

Summary
Sertoli cells are the major component of the spermatogonial stem cell (SSC) niche; however, regulatory mechanisms in Sertoli cells that dictate SSC fate decisions remain largely unknown. Here we revealed features of the N6-methyladenosine (m6A) mRNA modification in Sertoli cells and demonstrated the functions of WTAP, the key subunit of the m6A methyltransferase complex in spermatogenesis. m6A-sequencing analysis identified 21,909 m6A sites from 15,365 putative m6A-enriched transcripts within 6,122 genes, including many Sertoli cell-specific genes. Conditional deletion of Wtap in Sertoli cells resulted in sterility and the progressive loss of the SSC population. RNA sequencing and ribosome nascent-chain complex-bound mRNA sequencing analyses suggested that alternative splicing events of transcripts encoding SSC niche factors were sharply altered and translation of these transcripts were severely dysregulated by Wtap deletion. Collectively, this study uncovers a novel regulatory mechanism of the SSC niche and provide insights into molecular interactions between stem cells and their cognate niches in mammals.
Keywords: WTAP, Sertoli cell, m6A, spermatogonial stem cell, spermatogonial differentiation, alternative splicing, niche
Graphical Abstract
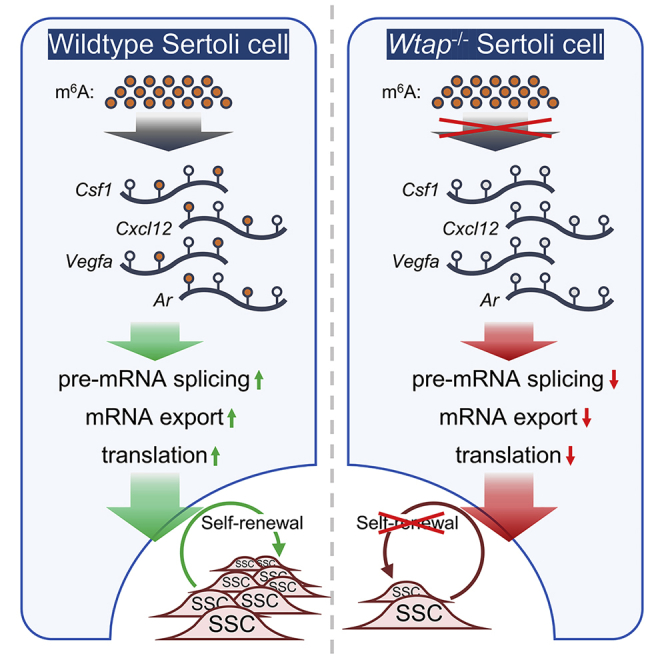
Highlights
-
•
WTAP is highly expressed in Sertoli cell and is essential in spermatogenesis
-
•
Wtap knockout in Sertoli cell causes defective spermatogonial stem cell maintenance
-
•
WTAP regulates transcription and translation of m6A-enriched genes in Sertoli cell
In this article, Yang and colleagues revealed features of m6A modification in Sertoli cells and showed that WTAP deficiency causes dysregulated transcripts encoding spermatogonial stem cell (SSC) niche factors and the progressive loss of SSCs. They conclude that WTAP-mediated m6A in Sertoli cell is indispensable for sustaining SSC niche.
Introduction
Adult stem cells provide foundation for the homeostasis, growth, and regeneration of most tissues in mammals. Stem cells rely on extrinsic signals from their specialized niches to balance self-renewal and lineage commitment (Jones and Wagers, 2008). The interactions between stem cells and niches also influence stem cell behaviors in aging and pathological conditions (Lukjanenko et al., 2016; Mesa et al., 2015). Identification and functional characterization of niche components and regulatory mechanisms are essential for elucidating the molecular basis of stem cell fate decisions.
Spermatogenesis is a classic stem cell-supported process that requires the activities of spermatogonial stem cells (SSCs). SSCs self-renew to maintain an undifferentiated state and differentiate to produce transient amplifying progenitors that are committed to enter meiosis after several rounds of mitotic divisions (De Rooij, 1988; Oatley and Brinster, 2008). Extrinsic cues from the SSC niche work in concert with intrinsic programs to dictate SSC proliferation, differentiation, and apoptosis (De Rooij, 2017). Sertoli cells serve as the major cellular component of the SSC niche by providing growth factors, extracellular matrix, and structural support (Griswold, 1998; Meng et al., 2000). Sertoli cells also regulate spermatogonial differentiation, meiosis progression, and spermatid development (Griswold, 2018). These specified functions of Sertoli cells are directed by precise gene expression regulation at the transcriptional, post-transcriptional, and translational levels; however, limited knowledge exists about how these regulations are conducted. Elucidating gene regulation programs mediating Sertoli cell-SSC interaction will enhance our understanding of stem cell niche formation and maintenance.
N6-methyladenosine (m6A), one of the most widespread modifications in eukaryotic mRNA, has been linked to mRNA stability, structure, splicing, processing, and translational efficiency (TE) (Gilbert et al., 2016; Zhao et al., 2017). The m6A modification is catalyzed by a multiprotein methyltransferase complex composed of methyltransferase-like 3 (METTL3), METTL14, and Wilms tumor 1-associated protein (WTAP) (Liu et al., 2014). METTL3 functions as the major catalytic subunit, while METTL14 forms a stable heterodimer with METTL3 to enhance methyltransferase activity (Wang et al., 2016). WTAP recruits METTL3 and METTL14 to mRNA targets (Ping et al., 2014). Findings from knockout mouse models uncovered pivotal roles of METTL3- and METTL14-mediated RNA methylation in embryogenesis (Horiuchi et al., 2006), neurogenesis (Wang et al., 2018), and adipogenesis (Kobayashi et al., 2018). In stem cell-dependent tissues, m6A modification has critical functions in controlling lineage specification and fate choice of stem and progenitor cells. METTL3-deficient hematopoietic stem cells fail to differentiate properly (Cheng et al., 2019) and Mettl14 deletion in neural progenitors causes defects in cell cycle progression (Yoon et al., 2017). Moreover, conditional deletion of Mettl3 or Mettl14 in germ cells using Ddx4-Cre leads to SSC depletion and male sterility (Lin et al., 2017). Despite these key findings, the roles of m6A modification in Sertoli cells are unknown and the functions of WTAP in establishing and maintaining the SSC niche remain to be determined.
Here, using spermatogenesis as a model system, we investigated features and functions of m6A modification in controlling the establishment and maintenance of stem cell niche. We illustrated the m6A methylome and examined the function of WTAP-dependent m6A in Sertoli cells. We showed that WTAP is essential for SSC maintenance and spermatogonial differentiation. Mechanistically, WTAP-mediated m6A modification controlled transcription and translation of a list of genes in Sertoli cells to sustain SSC niche and govern normal spermatogenesis.
Results
WTAP Is Highly Expressed in Murine Sertoli Cells
We firstly examined the expression of m6A methyltransferases WTAP, METTL3, and METTL14 in mouse testis. Immunofluorescent staining revealed that METTL3 or METTL14 was co-localized with WTAP in germ cells and Sertoli cells within seminiferous tubules (Figure S1A). Interestingly, we noticed that immunoreactive signal for WTAP appeared to be strong in Sertoli cells. Co-staining of WTAP, METTL3, or METTL14 with Sertoli cell markers showed that all these proteins were expressed in Sertoli cells and WTAP indeed was strongly enriched in Sertoli cells (Figure 1A). We then examined relative abundances of Wtap, Mettl3, and Mettl14 in fluorescence-activated cell sorting isolated Sertoli cells from Sox9Gfp transgenic mice. Wtap and Mettl3 transcript concentrations were increased by 5.23-fold and 3.47-fold in SOX9-GFP+ cells compared with those in Sertoli cell-depleted testicular cells (Figure S1B). Together, these data indicated that all three methyltransferases co-existed and likely catalyzed m6A modification in Sertoli cells.
Figure 1.

Function Analysis of m6A in Mouse Sertoli Cells
(A) Immunostaining for METTL3 and GATA4, METTL14 and GATA4, or WTAP and SOX9 with DAPI in mouse testes at P6. The arrows indicate Sertoli cells. Scale bars represent 40 μm.
(B) Average m6A sites per gene in distinct RNA sequence regions including 3′ UTR, CDS, and 5′ UTR.
(C and D) Average fold enrichment over input for m6A peaks of selected genes specially expressed in Sertoli cells (C) or related to spermatogenesis (D). Two independent Sertoli cell samples isolated from 110 mice were used (n = 2).
(E–G) Pie chart of molecular types (E), phenotype association analysis (F), and biological process GO analysis (G) of m6A-enriched genes in Sertoli cells. The bubble size indicates the number of matched genes.
Analysis Features of m6A Modification in Sertoli Cells
As expected, m6A signal was present in Sertoli cells (Figure 2A). To characterize features of m6A, we conducted affinity purification and m6A sequencing in isolated Sertoli cells from adult testis. A total of 21,909 m6A sites were identified from 15,365 putative m6A-enriched transcripts within 6,122 genes (Table S1), and these m6A sites were predominantly distributed in CDS (coding sequence) regions and less abundantly in 3′ UTR and 5′ UTR regions (Figures 1B and S1C). Two-hundred and seventy-eight of 369 previously identified Sertoli cell-specific transcripts (Green et al., 2018) contained m6A sites, such as Inhbb, Wt1, Arid4a, and Arid4b (Table S2). Etv5, Ar, Dmrt1, and Sin3a transcripts and other genes known to be essential for Sertoli cell function were also included in the m6A-enriched gene list (Figure 1C).
Figure 2.

Progressive Loss of Undifferentiated Spermatogonia Caused by Wtap Deletion in Sertoli Cells
(A) Immunostaining for GATA4 and m6A or WTAP and SOX9 in testis cross sections from the indicated genotypes at P6. The arrows indicate Sertoli cells. Scale bars represent 20 μm.
(B–E) Testes (B) of control and Wtap-sKO male mice at P180. Scale bar represents 2 mm. Testis/body weight ratios at P6, P14, P21, P28, P35, P60, P120, and P180 (C), litter sizes from P60 to P150 (D), and sperm counts of cauda epididymis at P180 (E) from the indicated genotypes. At least three mice were used for each time point (n = 3 different animals). Ten control or Wtap-sKO animals were used in fertility test (n = 10 different animals). ∗∗p < 0.01 and ∗∗∗p < 0.001.
(F–J) H&E staining of seminiferous tubules (F) from control and Wtap-sKO males at P6, P35, P60, and P180. Asterisks indicate SCO tubules. H&E staining and SOX9 and GCNA1 immunostaining (G) of SCO tubules from Wtap-sKO males. Quantification of seminiferous tubules from Wtap-sKO testes containing degenerated spermatogenesis and SCO phenotype (H) at P35, P60, P120, and P180. Immunostaining of LIN28A and SOX9 (I) in control and Wtap-sKO testes at P6, P35, P60, and P180. Statistics of the number of LIN28A+ cells per SOX9+ cell (J) from the indicated genotypes at P6, P21, P35, P60, and P180. At least 1500 SOX9+ cells were counted per genotype at each time point. Scale bars represent 40 μm. Three animals were used for control or Wtap-sKO at each time point (n = 3 different animals). ∗∗p < 0.01 and ∗∗∗p < 0.001.
See also Figures S2–S4.
To further decipher potential functions of m6A-enriched transcripts in Sertoli cells, we conducted molecular function classification, phenotype correlation analysis, and gene ontology (GO) analysis (Table S3). The results showed that these transcripts were preferentially associated with nucleic acid binding, hydrolase, enzyme modulator, transcription factor, transferase, and cytoskeleton (Figure 1E). Identified m6A-enriched transcripts were highly relevant to embryonic lethality, abnormal cell physiology, male infertility, abnormal spermatogenesis, and sperm abnormality (Figure 1F). GO analysis of biological process found that these transcripts were enriched in cell proliferation, DNA repair, mRNA export, 3′ end processing, mRNA stability regulation, regulation of translation, and transcription elongation (Figure 1G). In the top 100 of m6A-enriched genes (transcripts with fold enrichment ≥24.9), we found genes that have been reported to be essential for spermatogenesis, including Spaca1 (Fujihara et al., 2012), Rhox8 (Welborn et al., 2015), and H3f3b (Yuen et al., 2014), (Figure S1D). Interestingly, we noticed that a significant number of m6A-enriched transcripts were involved in SSC maintenance and spermatogonial proliferation. For example, Cxcl12, Bmp4, Fgf2, and Vegfa transcripts encoding SSC niche factors contained m6A sites (Figure 1D).
Sertoli Cell-Specific Deletion of Wtap Caused Male Infertility
Next, we hypothesized that m6A modification in Sertoli cells plays a functional role in regulating SSC niche. To test this hypothesis, we conditionally deleted the Wtap by crossing Wtap-floxed (Wtapfl/fl) and Amh-Cre mice (Figures S2A and S2B). Immunostaining confirmed a reduction of m6A and WTAP in Sertoli cells of Wtapfl/flAmh-Cre (hereafter referred to as Wtap-sKO) mice (Figure 2A). Testis/body weight ratio of Wtap-sKO mice was significantly reduced compared with that of the littermate controls (Figures 2B and 2C). Fertility test showed that Wtap-sKO mice were completely sterile (Figure 2D). Caudal epididymal sperm concentration in Wtap-sKO mice was only 20.20% of that in control mice (Figures 2E and S2C). H&E staining did not reveal obvious defects in sperm morphology of Wtap-sKO mice, and co-staining of the acrosome marker peanut agglutinin (PNA) with mitochondria marker mito-tracker showed that sperm of Wtap-sKO animals displayed normal heads and tails (Figures S2D and S2E). PNA staining of seminiferous tubules also demonstrated that morphology and number of round and elongated spermatids in Wtap-sKO mice were comparable with those of control littermates at postnatal days 28 (P28) (Figures S3A and S3B).
We examined meiotic progression using spermatocyte nuclear spreading and the results indicated percentage of germ cells in leptotene, zygotene, pachytene, diplotene, and diakinesis were similar in testes of Wtap-sKO and control animals (Figures S3C and S3D). Formation of synaptonemal complex and progression of meiotic prophase did not show defects. Furthermore, integrity of the blood-testis barrier, an important ultrastructure essential for spermatogenesis, was not affected by Wtap loss-of-function (Figure S3E). Reduced testis/body weight ratio was observed in Wtap-sKO males and began as early as P21 (Figure 2C), and total germ cell number was reduced from P21 (Figures S4A and S4B). Together, these data revealed that the sterile phenotype of Wtap-sKO mice was caused by reduced sperm concentration because of germ cell loss.
Sertoli Cell-Specific Deletion of Wtap Caused Progressive Loss of Undifferentiated Spermatogonia
To identify the underlying reasons for germ cell loss, we examined the process of germ cell development in Wtap-sKO and control testes. H&E analyses showed that formation of spermatogonial lineage and development of spermatocytes and round spermatids were similar in testes of Wtap-sKO and control animals. Degeneration of seminiferous tubules was detectable beginning at P35 and the condition became severe with aging (Figure 2F). Immunofluorescence staining confirmed that some of these tubules only contained Sertoli cells without GCNA1+ germ cells, suggesting the development of Sertoli cell-only (SCO) phenotype (Figure 2G). Quantitative analysis revealed that the percentages of degenerated tubules and SCO tubules increased with aging. Degenerated spermatogenesis was found in 83.0% of tubules and 49.3% of these tubules were SCO in Wtap-sKO mice at P180 (Figure 2H).
Next, we quantified the number of undifferentiated spermatogonia at different development stages. The ratio of LIN28A+ undifferentiated spermatogonia per SOX9+ cell did not change at P6, suggesting that formation of the undifferentiated spermatogonial pool was not affected by deleting Wtap in Sertoli cells. A significant reduction of undifferentiated spermatogonial population was found in Wtap-sKO mice at P21. Although the size of the Sertoli cell population did not change (Figure S4C), number of LIN28A+ spermatogonia continued to decrease from P21 to P180 (Figures 2I and 2J). The whole-mount staining of seminiferous tubules for GFRA1 and FOXO1 revealed a significant reduction of Asingle (As) spermatogonia, the primitive spermatogonia containing enriched SSCs in testes of Wtap-sKO mice (Figure 3A). Findings of these experiments indicated that deletion of Wtap in Sertoli cells resulted in a progressive loss of SSCs and depletion of germ cells.
Figure 3.

Impaired SSC Self-renewal and Proliferation Caused by Wtap Deletion in Mouse Sertoli Cells
(A–D) Whole-mount immunostaining of GFRA1 and FOXO1 (A) and TUNEL and LIN28A (B) in control and Wtap-sKO seminiferous tubules at P21. Whole-mount immunostaining of EdU and LIN28A (C) in control and Wtap-sKO seminiferous tubules after 2 h or 7 days post EdU injection at P14. Quantifications of EdU+ As, Apr, and Aal spermatogonia in Lin28A+ cells (D) of control and Wtap-sKO testes. At least 900 LIN28A+ cells were counted per genotype for each time point. Three animals were used for control or Wtap-sKO at each time point (n = 3 different animals). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Scale bars represent 40 μm.
(E–G) The schematic diagram (E) of germ cell transplantation experiment. Quantification of THY1+ cells obtained using MACS (F) and SSC numbers in THY1+ undifferentiated spermatogonia (G) from control and Wtap-sKO males. SSC numbers were derived from donor-derived colonies of spermatogenesis in recipient testes and normalized to 105 cells injected. THY1+ cells from three control or Wtap-sKO animals (4 weeks old) were isolated and transplanted into testes of six recipients (n = 3 different animals). ∗∗p < 0.01.
(H) qRT-PCR analysis of Thy1, Cxcr4, Gfra1, Id4, Lin28, Zbtb16, and Rb1 transcript abundances in THY1+ cells from control and Wtap-sKO testes. Four control or Wtap-sKO animals were used (n = 4 different animals). ∗p < 0.05.
See also Figures S4 and S5.
Sertoli Cell-Derived WTAP Is Essential for SSC Self-renewal and Proliferation
To further determine the fate decisions of undifferentiated spermatogonia, we examined apoptosis, proliferation, and differentiation of As, Apaired (Apr), and Aaligned (Aal) undifferentiated spermatogonia. Firstly, we ruled out the possibility that SSC loss was due to apoptosis, because TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling) assay confirmed that apoptotic spermatocytes increased modestly in testes of Wtap-sKO mice (Figures S4D–S4F); however, undifferentiated spermatogonia rarely underwent apoptosis either in control or Wtap-sKO mice (Figure 3B). Next, we labeled proliferative cells with 5-ethynyl-2′-deoxyuridine (EdU) in control and Wtap-sKO animals at P14 and quantified the number of EdU+ undifferentiated spermatogonia after 2 h or 7 days. The results uncovered that mitotic division was severely impaired in undifferentiated spermatogonia of Wtap-sKO mice. Particularly, EdU incorporation and retention rates in LIN28A+ As spermatogonia were significantly reduced in Wtap-sKO mice compared with those of controls (Figures 3C and 3D). Similarly, GFRA1+ As spermatogonia were sharply decreased in testis of Wtap-sKO mice (Figures S5A and S5B). To confirm that the SSC activity was indeed decreased, we performed a transplantation experiment to examine the SSC number in magnetic-activated cell sorted (MACS) THY1+ from 4-week-old control and Wtap-sKO donors. LacZ-expressing ROSA26 line (ROSA26-lacZ) was crossed with control or Wtap-sKO animals to label donor-derived spermatogenesis (Figure 3E). The numbers of THY1+ cells recovered from control and Wtap conditional knockout animals were comparable (Figure 3F) and a 10-μL single-cell suspension (containing 104 THY1+ cells) was transplanted into one testis of busulfan-treated recipients. The recipients were examined 8 weeks after transplantation for donor-derived spermatogenesis by X-Gal staining as previously described (Kubota et al., 2004; Yang et al., 2013). The average number of donor-cell-derived colonies was 56.7 colonies/105 cells injected for control animals; however, the same number of donor cells only generated 3.3 colonies for Wtap-sKO animals, indicating SSC activities were affected dramatically by Wtap deletion in Sertoli cells (Figure 3G). We next investigated relative abundance of factors related to SSC maintenance, and the data revealed that expressions of Cxcr4, Gfra1, Id4, Lin28a, Zbtb16, and Rb1 were greatly reduced in THY1+ germ cells from Wtap-sKO males; however, Thy1 expression did not show difference (Figure 3H). Lastly, we quantified number of differentiating spermatogonia by immunofluorescent staining for KIT or STRA8 and found that the number of KIT+ germ cells was decreased at P21 in testes of Wtap-sKO mice (Figures S5C and S5D). Interestingly, number of STRA8 and LIN28A double-positive cells (STRA8+ and LIN28A+) was significantly decreased in Wtap-sKO mice (Figures S5E–S5G), indicating that spermatogonial differentiation was impaired. From these data, we concluded that deletion of Wtap in Sertoli cells caused major defects in SSC self-renewal/proliferation. Wtap deletion also affected the spermatogonial differentiation. Together, findings of these studies demonstrated that, without critical roles of WTAP, Sertoli cells failed to provide a functional niche to support SSC fate decisions properly, leading to the exhaustion of SSC pool.
Wtap Deletion Altered Gene Transcription and Translation in Sertoli Cells
To understand how WTAP functions to maintain a healthy SSC niche, we conducted high-throughput RNA sequencing (RNA-seq) and ribosome nascent-chain complex-bound mRNA sequencing (RNC-seq) to evaluate changes in gene expression at transcriptional and translational levels in WTAP-null Sertoli cells. RNA-seq revealed that a total of 7,279 (2,646 up and 4,633 down) mRNA were differentially expressed genes (DEGs) in WTAP-deficient Sertoli cells. GO analysis indicated that downregulated transcripts were involved in transcription regulation, cellular response to DNA damage stimulus, cell cycle, chromatin organization, apoptosis process, MAPK cascade, methylation, WNT signaling pathway, DNA repair, and cell differentiation (Figure 4A and Table S4). RNC-seq data identified a list of 2,047 (1,065 up and 982 down) differentially translated genes (DTGs) evaluated by TE, normalized read count of ribosome-protected fragments (RPF)/mRNA fragments. Upregulated DTGs were involved in transcription regulation, WNT signaling pathway, H3K4 trimethylation, cell projection assembly, cell cycle, and mRNA processing (Figure 4B and Table S4). Notably, 81.97% (1,678/2,047) of DTGs were found in DEGs, and 789 transcriptionally downregulated genes were translationally upregulated while 880 transcriptionally upregulated genes were translationally downregulated (Figure 4C). Transcripts previously shown to be important for SSC maintenance, spermatogonial differentiation, and Sertoli cell fate decisions were dysregulated (Figure 4D). Of particular interest, we found that transcript abundances of Igf1r, Vegfa, Notch1, Csf1, Gdnf, Ptpn11, Rbpj, Gata4, Vegfb, Smad4, Inhbb, Ctnnb1, Kitl, Apc, Gja1, Dhh, Wt1, Wnt3, Insr, Arid4a, Ar, Rdh10, Cyp26b1, Cyp26a1, Rara, E2f1, Rad51, Brca2, E4f1, Sirt1, Ccnb1, Rb1, and Msh2 were altered. Among them, Vegfa, Rbpj, Arid4a, Ar, Rara, E2f1, Ccnb1, and Msh2 were translationally dysregulated (Figure 4E). Together, these data uncovered roles of WTAP in regulating gene expression in Sertoli cells at transcriptional and translational levels.
Figure 4.

Transcriptional and Translational Dysregulations in Sertoli Cells from Wtap-sKO Mice
(A and B) Heat map showing the DEGs (A) and DTGs (B) between control and Wtap-sKO animals and their function enrichment. Gene differences with |log2fold change| ≥ 1 and p < 0.01 are identified as significant. The enriched biological process GO terms in downregulated (blue boxes) and upregulated (red boxes) genes upon Wtap knockout are shown in the right panels.
(C) Scatterplots showing the relationship of fold changes between mRNA FPKM and RPF TE of genes in Sertoli cells upon Wtap knockout. Red, blue, green, and gray plots indicate genes belonging to both DEGs and DTGs, DEGs only, DTGs only, and neither DEGs nor DTGs.
(D) Heat maps showing the relative levels of mRNA FPKM and RPF TE of genes involving SSC maintenance and spermatogonial differentiation in Sertoli cells.
(E) Interaction network showing genes involved in spermatogenesis regulation. Two genes with regulatory relationships are connected directly, DEGs and DTGs are marked in blue and green respectively.
All analyses were performed on three different control or Wtap-sKO samples (n = 3 different biological replicates). ∗p < 0.05. See also Table S4.
Wtap Deletion Changed Transcription and Translation of m6A-Enriched Transcripts in Sertoli Cells
To further understand how WTAP-dependent m6A modifications were involved in sustaining the SSC niche, RNA-seq, RNC-seq, and m6A sequencing (m6A-seq) were comprehensively analyzed to decipher patterns of gene regulation in Sertoli cells. As expected, expressions of m6A-enriched transcripts were significantly affected by Wtap deletion; 37.59% (2,736/7,279) of DEGs and 37.13% (760/2047) of DTGs were m6A-enriched, and more m6A sites were enriched in transcriptionally downregulated and translationally upregulated genes (Figures 5A and 5B). It is noteworthy that DEGs with m6A showed higher expression levels than those without m6A in control Sertoli cells and exhibited sharper changes upon Wtap knockout (Figure 5C). Importantly, many transcripts that encode proteins to be important for m6A modifications were downregulated in WTAP-deficient Sertoli cells (Figure 5D), indicating a core role of WTAP in m6A formation in Sertoli cells. Reads distribution analyses of representative genes (Cxcl12 and Csf1) revealed remarkable transcriptional changes within m6A sites on mRNA reads, while translating mRNA was less affected (Figure 6A).
Figure 5.

Effects of WTAP-Mediated m6A Modification on Transcriptional and Post-transcriptional Regulation of Sertoli Cell Gene Expression
(A–D) Venn diagram showing numbers of DEGs and DTGs with or without m6A modification (A), proportions of m6A-enriched genes on differential transcriptional and translational changes (B), mRNA FPKM of DEGs and RPF TE of DTGs with or without m6A modification (C), and heat maps showing relative levels of mRNA FPKM and RPF TE of the main m6A regulators (D) in control and Wtap-sKO Sertoli cells.
(E–I) Statistics of five types of differential AS events (E) on mRNA transcripts in control and Wtap-sKO Sertoli cells. Venn diagram (F) showing numbers of DEGs and DTGs with or without differential AS events in Sertoli cells. Average m6A sites (G) of genes with or without differential AS events. Average inclusion levels (H) and cumulative frequency curves for inclusion levels (I) of A5SS and A3SS, MXE, RI, and SE in genes with or without m6A modification between control and Wtap-sKO animals.
(J) Interaction network showing genes involved in spermatogenesis regulation. Two genes with regulatory relationships are connected directly, genes with m6A modification and differential AS events are marked in red and yellow respectively.
All analyses were performed on three different control or Wtap-sKO samples (n = 3 different biological replicates). ∗p < 0.05. See also Table S5.
Figure 6.

Decreased Expression of SSC Niche Factors Caused by Wtap Deletion
(A) mRNA and RNC mRNA reads of Cxcl12 and Csf1 in control and Wtap-sKO samples. The m6A sites are marked as red triangles.
(B) qRT-PCR analysis of Wtap, Cxcl12, Ar, Csf1, Etv5, Wt1, Gata4, Fgf2, Bmp4, Cyp26b1, and Gdnf transcript abundances in control and Wtap-sKO Sertoli cells.
(C and D) Western blot (C) and quantitative data (D) showing CXCL12, GDNF, CSF1, and CYP26B1 expression in control and Wtap-sKO testes.
All quantitative data are presented as the mean ± SEM for n = 3 independent biological replicates. ∗p < 0.05 and ∗∗p < 0.01.
Several lines of evidences uncovered that m6A modification regulates pre-mRNA splicing (Adhikari et al., 2016), so we next examined alternative splicing (AS) events in control and Wtap-sKO Sertoli cells. In total, 5,003 differential AS events in 2,946 genes were detected, and skipped exon (SE) accounted for 57.87% (2,895/5,003) of the total (Figure 5E and Table S5). Venn diagrams showed that 1,214 DEGs and 367 DTGs contained splicing variants (Figure 5F). As expected, genes with differential AS events displayed abundant m6A enrichment (Figure 5G). The effect of m6A on AS events was further analyzed and the results showed that alternative 5′ splice site (A5SS) remained almost entirely unaffected by m6A modification, while differences in alternative 3′ splice site (A3SS) were apparent in mRNA with and without m6A modification. Interestingly, great influences of m6A modification on alternative exons including SE, mutually exclusive exons (MXEs), and retained introns (RIs) were evident (Figures 5H and 5I). Interaction network identified a list of genes that were showed to be important for Sertoli cell function. Most of them were m6A-enriched, including Igf1r, Vegfa, Notch1, Fgf2, Csf1, Cxcl12, Etv5, Ptpn11, Rbpj, Sin3a, Gata4, Vegfb, Smad4, Inhbb, Bmp4, Ctnnb1, Kitl, Apc, Gja1, Wt1, Arid4a, Ar, Cyp26b1, Aldh1a1, Aldh1a2, Aldh1a3, Rara, Pcna, E4f1, and Msh2. In addition, AS events were detected in Sin3a, Insr, Rara, Brca2, Sirt1, and Cdc25a (Figure 5J). These analyses indicated that changes in m6A modifications alter transcription and translation of genes that are important for fate decisions of SSCs and their progenitors; for example, Cxcl12 and Csf1 (Figure 6A). To validate these findings, we examined relative expression of Cxcl12, Csf1, Etv5, Wt1, Fgf2, Bmp4, and other downregulated transcripts using qRT-PCR (Figure 6B) and these results were consistent with RNA-seq. We also examined protein levels of CXCL12, GDNF, CSF1, and CYP26B1 in testis from control and Wtap-sKO animals and the results showed that CXCL12, GDNF, and CSF1 protein were reduced sharply by Wtap loss-of-function in Sertoli cells (Figures 6C and 6D). Together, these data demonstrated that WTAP-mediated m6A regulates AS events of genes regulating SSC niche and spermatogonial fate decisions.
Discussion
m6A is a highly conserved internal RNA modification that influences gene expression at transcriptional and post-transcriptional levels (Gilbert et al., 2016). Recent studies revealed crucial roles of m6A in stem cell fate decisions (Cheng et al., 2019; Geula et al., 2015); however, its dynamics and functions in stem cell and niche interactions remained largely unknown. Because WTAP is a key member of m6A modification complex (Ping et al., 2014) and highly expressed in Sertoli cells, here, we illustrate the m6A methylome landscape of Sertoli cells in murine testis and demonstrate that WTAP-mediated m6A modification is a key regulatory mechanism that instructs the expression of SSC niche factors at the transcriptional and translation levels in Sertoli cells.
We describe patterns of m6A methylome in mouse Sertoli cells and provide an important reference for further deciphering functions of transcripts with m6A modification in mediating molecular interactions between stem cells and their niches. Sertoli cells are major cellular components of the SSC niche (Oatley and Brinster, 2012); however, regulatory mechanisms that specify and maintain the SSC niche are not well understood. In this study, we found m6A modification in Sertoli cells is different from spermatogenic cells as previously described (Lin et al., 2017).The number of m6A peak sites of Sertoli cells was similar to that of spermatogonia and greater than that of spermatocytes or round spermatids, while the number of m6A peak sites of Sertoli cells identifeid in this study was similar to that of round spermatids and significantly less than that of spermatogonia. These finding support the conclusion that transcripts of Sertoli cells harbor intense m6A modification, and m6A-enriched transcripts serve different roles in governing normal spermatogenesis.
Transcripts of previously identified SSC niche factors and transcription regulators important for the development of spermatogenic lineage are m6A enriched. CXCL12 and CSF1 are important niche factors that enhance SSC self-renewal (Oatley et al., 2009; Yang et al., 2013). VEGFA, FGF2, and PDGFB are growth factors in the niche milieu that influence SSC fate decisions (Basciani et al., 2008; Ishii et al., 2012; Lu et al., 2013). Activation of NOTCH signaling in Sertoli cells increases the expression of GDNF and stimulates SSC expansion (Garcia et al., 2014). BMP4 and KITL direct spermatogonial differentiation (Ohta et al., 2000; Yang et al., 2016). ARID4A and ARID4B interact with RB1 to regulate Sertoli cell function and spermatogenesis (Wu et al., 2013); importantly, deletion of Arid4b in Sertoli cells affects the establishment of SSC niche in mouse (Wu et al., 2017). Wt1 deletion in neonatal Sertoli cells results in the accumulation of undifferentiated spermatogonia due to defects in spermatogonial differentiation and meiosis (Zheng et al., 2014). SIN3A-deficient Sertoli cells cannot support long-term maintenance of the spermatogonial population and normal development of spermatids (Payne et al., 2010). Functional roles of other transcripts in influencing the initial formation and maintenance of the SSC niche await further investigations.
In this study, we demonstrate that WTAP function in Sertoli cells serves specific roles in dictating SSC fate decisions. WTAP was first identified as a WT1-binding partner and WT1 mainly functions as a key regulator of somatic lineage specification in fetal gonad (Chen et al., 2017); in sharp contrast, Wtap deletion results in the exhaustion the SSC pool without affecting meiosis or spermiogenesis. We did not observe defects in the gonocyte to spermatogonial transition in the neonatal testis of Wtap conditional knockout animals, suggesting the initial establishment of the SSC niche is not affected. It is commonly observed that Sertoli cell dysfunctions affect development of meiotic and post-meiosis germ cells (O'Shaughnessy, 2014), and, to our knowledge, transcriptional or translational factor in Sertoli cells that only controls the fate of SSC has not been reported; therefore, our findings revealed a novel role of WTAP in determining the long-term maintenance of the SSC niche. Phenotypic and mechanistic analyses support the idea that WTAP machinery provides a microenvironment that ensures the balance between SSC self-renewal and differentiation.
WTAP regulates transcription and translation of niche factors by depositing the m6A marks directly on transcripts encoding the niche factors or indirectly on transcription regulators. Although WTAP deficiency dramatically induces transcriptional and translational changes of a large number of genes in Sertoli cells, we found that most significantly affected transcripts are involved in SSC maintenance, spermatogonial differentiation, retinol metabolism, and cell cycle. Gdnf, the essential factor required for SSC maintenance, is not marked by m6A modification; however, its expression level was downregulated upon Wtap deletion. Interestingly, several of the key transcription regulators that have been reported to be important for Gdnf transcription contained m6A sites and were dysregulated by Wtap knockout. Biogenesis of retinoic acid (RA) is strictly controlled in Sertoli cells (Teletin et al., 2019) and RA signaling interacts with the niche factors to influence the fate of undifferentiated spermatogonia (Yang et al., 2013); therefore, it is not surprising that transcripts involved in RA metabolism and functions are modified by m6A and dysregulated by Wtap deletion.
A major function of WTAP is to control mRNA splicing in Sertoli cells. m6A is established using S-adenosyl methionine as substrate, and catalyzed by m6A methyltransferases “writers” and recognized by m6A binding proteins “readers” to induce subsequent reactions (Balacco and Soller, 2019). In germ cells, deletion of m6A reader Ythdc1 leads to the loss of germ cells including mitotic spermatogonia (Kasowitz et al., 2018) and deletion of Ythdc2 causes defects in meiosis (Hsu et al., 2017; Wojtas et al., 2017). Deletion of m6A writer Mettl3 or Mettl14 inhibits SSC proliferation/differentiation and combined ablation of them in advanced germ cells disrupts spermiogenesis (Lin et al., 2017). Notably, these m6A methylases all affected mRNA splicing. WTAP is an evolutionarily conserved regulator of AS events that functions in post-transcriptional regulation, including mRNA splicing, stabilization, polyadenylation, and export (Horiuchi et al., 2013). WTAP interacts with METTL3 and METTL14 through the anchoring effect of ZC3H13 to control AS events and m6A formation (Wen et al., 2018). In the present study, we illustrate that loss of WTAP function in Sertoli cells produced a pronounced variation in splicing events and a great amount of A3SS, SE, MXE, and RI occurred in m6A-enriched genes. The alteration of splicing events in spermatogenesis-regulating genes caused by m6A deprivation could be one of the core reasons for defective spermatogenesis.
Collectively, our results demonstrate that WTAP-mediated m6A modification controls mRNA transcription and translation to orchestrate the gene expressions in Sertoli cells that are essential for SSC maintenance and spermatogonial differentiation. These findings provide new insights into molecular regulation of the SSC niche, and results of the present study may be helpful in deciphering interactions between stem cells and their cognate niches in other mammalian systems.
Experimental Procedures
Mice
The Wtap knockout first embryonic stem cells (C57BL/6N-Wtap<tm1a(EUCOMM)Hmgu>/H, hereafter Wtaptm1a) were obtained from MRC Harwell Institute (Oxford, UK). In the knockout first allele, a promoter-driven cassette (including LacZ and neo genes) flanked by FRT sites are inserted between exon 3 and exon 4, whereas the exon 4 and exon 5 of Wtap are flanked by LoxP sites. We converted the knockout first allele to a conditional allele by crossing Wtaptm1a mice with Flp deleter mice. The resulting floxed Wtap were crossed with Amh-Cre mice obtained from Jackson Laboratory (Maine, US) to allow specific knockout of Wtap in Sertoli cells. Obtained mice were genotyped with the tail DNA. All primers for genotyping are listed in the Table S6. The Sox9Gfp mice used for isolation of highly purified Sertoli cells were obtained from Mutant Mouse Resource & Research Centers (MMRRC_011019-UCD). The C57BL/6N mice used for isolation of a large number of Sertoli cells and 129S2/SvPasCrl mice used for transplantation experiments were purchased from Vital River Laboratory Animal Center (Beijing, China) and housed in the animal facilities of Northwest Institute of Plateau Biology, Chinese Academy of Sciences. All animal experiments were approved by the Animal Ethic and Welfare Committee at Northwest Institute of Plateau Biology, Chinese Academy of Sciences.
m6A-seq
m6A-seq was performed as previously described (Lin et al., 2017). Briefly, mRNA isolated from Sertoli cells was enriched using Seq-Star Poly(A) mRNA Isolation Kit. mRNA was fragmented to ~100 nt and mixed with affinity-purified anti-m6A antibody for immunoprecipitation (IP). The eluted RNA was resuspended and used for library preparation with KAPA Stranded RNA-seq Kit. Sequencing was carried out on Illumina HiSeq 4000 using an 8 pM template per sample for cluster generation with HiSeq 3000/4000 SBS Kit by KangChen Biotechnology Co., Ltd. (Shanghai, China).
After controlling quality by FastQC and removing adapters and low-quality reads by Trimmomatic (Bolger et al., 2014), sequencing reads were aligned to the reference genome (mm10) using HISAT2 (Kim et al., 2015). The longest isoform was used if the gene had multiple isoforms. The m6A peaks were detected by exomePeak (Meng et al., 2014) and windows with read counts <1/20 of the top window in both m6A IP and input samples were excluded. The differential windows were identified between m6A IP and input samples using edgeR (Robinson et al., 2010), and the enrichment score of each window was calculated as previously described (Lin et al., 2017). The window was called positive if enrichment score was >1 and p < 0.01.
RNA-seq and RNC-seq in Parallel
The isolated Sertoli cells were resolved in prechilled TRIzol for RNA-seq or fixed buffer (0.3 mg/mL antibiotic inhibitor and 50 mM MgCl2 in PBS) for RNC-seq and stored at −80°C immediately after liquid nitrogen freeze. RNC mRNA was enriched by sucrose density centrifugation to obtain full-length mRNA on ribosome as described (Wang et al., 2013). Then, total RNA and RNC mRNA were isolated respectively, equal amounts of total RNA or RNC mRNA from each sample were prepared for subsequent library construction, and RNA-seq or RNC-seq following the same procedure, including fragmentation, reverse transcription, synthesis, purification, adaptor ligation, and PCR amplification. Libraries successfully constructed were sequenced using Illumina HiSeq2500.
Clean reads without adapters and low-quality bases were further processed to remove rRNA reads by Bowtie2 (Langmead and Salzberg, 2012). TopHat2 was used for mapping reads to reference genome (mm10) (Kim et al., 2013), and Cufflinks was used together for the reconstruction of transcripts (Trapnell et al., 2012). Gene abundance was quantified using RSEM (RNA-seq by expectation maximization) (Li and Dewey, 2011), and the gene expression level was normalized by fragments per kilobase of exon model per million reads mapped (FPKM). Genes with no less than 0.5 FPKM in at least one sample were used for subsequent analysis. For identifying DEGs at transcription level, edgeR was used, and genes with |log2fold change| ≥ 1 and p < 0.01 between control and Wtap-sKO animals from RNA-seq were identified as significant DEGs. For digging DTGs between control and Wtap-sKO animals at translation level, TE was calculated based on normalized read count of RNC mRNA/mRNA and genes with |log2fold change| ≥ 1 and p < 0.01 between control and Wtap-sKO animals were identified as significant DTGs.
Data and Code Availability
The m6A-seq, RNA-seq, and RNC-seq data are available in the NCBI BioProject under accession numbers PRJNA661168 and PRJNA661226.
Author Contributions
Q.Y. devised and supervised this project. G.J., Z.L., R.Y., G.W., X.Z., and C.L. performed the experiments and bioinformatics analysis. Q.Y., G.J., and M.T. wrote the manuscript with contributions from all authors.
Acknowledgments
The work was supported by the National Natural Science Foundation of China (grants nos. 31571539, 31771656, and 31930034) and Natural Science Foundation of Qinghai Province (2020-ZJ-902). Q.Y. was supported by Chinese Academy of Sciences (CAS) “100 Talents” and Qinghai “1000 Talents” programs. G.J. was supported by CPSF-CAS Joint foundation for Postdoctoral Fellows and “CAS Light of West” programs.
Published: October 13, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.09.001.
Supplemental Information
References
- Adhikari S., Xiao W., Zhao Y.L., Yang Y.G. m6A: signaling for mRNA splicing. RNA Biol. 2016;13:756–759. doi: 10.1080/15476286.2016.1201628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balacco D.L., Soller M. The m6A writer: rise of a machine for growing tasks. Biochemistry. 2019;58:363–378. doi: 10.1021/acs.biochem.8b01166. [DOI] [PubMed] [Google Scholar]
- Basciani S., De Luca G., Dolci S., Brama M., Arizzi M., Mariani S., Rosano G., Spera G., Gnessi L. Platelet-derived growth factor receptor β-subtype regulates proliferation and migration of gonocytes. Endocrinology. 2008;149:6226–6235. doi: 10.1210/en.2008-0349. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Zhang L.J., Cui X.H., Lin X.W., Li Y.Q., Wang Y.Q., Wang Y.B., Qin Y., Chen D.H., Han C.S. Wt1 directs the lineage specification of Sertoli and granulosa cells by repressing Sf1 expression. Development. 2017;144:44–53. doi: 10.1242/dev.144105. [DOI] [PubMed] [Google Scholar]
- Cheng Y.M., Luo H.Z., Izzo F., Pickering B.F., Nguyen D., Myers R., Schurer A., Gourkanti S., Bruning J.C., Vu L.P. m6A RNA methylation maintains hematopoietic stem cell identity and symmetric commitment. Cell Rep. 2019;28:1703–1716. doi: 10.1016/j.celrep.2019.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rooij D.G. The nature and dynamics of spermatogonial stem cells. Development. 2017;144:3022–3030. doi: 10.1242/dev.146571. [DOI] [PubMed] [Google Scholar]
- De Rooij D.G. Regulation of the proliferation of spermatogonial stem cells. J. Cell Sci. 1988;10:181–194. doi: 10.1242/jcs.1988.supplement_10.14. [DOI] [PubMed] [Google Scholar]
- Fujihara Y., Satouh Y., Inoue N., Isotani A., Ikawa M., Okabe M. SPACA1-deficient male mice are infertile with abnormally shaped sperm heads reminiscent of globozoospermia. Development. 2012;139:3583–3589. doi: 10.1242/dev.081778. [DOI] [PubMed] [Google Scholar]
- Garcia T.X., Farmaha J.K., Kow S., Hofmann M.C. RBPJ in mouse Sertoli cells is required for proper regulation of the testis stem cell niche. Development. 2014;141:4468–4478. doi: 10.1242/dev.113969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A.A., Kol N., Salmon-Divon M., Hershkovitz V., Peer E., Mor N., Manor Y.S. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- Gilbert W.V., Bell T.A., Schaening C. Messenger RNA modifications: form, distribution, and function. Science. 2016;352:1408–1412. doi: 10.1126/science.aad8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green C.D., Ma Q.Y., Manske G.L., Shami A.N., Zheng X.N., Marini S., Moritz L., Sultan C., Gurczynski S.J., Moore B.B. A comprehensive roadmap of murine spermatogenesis defined by single-cell RNA-seq. Dev. Cell. 2018;46:651–667. doi: 10.1016/j.devcel.2018.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold M.D. The central role of Sertoli cells in spermatogenesis. Semin. Cell Dev. Biol. 1998;9:411–416. doi: 10.1006/scdb.1998.0203. [DOI] [PubMed] [Google Scholar]
- Griswold M.D. 50 years of spermatogenesis: Sertoli cells and their interactions with germ cells. Biol. Reprod. 2018;99:87–100. doi: 10.1093/biolre/ioy027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K., Umetani M., Minami T., Okayama H., Takada S., Yamamoto M., Aburatani H., Reid P.C., Housman D.E., Hamakubo T. Wilms' tumor 1-associating protein regulates G2/M transition through stabilization of cyclin A2 mRNA. Proc. Natl. Acad. Sci. U S A. 2006;103:17278–17283. doi: 10.1073/pnas.0608357103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K., Kawamura T., Iwanari H., Ohashi R., Naito M., Kodama T., Hamakubo T. Identification of Wilms' tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J. Biol. Chem. 2013;288:33292–33302. doi: 10.1074/jbc.M113.500397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P.J., Zhu Y.F., Ma H.H., Guo Y.H., Shi X.D., Liu Y.Y., Qi M.J., Lu Z.K., Shi H.L., Wang J.Y. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115–1127. doi: 10.1038/cr.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K., Kanatsu-Shinohara M., Toyokuni S., Shinohara T. FGF2 mediates mouse spermatogonial stem cell self-renewal via upregulation of Etv5 and Bcl6b through MAP2K1 activation. Development. 2012;139:1734–1743. doi: 10.1242/dev.076539. [DOI] [PubMed] [Google Scholar]
- Jones D.L., Wagers A.J. No place like home: anatomy and function of the stem cell niche. Nat. Rev. Mol. Cell Biol. 2008;9:11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- Kasowitz S.D., Ma J., Anderson S.J., Leu N.A., Xu Y., Gregory B.D., Schultz R.M., Wang P.J. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. 2018;14:e1007412. doi: 10.1371/journal.pgen.1007412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M., Ohsugi M., Sasako T., Awazawa M., Umehara T., Iwane A., Kobayashi N., Okazaki Y., Kubota N., Suzuki R. The RNA methyltransferase complex of WTAP, METTL3, and METTL14 regulates mitotic clonal expansion in adipogenesis. Mol. Cell. Biol. 2018;38:e00116-18. doi: 10.1128/MCB.00116-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota H., Avarbock M.R., Brinster R.L. Culture conditions and single growth factors affect fate determination of mouse spermatogonial stem cells. Biol. Reprod. 2004;71:722–731. doi: 10.1095/biolreprod.104.029207. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z., Hsu P.J., Xing X.D., Fang J.H., Lu Z.K., Zou Q., Zhang K.J., Zhang X., Zhou Y.C., Zhang T. Mettl3-/Mettl14-mediated mRNA N6-methyladenosine modulates murine spermatogenesis. Cell Res. 2017;27:1216–1230. doi: 10.1038/cr.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.Z., Yue Y.N., Han D.L., Wang X., Fu Y., Zhang L., Jia G.F., Yu M., Lu Z.K., Deng X. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu N.X., Sargent K.M., Clopton D.T., Pohlmeier W.E., Brauer V.M., McFee R.M., Weber J.S., Ferrara N., Silversides D.W., Cupp A.S. Loss of vascular endothelial growth factor A (VEGFA) isoforms in the testes of male mice causes subfertility, reduces sperm numbers, and alters expression of genes that regulate undifferentiated spermatogonia. Endocrinology. 2013;154:4790–4802. doi: 10.1210/en.2013-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukjanenko L., Jung M.J., Hegde N., Perruisseau-Carrier C., Migliavacca E., Rozo M., Karaz S., Jacot G., Schmidt M., Li L.J. Loss of fibronectin from the aged stem cell niche affects the regenerative capacity of skeletal muscle in mice. Nat. Med. 2016;22:897–905. doi: 10.1038/nm.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X., Lindahl M., Hyvonen M.E., Parvinen M., De Rooij D.G., Hess M.W., Raatikainen-Ahokas A., Sainio K., Rauvala H., Lakso M. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 2000;287:1489–1493. doi: 10.1126/science.287.5457.1489. [DOI] [PubMed] [Google Scholar]
- Meng J., Lu Z.L., Liu H., Zhang L., Zhang S.W., Chen Y.D., Rao M.K., Huang Y.F. A protocol for RNA methylation differential analysis with MeRIP-seq data and exomePeak R/Bioconductor package. Methods. 2014;69:274–281. doi: 10.1016/j.ymeth.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa K.R., Rompolas P., Greco V. The dynamic duo: niche/stem cell interdependency. Stem Cell Reports. 2015;4:961–966. doi: 10.1016/j.stemcr.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shaughnessy P.J. Hormonal control of germ cell development and spermatogenesis. Semin. Cell Dev. Biol. 2014;29:55–65. doi: 10.1016/j.semcdb.2014.02.010. [DOI] [PubMed] [Google Scholar]
- Oatley J.M., Brinster R.L. Regulation of spermatogonial stem cell self-renewal in mammals. Annu. Rev. Cell Dev. Biol. 2008;24:263–286. doi: 10.1146/annurev.cellbio.24.110707.175355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oatley J.M., Brinster R.L. The germline stem cell niche unit in mammalian testes. Physiol. Rev. 2012;92:577–595. doi: 10.1152/physrev.00025.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oatley J.M., Oatley M.J., Avarbock M.R., Tobias J.W., Brinster R.L. Colony stimulating factor 1 is an extrinsic stimulator of mouse spermatogonial stem cell self-renewal. Development. 2009;136:1191–1199. doi: 10.1242/dev.032243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta H., Yomogida K., Dohmae K., Nishimune Y. Regulation of proliferation and differentiation in spermatogonial stem cells: the role of c-kit and its ligand SCF. Development. 2000;127:2125–2131. doi: 10.1242/dev.127.10.2125. [DOI] [PubMed] [Google Scholar]
- Payne C.J., Gallagher S.J., Foreman O., Dannenberg J.H., Depinho R.A., Braun R.E. Sin3a is required by Sertoli cells to establish a niche for undifferentiated spermatogonia, germ cell tumors, and spermatid elongation. Stem Cells. 2010;28:1424–1434. doi: 10.1002/stem.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping X.L., Sun B.F., Wang L., Xiao W., Yang X., Wang W.J., Adhikari S., Shi Y., Lv Y., Chen Y.S. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teletin M., Vernet N., Yu J.S., Klopfenstein M., Jones J.W., Feret B., Kane M.A., Ghyselinck N.B., Mark M. Two functionally redundant sources of retinoic acid secure spermatogonia differentiation in the seminiferous epithelium. Development. 2019;146:dev170225. doi: 10.1242/dev.170225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Cui Y.Z., Jin J.J., Guo J.H., Wang G.B., Yin X.F., He Q.Y., Zhang G. Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic Acids Res. 2013;41:4743–4754. doi: 10.1093/nar/gkt178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Feng J., Xue Y., Guan Z.Y., Zhang D.L., Liu Z., Gong Z., Wang Q., Huang J.B., Tang C. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534:575–578. doi: 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- Wang Y., Li Y., Yue M.H., Wang J., Kumar S., Wechsler-Reya R.J., Zhang Z.L., Ogawa Y., Kellis M., Duester G. N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci. 2018;21:195–206. doi: 10.1038/s41593-017-0057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welborn J.P., Davis M.G., Ebers S.D., Stodden G.R., Hayashi K., Cheatwood J.L., Rao M.K., MacLean J.A., 2nd Rhox8 ablation in the Sertoli cells using a tissue-specific RNAi approach results in impaired male fertility in mice. Biol. Reprod. 2015;93:8. doi: 10.1095/biolreprod.114.124834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J., Lv R.T., Ma H.H., Shen H.J., He C.X., Wang J.H., Jiao F.F., Liu H., Yang P.Y., Tan L. Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol. Cell. 2018;69:1028–1038. doi: 10.1016/j.molcel.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtas M.N., Pandey R.R., Mendel M., Homolka D., Sachidanandam R., Pillai R.S. Regulation of m6A transcripts by the 3'→5' RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol. Cell. 2017;68:374–387. doi: 10.1016/j.molcel.2017.09.021. [DOI] [PubMed] [Google Scholar]
- Wu R.C., Jiang M., Beaudet A.L., Wu M.Y. ARID4A and ARID4B regulate male fertility, a functional link to the AR and RB pathways. Proc. Natl. Acad. Sci. U S A. 2013;110:4616–4621. doi: 10.1073/pnas.1218318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.C., Zeng Y., Chen Y.F., Lanz R.B., Wu M.Y. Temporal-spatial establishment of initial niche for the primary spermatogonial stem cell formation is determined by an ARID4B regulatory network. Stem Cells. 2017;35:1554–1565. doi: 10.1002/stem.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q.E., Kim D., Kaucher A., Oatley M.J., Oatley J.M. CXCL12-CXCR4 signaling is required for the maintenance of mouse spermatogonial stem cells. J. Cell Sci. 2013;126:1009–1020. doi: 10.1242/jcs.119826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.G., Feng Y.M., Feng X., Liao S.Y., Wang X.X., Gan H.Y., Wang L.X., Lin X.W., Han C.S. BMP4 cooperates with retinoic acid to induce the expression of differentiation markers in cultured mouse spermatogonia. Stem Cells Int. 2016;2016:9536192. doi: 10.1155/2016/9536192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K.J., Ringeling F.R., Vissers C., Jacob F., Pokrass M., Jimenez-Cyrus D., Su Y.J., Kim N.S., Zhu Y.H., Zheng L. Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell. 2017;171:877–889. doi: 10.1016/j.cell.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen B.T.K., Bush K.M., Barrilleaux B.L., Cotterman R., Knoepfler P.S. Histone H3.3 regulates dynamic chromatin states during spermatogenesis. Development. 2014;141:3483–3494. doi: 10.1242/dev.106450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B.S., Roundtree I.A., He C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q.S., Wang X.N., Wen Q., Zhang Y., Chen S.R., Zhang J., Li X.X., Sha R.N., Hu Z.Y., Gao F. Wt1 deficiency causes undifferentiated spermatogonia accumulation and meiotic progression disruption in neonatal mice. Reproduction. 2014;147:45–52. doi: 10.1530/REP-13-0299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The m6A-seq, RNA-seq, and RNC-seq data are available in the NCBI BioProject under accession numbers PRJNA661168 and PRJNA661226.