

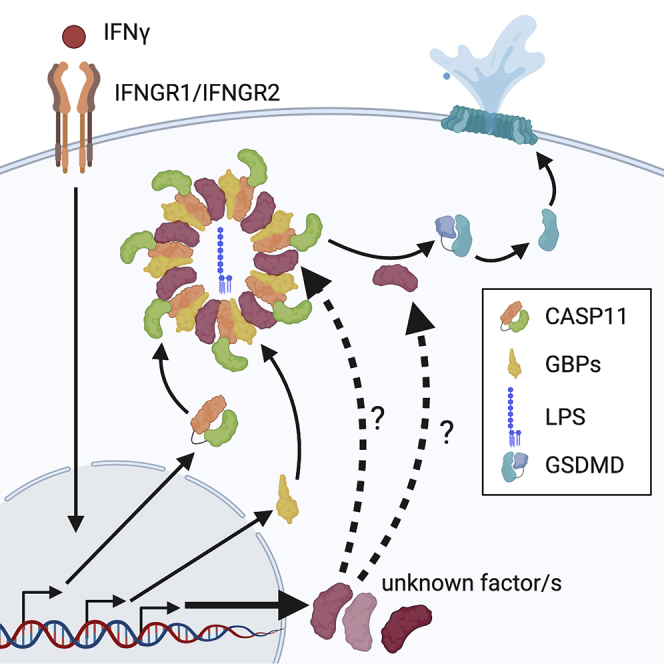
Summary
In mammalian cells, inflammatory caspases detect Gram-negative bacterial invasion by binding lipopolysaccharides (LPS). Murine caspase-11 binds cytosolic LPS, stimulates pyroptotic cell death, and drives sepsis pathogenesis. Extracellular priming factors enhance caspase-11-dependent pyroptosis. Herein we compare priming agents and demonstrate that IFNγ priming elicits the most rapid and amplified macrophage response to cytosolic LPS. Previous studies indicate that IFN-induced expression of caspase-11 and guanylate binding proteins (GBPs) are causal events explaining the effects of priming on cytosolic LPS sensing. We demonstrate that these events cannot fully account for the increased response triggered by IFNγ treatment. Indeed, IFNγ priming elicits higher pyroptosis levels in response to cytosolic LPS when macrophages stably express caspase-11. In macrophages lacking GBPs encoded on chromosome 3, IFNγ priming enhanced pyroptosis in response to cytosolic LPS as compared with other priming agents. These results suggest an unknown regulator of caspase-11-dependent pyroptosis exists, whose activity is upregulated by IFNγ.
Subject Areas: Immunology, Cell Biology
Graphical Abstract
Highlights
-
•
IFNγ priming elicits the most rapid and amplified response to cytosolic LPS
-
•
The enhanced IFNγ-triggered response is separable from CASP11 expression
-
•
The enhanced IFNγ-triggered response is independent of GBPs encoded on chromosome 3
-
•
We propose an unknown IFNγ-induced regulator of CASP11-dependent pyroptosis exists
Immunology; Cell Biology
Introduction
The innate immune response to lipopolysaccharide (LPS) at a cellular level is compartmentalized, allowing for distinct responses to Gram-negative pathogens based on subcellular localization (Brubaker et al., 2015). Extracellular LPS engages Toll-like receptor 4 (TLR4) at the plasma membrane and induces the transcription of pro-inflammatory cytokines such as IL-6, pro-IL-1β, pro-IL-18, as well as type-I interferons (IFNα/β) (Akira and Takeda, 2004). In contrast, the response to cytosolic LPS triggers an inflammatory form of programmed cell death termed pyroptosis by binding to murine Caspase-11 (CASP11) or either human Caspase-4 or Caspase-5 (CASP4, CASP5) (Kayagaki et al., 2013; Hagar et al., 2013; Shi et al., 2014). These inflammatory caspases, as well as Caspase-1 (CASP1), are cysteine proteases that control pyroptosis by enzymatically cleaving gasdermin D (GSDMD) (Shi et al., 2015; Kayagaki et al., 2015). Following GSDMD cleavage, N-terminal “p30” GSDMD domain subunits (NT-GSDMD) oligomerize and form pores in the plasma membrane (Ding et al., 2016; Liu et al., 2016; Aglietti et al., 2016). These pores result in cell membrane damage and ultimately death. The response to cytosolic LPS is also associated with release of bioactive IL-18 and IL-1β, which requires active CASP1 for processing (Kayagaki et al., 2011). Mechanistically CASP1 is activated downstream of CASP11 through NLRP3-dependent sensing of K+ efflux that results from NT-GSDMD pore formation (Ruhl and Broz, 2015).
Increasingly, evidence supports the idea that the macrophage response to cytosolic LPS is regulated by extracellular stimuli. For example, members of the IFN family of cytokines influence the macrophage response to cytosolic LPS. Several reports indicate that IFNs upregulate the expression of CASP11, which is believed to increase sensitivity to cytosolic LPS by facilitating the chance of interaction (Rathinam et al., 2012; Broz et al., 2012; Meunier et al., 2014). Indeed, a set of TLRs trigger the production of IFNα/β and thus TLR ligands are commonly used to enhance the macrophage response to cytosolic LPS. Additionally IFNs promote the expression of a set of GTPases known as the guanylate binding proteins (GBPs), which are recruited to microbe-associated membranes including endosomes, pathogen-containing vacuoles, as well as microbial membranes (Tretina et al., 2019). At these sites, and in the context of Gram-negative bacterial infections, murine GBPs encoded on chromosome 3 facilitate CASP11/4/5-dependent responses to LPS (Meunier et al., 2014; Pilla et al., 2014; Finethy et al., 2015). Recent studies in human cells have further elucidated the role of GBPs in this process by providing evidence that a GBP signaling platform is assembled with LPS to facilitate CASP4 activation (Wandel et al., 2020; Santos et al., 2020). Both studies provide evidence that human GBP1 is critically required for assembling a complex with LPS, GBP2, GBP3, GBP4, and CASP4 for inflammasome activation. Thus, IFNs promote the transcriptional upregulation of factors that enhance pyroptosis in response to LPS in the cytosol.
Despite many insights into the transcriptional control of pyroptosis regulators by these extracellular signals, the functional differences between IFNs and TLR ligands when comparing inflammasome activation remains poorly defined. One reason for this lack of clarity derives from the fact that many studies have examined the distinct activities of IFNs or TLR ligands individually on the effects of pyroptosis. These events are hereafter referred to as “priming” events and represent activities that increase the rate or magnitude of pyroptosis. In this study, we compared the effects of IFN or TLR4 priming on CASP11 inflammasome activation to characterize differences in how these extracellular stimuli regulate the response to cytosolic LPS.
We demonstrate that type II IFN (IFNγ), as compared with IFNβ or LPS, is the most potent at enhancing CASP11-dependent pyroptosis induced by cytosolic LPS in murine macrophages. This enhancement may be relevant during infection with Gram-negative bacteria given that IFNγ production in vivo is critical for driving CASP11-dependent protective responses against Burkholderia thailandensis (Aachoui et al., 2015). The amplified IFNγ-mediated response in macrophages is specific to CASP11 inflammasome activation as IFNγ priming has no effect on NLRP3 activity. We provide evidence that, although IFNγ promotes the expression of CASP11, the priming effects of IFNγ extend beyond CASP11 upregulation. Moreover, IFNγ priming significantly enhances CASP11-dependent responses compared with IFNβ or LPS priming in BMDMs lacking CASP1 or all GBPs encoded on chromosome 3, which includes the homolog of human GBP1. Overall, we conclude that IFNγ priming specifically promotes rapid CASP11-dependent pyroptosis in response to cytosolic LPS by a process that is independent of GBPs encoded on chromosome 3 or its ability to upregulate CASP11 expression. An unknown IFNγ-inducible factor likely contributes to CASP11 activation and pyroptosis in response to cytosolic LPS.
Results
IFNγ Is a Potent Priming Agent for the Response to Cytosolic LPS in Macrophages
To study inflammasome-dependent pyroptosis, researchers have historically primed macrophages with a variety of TLR agonists, recombinant IFNs or cytokines (Latz et al., 2013). Priming is understood to transcriptionally upregulate the expression of critical inflammasome components. For example, the expression of NLRP3 and IL-1β are transcriptionally upregulated by TLR ligands or TNF, a key step for NLRP3 inflammasome activation (Bauernfeind et al., 2009, 2016). Priming for CASP11-dependent pyroptosis can be achieved with TLR agonists, such as LPS, or recombinant IFNs, which induce the expression of CASP11. Mechanistically, LPS upregulates CASP11 expression indirectly via TLR4-dependent TRIF signaling and the production of type I IFNs, highlighting the importance of IFN signaling in CASP11 expression (Rathinam et al., 2012; Broz et al., 2012). IFNγ, the sole type II IFN, is a well-established activator of macrophages and also primes bone marrow-derived macrophages (BMDMs) for the CASP11-dependent response to cytosolic LPS(Schauvliege et al., 2002; Rathinam et al., 2012; Meunier et al., 2014). Despite our understanding that priming is important for both NLRP3 and CASP11-dependent inflammasome activation and pyroptosis, little is known about how these priming agents differ in their ability to promote these distinct processes.
To further characterize differences between priming conditions, we set out to determine the relative abilities of IFNs and TLR ligands to promote NLRP3- and CASP11-dependent inflammasome activation and pyroptosis. Based on previously published priming conditions, we treated primary BMDMs for 16 h with IFNγ (100 U/mL), IFNβ (100 U/mL), or LPS (10 ng/mL) and monitored NLRP3- or CASP11-dependent inflammasome activation. As expected, LPS priming significantly increased NLRP3-induced BMDM cell death (measured by release of lactate dehydrogenase; LDH) 2 h after treatment with the classical NLRP3 activators, ATP or Nigericin (Figure 1A). IL-18 and IL-1β cytokine release, which are associated with inflammasome activation, were also significantly higher 2 h after NLRP3 activation in BMDMs primed with LPS (Figures 1B and 1C). These results are in line with previous publications demonstrating that LPS is a potent priming agent for NLRP3 inflammasome activation. However, neither IFNγ nor IFNβ promoted NLRP3 inflammasome activation as determined by cell death or cytokine release 2 h after activation (Figures 1A–1C). The kinetics of cell death monitored by the incorporation of SYTOX Green further support the conclusion that LPS, but not IFNγ or IFNβ, primes BMDMs for NLRP3 inflammasome activation (Figure 1D). Interestingly, we observed a different pattern of cell death and cytokine release using the same priming conditions for CASP11 inflammasome activation by transfected LPS. IFNγ priming significantly increased cell death and IL-18 release following LPS transfection compared with BMDMs primed with LPS or IFNβ or left unprimed (Figures 1A and 1B). However, IL-1β release following LPS transfection was only detected in BMDMs primed with LPS (Figure 1C). Monitoring cell death at later time points demonstrates that priming with IFNβ or LPS also promotes cell death in response to transfected LPS, which is in line with previous reports (Figure 1D). However, BMDMs primed with IFNγ and subsequently transfected with LPS die at a faster rate and to a greater extent than BMDMs primed with IFNβ or LPS (Figures 1A and 1D). These data demonstrate that inflammasome complex activation is differentially regulated by the macrophage priming agents IFNγ, IFNβ, and LPS. For example, NLRP3 inflammasome activation could only be promoted with LPS, whereas all three priming agents promoted CASP11 inflammasome activation. Interestingly, these priming conditions promoted CASP11 inflammasome activation to different extents, with IFNγ priming inducing the most potent response.
Figure 1.

IFNγ is a Potent Priming Agent for the Response to Cytosolic LPS in Macrophages
(A–D) WT BMDMs were primed for 16 h overnight with the following treatments: unprimed (N/A), IFNγ (100 U/mL), IFNβ (100 U/mL), or LPS (10 ng/mL). NLRP3 and CASP11 inflammasome activation was triggered with ATP (5 mM), Nigericin (10 μM), or by LPS (E. coli 0111:B4, 25 μg/mL) transfection with FuGENE HD (Fu/LPS). At 2 h following inflammasome activation, supernatants were collected to measure release of (A) lactate dehydrogenase (LDH) for percent cell death calculations, (B) IL-18, and/or (C) IL-1β. (D) Cell death kinetics were monitored over time following inflammasome activation by measuring the incorporation of SYTOX Green.
(E) Cell lysates were collected from BMDMs treated for 16 h of priming as described above and separated by SDS-PAGE. Western blot analysis was performed to determine protein expression of critical inflammasome components: NLRP3, CASP11, GSDMD, pro-IL-18, pro-IL-1β, and the loading control ACTIN.
(F) Quantitation of CASP11 expression was compared with CASP11-induced cell death by LPS transfection (Fu/LPS). Bar graphs in black correspond to the left axis, which represents CASP11 expression as band intensity normalized to ACTIN. The mean percent cell death at 2 h following LPS transfection (from the three independent experiments shown in A) are plotted in pink and correspond to the right axis.
(G) WT BMDMs were primed and transfected with LPS (E. coli 0111:B4, 25 μg/mL) as described for (A–D), then supernatants and lysates were collected at 1, 2, 3 h post transfection to monitor for the cleavage and release of inflammasome-related proteins by SDS-PAGE and western blot. Molecular weight marker positions are shown to the left of each blot, and arrows indicate a cleavage product. Bar graphs show the mean value +/− SEM along with individual data points pooled from independent experiments depicted with different shapes (A–C and F). Line graphs show the mean ± SD of three technical replicates (D). Data were pooled from three (A and F) or two (B and C) independent experiments or are representative of three (D and E) or two (G) independent experiments.
Statistical analysis performed using a two-way ANOVA and Tukey's multiple comparisons test; ∗∗∗∗ <0.0001; ∗∗∗ = 0.0002-0.0001; ∗ = 0.0463. See also Figure S1.
Since priming is known to induce the expression of critical inflammasome components, we sought to determine how IFNγ, IFNβ, and LPS differentially regulate these proteins as a way to mechanistically explain inflammasome activation differences. To this end, primary BMDMs were treated for 16 h with IFNγ, IFNβ, or LPS as described above and cell lysates were separated by SDS-PAGE for western blot. As expected, LPS priming increased the expression of NLRP3 compared with priming with IFNγ or IFNβ (Figure 1E). Therefore, the difference between IFNγ, IFNβ, and LPS priming on NLRP3 inflammasome activation can in part be attributed to the ability or inability of a priming agent to induce the expression of NLRP3. Furthermore, the expression of pro-IL-1β is highly induced by LPS priming, demonstrating that release of IL-1β is not a good marker of inflammasome activation when BMDMs are primed with IFNγ or IFNβ (Figures 1C and 1E). Although the expression of pro-IL-18 is increased by priming with IFNβ or LPS, expression levels between priming conditions are more similar making the release of IL-18 a better marker of inflammasome activation when comparing conditions of priming (Figures 1B and 1E, S1A, and S1C). As expected, the expression of CASP11 was induced by IFNγ, IFNβ, and LPS priming (Figure 1E). The two CASP11 proteins detected by western blot represent the full-length protein and an N-terminally truncated isoform generated by alternative translation similar to isoforms generated by the transcript encoding MAVS (data not shown) (Brubaker et al., 2014; Kang et al., 2000). In contrast to the induction of CASP11 by priming, GSDMD expression remained constant as described previously (Kayagaki et al., 2019). By comparing CASP11 expression with cell death 2 h following LPS transfection (Fu/LPS), we found that the percentage of cell death correlated with the level of CASP11 expression for IFNγ-primed BMDMs (Figure 1F). However, the percentage of cell death at 2 h following LPS transfection was barely elevated for IFNβ- or LPS-primed BMDMs despite higher levels of CASP11 expression (Figure 1F). These data suggest that other factors in addition to CASP11 regulate the response to cytosolic LPS. We also conducted a dose titration of priming agents to compare CASP11 expression and cell death at 3 h following LPS transfection (Figures S1A–S1D). LPS induced high levels of CASP11 expression; however, LPS-induced cell death by transfection was much lower compared with similar levels of CASP11 priming by IFNγ (Figures S1A and S1B). In contrast, priming with IFNβ required much higher concentrations compared with IFNγ to induce similar levels of CASP11 expression and to induce equivalent levels of LPS-induced cell death (Figures S1C and S1D).
Further supporting the notion that IFNγ is the most potent promoter of CASP11 inflammasome activation, we found that increased cell death by IFNγ priming correlated with GSDMD cleavage. Cleaved NT-GSDMD accumulated more rapidly in cell lysates from IFNγ-primed BMDMs transfected with LPS compared with IFNβ or LPS primed BMDMs (Figure 1G). In addition, the release of CASP11, NT-GSDMD, HMGB1, cleaved CASP1, and cleaved IL-18 was higher in supernatants from BMDMs primed with IFNγ compared with IFNβ or LPS (Figure 1G). Release of cleaved IL-1β into the supernatants was highest in LPS-primed BMDMs; however, this can be explained by the dramatic difference in pro-IL-1β expression between priming agents (Figures 1E and 1G). Collectively, these data demonstrate that IFNγ is a potent priming agent for the response to cytosolic LPS in BMDMs.
IFNγ Priming Specifically Promotes CASP11 Inflammasome Activation
Previous publications have demonstrated that the murine BMDM inflammasome response to cytosolic LPS requires CASP11 activity and GSDMD. Once cleaved by CASP11, the NT-GSDMD fragment generates pores in the plasma membrane causing a K+ efflux that triggers NLRP3-dependent CASP1 inflammasome activation (Ruhl and Broz, 2015). Thus, the response to cytosolic LPS is associated with downstream CASP1 activity, which contributes to cell death as well as the release of bioactive IL-18 and IL-1β (Kayagaki et al., 2011; Ruhl and Broz, 2015). Since IFNγ priming did not promote NLRP3 inflammasome activation with classic NLRP3 agonists (Figures 1A–1D), we speculated that the effect of IFNγ may specifically promote CASP11 inflammasome activation rather than promoting downstream NLRP3/CASP1 inflammasome activation. To test this hypothesis, we compared CASP11 inflammasome activation by transfected LPS in WT, Casp1−/−, and Casp11−/− BMDMs. In line with our previous results, IFNγ priming significantly increased WT BMDM cell death (measured by release of LDH) and release of IL-18 3 h after LPS transfection compared with unprimed, IFNβ-primed, or LPS primed WT BMDMs (Figures 2A and 2B). The release of IL-1β was only detected in WT BMDMs primed with LPS (Figure 2C). However, as described earlier, IFNγ and IFNβ do not induce the expression of pro-IL-1β, making release of IL-1β an unreliable marker of inflammasome activation when comparing these priming conditions (Figures 1E and 2E). As expected, cell death and IL-18 release following LPS transfection was completely dependent on CASP11 regardless of the priming condition used (Figures 2A and 2B). Furthermore, IL-18 and IL-1β release were completely dependent on CASP1, which is consistent with its role in cleaving these cytokines and promoting their secretion (Kayagaki et al., 2011; Ruhl and Broz, 2015). There was a significant decrease in the level of cell death following LPS transfection when comparing WT with Casp1−/− BMDMs primed with IFNγ (Figure 2A). This result is consistent with a role for NLRP3-dependent sensing of K+ efflux contributing to the cytosolic LPS response observed previously (Ruhl and Broz, 2015). However, there were significant differences in the cell death response to cytosolic LPS when CASP1-deficient BMDMs were primed with IFNγ, IFNβ, or LPS (Figure 2A). Similar to WT BMDMs, IFNγ priming in Casp1−/− BMDMs promoted a faster rate and greater magnitude of cell death compared with priming with IFNβ or LPS (Figures 2A and 2D). Importantly, the levels of CASP11 expression in CASP1-deficient BMDMs primed with IFNγ and LPS were similar, suggesting that a factor other than CASP11 controls the differential response seen between IFNγ and LPS priming (Figure 2E). Collectively, these data support the conclusion that IFNγ priming specifically promotes CASP11 activation in response to transfected LPS independent of NLRP3 and CASP1.
Figure 2.

IFNγ Priming Specifically Promotes CASP11 Inflammasome Activation
(A–D) WT, Casp1−/−, and Casp11−/− BMDMs were primed for 16 h overnight with the following treatments: unprimed (N/A), IFNγ (100 U/mL), IFNβ (100 U/mL), or LPS (10 ng/mL). CASP11 inflammasome activation was triggered by LPS (E. coli 0111:B4, 25ug/mL) transfection with FuGENE HD. At 3 h following inflammasome activation, supernatants were collected to measure release of (A) LDH for percent cell death calculations, (B) IL-18, and/or (C) IL-1β. (D) Cell death kinetics were monitored over time following inflammasome activation by measuring the incorporation of SYTOX Green.
(E) Cell lysates were collected from WT, Casp1−/−, or Casp11−/− BMDMs treated for 16 h of priming as described above and separated by SDS-PAGE. Western blot analysis was performed to determine protein expression of critical inflammasome components: CASP11, CASP1, GSDMD, pro-IL-18, pro-IL-1β, and the loading control ACTIN. Molecular weight marker positions are shown to the left of each blot. Bar graphs show the mean value +/− SEM along with individual data points pooled from independent experiments depicted with different shapes (A–C). Line graphs show the mean value +/− SEM from pooled independent experiments with technical triplicates (D). Data were pooled from three (A and D) or two (B and C) independent experiments or are representative of three independent experiments (E).
Statistical analysis performed using a two-way ANOVA and Tukey's multiple comparisons test; ∗∗∗∗ <0.0001. See also Figure S2.
We considered whether IFNγ priming might specifically promote delivery of LPS to the cytosol during transfection. Using a FITC-labeled LPS conjugate, we found that IFNγ priming did not increase the amount of FITC-LPS recovered from transfected BMDMs (Figure S2A). Therefore, IFNγ does not simply promote ligand binding by increasing the efficiency of LPS transfection.
NT-GSDMD pore formation occurs downstream of GSDMD cleavage but upstream of NLRP3 inflammasome activation (Kayagaki et al., 2015). Comparing the kinetics of GSDMD cleavage between differentially primed BMDMs (Figure 1G) suggested to us that IFNγ promotes a mechanism upstream of NT-GSDMD rather than pore formation itself. To directly test if IFNγ treatment regulates NT-GSDMD pore formation, we generated a cell line in Gsdmd-CRISPR/Cas9 KO RAW cells that expresses the NT-GSDMD fragment under a Doxycycline-inducible promoter. These NT-GSDMD inducible cells were either primed with IFNγ or left unprimed for 16 h, then treated with increasing concentrations of Doxycycline to induce the expression of NT-GSDMD and monitored for cell death (Figure S2B). IFNγ had no effect on cell death following Doxycycline treatment, demonstrating that NT-GSDMD pore formation proceeds independently of IFNγ priming. These data suggest that IFNγ promotes CASP11-dependent pyroptosis upstream of NT-GSDMD pore formation.
Priming Promotes Cell Death in Response to Cytosolic LPS Independently of CASP11 Expression
We observed that the kinetics and extent of pyroptosis in WT BMDMs in response to cytosolic LPS did not always correlate directly with the level of CASP11 expression following treatment with IFNγ, IFNβ, or LPS (Figures 1A, 1B, 1D–1F, and S1A–S1D). These findings suggest that another factor or factors dependent on priming may promote the response to cytosolic LPS independently of CASP11 expression. To test this hypothesis directly, we set out to generate a cell line that constitutively expresses CASP11 from a retroviral expression vector to uncouple the expression of CASP11 from priming. To this end, we chose to use RAW 264.7 cells, which are more amenable to genetic manipulations. Importantly, this immortalized cell line behaves similarly to WT BMDMs in our assays despite requiring a higher concentration of IFNβ to induce comparable levels of CASP11 expression (Figures S3A–S3E). IFNγ priming of RAW 264.7 cells promoted faster kinetics and higher levels of cell death in response to cytosolic LPS delivered by transfection or with the Cholera Toxin B (CTB) subunit as compared with unprimed, IFNβ-primed, or LPS-primed cells (Figures S3B and S3C). CTB-mediated delivery of LPS to activate CASP11 has been described previously (Kayagaki et al., 2013; Hagar et al., 2013). In addition, similar to observations in WT BMDMs (Figure S2A), IFNγ did not appear to increase the efficiency of LPS transfection into RAW 264.7 as measured with a FITC-labeled LPS conjugate capable of inducing CASP11-dependent inflammasome activation (Figures S3D and S3E). Thus, the immortalized RAW 264.7 cell line is a suitable background to uncouple the expression of CASP11 from priming and test if priming can still influence the response to cytosolic LPS.
To generate the CASP11 constitutive cell line, we transduced Casp1,Casp11-CRISPR/Cas9 DKO RAW macrophages with a construct that constitutively drives CASP11 expression and selected for a clone with stable expression similar to that observed in WT RAW 264.7 cells primed with IFNγ (Figure 3A). There were minimal changes to CASP11 and GSDMD expression when this cell line was primed with IFNγ, IFNβ, or LPS (Figure 3B). Despite this, each of the priming agents promoted cell death following LPS transfection as observed by the release of LDH and GSDMD cleavage (Figures 3C and 3D). In support of our hypothesis that IFNγ is the most potent promoter of CASP11 inflammasome activation, IFNγ priming resulted in significantly higher levels of cell death in this cell line compared with IFNβ or LPS priming (Figure 3C). In addition, higher amounts of HMGB1 were released from IFNγ-primed cells compared with IFNβ or LPS priming (Figure 3D). Similar results were obtained when cytosolic LPS was delivered with CTB (Figures S3F and S3G). Specifically, IFNγ priming promoted higher levels of cell death, GSDMD cleavage, and release of HMGB1 (Figures S3F and S3G). Because this constitutive CASP11-expressing cell line is CASP1 deficient, these data further support our conclusion that IFNγ specifically promotes CASP11-dependent cell death independently of CASP1. By genetically uncoupling the ability of priming agents to induce CASP11 expression, these data demonstrate that IFNγ promotes rapid pyroptosis in response to cytosolic LPS through an unknown mechanism that is separate from its ability to induce CASP11 expression.
Figure 3.

Priming Promotes Cell Death in Response to Cytosolic LPS Independently of CASP11 Expression
A constitutive CASP11-expressing cell line was generated by transducing a CASP11 expression vector into Casp1,Casp11-CRISPR/Cas9 DKO RAW cells (Casp1/11 DKO).
(A) Cell lysates were collected from WT, Casp1/11 DKO, and the constitutive cell line (Casp1/11 DKO + Casp11) and analyzed by western blot to determine CASP1 and CASP11 expression. WT and Casp1/11 DKO were treated with IFNγ (100 U/mL) for 16 h.
(B) The constitutive CASP11-expressing cell line was primed for 16 h overnight with the following treatments; unprimed (N/A), IFNγ (100 U/mL), IFNβ (1,000 U/mL), or LPS (10 ng/mL). Lysates were collected to determine the effects of priming on CASP11 expression by western blot.
(C and D) The constitutive CASP11-expressing cell line was primed as described above and CASP11 inflammasome activation was triggered by LPS (E. coli 0111:B4, 50 μg/mL) transfection with FuGENE HD. (C) At 4 and 8 h following inflammasome activation, supernatants were collected to measure release of LDH for percent cell death calculations. (D) Alternatively, supernatants and lysates were collected 3 h post transfection to monitor for the cleavage and release of inflammasome-related proteins by SDS-PAGE and western blot. Molecular weight marker positions are shown to the left of each blot, and arrows indicate a cleavage product. Bar graphs show the mean value +/− SEM along with individual data points pooled from independent experiments depicted with different shapes (C). Data were pooled from two (C) independent experiments or are representative of three (A and B) or two (D) independent experiments.
Statistical analysis performed using a two-way ANOVA and Tukey's multiple comparisons test; ∗∗∗∗ <0.0001. See also Figure S3.
IFNγ Receptor Signaling Enhances but Is Not Required for CASP11 Inflammasome Activation
Our data demonstrate that IFNγ priming enhances CASP11 inflammasome activation in response to cytosolic LPS. To identify genes required for this enhanced response to cytosolic LPS, we utilized the GeCKOv2 pooled gRNA library to generate and screen a genome-wide CRISPR knockout library in the constitutive CASP11-expressing cell line described in Figure 3 (Sanjana et al., 2014). We conducted survival screens by activating the cell death response to cytosolic LPS with or without IFNγ priming. Enrichment of gRNAs in the surviving cell populations was determined using the MAGeCK algorithm (Li et al., 2014). Cells containing gRNAs that target the IFNγ receptor-encoding genes, Ifngr1 and Ifngr2, were highly enriched with IFNγ priming compared with the unprimed condition (Figure S4). More specifically, there was no enrichment of Ifngr1 and Ifngr2 gRNA-containing cells following CASP11 inflammasome activation without IFNγ priming. These data suggest that IFNγ receptor signaling is not required for CASP11 inflammasome activation but is required for the IFNγ enhanced response against cytosolic LPS.
IFNγ induces a pleiotropic set of transcriptional, translational, and metabolic changes in macrophages by binding and signaling through a heterodimer receptor complex composed of IFNγR1 and IFNγR2 (Schroder et al., 2004; Su et al., 2015). Based on our CRISPR screen results, we suspected that signaling through this receptor complex enhances but is not absolutely critical for the response to cytosolic LPS. To formally test this hypothesis, we compared the response to cytosolic LPS between WT and Ifngr1−/− BMDMs (Huang et al., 1993). Compared with WT BMDMs, there was a significant reduction in cell death following LPS transfection in Ifngr1−/− BMDMs primed with IFNγ (Figure 4A). This result is not surprising, given that CASP11 expression was not induced in Ifngr1−/− BMDMs primed with IFNγ as compared with WT BMDMs (Figure 4B). If we primed Ifngr1−/− BMDMs with IFNβ or LPS, however, CASP11 expression was induced and there was a significant increase in the cell death response to cytosolic LPS compared with unprimed Ifngr1−/− BMDMs (Figures 4A and 4B). Furthermore, there was no significant difference in the cell death response to cytosolic LPS between WT and Ifngr1−/− BMDMs when cells were primed with IFNβ or LPS (Figure 4A). These data provide evidence that IFNγ receptor signaling is not critical for the response to cytosolic LPS but rather enhances it.
Figure 4.

IFNγ Receptor Signaling Enhances but is not Required for CASP11 Inflammasome Activation
(A, C, and D) WT and Ifngr1−/− BMDMs were primed for 16 h overnight with the following treatments: unprimed (N/A), IFNγ (100 U/mL), IFNβ (100 U/mL), or LPS (10 ng/mL). CASP11 inflammasome activation was triggered by LPS (E. coli 0111:B4, 25 μg/mL) transfection with FuGENE HD.
(A) At 2 h following inflammasome activation, supernatants were collected to measure release of LDH for percent cell death calculations.
(B) Cell lysates were collected from BMDMs treated for 16 h of priming as described above and separated by SDS-PAGE. Western blot analysis was performed to determine protein expression of CASP11, GSDMD, and the loading control ACTIN. Molecular weight marker positions are shown to the left of each blot.
(C and D) Cell death kinetics were monitored over time following inflammasome activation by measuring the incorporation of SYTOX Green. (C) Comparisons between different priming conditions are shown for BMDMs transfected with LPS. (D) Additionally, a comparison between unprimed WT and Ifngr1−/− BMDMs treated with or without transfection reagent are shown. Bar graphs show the mean value +/− SEM along with individual data points pooled from independent experiments depicted with different shapes (A). Line graphs show the mean ± SD of three technical replicates (C and D). Data were pooled from two (A) independent experiments or are representative of two (B–D) independent experiments.
Statistical analysis performed using a two-way ANOVA and Tukey's multiple comparisons test; ∗∗∗∗ <0.0001; ∗∗∗ <0.002. See also Figure S4.
Monitoring the kinetics of cell death following LPS transfection also supports the conclusion that IFNγ receptor signaling is not critical for the response to cytosolic LPS. As we observed previously, WT BMDMs primed with IFNγ exhibited the most rapid cell death response to transfected LPS compared with IFNβ-, LPS-, or unprimed BMDMs (Figure 4C). The kinetics of cell death for IFNγ receptor-deficient BMDMs were similar to WT BMDMs when primed with IFNβ or LPS, whereas Ifngr1−/− BMDMs primed with IFNγ died at a rate similar to unprimed BMDMs. These data demonstrate that IFNγ receptor signaling is only required to promote rapid cell death in response to cytosolic LPS when BMDMs are primed with IFNγ (Figure 4C). Finally, unprimed WT and Ifngr1−/− BMDMs undergo similar cell death kinetics in response to transfected LPS compared with LPS treatment alone (Figure 4D). Thus, IFNγ receptor signaling is not required for basal responses to cytosolic LPS in the absence of priming. Collectively, our data demonstrate that IFNγ receptor signaling is not critical for CASP11 activation but suggest that IFNγ priming induces the expression or activation of an unknown factor that enhances the response to cytosolic LPS.
IFNγ Enhancement of CASP11-Dependent Pyroptosis Requires GSDMD and Is Independent of GSDME
Pyroptosis as an outcome of inflammasome activation has primarily been attributed to cleavage of GSDMD by CASP11 or CASP1(Shi et al., 2015; Kayagaki et al., 2015). Following cleavage, the N-terminal fragment of GSDMD forms pores in the plasma membrane resulting in pyroptotic cell death, and thus, BMDMs deficient for GSDMD are refractory to CASP11- or CASP1-induced pyroptosis (Shi et al., 2015; Kayagaki et al., 2015; Ding et al., 2016; Liu et al., 2016; Aglietti et al., 2016). Recent publications have now revealed that GSDME, a homolog of GSDMD, is cleaved by alternative cell death pathways and the GSDME N-terminal fragment similarly causes a necrotic form of rapid cell death (Rogers et al., 2017; Wang et al., 2017; Sarhan et al., 2018; Aizawa et al., 2020). Based on these previous findings, we set out to determine whether the enhanced response to cytosolic LPS induced by IFNγ priming strictly requires GSDMD or whether it may also depend on GSDME.
We began by comparing the cell death response to cytosolic LPS in WT, Gsdmd−/−, and Gsdme−/− BMDMs primed with or without IFNγ. As expected, there were significantly lower levels of cell death at 4 h following LPS transfection in IFNγ-primed BMDMs derived from Gsdmd−/− mice (Figure 5A). Monitoring the kinetics of cell death by SYTOX Green incorporation over time also demonstrated the absolute requirement of GSDMD for the response to IFNγ priming and cytosolic LPS (Figure 5B). Although GSDMD was absolutely required, we observed no difference in cell death between WT and Gsdme−/− BMDMs primed with IFNγ and transfected with LPS (Figures 5A and 5B). Importantly, CASP11 expression was similarly induced across all genotypes, eliminating the possibility that GSDMD and GSDME might control CASP11 expression in response to IFNγ (Figure 5C). These data provide evidence that the enhanced CASP11-dependent response to cytosolic LPS induced by IFNγ strictly requires GSDMD and is independent of GSDME.
Figure 5.

IFNγ Enhancement of CASP11-Dependent Cell Death Requires GSDMD and Is Independent of GSDME
(A and B) WT, Gsdmd−/−, and Gsdme−/− BMDMs were primed for 16 h overnight with or without IFNγ (100 U/mL). CASP11 inflammasome activation was triggered by LPS (E. coli 0111:B4, 25 μg/mL) transfection with FuGENE HD. (A) At 4 h following inflammasome activation, supernatants were collected to measure release of LDH for percent cell death calculations. (B) Cell death kinetics were monitored over time following inflammasome activation by measuring the incorporation of SYTOX Green.
(C) Cell lysates were collected from WT, Gsdmd−/−, or Gsdme−/− BMDMs treated for 16 h of priming as described above and separated by SDS-PAGE. Western blot analysis was performed to determine protein expression of CASP11, GSDMD, GSDME, and the loading control ACTIN. Molecular weight marker positions are shown to the left of each blot. Bar graphs show the mean value +/− SEM along with individual data points pooled from independent experiments depicted with different shapes (A). Line graphs show the mean ± SEM from pooled independent experiments (B). Data were pooled from two (A and B) independent experiments or are representative of two independent experiments (C).
Statistical analysis performed using a two-way ANOVA and Tukey's multiple comparisons test; ∗∗∗∗ <0.0001. See also Figure S5.
As mentioned above, GSDME can be cleaved by alternative cell death pathways. For example, TAK1 inhibition in conjunction with TLR4 activation triggers an alternative CASP8/CASP3 cell death pathway in BMDMs that results in both GSDMD and GSDME cleavage (Sarhan et al., 2018; Orning et al., 2018). NLRP3 inflammasome activation in the absence of CASP1 activity provides another example in which macrophages switch to an alternative cell death pathway that triggers CASP8/CASP3 cleavage and relies on GSDMD as well as GSDME cleavage for cell death (Sagulenko et al., 2013; Schneider et al., 2017; Aizawa et al., 2020). Based on our results from experiments using Gsdme−/− BMDMs (Figures 5A–5C), we did not expect IFNγ priming to promote cell death through the CASP8/CASP3/GSDME alternative cell death pathway. In support of this hypothesis, we found no evidence of CASP8, CASP3, or GSDME cleavage under IFNγ priming conditions in our constitutively expressing CASP11 RAW 264.7 cell line or in WT BMDMs (Figures S5A and S5B). TAK1 inhibition by 5z7 or CASP1-deficient BMDMs were used as positive controls in these experiments to observe CASP8, CASP3, GSDME cleavage. In conditions where CASP8 and CASP3 were cleaved, we also detected the smaller p20 fragment of GSDMD, a product of CASP3 activity (Sarhan et al., 2018; Orning et al., 2018; Chen et al., 2019). Since IFNγ priming does not directly promote NLRP3 inflammasome activation (Figure 1), cleavage of CASP8, CASP3, and GSDME in Casp1−/− BMDMs primed with IFNγ and transfected with LPS is likely the result of downstream K+ efflux-mediated NLRP3 activation, which switches to an alternative necrotic cell death pathway in the absence of CASP1(Ruhl and Broz, 2015; Schneider et al., 2017; Aizawa et al., 2020).
Collectively, these data suggest that the enhanced CASP11-dependent response to cytosolic LPS that is triggered by IFNγ priming strictly requires GSDMD for pyroptosis. This enhanced cell death response to IFNγ priming and LPS transfection did not rely on GSDME and we found no evidence of GSDME cleavage that would suggest it plays a role in the process. Furthermore, the enhanced response to cytosolic LPS triggered by IFNγ priming cannot be attributed to cleavage of CASP8 or CASP3, which constitute an alternative cell death pathway under certain conditions. Although it is possible that IFNγ priming enhances the response to cytosolic LPS through a cell death pathway that has yet to be determined, our findings are consistent with the idea that IFNγ specifically primes and enhances the CASP11-GSDMD-pyroptosis pathway.
GBPs Encoded on Chromosome 3 Do Not Fully Account for Enhanced CASP11-Dependent Cell Death Triggered by IFNγ Priming
The guanylate-binding proteins (GBPs) have gained attention for their role during inflammasome activation in response to bacterial infection or in response to OMVs shed by Gram-negative bacteria (Pilla et al., 2014; Meunier et al., 2014; Finethy et al., 2015, 2017; Man et al., 2016; Santos et al., 2018; Cerqueira et al., 2018; Tang et al., 2018; Fisch et al., 2019; Gu et al., 2019). These proteins are highly upregulated in response to IFNγ and are recruited to pathogen containing vacuoles and/or directly to bacteria during infection (Tretina et al., 2019). A subset of GBPs are required for CASP11 inflammasome activation in murine-derived BMDMs and are thought to play a role in facilitating CASP11-LPS interaction (Santos et al., 2018; Cerqueira et al., 2018). Recent evidence in human cells demonstrates that GBP1 binds to LPS and assembles a signaling platform with several other GBPs for CASP4 activation (Wandel et al., 2020; Santos et al., 2020). Based on these findings, we speculated that inducible GBPs could account for the increased level of CASP11-dependent pyroptosis we observe with IFNγ priming (Figures 1, 2, and 3).
There are eleven highly conserved GBPs encoded in mice. Six of these are tandemly encoded on chromosome 3 and were collectively deleted to generate Gbpchr3−/- mice (Yamamoto et al., 2012). These mice have been used previously to demonstrate the importance of GBPs for inflammasome activation during bacterial infection (Pilla et al., 2014; Meunier et al., 2014, 2015; Finethy et al., 2015; Man et al., 2016). Importantly, the closest homolog of human GBP1 is encoded within the chromosome 3 locus. Given the importance of human GBP1 in activating CASP4 (Wandel et al., 2020; Santos et al., 2020), this may help to explain defects in CASP11 inflammasome activation during bacterial infection using Gbpchr3−/- BMDMs (Pilla et al., 2014; Meunier et al., 2014; Finethy et al., 2015). In line with previous reports, IFNγ primed Gbpchr3−/- BMDMs displayed a partial defect in the cell death response to cytosolic LPS as triggered by treatment with E. coli-derived OMVs, LPS transfection, or LPS complexed with CTB (Figure 6A and S6A) (Pilla et al., 2014; Finethy et al., 2017; Santos et al., 2018; Cerqueira et al., 2018; Tang et al., 2018; Gu et al., 2019). BMDMs derived from Gbpchr3−/- mice failed to upregulate Gbp2 when treated with IFNγ but importantly express equivalent levels of CASP11 as compared with WT controls (Figure 6B). These data demonstrate that the interferon inducible GBPs encoded on chromosome 3 are partially required for CASP11 inflammasome activation when BMDMs are primed with IFNγ. However, it remained to be determined whether the enhanced IFNγ response to cytosolic LPS can be fully attributed to the function of these GBPs encoded on chromosome 3.
Figure 6.

GBPs Encoded on Chromosome 3 Do Not Fully Account for Enhanced CASP11-Dependent Cell Death Triggered by IFNγ Priming
(A and B) WT, Gbpchr3−/−, and Casp11−/− BMDMs were primed for 16 h overnight with or without IFNγ (100U/mL). CASP11 inflammasome activation was triggered by treating cells with OMVs (E. coli DH5α), LPS (E. coli 0111:B4, 25 μg/mL) transfection with FuGENE HD, or LPS (E. coli 0111:B4, 25 μg/mL) mixed with CTB (Cholera Toxin B Subunit, 20 μg/mL). (A) At the indicated time points following inflammasome activation, supernatants were collected to measure release of LDH for percent cell death calculations. (B) Cell lysates were collected from WT, Gbpchr3−/−, and Casp11−/− BMDMs treated with or without IFNγ as described above and separated by SDS-PAGE. Western blot analysis was performed to determine protein expression of GBP2, CASP11, GSDMD, and the loading control ACTIN.
(C–F) WT, Gbpchr3−/−, and Casp11−/− BMDMs were primed for 16 h overnight with the following treatments: unprimed (N/A), IFNγ (100 U/mL), IFNβ (100 U/mL), or LPS (10 ng/mL). CASP11 inflammasome activation was triggered by LPS transfection (C and E) or with CTB and LPS (D and F) as described above. Cell death was determined following inflammasome activation by collecting supernatants at 3 h to monitor LDH release (C and D), or cell death kinetics were monitored over time by measuring the incorporation of SYTOX Green (E and F).
(G) Cell lysates were collected from WT and Gbpchr3−/− BMDMs treated for 16 h of priming as described above and separated by SDS-PAGE. Western blot analysis was performed to determine protein expression of GBP2, CASP11, GSDMD, pro-IL-18, and the loading control ACTIN.
(H) WT and Gbpchr3−/− BMDMs were primed and transfected with LPS as described above. Supernatants and lysates were collected from untransfected, 1, 2, or 3 h post-transfection to monitor for the cleavage and release of inflammasome-related proteins by SDS-PAGE and western blot. Molecular weight marker positions are shown to the left of each blot, and arrows indicate a cleavage product. Bar graphs show the mean value +/− SEM along with individual data points pooled from independent experiments depicted with different shapes (A, C, and D). Line graphs show the mean value +/− SEM from pooled independent experiments with technical replicates (E and F). Data were pooled from four (A - Fu/LPS and CTB/LPS) or two (A - OMV, C, D, E, F) independent experiments or are representative of two (B and G) or one (H) independent experiment.
Statistical analysis performed using a two-way ANOVA and Tukey's multiple comparisons test; ∗∗∗∗ <0.0001; ∗∗∗ for (C) = 0.0005; ∗ for (A) = 0.0453; ∗ for (D) = 0.0294. See also Figure S6.
An important aspect of our current study lies in the comparison between different priming agents demonstrating that IFNγ priming elicits the most rapid and amplified macrophage response to cytosolic LPS. For example, in testing Casp1−/− BMDMs we observed a partial defect in the cell death response following IFNγ priming and LPS transfection (Figures 2A and 2D). However, we also compared the cell death response in Casp1−/− BMDMs using different priming agents and found significant differences between IFNγ priming compared with IFNβ or LPS. Together these data demonstrate that CASP1 is involved in the response to cytosolic LPS (likely downstream of K+ efflux as discussed above) but that CASP1 is not required for the differences observed between priming agents. Based on these results, we reasoned it would also be important to compare the response to cytosolic LPS using different priming agents in Gbpchr3−/- BMDMs to determine if the GBPs encoded on chromosome 3 are required for the enhanced cell death response observed with IFNγ priming.
When comparing the effect of different priming agents, we found significant differences in the cell death response to cytosolic LPS in Gbpchr3−/- BMDMs. In line with our previous results, IFNγ priming significantly increased CASP11-dependent cell death in WT BMDMs transfected with LPS or treated with CTB/LPS (Figures 6C and 6D). Similarly, IFNγ priming promoted significantly higher levels of CASP11-dependent cell death in Gbpchr3−/- BMDMs compared with priming with IFNβ or LPS (Figures 6C and 6D). IFNγ priming also promotes a more rapid CASP11-dependent response in Gbpchr3−/- BMDMs compared with other priming agents (Figures 6E and 6F). These data suggest that GBPs encoded on chromosome 3 are not required for the differential outcome observed between priming conditions. In line with results from WT and Casp1−/− BMDMs, the greatest difference in priming responses for CASP11 inflammasome activation in Gbpchr3−/- BMDMs lies between IFNγ and LPS priming. Importantly, LPS priming induces equivalent levels of CASP11 expression, if not more, than IFNγ priming in Gbpchr3−/- BMDMs (Figure 6G). Thus, the defect in CASP11-dependent responses using LPS priming conditions cannot be attributed to defects in CASP11 expression.
To confirm that IFNγ priming promotes a more rapid and robust CASP11-dependent response independently of GBPs encoded on chromosome 3, we compared GSDMD cleavage over time and the release of cytokines from WT and Gbpchr3−/- BMDMs under different priming conditions. For the GSDMD cleavage experiment, we chose to focus on the difference between IFNγ and LPS priming based on the greater phenotypic range between these conditions. Following LPS transfection, the N-terminal cleaved fragment of GSDMD accumulated more rapidly in WT BMDM lysates primed with IFNγ compared with LPS priming, which confirms our previous results (Figures 6H and 1G). In support of our hypothesis, cleaved NT-GSDMD also accumulated more rapidly in the lysates of Gbpchr3−/- BMDMs primed with IFNγ compared with LPS priming. Furthermore, the release of cleaved NT-GSDMD and HMGB1 into the supernatant was higher in Gbpchr3−/- BMDMs primed with IFNγ compared with LPS priming (Figure 6H). An increased release of cleaved IL-18 from IFNγ-primed Gbpchr3−/- BMDMs was more subtle but could be confirmed by an ELISA on supernatants collected at 3 h following CASP11 activation (Figures 6H and S6B).
Our data provide support to a growing body of research that demonstrate an important role for GBPs encoded on chromosome 3 in the response to cytosolic LPS by CASP11. However, we show that the enhanced CASP11-dependent response triggered by IFNγ priming cannot solely be attributed to the function of GBPs encoded on chromosome 3. Interestingly, GBP1 is encoded within the chromosome 3 locus, which is of interest because its human homolog was recently shown to play an important role in assembling a GBP signaling platform with CASP4 and LPS (Wandel et al., 2020; Santos et al., 2020). Because Gbpchr3 deficiency does not account for differences in CASP11-dependent responses between IFNγ- and LPS-priming, our data suggest that an unknown factor(s) controls the differential response seen between these priming conditions.
Discussion
By comparing CASP11-dependent responses under different priming conditions, our study reveals specificity between these external cues that dictate inflammasome activation outcome. A key finding from our research is the demonstration that IFNγ priming promotes the most rapid and robust CASP11-dependent response compared with IFNβ or LPS priming. Our results suggest that IFNγ induces the activity of an unidentified CASP11 regulator that enhances pyroptosis in response to cytosolic LPS. This finding justifies a renewed search for unknown regulators of cytosolic LPS sensing, as the known regulators of this process cannot fully explain the priming effects of IFNγ. Several lines of evidence support this conclusion. First, with the exception of complete CASP11 deficiency, we did not observe a direct correlation between the extent of CASP11 expression and the responsiveness of cells to different priming agents. Indeed, IFNγ retained the ability to prime and increase pyroptosis levels in a cell line constitutively expressing CASP11. Furthermore, we provide evidence that GBPs encoded on chromosome 3 do not fully account for the enhanced effects of IFNγ priming compared with IFNβ or LPS priming. We note, however, that we verify a role for GBPs encoded on chromosome 3 in CASP11 inflammasome activation as demonstrated by a partial defect in Gbpchr3−/- BMDMs response to cytosolic LPS (Pilla et al., 2014; Finethy et al., 2017; Santos et al., 2018; Cerqueira et al., 2018; Tang et al., 2018; Gu et al., 2019). Our results do not exclude the possibility that other interferon inducible GBPs (outside the chromosome 3 locus) are responsible for the enhancement of CASP11 responses driven by IFNγ priming. There is no evidence to date, however, indicating a role in CASP11 function for these GBPs encoded outside chromosome 3. In conclusion, our data demonstrate that the observed IFNγ-dependent increased response to transfected LPS must be controlled by a mechanism independent of its ability to control the expression of CASP11 or GBPs encoded on chromosome 3. Based on these findings we propose that IFNγ priming induces the activity of an unknown CASP11 regulator(s).
Often with little regard to the priming step, the field of inflammasome research has primarily focused on the activation step and downstream effector functions, such as pyroptosis and cytokine release. Our data support previous research that demonstrates that priming agents differentially dictate inflammasome outcome (Figure 1) (Bauernfeind et al., 2009, 2016). To understand the biological relevance of these variable outcomes, future studies will likely require the use of in vivo models of infection. For example, Aachoui and colleagues provide compelling evidence that IFNγ produced by NK and T cells in vivo is critically important to prime CASP11 and protect against infection using the model pathogen Burkholderia thailandensis (Aachoui et al., 2013, 2015; Kovacs et al., 2020). However, in vivo CASP11 responses can also be deleterious, causing higher rates of mortality in models of septic shock (Kayagaki et al., 2013; Hagar et al., 2013). Based on these diametrically opposed outcomes, the activation of CASP11 in vivo must be tightly regulated to provide protection when needed while simultaneously limiting collateral damage. Therefore, we speculate that the CASP11 response may have evolved a sensitivity to IFNγ priming as a mechanism for gauging the level of infectious threat in vivo.
Transcription of Casp11 is undoubtedly an important factor in tightly regulating the effects of CASP11, and yet our understanding of the promoter elements and transcription factors controlling expression is somewhat limited. Previous work demonstrates that CASP11 induced by LPS priming requires the nuclear factor κB (NF-κB), the C/EBP homologous protein (CHOP), and Poly (ADP-ribose) polymerase-1 (PARP-1) (Schauvliege et al., 2002; Endo et al., 2006; Yoo et al., 2011). The only identified transcription factor known to control CASP11 expression following IFNγ priming is STAT1 (Schauvliege et al., 2002). Future research that expands our understanding of the transcription factors regulating CASP11 expression under different priming conditions may help to clarify the functional differences we observe between these conditions.
The effects of IFNγ treatment on macrophages are pleiotropic but generally understood to drive antimicrobial responses (Schroder et al., 2004). Hundreds of genes are transcriptionally induced, rates of mRNA translation are shifted, and changes to metabolism highlight just some of the changes that occur following IFNγ treatment (Su et al., 2015). Thus, the phenotype described in this manuscript could be regulated transcriptionally, translationally, or enzymatically. The pleiotropic effects of IFNγ treatment are akin to our understanding of IFN-driven mechanisms that are broadly antiviral. For example, hundreds of interferon stimulated genes (ISGs) contribute to antiviral mechanisms that protect the host, yet our understanding of their functional relevance individually remains limited (Schoggins, 2019).
As mentioned previously, a strength of our study is derived from the comparison between different priming agents on inflammasome outcome. The greatest difference in response was observed between IFNγ and LPS priming, which could not be explained by differential CASP11 or GBP expression. Our interpretation of these results led us to hypothesize that IFNγ priming may induce the expression or activity of an enhancer(s) that promotes CASP11 inflammasome activation. Alternatively, LPS may induce the expression or activity of a negative regulator(s) that inhibits CASP11 inflammasome activation. This alternative hypothesis would fit with most of the data that we present in this manuscript. However, experiments utilizing the constitutive CASP11 expressing cell line suggest this may not be the case. Specifically, LPS priming in this cell line promoted higher levels of cell death following CASP11 activation compared with unprimed cells (Figure 3C). If LPS priming induced the expression or activity of an inhibitor, we might expect to see lower levels of CASP11-induced cell death in this cell line as compared with unprimed cells.
Future studies are required to test these hypotheses and may lead to the identification of our proposed IFNγ-inducible enhancer(s) of CASP11 activation or an LPS-inducible inhibitor(s) of the same response. We speculate that unknown CASP11 regulators that either enhance or inhibit the response to cytosolic LPS may also play a role in CASP4/CASP5-dependent responses in human cells. Furthermore, the identification of unknown enhancers or inhibitors to these responses may aid in drug development for treating bacterial infections or sepsis.
Limitations of the Study
Several publications have highlighted inflammasome activation differences between mouse and human systems. Our study was limited to experiments in mouse macrophages, which provided us with the specific advantage of utilizing primary cells (BMDMs) for the majority of our experiments. Therefore, it will be of importance to compare different priming agents in the context of human CASP4/CASP5 inflammasome activation. Given that CASP4 is constitutively expressed in many cell types, inducible regulators of the response to cytosolic LPS may be even more important in this context. Additionally, it was beyond the scope of this manuscript to compare priming conditions in other cell types. It will be interesting to determine if IFNγ priming enhances CASP11 inflammasome activation in other cell types or if this response is specific to macrophages.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Denise Monack (dmonack@stanford.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
The dataset supporting the current study is available from the lead contact upon reasonable request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank Manuel Amieva as well as members of the Monack and Amieva laboratories for valuable discussion and technical support. The following funding sources supported the research reported in this manuscript: NIAID, United States 1R01 AI109539, NIAID, United States 2R01 AI095396-06, NIGMS, United States T32GM007276, NIAID, United States 5T32AI00729035, Dean's Postdoctoral Fellowship—Stanford School of Medicine, Stanford Graduate Fellowship—Gabilan Fellow, Allen Stanford Discovery Center Grant to D.M.M. from the Paul Allen Family Foundation. The graphical abstract was created with BioRender.com.
Author Contributions
Conceptualization, S.W.B. and D.M.M.; Investigation S.W.B., S.M.B., and L.M.M.; B.A.N. made Casp1,Casp11-CRISPR/Cas9 DKO RAW macrophages utilized in this study; Writing – Original Draft, S.W.B.; Writing – Review & Editing, S.W.B., S.M.B., and D.M.M.; Funding Acquisition, S.W.B., S.M.B., and D.M.M.
Declaration of Interests
The authors declare no competing interests.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101612.
Supplemental Information
References
- Aachoui Y., Kajiwara Y., Leaf I.A., Mao D., Ting J.P., Coers J., Aderem A., Buxbaum J.D., Miao E.A. Canonical inflammasomes drive IFN-gamma to Prime Caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe. 2015;18:320–332. doi: 10.1016/j.chom.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aachoui Y., Leaf I.A., Hagar J.A., Fontana M.F., Campos C.G., Zak D.E., Tan M.H., Cotter P.A., Vance R.E., Aderem A., Miao E.A. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aglietti R.A., Estevez A., Gupta A., Ramirez M.G., Liu P.S., Kayagaki N., Ciferri C., Dixit V.M., Dueber E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. U S A. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizawa E., Karasawa T., Watanabe S., Komada T., Kimura H., Kamata R., Ito H., Hishida E., Yamada N., Kasahara T. GSDME-dependent incomplete pyroptosis permits selective IL-1alpha release under caspase-1 inhibition. iScience. 2020;23:101070. doi: 10.1016/j.isci.2020.101070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S., Takeda K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Bauernfeind F., Niepmann S., Knolle P.A., Hornung V. Aging-associated TNF production primes inflammasome activation and NLRP3-related metabolic disturbances. J. Immunol. 2016;197:2900–2908. doi: 10.4049/jimmunol.1501336. [DOI] [PubMed] [Google Scholar]
- Bauernfeind F.G., Horvath G., Stutz A., Alnemri E.S., Macdonald K., Speert D., Fernandes-Alnemri T., Wu J., Monks B.G., Fitzgerald K.A. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P., Ruby T., Belhocine K., Bouley D.M., Kayagaki N., Dixit V.M., Monack D.M. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. 2012;490:288–291. doi: 10.1038/nature11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker S.W., Bonham K.S., Zanoni I., Kagan J.C. Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker S.W., Gauthier A.E., Mills E.W., Ingolia N.T., Kagan J.C. A bicistronic MAVS transcript highlights a class of truncated variants in antiviral immunity. Cell. 2014;156:800–811. doi: 10.1016/j.cell.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira D.M., Gomes M.T.R., Silva A.L.N., Rungue M., Assis N.R.G., Guimaraes E.S., Morais S.B., Broz P., Zamboni D.S., Oliveira S.C. Guanylate-binding protein 5 licenses caspase-11 for gasdermin-D mediated host resistance to brucella abortus infection. PLoS Pathog. 2018;14:e1007519. doi: 10.1371/journal.ppat.1007519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.W., Demarco B., Heilig R., Shkarina K., Boettcher A., Farady C.J., Pelczar P., Broz P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019;38:e101638. doi: 10.15252/embj.2019101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J., Wang K., Liu W., She Y., Sun Q., Shi J., Sun H., Wang D.C., Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- Endo M., Mori M., Akira S., Gotoh T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J. Immunol. 2006;176:6245–6253. doi: 10.4049/jimmunol.176.10.6245. [DOI] [PubMed] [Google Scholar]
- Finethy R., Jorgensen I., Haldar A.K., De Zoete M.R., Strowig T., Flavell R.A., Yamamoto M., Nagarajan U.M., Miao E.A., Coers J. Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in chlamydia-infected macrophages. Infect. Immun. 2015;83:4740–4749. doi: 10.1128/IAI.00856-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finethy R., Luoma S., Orench-Rivera N., Feeley E.M., Haldar A.K., Yamamoto M., Kanneganti T.D., Kuehn M.J., Coers J. Inflammasome activation by bacterial outer membrane vesicles requires guanylate binding proteins. mBio. 2017;8:e01188-17. doi: 10.1128/mBio.01188-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisch D., Bando H., Clough B., Hornung V., Yamamoto M., Shenoy A.R., Frickel E.M. Human GBP1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J. 2019;38:e100926. doi: 10.15252/embj.2018100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L., Meng R., Tang Y., Zhao K., Liang F., Zhang R., Xue Q., Chen F., Xiao X., Wang H. Toll-like receptor 4 signaling licenses the cytosolic transport of lipopolysaccharide from bacterial outer membrane vesicles. Shock. 2019;51:256–265. doi: 10.1097/SHK.0000000000001129. [DOI] [PubMed] [Google Scholar]
- Hagar J.A., Powell D.A., Aachoui Y., Ernst R.K., Miao E.A. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Hendriks W., Althage A., Hemmi S., Bluethmann H., Kamijo R., Vilcek J., Zinkernagel R.M., Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- Kang S.J., Wang S., Hara H., Peterson E.P., Namura S., Amin-Hanjani S., Huang Z., Srinivasan A., Tomaselli K.J., Thornberry N.A. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J. Cell Biol. 2000;149:613–622. doi: 10.1083/jcb.149.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N., Lee B.L., Stowe I.B., Kornfeld O.S., O'rourke K., Mirrashidi K.M., Haley B., Watanabe C., Roose-Girma M., Modrusan Z. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci. Signal. 2019;12:eaax4917. doi: 10.1126/scisignal.aax4917. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Stowe I.B., Lee B.L., O'rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q.T. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J., Newton K., Qu Y., Liu J., Heldens S. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- Kayagaki N., Wong M.T., Stowe I.B., Ramani S.R., Gonzalez L.C., Akashi-Takamura S., Miyake K., Zhang J., Lee W.P., Muszynski A. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- Kovacs S.B., Oh C., Maltez V.I., Mcglaughon B.D., Verma A., Miao E.A., Aachoui Y. Neutrophil caspase-11 is essential to defend against a cytosol-invasive bacterium. Cell Rep. 2020;32:107967. doi: 10.1016/j.celrep.2020.107967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E., Xiao T.S., Stutz A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V.G., Wu H., Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man S.M., Karki R., Sasai M., Place D.E., Kesavardhana S., Temirov J., Frase S., Zhu Q., Malireddi R.K.S., Kuriakose T. IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase-11-NLRP3 inflammasomes. Cell. 2016;167:382–396 e17. doi: 10.1016/j.cell.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier E., Dick M.S., Dreier R.F., Schurmann N., Kenzelmann Broz D., Warming S., Roose-Girma M., Bumann D., Kayagaki N., Takeda K. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature. 2014;509:366–370. doi: 10.1038/nature13157. [DOI] [PubMed] [Google Scholar]
- Meunier E., Wallet P., Dreier R.F., Costanzo S., Anton L., Ruhl S., Dussurgey S., Dick M.S., Kistner A., Rigard M. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat. Immunol. 2015;16:476–484. doi: 10.1038/ni.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning P., Weng D., Starheim K., Ratner D., Best Z., Lee B., Brooks A., Xia S., Wu H., Kelliher M.A. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018;362:1064–1069. doi: 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla D.M., Hagar J.A., Haldar A.K., Mason A.K., Degrandi D., Pfeffer K., Ernst R.K., Yamamoto M., Miao E.A., Coers J. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc. Natl. Acad. Sci. U S A. 2014;111:6046–6051. doi: 10.1073/pnas.1321700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam V.A., Vanaja S.K., Waggoner L., Sokolovska A., Becker C., Stuart L.M., Leong J.M., Fitzgerald K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C., Fernandes-Alnemri T., Mayes L., Alnemri D., Cingolani G., Alnemri E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017;8:14128. doi: 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhl S., Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 2015;45:2927–2936. doi: 10.1002/eji.201545772. [DOI] [PubMed] [Google Scholar]
- Sagulenko V., Thygesen S.J., Sester D.P., Idris A., Cridland J.A., Vajjhala P.R., Roberts T.L., Schroder K., Vince J.E., Hill J.M. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20:1149–1160. doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos J.C., Boucher D., Schneider L.K., Demarco B., Dilucca M., Shkarina K., Heilig R., Chen K.W., Lim R.Y.H., Broz P. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat. Commun. 2020;11:3276. doi: 10.1038/s41467-020-16889-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos J.C., Dick M.S., Lagrange B., Degrandi D., Pfeffer K., Yamamoto M., Meunier E., Pelczar P., Henry T., Broz P. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J. 2018;37:e98089. doi: 10.15252/embj.201798089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarhan J., Liu B.C., Muendlein H.I., Li P., Nilson R., Tang A.Y., Rongvaux A., Bunnell S.C., Shao F., Green D.R., Poltorak A. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. U S A. 2018;115:E10888–E10897. doi: 10.1073/pnas.1809548115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauvliege R., Vanrobaeys J., Schotte P., Beyaert R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J. Biol. Chem. 2002;277:41624–41630. doi: 10.1074/jbc.M207852200. [DOI] [PubMed] [Google Scholar]
- Schneider K.S., Gross C.J., Dreier R.F., Saller B.S., Mishra R., Gorka O., Heilig R., Meunier E., Dick M.S., Cikovic T. The inflammasome drives GSDMD-independent secondary pyroptosis and IL-1 release in the absence of caspase-1 protease activity. Cell Rep. 2017;21:3846–3859. doi: 10.1016/j.celrep.2017.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W. interferon-stimulated genes: what do they all do? Annu. Rev. Virol. 2019;6:567–584. doi: 10.1146/annurev-virology-092818-015756. [DOI] [PubMed] [Google Scholar]
- Schroder K., Hertzog P.J., Ravasi T., Hume D.A. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Shi J., Zhao Y., Wang Y., Gao W., Ding J., Li P., Hu L., Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- Su X., Yu Y., Zhong Y., Giannopoulou E.G., Hu X., Liu H., Cross J.R., Ratsch G., Rice C.M., Ivashkiv L.B. Interferon-gamma regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat. Immunol. 2015;16:838–849. doi: 10.1038/ni.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y., Zhang R., Xue Q., Meng R., Wang X., Yang Y., Xie L., Xiao X., Billiar T.R., Lu B. TRIF signaling is required for caspase-11-dependent immune responses and lethality in sepsis. Mol. Med. 2018;24:66. doi: 10.1186/s10020-018-0065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretina K., Park E.S., Maminska A., Macmicking J.D. Interferon-induced guanylate-binding proteins: guardians of host defense in health and disease. J. Exp. Med. 2019;216:482–500. doi: 10.1084/jem.20182031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandel M.P., Kim B.H., Park E.S., Boyle K.B., Nayak K., Lagrange B., Herod A., Henry T., Zilbauer M., Rohde J. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat. Immunol. 2020;21:880–891. doi: 10.1038/s41590-020-0697-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Gao W., Shi X., Ding J., Liu W., He H., Wang K., Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Okuyama M., Ma J.S., Kimura T., Kamiyama N., Saiga H., Ohshima J., Sasai M., Kayama H., Okamoto T. A cluster of interferon-gamma-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity. 2012;37:302–313. doi: 10.1016/j.immuni.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Yoo L., Hong S., Shin K.S., Kang S.J. PARP-1 regulates the expression of caspase-11. Biochem. Biophys. Res. Commun. 2011;408:489–493. doi: 10.1016/j.bbrc.2011.04.070. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset supporting the current study is available from the lead contact upon reasonable request.