have a direct role in fusion, at least in B cells (30). Previously, we demonstrated that the length of the EBV gH tail is important for fusion function (31). Thus, we investigated in our current studies whether the length of KSHV and EBV gL might contribute to the differential fusion activity of their gH/gL complexes. Our data showed that if we truncate KSHV gL to the same length as EBV gL, the fusion activity is unchanged, indicating that the length of gL does not have a role in fusion (Fig. 4).
Previous studies indicated that EBV gL is glycosylated at NGSN69-72 (20, 32). The gL-N69L/S71V mutant enhances fusion only in epithelial cells but not in B cells (20, 32). There are several possibilities for why this may be the case. The gL-N69L/S71V mutant may alter the gL conformation and/or alter gH/gL complex formation since gL is the chaperone protein for gH (28). The gL-N69L/S71V mutant might also alter the interaction of gH/gL with gB. Previous studies in our laboratory showing a species-specific dependence between gB and gL indicated that that there is a direct role of gL in gB-induced membrane fusion (30). Functional analysis of EBV gL and rhesus lymphocryptovirus gL mapped a domain with residues 54 and 94 as interaction sites for EBV-mediated fusion on gL and EBV gB from residues 456 to 807 as the gB-gH/gL interaction region in EBV-mediated fusion (30, 33). The final possibility is that mutation of the gL-N69/S71 glycosylation site may facilitate better interaction with a cellular protein that is important for fusion. Recently, our lab and the Zeng lab identified that EphA2 functions as an EBV entry receptor (7, 8). Overexpression of EphA2 in target cells increases EBV fusion activity. Interestingly, when EphA2 is present in the target cells, the gL-N69L/S71V mutant does not have its typical hyperfusogenic phenotype (Fig. 5A). This may be because the N69/S71 N glycosylation site is the binding site of gL for EphA2. Glycosylation of this region could prevent gL binding access to EphA2; therefore, mutation of gL NGSN69-72 to gL-N69L/S71V removes the steric hindrance. One possibility for the loss of the hyperfusogenic phenotype in the presence of EphA2 could be that overexpression of EphA2 saturates the fusion activity. To exclude this possibility, we used a gB hyperfusogenic mutant, gB843. Interestingly, we still observed the hyperfusogenic phenomenon in the presence of EphA2 (Fig. 5B). These data indicate that the EBV gL glycosylation site might be the EphA2 binding site. These data also further confirmed our previous observation that the regulatory mechanisms of gB-driven fusion by the CTD of gB and the glycosylation site of gL are different (32). We further examined whether this glycosylation site negatively regulates receptor binding. Posttranslational modifications such as the N-linked glycosylation of viral envelope glycoproteins are involved in a variety of functions: correct expression and transport, receptor binding, fusion activity, and shielding against neutralizing antibodies, for example (34). Nipah virus is an example of a virus that uses N-linked glycosylation of the viral envelope to shield against neutralizing antibodies and also to negatively regulate fusion activity (35). This may be the case for EBV gL NGSN69-72 glycosylation site, but further investigation will be required. For example, in avian coronavirus and influenza A virus, the effect of glycosylation on receptor binding has shown both positive and negative roles. (36, 37). Our study found that the gL-N69L/S71V mutant has increased binding affinity with EphA2 compared to WT EBV gL, indicating that N-linked glycosylation of this site reduces receptor binding (Fig. 7). Interestingly, KSHV gH ELEFN50-54 is also a potential glycosylation site which is located in a putative beta-hairpin at the KSHV gH/gL interaction site (18). Mutation of the ELEFN50-54 to ELAAN decreased fusion activity (Fig. 3A). These data may indicate that reduced glycosylation of the KSHV gH ELEFN50-54 site decreases EphA2 binding and reduces subsequent fusion. The corresponding sequence to KSHV ELEFN50-54 on EBV is DIEGH28-32. The DIEGH28-32 sequence is located on the beta-hairpin of EBV, and the location is symmetrical to the site of EBV gL N69/S71 (Fig. 6B). The distance to the receptor binding site is different between the KSHV gH ELEFN50-54 and EBV gL N69/S71 glycosylation sites, which might explain their different effects on fusion activity. However, when we mutated the KSHV gL corresponding site NG (EBV gL N69/S71) to NGSN66-69, the fusion activity was comparable to WT KSHV gL (Fig. 2B). In contrast to KSHV, EBV, in addition to DI, also uses the large groove between gH/gL DI and DII for receptor binding for epithelial fusion since the mutation of this region specifically decreases epithelial cell fusion but not B cell fusion (22). Moreover, mutations in this region also decrease the binding with EphA2 (Fig. 8). Taking together, these data suggest that KSHV and EBV use different regions for EphA2 binding.
In the present study, we show that the N-linked glycosylation site NGSN69-72 on EBV gL and the large groove between EBV gH/gL are important for the association of EBV gH/gL and EphA2. The results indicate that the mechanism of EphA2 binding to KSHV gH/gL and EBV gH/gL is different (Fig. 9). Interestingly, this difference may also indicate why gp42 is such an efficient inhibitor of epithelial cell infection since the binding of gp42 to gH/gL could block EphA2 interactions with the gH/gL binding regions that we found important for epithelial fusion (Fig. 9). The detailed mechanism of binding both EBV and KSHV gH/gL with EphA2 will require additional studies using Cryo-EM or crystallography. The interaction site may serve as an intervention target for small molecule or monoclonal antibody development to prevent EBV and infection. EphA2 serves as a host receptor not only for gammaherpesviruses but also other pathogens. Our study may also provide a better understanding of the interaction mechanism of EphA2 with the other pathogens.
MATERIALS AND METHODS
Cell culture.
Chinese hamster ovary (CHO-K1) cells (ATCC CCL-61) were grown in Ham’s F-12 medium (Corning) containing 10% heat-inactivated fetal bovine serum (FBS; Corning) and 1% penicillin-streptomycin (100 U penicillin/ml, 100 μg streptomycin/ml; Sigma). Human embryonic kidney 293T (HEK293T) cells (ATCC CRL-3216) (38) were grown in Dulbecco modified Eagle medium (Corning) containing 10% heat-inactivated FBS and 1% penicillin-streptomycin.
Constructs.
The EphA2 and EphA4 constructs (7) were a gift from Spiro Getsios (Northwestern University). The construction of the EphA2 and EphA4 LBD chimeras (EphA2A4 or EphA4A2) was as described previously (7). The construction of the EphA2A4 CYS and EphA2A4 FBN chimeras was performed using the forward primer EphA2A4 F AfeI (5′-ACGTGAAGCTGAACGTGGAGG-3′) and the reverse primer EphA2A4 Xho I R (5′-AGACTCGAGTCACCCTTTGTCGTCGTCATCCT-3′). The overlapping primers for EphA2A4 CYS were EphA2A4 CYS F (5′-ATGCCTTGCACACGACCCCCCTCTGCTCCCCTGAACTTGATT-3) and EphA2A4 CYS R (5′-AATCAAGTTCAGGGGAGCAGAGGGGGGTCGTGTGCAAGGCAT-3′), and the overlapping primers for EphA2A4 FBN were EphA2A4 FBN F (5′-GAGGGCCAGGGGGCCGGCAGCGAGCCCTTGGAGGTTACAACC-3′) and EphA2A4 FBN R (5′-GGTTGTAACCTCCAAGGGCTCGCTGCCGGCCCCCTGGCCCTC-3′). Soluble EphA2-Fc and EphA4-Fc were cloned in an Fc construct, as previously described (10). KSHV His-tagged gL and gH were subcloned into the pSG5 vector using the following primers: KSHV gL EcoRI F, 5′-GCGAATTCCATGGGGATCTTTGCGCTATTT-3′; KSHV His gL BglII R, 5′-TAAGATCTGTTTAGTGGTGATGGTGATGATGTTTTCCCTTTTGACCTGCGTG-3′; KSHV gH BamHI F, 5′-TTGGATCCATGCAGGGTCTAGCCTTCTTGGC-3′; and KSHV His gH DraI R, 5′-ATGTTTAAACTAGTGGTGATGGTGATGATGATAAAGGATGGAAAACAGTCT-3′.
KSHV His gH and KSHV His gL point mutation mutants were generated using a QuikChange site-directed mutagenesis kit (Stratagene). The primers used were as follows: KSHV His gL NGSN F, 5′-ATAGCCAAGCTGCGAAGCAAAAATGGCAGCAACGACATTACCGTGGAAACATGC-3′; KSHV His gL NGSN R, 5′-GCATGTTTCCACGGTAATGTCGTTGCTGCCATTTTTGCTTCGCAGCTTGGCTAT-3′; KSHV gL 131stop F, 5′-TCTATTCACAACGTAAACTGAATTATAATAAGCGTGGGA-3′; KSHV gL 131stop R, 5′-TCCCACGCTTATTATAATTCAGTTTACGTTGTGAATAGA-3′; KSHV His gH ELAAN F, 5′-ACCAACCTCTCCATAGAACTGGCCGCCAACGGCACTAGTTTTTTTCTA-3′; KSHV His gH ELAAN R, 5′-TAGAAAAAAACTAGTGCCGTTGGCGGCCAGTTCTATGGAGAGGTTGGT-3′; EBV gH ELEFN F, 5′-AGCGAGGTTAAGCTGCACCTGGAACTGGAATTCAACGCTTCGCATTACACCATCCCA-3′; and EBV gH ELEFN R, 5′-TGGGATGGTGTAATGCGAAGCGTTGAATTCCAGTTCCAGGTGCAGCTTAACCTCGCT-3′. Sequencing was performed for all constructs to confirm the correct sequence.
Fusion assay.
The virus-free cell-based fusion assay was performed as previously described (39). Briefly, CHO-K1 cells grown to approximately 80% confluence in a 6-well plate were transiently transfected with T7 luciferase reporter plasmid with a T7 promoter (1.5 μg) or green fluorescent protein (GFP; 1.5 μg) for the syncytium formation assay, and the essential glycoproteins used for EBV fusion (EBV gB [0.8 μg], EBV gH [0.5 μg], and EBV gL [0.5 μg]) or for KSHV fusion (EBV gB [0.8 μg], KSHV gH [0.5 μg], and KSHV gL [0.5 μg]) with Lipofectamine 2000 transfection reagent (Invitrogen) in Opti-MEM (Gibco/Life Technology) were as previously described (19). HEK293T cells were transfected with T7 polymerase (1.5 μg) plus 1.5 μg of pcDNA 3.1, EphA2, or EphA4 for the fusion assay. By 24 h posttransfection, the transfected CHO-K1 cells were detached, counted, and mixed in a 1:1 ratio with target cells (HEK293T cells, 2.0 × 105 per sample) in a 48-well plate in 0.5 ml of Ham’s F-12 medium with 10% heat-inactivated FBS. After 24 h, the cells were washed once with PBS and lysed with 50 μl of passive lysis buffer (Promega). The luciferase activity was quantified by transferring 20 μl of lysed cells to a 96-well plate and adding 50 μl of luciferase assay reagent (Promega). Luminescence was measured on a Perkin-Elmer Victor II plate reader.
Cell enzyme-linked immunosorbent assay.
The association of EphA2-Fc with EBV gH/gL or EBV gH/gL-N69L/S71V was determined by CELISA. CHO-K1 cells were transiently transfected with EphA2 Fc only or together with control plasmid pcDNA 3.1, EBV gH/gL, or EBV gH/gL-N69L/S71V. At 24 h after transfection, the cells were incubated with anti-human IgG(H&L) (HRP, ab6759; Abcam, 1:1,000) against the Fc region or E1D1 against EBV gH/gL for 30 min and fixed with 2% formaldehyde and 0.2% glutaraldehyde in PBS for 15 min, followed by three PBS washes. TMB one-component HRP microwell substrate was added, and the amount of bound EphA2-Fc or gH/gL on the cell surface was determined by measuring the absorbance at 380 nm with a Perkin-Elmer Victor II plate reader. Binding activity was standardized in comparison to EphA2-Fc binding to EBV gH/gL, which was set to 100%.
Western blotting.
The expression of His tagged KSHV gL and KSHV gL NGSN66-69 was examined by Western blotting. CHO-K1 cells in 6-well plates were transfected with His-tagged KSHV gL and KSHV gL NGSN66-69, together with KSHV gH. At 24 h after transfection, the cells were collected and resuspended in 50 μl of PBS and then mixed with 50 μl of 2× SDS loading buffer (60 mM Tris-Cl [pH 6.8], 0.2% SDS, 25% glycerol, 0.01% bromophenol blue). Samples were boiled for 3 min and loaded onto a Bio-Rad 4 to 20% mini-Protean-TGX gel for Western blotting. After electrophoresis, the proteins were transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). The blots were blocked with 5% nonfat dry milk in PBS buffer (20 mM Tris-HCl [pH 7.6], 137 mM NaCl,) for 2 h at room temperature. The blots were washed with PBS and incubated with primary anti-His tag antibody (OB05; Calbiochem, 1:1,000) for His-tagged KSHV gL overnight at 4°C. Anti-mouse IRDye800 secondary antibody (LI-COR Biosciences, Lincoln, NE) was added to the membranes at a dilution ratio of 1:10,000, followed by incubation for 1 h at room temperature. For detection of EphA2-Fc, the membrane with transferred proteins was incubated with anti-human IgG(H&L) (HRP, ab6759; Abcam, 1:1,000) against the Fc region. The membrane was then incubated with 1 ml of SuperSignal chemiluminescent substrate (Thermo-Fisher Scientific) prior to imaging. Protein bands on the membrane were visualized with an Odyssey Fc Western blotting imager using Image Studio (v2.0; LI-COR Biosciences).
Statistical analysis.
Data were collected from three independent experiments. Statistical differences between multiple groups were determined by one-way analysis of variance with post hoc Tukey’s multiple-comparison test. Two-group comparisons were analyzed by the two-tailed unpaired Student t test. A P value of <0.05 denotes the presence of a statistically significant difference. Data are expressed as means ± the standard errors of the mean (SEM). The analysis was performed using GraphPad Prism (v6.0c for Mac; GraphPad Software, San Diego, CA). Microscopy images are representative of at least two independent experiments.
ACKNOWLEDGMENTS
We appreciate the help and advice from members of the Longnecker and Jardetzky laboratories, especially Nanette Susmarski.
This research was supported by grants AI076183 (R.L. and T.S.J.) and AI137267 (R.L. and T.S.J.) from the National Institute of Allergy and Infectious Diseases, grant IRG-15-173-21 (J.C.) from the American Cancer Society, a supplement from P30CA060553 (R.L.), and the Third Coast Center for AIDS Research pilot award (J.C.).
J.C. and R.L. designed the overall study with input from the coauthors. J.C. performed the key experiments. S.S. helped with cell cultures and fusion assays and the generation of EphA2/A4 chimeras. J.C. and R.L. wrote the manuscript. S.S. and T.S.J. contributed expertise and helped write the paper. All authors analyzed the results, read, and approved the manuscript for submission.
We declare there are no competing financial interests.
REFERENCES
- 1.Pellett PE, Roizman B. 2013. Herpesviridae, p 1802–1822. In Fields virology, 6th ed, vol 2 Lippincott/Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Damania BA, Cesarman E. 2013. Kaposi’s sarcoma-associated herpesvirus, p 2080–2128. In Fields virology, 6th ed, vol 2 Lippincott/Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Longnecker R, Kieff E, Cohen JI. 2013. Epstein-Barr virus, p 1898–1959. In Fields virology, 6th ed, vol 2 Lippincott/Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Kumar B, Chandran B. 2016. KSHV entry and trafficking in target cells: hijacking of cell signal pathways, actin and membrane dynamics. Viruses 8:305. doi: 10.3390/v8110305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci U S A 106:2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Sathiyamoorthy K, Zhang X, Schaller S, Perez White BE, Jardetzky TS, Longnecker R. 2018. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat Microbiol 3:172–180. doi: 10.1038/s41564-017-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H, Li Y, Wang HB, Zhang A, Chen ML, Fang ZX, Dong XD, Li SB, Du Y, Xiong D, He JY, Li MZ, Liu YM, Zhou AJ, Zhong Q, Zeng YX, Kieff E, Zhang Z, Gewurz BE, Zhao B, Zeng MS. 2018. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat Microbiol 3:1–171. doi: 10.1038/s41564-017-0080-8. [DOI] [PubMed] [Google Scholar]
- 9.Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, Holzer A, Schmidt M, Chen J, Konig S, Ensser A, Myoung J, Brockmeyer NH, Sturzl M, Fleckenstein B, Neipel F. 2012. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nat Med 18:961–966. doi: 10.1038/nm.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Zhang X, Schaller S, Jardetzky TS, Longnecker R. 2019. Ephrin receptor A4 is a new Kaposi’s sarcoma-associated herpesvirus virus entry receptor. mBio 10:e02892-18. doi: 10.1128/mBio.02892-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grosskopf AK, Schlagowski S, Hörnich BF, Fricke T, Desrosiers RC, Hahn AS. 2019. EphA7 functions as receptor on BJAB cells for cell-to-cell transmission of the Kaposi’s sarcoma-associated herpesvirus and for cell-free infection by the related rhesus monkey rhadinovirus. J Virol 93:e00064-19. doi: 10.1128/JVI.00064-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.TerBush AA, Hafkamp F, Lee HJ, Coscoy L. 2018. A Kaposi’s sarcoma-associated herpesvirus infection mechanism is independent of integrins α3β1, αVβ3, and αVβ5. J Virol 92:e00803-18. doi: 10.1128/JVI.00803-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kania A, Klein R. 2016. Mechanisms of ephrin-Eph signaling in development, physiology, and disease. Nat Rev Mol Cell Biol 17:240–256. doi: 10.1038/nrm.2015.16. [DOI] [PubMed] [Google Scholar]
- 14.Chakraborty S, Veettil MV, Bottero V, Chandran B. 2012. Kaposi’s sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc Natl Acad Sci U S A 109:E1163–E1172. doi: 10.1073/pnas.1119592109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aaron PA, Jamklang M, Uhrig JP, Gelli A. 2018. The blood-brain barrier internalizes Cryptococcus neoformans via the EphA2-tyrosine kinase receptor. Cell Microbiol 20:e12811. doi: 10.1111/cmi.12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hahn AS, Desrosiers RC. 2014. Binding of the Kaposi’s sarcoma-associated herpesvirus to the ephrin binding surface of the EphA2 receptor and its inhibition by a small molecule. J Virol 88:8724–8734. doi: 10.1128/JVI.01392-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grosskopf AK, Ensser A, Neipel F, Jungnickl D, Schlagowski S, Desrosiers RC, Hahn AS. 2018. A conserved Eph family receptor-binding motif on the gH/gL complex of Kaposi’s sarcoma-associated herpesvirus and rhesus monkey rhadinovirus. PLoS Pathog 14:e1006912. doi: 10.1371/journal.ppat.1006912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Omerovic J, Longnecker R. 2007. Functional homology of gHs and gLs from EBV-related gammaherpesviruses for EBV-induced membrane fusion. Virology 365:157–165. doi: 10.1016/j.virol.2007.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sathiyamoorthy K, Hu YX, Mohl BS, Chen J, Longnecker R, Jardetzky TS. 2016. Structural basis for Epstein-Barr virus host cell tropism mediated by gp42 and gHgL entry glycoproteins. Nat Commun 7:13557. doi: 10.1038/ncomms13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia NJ, Chen J, Longnecker R. 2013. Modulation of Epstein-Barr virus glycoprotein B (gB) fusion activity by the gB cytoplasmic tail domain. mBio 4:e00571-12. doi: 10.1128/mBio.00571-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Jardetzky TS, Longnecker R. 2013. The large groove found in the gH/gL structure is an important functional domain for Epstein-Barr virus fusion. J Virol 87:3620–3627. doi: 10.1128/JVI.03245-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107:22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY. 2010. An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nat Struct Mol Biol 17:398–402. doi: 10.1038/nsmb.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subbarayal P, Karunakaran K, Winkler AC, Rother M, Gonzalez E, Meyer TF, Rudel T. 2015. EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog 11:e1004846. doi: 10.1371/journal.ppat.1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, Tajyar S, Lee B. 2005. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 436:401–405. doi: 10.1038/nature03838. [DOI] [PubMed] [Google Scholar]
- 27.Peng T, Ponce de Leon M, Novotny MJ, Jiang H, Lambris JD, Dubin G, Spear PG, Cohen GH, Eisenberg RJ. 1998. Structural and antigenic analysis of a truncated form of the herpes simplex virus glycoprotein gH-gL complex. J Virol 72:6092–6103. doi: 10.1128/JVI.72.7.6092-6103.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Q, Buranathai C, Grose C, Hutt-Fletcher LM. 1997. Chaperone functions common to nonhomologous Epstein-Barr virus gL and varicella-zoster virus gL proteins. J Virol 71:1667–1670. doi: 10.1128/JVI.71.2.1667-1670.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hahn A, Birkmann A, Wies E, Dorer D, Mahr K, Sturzl M, Titgemeyer F, Neipel F. 2009. Kaposi’s sarcoma-associated herpesvirus gH/gL: glycoprotein export and interaction with cellular receptors. J Virol 83:396–407. doi: 10.1128/JVI.01170-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plate AE, Smajlovic J, Jardetzky TS, Longnecker R. 2009. Functional analysis of glycoprotein L (gL) from rhesus lymphocryptovirus in Epstein-Barr virus-mediated cell fusion indicates a direct role of gL in gB-induced membrane fusion. J Virol 83:7678–7689. doi: 10.1128/JVI.00457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, Jardetzky TS, Longnecker R. 2016. The cytoplasmic tail domain of Epstein-Barr virus gH regulates membrane fusion activity through altering gH binding to gp42 and epithelial cell attachment. mBio 7:e01871-16. doi: 10.1128/mBio.01871-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohl BS, Chen J, Park SJ, Jardetzky TS, Longnecker R. 2017. Epstein-Barr virus fusion with epithelial cells triggered by gB is restricted by a gL glycosylation site. J Virol 91:e01255-17. doi: 10.1128/JVI.01255-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plate AE, Reimer JJ, Jardetzky TS, Longnecker R. 2011. Mapping regions of Epstein-Barr virus (EBV) glycoprotein B (gB) important for fusion function with gH/gL. Virology 413:26–38. doi: 10.1016/j.virol.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vigerust DJ, Shepherd VL. 2007. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol 15:211–218. doi: 10.1016/j.tim.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aguilar HC, Matreyek KA, Filone CM, Hashimi ST, Levroney EL, Negrete OA, Bertolotti-Ciarlet A, Choi DY, McHardy I, Fulcher JA, Su SV, Wolf MC, Kohatsu L, Baum LG, Lee B. 2006. N-glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J Virol 80:4878–4889. doi: 10.1128/JVI.80.10.4878-4889.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parsons LM, Bouwman KM, Azurmendi H, de Vries RP, Cipollo JF, Verheije MH. 2019. Glycosylation of the viral attachment protein of avian coronavirus is essential for host cell and receptor binding. J Biol Chem 294:7797–7809. doi: 10.1074/jbc.RA119.007532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mir-Shekari SY, Ashford DA, Harvey DJ, Dwek RA, Schulze IT. 1997. The glycosylation of the influenza A virus hemagglutinin by mammalian cells. A site-specific study. J Biol Chem 272:4027–4036. doi: 10.1074/jbc.272.7.4027. [DOI] [PubMed] [Google Scholar]
- 38.McShane MP, Longnecker R. 2004. Cell-surface expression of a mutated Epstein-Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc Natl Acad Sci U S A 101:17474–17479. doi: 10.1073/pnas.0404535101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McShane MP, Longnecker R. 2005. Analysis of fusion using a virus-free cell fusion assay. Methods Mol Biol 292:187–196. doi: 10.1385/1-59259-848-x:187. [DOI] [PubMed] [Google Scholar]