

Supplemental Digital Content is Available in the Text.
Patients diagnosed with Christianson syndrome have intellectual disability, epilepsy, ataxia, and mutism, as well as hyposensitivity to pain. In this study, we use a mouse model of Christianson syndrome to demonstrate that this pain hyposensitivity is due in part to a decrease in excitability of nociceptors.
Keywords: Pain, Nociception, Neurodevelopmental disorder, Sodium proton exchanger, Trpv1, Nociceptor, Pain tolerance, Christianson syndrome, NHE6
Abstract
Children diagnosed with Christianson syndrome (CS), a rare X-linked neurodevelopmental disorder characterized by intellectual disability, epilepsy, ataxia, and mutism, also suffer from hyposensitivity to pain. This places them at risk of sustaining serious injuries that often go unattended. Christianson syndrome is caused by mutations in the alkali cation/proton exchanger SLC9A6/NHE6 that regulates recycling endosomal pH homeostasis and trafficking. Yet, it remains unclear how defects in this transporter lead to altered somatosensory functions. In this study, we validated a Nhe6 knockout (KO) mouse as a model of CS and used it to identify the cellular mechanisms underlying the elevated pain tolerance observed in CS patients. Within the central nervous system, NHE6 immunolabelling is detected in a small percentage of cortical neurons involved in pain processing, including those within the primary somatosensory and the anterior cingulate cortices as well as the periaqueductal gray. Interestingly, it is expressed in a larger percentage of nociceptors. Behaviourally, Nhe6 KO mice have decreased nocifensive responses to acute noxious thermal, mechanical, and chemical (ie, capsaicin) stimuli. The reduced capsaicin sensitivity in the KO mice correlates with a decreased expression of the transient receptor potential channel TRPV1 at the plasma membrane and capsaicin-induced Ca2+ influx in primary cultures of nociceptors. These data indicate that NHE6 is a significant determinant of nociceptor function and pain behaviours, vital sensory processes that are impaired in CS.
1. Introduction
Nociception is a critical sensory signal that alerts the organism to potential or actual injury. The cellular and molecular mechanisms of nociceptor activation are intricate and strictly regulated.1 Consequently, genetic conditions causing heightened pain sensitivity are detrimental to an individual's quality of life. For example, patients with erythromelalgia or paroxysmal extreme pain disorder suffer recurrent episodes of burning pain and skin redness due to gain-of-function mutations in the voltage-gated sodium channel Nav1.7.11,33,35 Conversely, patients with congenital insensitivity to pain experience frequent injuries, self-mutilation, and shortened life expectancy.23,29,39 Hyposensitivity to pain is also observed in children diagnosed with Christianson syndrome (CS),7,25 a rare but increasingly diagnosed neurodevelopmental and regressive form of X-linked intellectual disability.7,13,14,25 Children with CS also exhibit a range of other neurological, somatic, and behavioural abnormalities, such as autistic behaviours, early-onset seizures, truncal ataxia, hyperkinesis, hypotonia, strabismus, and mutism.21,36,42 This type of pain tolerance predisposes CS patients to sustaining serious physical injuries that often go unnoticed because of their inability to sense and verbally communicate such events.
Christianson syndrome is caused by various inherited and de novo mutations (>60) in the SLC9A6 gene (solute carrier family 9, member A6)25 (also see databases ClinVar [https://www.ncbi.nlm.nih.gov/clinvar/] and DECIPHER [https://decipher.sanger.ac.uk/]), many of which are predicted to cause complete loss-of-function. This ion transporter is widely expressed but is especially abundant in the central nervous system (CNS), which likely accounts for the prominent neurological phenotype of CS (https://www.proteinatlas.org/ENSG00000198689-SLC9A6/tissue).10 Within cells, NHE6 is present at the plasma membrane, but resides predominantly in a subpopulation of early/sorting and recycling endosomes where it functions as a H+ efflux pathway to counterbalance the acidifying effects of the vacuolar H+-ATPase, achieving a steady-state intraluminal pH of ∼6.4 ± 0.2.15,37,40,41 This relatively mild acidification facilitates dissociation of many ligand/receptor complexes and allows the freed receptors to recycle back to the plasma membrane for reuse. This compartment is distinct from more acidic endocytic vesicles (ie, late endosomes/multivesicular bodies, pH 5.0-5.5), which contain sorted cargoes, such as dissociated ligands or damaged receptors, that are destined for degradation in lysosomes (pH 4.6-5.0).20 In both non‐neuronal and neuronal cells, loss of NHE6 function leads to excessive acidification of recycling endosomes, and impairs both internalization and delivery of cargo to the cell surface.15,24,40 This disrupted trafficking also seems to compromise late endosomal–lysosomal function comparable to lysosomal storage diseases,32,34 as well as reducing dendritic arborization and neurodegeneration of cerebellar Purkinje,32,34 cortical, and hippocampal pyramidal neurons.15,24
Whether these impairments also take place in the somatosensory system, and how they contribute to the elevated nociceptive thresholds of CS patients are yet to be determined. These questions can now be addressed due to the development of a Nhe6 knockout (KO) mouse that manifests CS-like symptoms.24,32,34 In the current study, we examine the expression of NHE6 in the CNS, with a focus on pain-processing centers, and explore the somatosensory consequences of NHE6 loss-of-function in a mouse model of CS.
2. Methods
2.1. Animals
All animal use protocols were approved by the Comparative Medicine and Animal Resources Centre of McGill University. Mice were housed in 12-hour light/dark cycles with free access to standard rodent chow and water. Immunohistochemistry experiments examining the expression of NHE6 in wild-type (WT) tissue were performed on C57BL/6 mice. The NHE6 KO mice were purchased from Jackson Laboratories (B6.129P2-Slc9a6tm1Dgen/J, Stock No. 005843). The Slc9a6 gene was inactivated in these mice by inserting a LacZ-Neo cassette into exon 6 of the gene. Mice were genotyped by PCR using the forward primers 5′-GGG TGG GAT TAG ATA AAT GCC TGC TCT-3′ and 5′-AAC AGC TGT GGA GGG ATA TGT GCT-3′ for mutant and WT, respectively, and the reverse primer 5′-AGC TGG CTT TGC GCA TGG AGC ATT C-3′. The PCR product shows a band at 432 bp for mutants and 224 bp for WT with heterozygotes showing both bands. Breedings were made with a WT male C57BL/6 mouse and 2 NHE6−/+ females to obtain NHE6-/Y and NHE6+/Y male mice littermates for behaviour and immunohistochemistry experiments. Males were used because CS is an X-linked disorder and the disease predominantly affects young males, whereas females display a mild and mosaic phenotype.25 Mice were backcrossed to C57BL/6 line before experimentation. To visualize neuronal activity in nociceptors, we crossed mice expressing the Cre-dependent genetically encoded calcium sensor, GCaMP6f, to mice expressing the Cre recombinase under the control of a nociceptor-specific driver: Trpv1-Cre. The resulting TRPV1-Cre::GCaMP6f mice express the sensor in all nociceptors due to its broad embryonic expression.4 NHE6−/+ females were crossed with TRPV1-Cre::GCaMP6f mice to obtain male mice NHE6−/Y (Nhe6−/Y:Trpv1-Cre GCaMP6f) and NHE6+/Y (Nhe6+/Y:Trpv1-Cre GCaMP6f) expressing GCaMP6f in nociceptors.
2.2. Immunohistochemistry and imaging
Mice were deeply anesthetized with a mixture of ketamine and xylazine before being transcardially perfused with 10 mL of phosphate-buffered saline (PBS) followed by 25 to 30 mL of 4% paraformaldehyde. Their brains, spinal cords, and dorsal root ganglia (DRG) were dissected and postfixed in 4% paraformaldehyde for 1 hour, followed by cryoprotection in PBS containing 30% sucrose for 72 hours. The tissues were then embedded in optimal cutting temperature compound and cut into 25-µm (brains and spinal cords) and 14-μm (DRG) sections with a cryostat (Microm HM525 NX Cryostat; Thermo Scientific, Rockford, IL). Brain and spinal cord sections were kept floating in PBS at 4°C, whereas DRG sections were mounted directly on Fisherbrand Superfrost Plus microscope slides (Thermo Fisher Scientific, Rockford, IL) for immunofluorescence staining. Tissue sections were blocked in 10% normal goat serum with 0.33% triton X-100 (NGST) in PBS for 1 hour at room temperature, followed by incubation with primary antibodies diluted in 2.5% NGST for 48 hours at 4°C. The following primary antibodies were used: rabbit anti-NHE6 (1:250),8 mouse anti-NF200 (1:500; Sigma-Aldrich, Oakville, Canada), sheep anti-tyrosine hydroxylase (TH) antibody (1:400; Millipore, Oakville, Canada), mouse anti-calcitonin gene-related peptide (CGRP) (1:500; Sigma-Aldrich), biotinylated-IB4 (1:1000; Sigma-Aldrich), and biotinylated mouse anti-NeuN (1:500; Millipore). The primary antibody reaction was stopped by 3 × 10-minute rinses in 1% NGST at room temperature. The tissue sections were then incubated with the secondary antibodies (1:500 in 1% NGST for 1 hour at room temperature). Alexa Fluor-conjugated antibodies raised in goat against rabbit, mouse, or rat were used as secondary antibodies. Streptavidin-conjugated to Alexa Fluor 647 was used to detect biotinylated primary antibodies. The tissue was then rinsed 3 × 10 minutes with PBS. Brain and spinal cord sections were then mounted on microscope slides and cover-slipped using Aqua-Poly/Mount mounting medium (Polysciences, Inc, Warrington, PA). Immunohistochemistry experiments on DRG tissue involving costaining for TH were performed sequentially. Stains were imaged using an inverted fluorescent Zeiss (Oberkochen, Germany) LSM 710 Confocal Microscope. Images were analyzed using Image J software.
For imaging sessions, all images were taken using identical acquisition parameters and were quantified following frameworks used by Li et al.18 In each section analyzed, cells were quantified by manually tracing the cell membrane (after NeuN staining) and counting the number of cells within a region of the optic field in each slice using a computerized image analysis system (ImageJ, National Institutes of Health). For each stain, colocalization between NHE6 and the following markers was counted: NeuN, CGRP, TH, and IB4. Immunolabeled neurons were quantified as a percentage of the total number of neurons.
2.3. Nociceptive behavior experiments
Behaviour tests were performed on KO mice using their WT littermates as controls. Mice were tested 3 times within 1 week every 4 weeks starting at 8 weeks of age until 24 weeks. Tester was blind to genotype of mice during the tests. Mice were acclimatized to each behavioural apparatus for 3 sessions of 30 minutes before testing.
2.3.1. Thermal sensitivity
To assess nociceptive heat sensitivity, we used a conventional hot-plate test or a radiant heat paw withdrawal test (Hargreaves method). The temperature of the hot plate was adjusted to 42 or 52 ± 0.1°C. The latency to the first hind paw licking or withdrawal was taken as an index of nociceptive threshold. The cutoff time was set at 120 seconds at 42°C and 20 seconds at 52°C, to avoid damage to the paw. For the Hargreaves method, the mice were placed in clear Plexiglas boxes (22 × 19 × 14 cm) on a glass surface and allowed to habituate for 15 minutes before testing. A thermal stimulus was pointed at the plantar surface of each hind paw through the glass using a Hargreaves apparatus (Stoelting, Wood Dale, IL) until the animal lifted its paw away from the heat source. The latency to their paw withdrawal was automatically recorded with the apparatus to the nearest 0.1 seconds, and a cutoff latency of 20 seconds was used to avoid tissue damage. Three to 5 trials were conducted with at least 5 minutes between each trial. Mean values per paw were calculated and used for statistical analysis.
2.3.2. Mechanical sensitivity
Two readouts of mechanical sensitivity were assessed with the von Frey test: the mechanical withdrawal threshold and the mechanical behavioral response. For both types, mice were placed in Plexiglas restrainers on an elevated wire mesh platform and von Frey filaments were used to apply varying weights (starting with 40 mg) to the plantar hind paw in an ascending order. Each filament was applied 5 times against the hind paw in a static manner with an interval of at least 30 seconds. The mechanical withdrawal threshold was measured as the first filament that elicited 3 paw withdrawals (regardless of whether they were reflex-like or nocifensive-like responses) out of 5 applications. The mechanical behavior response was assessed according to the following scheme: no response, a brief stretching of the digits without a withdrawal or a reflex-like paw withdrawal, or nocifensive reaction (intense grooming, paw licking, or flicking combined with escape behaviors).
2.3.3. Capsaicin test and edema
The intraplantar injection of capsaicin produces spontaneous nocifensive behavioral responses. Briefly, capsaicin (5 μL at 0.5% in saline:ethanol:tween20 at ratios of 80:10:10) was injected subcutaneously in the left heel of the mouse. The duration of nociceptive responses (licking and paw guarding) after injection were measured for 5 minutes. Edema was quantified by measuring the width of the dorsoplantar aspect of the hind paw before and after the injection of capsaicin, and represented as a percentage of the preinjection width. The carprofen treatment consisted of an intraperitoneal injection of a carprofen solution (2 mg/kg) 30 minutes before initiation of the nociceptive behavior experiments.
2.3.4. Dorsal root ganglion cell culture
Nhe6−/Y:Trpv1-Cre GCaMP6f and Nhe6+/Y:Trpv1-Cre GCaMP6f mice were used. The culture of dorsal root ganglion (DRG) neurons was performed as previously described.22 Briefly, DRG were dissected from 3-month-old male mice, and placed in sterile HBSS at 4°C. Enzymatic dissociation was performed with 0.2% type IV collagenase and 0.2% dispase for 105 minutes, followed by a 5-minute treatment with 0.25 mg/mL trypsin. DRG were dissociated mechanically by trituration and plated at low density on glass-bottom dishes with DMEM/F12 supplemented with 1% fetal bovine serum and 1% penicillin/streptomycin maintained at 37°C with 5% CO2, with media changes every other day. Neurons were tested 3 to 5 days after plating.
To quantify the expression of the TRPV1 channel at the membrane of DRG neurons, dissociated neurons from NHE6+/Y:Trpv1-Cre GCaMP6f (WT) and NHE6-/Y:Trpv1-Cre GCaMP6f (KO) mice were fixed using the flowing protocol: after a first rinse in PBS (10 minutes), ice-cold pure acetone was added for 10 minutes. Cells were rinsed 3 times with ice-cold PBS, then ice-cold methanol was added for 10 minutes. Cells were rinsed again 3 times with PBS and then incubated with 10% normal goat serum with 0.33% triton x-100 (NGST) in PBS for 1 hour at room temperature, followed by incubation with a rabbit antibody against TRPV1 (1:1000). The primary antibody reaction was stopped by 3 × 10-minute rinses in 1% NGST at room temperature. The cells were then incubated with a secondary antibody, an Alexa Fluor-conjugated antibody raised in goat against rabbit (Alexa Fluor 647 1:500 in 1% NGST for 1 hour at room temperature) and a wheat germ agglutinin (WGA) conjugated with an Alexa Fluor 555 (WGA-555 1:1000; Invitrogen). Wheat germ agglutinin is a plant lectin that is used to stain the cell membrane.6,31 Images were collected as described in paragraph 2.2. The WGA-555 signal from the membrane is used to restrict the quantification of TRPV1 immunoreactivity (TRPV1-iR) from DRG cell plasmid membrane. The TRPV1-IR/WGA-555 ratios from WT and KO DRG cells were compared.
2.3.5. Calcium imaging
For calcium imaging experiments, neurons were continuously perfused with a bath solution composed of 150 mM NaCl, 3 mM KCl, 10 mM Glucose, 10 mM HEPES, 2 mM CaCl2, and 1 mM MgCl2 in distilled water, buffered to a pH of 7.3 with 1 N NaOH. Compounds tested were prepared in bath solution and applied locally to the cells using a single-tip multichannel gravity-fed system, which allowed for rapid switching between solutions. Cells were excited at a wavelength of 488 nm and fluorescence measurements were performed with an inverted Olympus IX71 microscope. Metafluor software (Molecular Devices Version 7.7) was used for data acquisition and analysis. In every recording event, KCl was applied to neurons at the end of the recording session and only those that responded to KCl were analyzed for their responses to 5-μM capsaicin.27
2.3.6. Conditioned place aversion
Conditioned place aversion (CPA) experiments were performed as described in Ref. 26. On the first testing day, mice were enclosed in the central chamber for 1 minute and then allowed to roam freely in the entire testing apparatus for 30 minutes to explore the 2 testing chambers. On the second day, this process was repeated and the duration of time they spent in each chamber was recorded. On the third and fourth testing days, mice were restricted to one chamber for 2 hours after receiving an injection of capsaicin or vehicle; this was done to pair the painful stimulus with one testing chamber. On the fifth testing day, mice were enclosed in the central chamber for 1 minute and then allowed to roam the 2 chambers freely for 30 minutes, with the time they spent in each chamber recorded as on the second day. The amount of time that mice spent in each chamber was compared between the second and fifth day of testing to determine whether there was a difference in their aversion of the paired chamber.
2.4. Real time quantitative polymerase chain reaction
DRG from Nhe6 KO (5 DRG per mouse, n = 3 mice) and WT (5 DRG per mouse, n = 3 mice) mice were extracted, flash frozen in liquid N2, and stored at −80°C. Total RNA was extracted using Promega Relia Prep Tisue RNA System (Cat# Z6111) and converted to cDNA using Applied Biological Materials Inc., Richmond, Canada, 5X All-In-One RT Master Mix (Cat# G490). For each sample, quantitative polymerase chain reaction (qPCR) reactions were performed in triplicate on Applied Biosystems Step One Plus Real-Time qPCR System using Applied Biological Materials, Inc., Richmond, Canada, TaqProbe 2X qPCR MasterMix (Cat# MasterMix-P) and Taqman Gene expression primer probe mixes for Trpv1 (assay number Mm01246300_m1). Trpv1 gene expression levels were quantified by the delta-delta Ct analysis using β−actin (assay number Mm00607939_s1) as a housekeeping reference gene. Values were then normalized to WT and expressed as a proportion of WT gene expression.
2.5. Statistical analysis
Data are expressed as the mean ± SE of the mean. Statistical analyses were performed with Kyplot 2.15 (Kyenslab, Inc, Tokyo, Japan). Means were compared by use of a parametric Student t-test for unpaired data or other tests as indicated. Proportions in contingency tables were compared by use of a Fisher exact test. For all statistical tests, the significance level was set at 0.05.
3. Results
3.1. NHE6 is expressed in central nervous system regions associated with pain processing
To understand how NHE6 can contribute to pain sensation, we first examined whether it is expressed in areas associated with the processing and perception of pain. Using a previously validated NHE6 antibody,8 we examined the expression of the exchanger in the primary somatosensory (S1) and anterior cingulate (ACC) cortices, which are involved in the localization and emotional valence of the painful stimulus, respectively, as well as the periaqueductal gray region, involved in the modulation of the pain response.1 Our data indicate that the exchanger is expressed in a small percentage of the neurons in these regions (20.1% of NeuN expressing neurons coexpressed NHE6 in the anterior cingulate cortex, 20.8% in S1 and 10.7% in the periaqueductal gray; n = 3 mice, Figs. 1A and B, and virtually absent from rostral ventromedial medulla, Fig. S1, available at http://links.lww.com/PAIN/B77).
Figure 1.
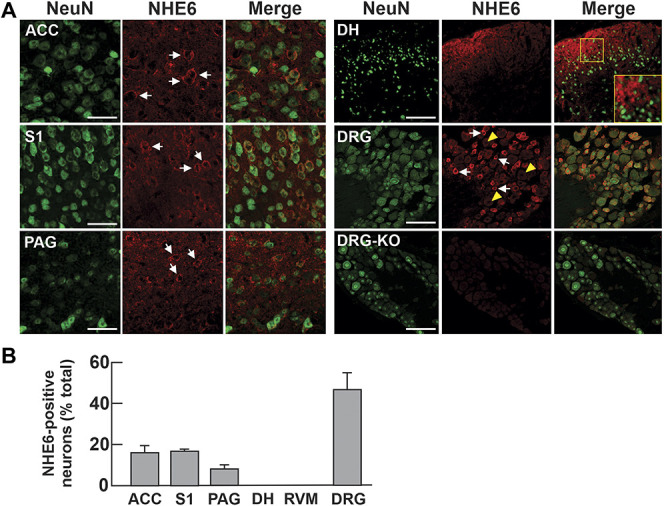
NHE6 is moderately expressed in pain centers in the CNS and highly expressed in sensory neurons. (A) Brain, spinal cord, and DRG tissues from 8-week-old WT mice stained for NeuN and NHE6. Representative images are shown (n = 3 mice, scale bar: 50 µm). White arrows and yellow arrowheads indicate cells positive and negative for NHE6, respectively. (B) Number of NHE6-positive neurons presented as a percentage of NeuN-positive neurons in each structure (ACC, n = 674/3288; S1, primary somatosensory cortex 1, n = 727/3556; PAG, n = 111/938; DH of the spinal cord, n = 6/701; RVM, rostral ventromedial medulla, n = 5/602; n = 474/989). ACC, anterior cingulate cortex; CNS, central nervous system; DH, dorsal horn; DRG, dorsal root ganglia; PAG, periaqueductal gray.
We next examined the expression of NHE6 in the dorsal horn (DH) of the spinal cord, which constitutes the first relay station where peripheral sensory inputs are processed.1,38 Although no neuronal cell bodies were positive for NHE6 (Figs. 1A and B), we found a strong signal in the superficial regions of the DH. The signal presented a crest-like profile within the superficial laminae of the DH resembling the innervation by the central terminals of primary afferents from C and Aδ fibers.38 This suggests that the signal might originate from sensory neurons in the DRG. Indeed, when we stained the DRG, we found that more than half of the neurons in the sensory ganglia were immunopositive for NHE6 (Figs. 1A and B), suggesting it might play a significant role in sensory transmission. DRG from Nhe6 KO mice did not exhibit an immunoreactive signal for NHE6, reaffirming the specificity of the NHE6 antibody (Fig. 1A, lower right panels). Also, it is essential to underline that there is no loss of neurons in Nhe6 KO mice (Fig. S2, available at http://links.lww.com/PAIN/B77).
3.2. NHE6 is highly expressed in small-diameter sensory neurons
To identify the population of sensory neurons expressing NHE6, we compared the expression pattern of NHE6 to that of subset-specific biomarkers in the DRG (Fig. 2A, quantified in Fig. 2B). Of the NHE6-positive neurons, only 5.2% expressed parvalbumin, a marker of proprioceptive neurons. Neurofilament-200 (NF200), a marker of myelinated neurons, was found in 29% of NHE6-expressing neurons. Tyrosine hydroxylase, which labels C-type low-threshold mechanoreceptors, was expressed by 12.5% of NHE6-positive neurons. Calcitonin gene-related peptide, a marker of peptidergic C fibres, was expressed by 18.7% of NHE6 neurons. Finally, nonpeptidergic nociceptive fibres that bind isolectin B4 (IB4) accounted for 34.6% of NHE6 neurons. To determine whether the NF200-positive subset of NHE6-expressing neurons clustered with large-diameter low-threshold mechanoreceptors or small-diameter high-threshold thinly myelinated nociceptors, we examined the cell size distribution of NHE6-expressing neurons (Fig. 2C). When compared to the cell size distributions of a large-diameter subset such as the parvalbumin-expressing proprioceptors and a small-diameter subset such as the nonpeptidergic IB4-binding nociceptors, the NHE6-positive neurons tended to cluster in the small size range. Taken together, our data indicate that NHE6 expression is predominantly in small-diameter nociceptors.
Figure 2.
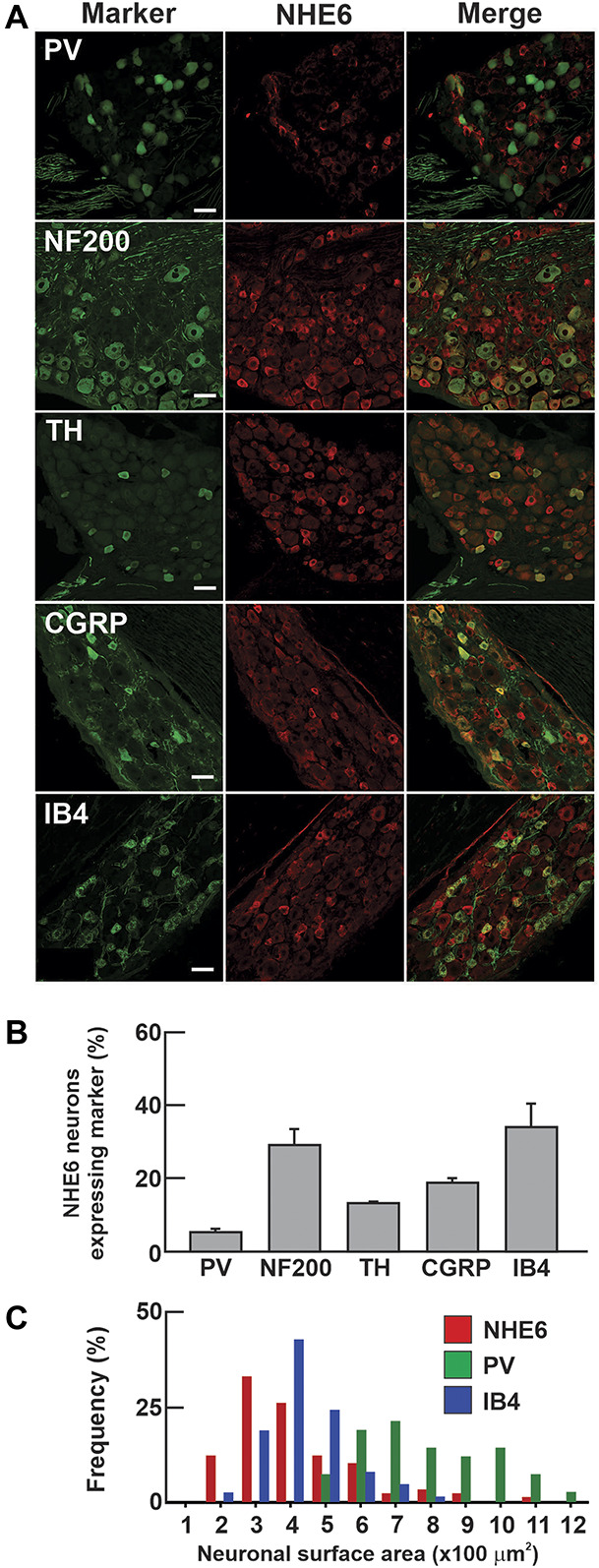
NHE6 is expressed predominantly in small-diameter neurons of the DRG. (A) Double immunofluorescent stains for NHE6 and markers of sensory neuron subsets on DRG from 8-week-old mice. Representative images are shown (n = 3 mice, scale bar: 50 µm). (B) Percentage of NHE6 neurons colabeled for the different markers, (PV, n = 41/536; NF200, n = 55/211; TH, n = 27/117; CGRP, n = 56/210; IB4, n = 67/198). (C) Size distribution of NHE6-expressing neurons alongside IB4-binding nociceptors and PV-expressing proprioceptors (n = 3 mice). CGRP, calcitonin gene-related peptide; DRG, dorsal root ganglia; IB4, isolectin-B4; NF200, neurofilament-200; PV, parvalbumin; TH, tyrosine hydroxylase.
3.3. NHE6 contributes to nociception in mice
To gain a better understanding of the function of NHE6 in pain transmission and processing, we performed behavioural tests on male Nhe6 KO mice and compared them to their WT littermates. The radiant heat (Hargreaves) test was used to examine their thermal sensitivity. At 8 weeks of age, KO mice had paw withdrawal latencies that were comparable to WT (KO: 10.04 ± 0.86 seconds; n = 5; WT: 9.18 ± 0.89 seconds, n = 10, paired t test, P = 0.708), suggesting similar sensitivities to noxious thermal stimuli. However, when tested at 24 weeks, the KO mice had significantly increased paw withdrawal latencies (13.81 ± 1.33 seconds) compared to 8 weeks (paired t test, P = 0.015) and to their age-matched WT littermates (8.97 ± 1.03 seconds, t test; P = 0.012) (Fig. 3A). The withdrawal latency of WT mice did not change between 8 and 24 weeks (paired t test, P = 0.086). The deficit in noxious heat sensitivity in aged KO mice was also observed in the hot-plate test, where a clear difference in their withdrawal latencies from WT littermates was observed at 42°C (44.9 ± 4.2 seconds in WT [n = 4] vs 110.2 ± 3.5 seconds in KO [n = 7] mice, P < 0.001) and at 52°C (10.1 ± 0.8 seconds in WT [n = 4] vs 12.9 ± 0.8 in KO [n = 7] mice; P = 0.027). This suggests that Nhe6−/Y mice become progressively less responsive to noxious thermal stimuli as a function of age.
Figure 3.
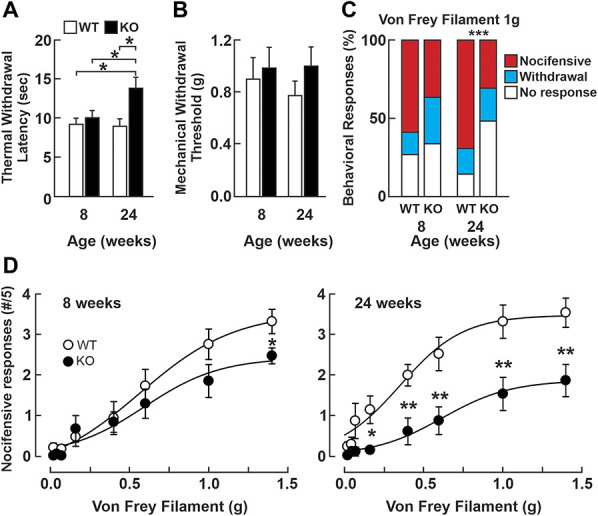
Mice lacking the NHE6 exchanger have decreased responses to noxious thermal and mechanical stimuli. (A) Mean (±SEM) paw withdrawal latency measured in the Hargreaves test for heat sensitivity in NHE6 WT (n = 10) and KO (n = 5) mice. *P < 0.05, paired t test. (B) Mean (±SEM) mechanical withdrawal thresholds measured with von Frey filaments. No significant differences are observed between the genotypes or over time. One-way ANOVA, P = 0.619 (8-week-old NHE6 WT (n = 10) and KO (n = 8); 24-week-old NHE6 WT (n = 10) and KO (n = 12). (C) Distribution (%) of behavioral responses to von Frey filament stimuli. The distributions of the behavioral responses to a 1.0 g mechanical stimulus are not significantly different between in 8-week-old WT (n = 10) and KO (n = 8) mice. Fisher exact test, P = 0.064. When assessed in 24-week-old mice, there is a significant change in the distribution of the responses of NHE6 KO mice, with an increase in the “no response” group (14.5% in WT, n = 10 vs 49.3% in KO, n = 12), and a decrease in the “nocifensive response” group (69.1% in WT vs 30.7% in KO), contingency table P < 0.001. (D) Nocifensive responses to von Frey filaments in 8-week-old NHE6 WT (n = 10) and KO (n = 8) mice (left panel) and 24-week-old NHE6 WT (n = 10) and KO (n = 12) mice (right panel). Student t-test; *P < 0.05; **P < 0.01. ANOVA, analysis of variance; KO, knockout; WT, wild-type.
Next, we measured their mechanical withdrawal threshold with static indentations of the skin using the von Frey assay,9 a stimulus that activates touch-sensing fibers at low intensity and nociceptors at higher intensity. No significant difference was observed between the 2 genotypes or between the 2 ages tested (Fig. 3B). Although von Frey measurements of mechanical withdrawal thresholds are indicative of the animal's mechanosensitivity, the behavior they elicit may be restricted to a spinal reflex. We therefore examined the behaviors elicited by the mechanical stimuli. Interestingly, despite the similarity in the withdrawal threshold, the KO mice displayed significantly less nocifensive-like behaviors such as flutter/guarding and licking of the paw when the intensity of the mechanical stimulus was close to the withdrawal threshold (ie, 1.0 g) compared to WT (Fig. 3C). Furthermore, when we assessed the occurrence of nocifensive responses to a range of mechanical stimuli, we observe a significant difference in 8-week-old mice at high-intensity stimuli (Fig. 3D, left panel). When tested in older mice, the difference between the genotypes was visible over a wider range of stimulus intensities (Fig. 3D, right panel). Taken together, these observations indicate that the absence of NHE6 induces a deficit in nociceptive processing in aged mice through impairment in thermal heat sensitivity and in nocifensive reactions to high-intensity mechanical stimuli.
3.4. Absence of NHE6 impairs nociceptor function
The expression of NHE6 in nociceptors and the hyposensitivity of aged KO mice to noxious stimuli led us to examine whether the exchanger is involved in the function of these neurons. In mice, peptidergic nociceptors can be recruited by capsaicin and therefore induce nocifensive reactions.30 To examine their activity, aged WT and KO mice (24 weeks) received an intraplantar injection of capsaicin and the time they spent licking the injected paw was recorded (Fig. 4A). We observed that most of the nocifensive behavior occurred in the first minute after the injection. Furthermore, the KO mice spent significantly less time licking the injected paw compared to WT littermates (8.0 ± 2.8 seconds in KO mice (n = 11) compared to 20.1 ± 2.8 seconds in WT mice (n = 15), t test, P = 0.009). However, treatment with the analgesic carprofen, a nonsteroidal anti-inflammatory drug (2 mg/kg), 30 minutes before capsaicin injection abolishes the spontaneous nocifensive reactions in both genomes (Fig. 4A), suggesting that KO mice remain sensitive to analgesic drugs.
Figure 4.
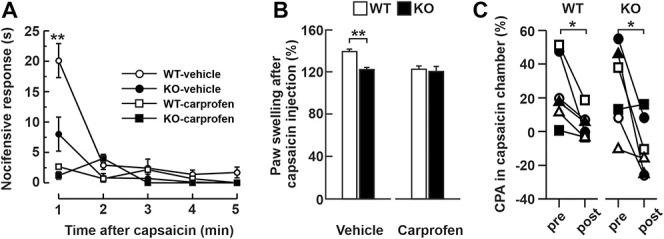
Nociceptor hypoactivity in aged NHE6 KO mice. (A) Mean (±S.E.M.) duration of nocifensive behaviors in NHE6 WT and KO mice injected intraplantarly with capsaicin (0.5%) after treatment with carprofen (2 mg/kg, white squares for WT black squares for KO, n = 6 mice per conditions) or vehicle (white circles for WT, n = 15, black circles for KO, n = 11). Student t-test, **P < 0.01. (B) Percentage (±S.E.M.) change in paw edema after intraplantar capsaicin injection after treatment with carprofen (2 mg/kg) or with vehicle (for WT, n = 8 and KO for, n = 7 mice). Student t-test, *P < 0.05. (C) Conditioned place aversion (CPA) test. Relative percentage of time spent in the capsaicin-paired chamber before (pre) or after (post) the conditioning phases for WT (left panel, n = 6) and KO (right panel, n = 6) mice. Paired Student t-test **P < 0.01. Each symbol represents the value for one mouse. KO, knockout; WT, wild-type.
An important question in CS is whether the elevated pain threshold results from impaired cortical functions that affect pain perception (ie, indifference to pain) or an impaired function of nociceptors (ie, insensitivity to pain) that prevents the pain signal from being generated or reaching the brain. One behavior-independent approach to quantify the activation of nociceptors is to assess neurogenic inflammation, which was measured by paw edema.2,16,19 Thirty minutes after capsaicin injection, both groups exhibited an increase in paw thickness after injection (Fig. 4B), but that increase was significantly lower in KO mice (123.1 ± 3.1% in KO mice (n = 7); 142.1 ± 4.1% in WT mice (n = 8); t test, P = 0.029). These results suggest that the elevated pain threshold in the KO mice is due, at least in part, to impairment in nociceptor function. Furthermore, treatment with the analgesic carprofen (2 mg/kg) 30 minutes before capsaicin injection reduced paw edema values in WT mice to those of KO mice (Fig. 4B).
3.5. Activation of nociceptors in Nhe6−/Y mice generates an aversive pain memory
To determine whether activating nociceptors in KO mice contributes to the emotive aspect of pain, we used a CPA assay according to published protocols.28 Mice were injected with capsaicin to pair activation of nociceptors with one chamber of the device (capsaicin chamber) and vehicle the following day to pair with the control chamber. We then compared the time spent in the capsaicin or control chamber before and after conditioning. Our results indicate that WT mice displayed a significant reduction in occupancy (−15.3% ± 0.07%) in the capsaicin chamber (747.6 ± 49.8 seconds before vs 624.1 ± 20.6 seconds after the capsaicin conditioning, P < 0.01, n = 6) (Fig. 4C). Likewise, KO mice also displayed a significant reduction in occupancy (−24.8% ± 0.04%) in the capsaicin chamber (749.1 ± 61.7 seconds before vs 546.1 ± 42.8 seconds after the capsaicin conditioning, P < 0.01, n = 6) (Fig. 4C). These findings suggest that despite the reduced nocifensive behavior of the KO mice, they are still able to exhibit an aversive pain memory, which suggests that their central emotive processing of painful stimuli remains intact.
3.6. NHE6−/Y knockout mice show a functional reduction in expression of Trpv1
The observed reduction in sensitivity of KO mice to capsaicin led us to examine whether the expression of its target receptor, the transient receptor potential cation channel subfamily V member 1 (TRPV1), was affected. This is particularly relevant in primary afferent sensory neurons because TRPV1 is activated by noxious stimuli including capsaicin and also by noxious heat temperature.3 In cultured DRG neurons from KO and WT littermate mice, we observed a reduction in expression of TRPV1 at the plasma membrane, as well as a reduction in expression of Trpv1 mRNA (Figs. 5A–C)). Furthermore, functional examination of TRPV1 activity in these neurons using calcium imaging indicated that nociceptors from KO mice displayed a markedly smaller calcium response after bath application of capsaicin than nociceptors from their WT littermates. However, the amplitudes of calcium responses after bath application of 50 mM of KCl to depolarize the nociceptors were similar, indicating proper calcium dynamics within these cells (Figs. 5D and E). Thus, it seems that there is a reduction of expression of TRPV1 in Nhe6−/Y sensory neurons that is responsible, at least in part, for the animal's behavioural insensitivity to pain.
Figure 5.
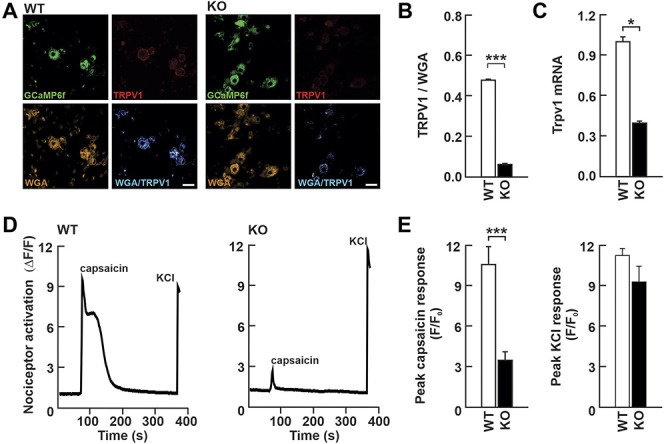
Functional impairment of TRPV1 activity in cultured dorsal root ganglion neurons from Nhe6 KO mice. (A) Epifluorescence image of cultured dorsal root ganglion neurons from WT and KO mice showing the TRPV1-GCaMP6f signal (top left), TRPV1 immunoreactivity (top right), wheat germ agglutinin (WGA) immunoreactivity (bottom left), and the merged WGA/TRPV1 image, represented with a false color. Blue color indicates the presence of TRPV1 at the plasma membrane of nociceptors (bottom right) Scale bar: 20 μm. (B) Quantification of TRPV1 signal intensities in panel A shows decreased expression of TRPV1 at the cell membrane in KO mice (n = 59 cells for WT and n = 134 cells for KO) (unpaired Student t‐test, ***P < 0.001). (C) Results of real-time-qPCR on whole DRG from WT (n = 5 DRG from 3 mice) and KO mice (n = 5 DRG from 3 mice) showing decreased presence of Trpv1 mRNA in KO DRG. Unpaired Student t-test, *P < 0.05 (D) Calcium imaging traces of cultured DRG neurons prepared from WT and KO mice in response to capsaicin (5 μM) and KCl (60 mM). (E) Quantification of peak responses to capsaicin (5 μM) (n = 50 cells for WT and n = 50 cells for KO; unpaired Student t‐test, ***P < 0.001) and KCl (60 mM) (n = 50 cells for WT and n = 50 cells for KO; unpaired Student t‐test, not significant). DRG, dorsal root ganglia; KO, knockout; WT, wild-type.
4. Discussion
Children suffering from CS exhibit not only severe limitations in cognitive, motor, and communication skills, but also display a high tolerance to pain,25 which puts them at risk of sustaining unreported injuries. However, a thorough characterization of the sensory deficits in CS children is presently lacking, which prevents us from understanding how mutations in NHE6 cause an elevated pain tolerance and whether the pain impairments result from central or peripheral pain-processing deficits. A Nhe6−/Y null mouse has been used to understand some of the defects in cerebellar32,34 and hippocampal24 neuronal morphology and circuit function in CS patients. Recent exploratory analyses indicated that these mice also display somatosensory deficits that could be used to understand pain tolerance in CS patients,17 although the studies were limited in scope. In this study, we examined in greater detail whether the Nhe6−/Y KO mouse displays sensory impairments similar to those seen in CS patients and whether those impairments result from central or peripheral dysfunctions.
To better understand the contribution of NHE6 to pain processing, we examined its expression in areas of the nervous system involved in the generation and interpretation of pain signals. Our results demonstrate that NHE6 is moderately expressed in CNS areas involved in the perception, valence, and modulation of pain signals, and is highly expressed in the peripheral nervous system, predominantly by nociceptors. These sensory neurons can be divided in 2 main groups: the peptidergic and the nonpeptidergic nociceptors, both previously proposed to contribute to the detection of painful heat and mechanical stimuli, respectively.5 Because NHE6 is expressed in both subsets of nociceptors, we hypothesized that its absence would impair the responses to these stimuli. Indeed, we demonstrate that Nhe6−/Y KO mice have decreased responses to noxious thermal and mechanical stimuli.
These sensory impairments may result from defects at the nociceptor level, the spinal cord, or at supraspinal sites. Indeed, even in the spinal cord, absence of NHE6-immunoreactivity in DH neurons does not mean this zone is not affected in the Nhe6−/Y KO mice. A recent study demonstrated evidence of lysosomal storage pathology in superficial DH of Nhe6−/Y KO mice, as well as increased astrocytic and microglial immunoreactivity in the dorsal and ventral horns, as well as near the pericentral canal.17 Nonetheless, given the predominant expression of NHE6 in nociceptors, we examined their ability to elicit peripheral neurogenic inflammation. Our findings demonstrate that neurogenic inflammation is significantly lower in Nhe6−/Y KO mice, indicating that part of the tolerance to pain results from a dysfunction in nociceptor activity. The mechanisms responsible for the reduced nociceptor activity in KO mice are currently unknown. However, because of its role in clathrin-mediated endocytosis and recycling of cargo, we postulate that KO mice may have defects in the plasma membrane targeting of proteins such as ion channels that are responsible for the excitability of nociceptors. In this regard, NHE6 has been implicated in the recruitment of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors to synapses during long-term potentiation in the hippocampus10,12 as well as the plasma membrane abundance of the neurotrophic tropomyosin receptor kinase B receptor.24 Our results demonstrate that this mechanism may also occur in KO sensory neurons where there is less expression of the TRPV1 channel at the plasma membrane. This deficit directly contributed to reduced response to capsaicin and thereby spontaneous pain. However, the KO mice still exhibited aversive behavior to this painful stimulus, as defined by the CPA experiments. This suggests that their central ability to remember the emotional valence a painful stimulus, however diminished, remains relatively intact. Future electrophysiological characterization of nociceptors isolated from NHE6 WT and KO mice will shed light on the role of this exchanger in neuronal excitability and ion channel function. Importantly, our observations do not rule out the possibility that defective pain sensitivity may also result from dysfunctions at supraspinal sites. Indeed, it is possible that despite the moderate expression of NHE6 in these areas, absence of the exchanger may significantly affect pain processing and perception.
Christianson syndrome is a neurodevelopment and regressive disorder with some symptoms appearing early, whereas others later in development. Mice with a global knockout for the Slc9a6 gene present also these observations. It was reported in the mice brain that Purkinje cell loss is quite noticeable at 15 weeks, whereas endolysosomal dysfunction is already evident at 3 weeks in the amygdala and 8 weeks in the hippocampus.34 In our study, the deficit in nociception functions appears after 24 weeks, although already noticeable by 16 weeks. We can hypothesise that rodent nociceptors progressively accumulate lysosomal damage,17 which will impact only their activity in the adult stage. It is also possible that deficits do appear much earlier, but that our experimental approaches only allow us to detect significant differences in older mice. Thus, despite our findings, additional experiments will be required to better understand the CS pathology, including an electrophysiological analysis of the excitability of the nociceptors at different developmental time points.
In conclusion, better understanding of the regulatory mechanisms behind nociceptive excitability is essential to improve treatment for pain disorders. Hyposensitivity to pain, as observed in CS patients, presents an urgent physiological issue due to its negative impact on the quality of life of those afflicted. Our work characterizes the sensory impairments in a mouse model of CS and suggests that part of the deficits is due to decreased nociceptor excitability.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B77.
Supplemental video content
A video abstract associated with this article can be found at http://links.lww.com/PAIN/B126.
Acknowledgments
This work was supported by operating grants to R. Sharif-Naeini (CIHR MOP-130471 and PJT-166165, NSERC Discovery Grant 436091) and J. Orlowski (CIHR PJT-166165). S. Mouchbahani-Constance is supported by a CIHR Canada Graduate Scholarship (CGS) Master's award.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
H. Petitjean and T. Fatima contributed equally to the work.
References
- [1].Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell 2009;139:267–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Brain SD, Williams TJ. Inflammatory oedema induced by synergism between calcitonin gene-related peptide (CGRP) and mediators of increased vascular permeability. Br J Pharmacol 1985;86:855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997;389:816–24. [DOI] [PubMed] [Google Scholar]
- [4].Cavanaugh DJ, Chesler AT, Braz JM, Shah NM, Julius D, Basbaum AI. Restriction of transient receptor potential vanilloid-1 to the peptidergic subset of primary afferent neurons follows its developmental downregulation in nonpeptidergic neurons. J Neurosci 2011;31:10119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cavanaugh DJ, Lee H, Lo L, Shields SD, Zylka MJ, Basbaum AI, Anderson DJ. Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc Natl Acad Sci U S A 2009;106:9075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chang KJ, Bennett V, Cuatrecasas P. Membrane receptors as general markers for plasma membrane isolation procedures: the use of 125-I-labeled wheat germ agglutinin, insulin, and cholera toxin. J Biol Chem 1975;250:488–500. [PubMed] [Google Scholar]
- [7].Christianson AL, Stevenson RE, van der Meyden CH, Pelser J, Theron FW, van Rensburg PL, Chandler M, Schwartz CE. X linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large South African kindred is localised to Xq24-q27. J Med Genet 1999;36:759–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Deane EC, Ilie AE, Sizdahkhani S, Das Gupta M, Orlowski J, McKinney RA. Enhanced recruitment of endosomal Na+/H+ Exchanger NHE6 into dendritic spines of hippocampal pyramidal neurons during NMDA receptor-dependent long-term potentiation. J Neurosci 2013;33:595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Deuis JR, Dvorakova LS, Vetter I. Methods used to evaluate pain behaviors in rodents. Front Mol Neurosci 2017;10:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, Asplund A, Sjostedt E, Lundberg E, Szigyarto CA, Skogs M, Takanen JO, Berling H, Tegel H, Mulder J, Nilsson P, Schwenk JM, Lindskog C, Danielsson F, Mardinoglu A, Sivertsson A, von Feilitzen K, Forsberg M, Zwahlen M, Olsson I, Navani S, Huss M, Nielsen J, Ponten F, Uhlen M. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 2014;13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fertleman CR, Ferrie CD, Aicardi J, Bednarek NA, Eeg-Olofsson O, Elmslie FV, Griesemer DA, Goutieres F, Kirkpatrick M, Malmros IN, Pollitzer M, Rossiter M, Roulet-Perez E, Schubert R, Smith VV, Testard H, Wong V, Stephenson JB. Paroxysmal extreme pain disorder (previously familial rectal pain syndrome). Neurology 2007;69:586–95. [DOI] [PubMed] [Google Scholar]
- [12].Gao AYL, Ilie A, Chang PKY, Orlowski J, McKinney RA. A Christianson syndrome-linked deletion mutation (Delta287ES288) in SLC9A6 impairs hippocampal neuronal plasticity. Neurobiol Dis 2019;130:104490. [DOI] [PubMed] [Google Scholar]
- [13].Garbern JY, Neumann M, Trojanowski JQ, Lee VM, Feldman G, Norris JW, Friez MJ, Schwartz CE, Stevenson R, Sima AA. A mutation affecting the sodium/proton exchanger, SLC9A6, causes mental retardation with tau deposition. Brain 2010;133(pt 5):1391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K, Kroken M, Mattingsdal M, Egeland T, Stenmark H, Sjoholm H, Server A, Samuelsson L, Christianson A, Tarpey P, Whibley A, Stratton MR, Futreal PA, Teague J, Edkins S, Gecz J, Turner G, Raymond FL, Schwartz C, Stevenson RE, Undlien DE, Stromme P. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am J Hum Genet 2008;82:1003–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ilie A, Gao AYL, Reid J, Boucher A, McEwan C, Barriere H, Lukacs GL, McKinney RA, Orlowski J. A Christianson syndrome-linked deletion mutation (Δ287ES288) in SLC9A6 disrupts recycling endosomal function and elicits neurodegeneration and cell death. Mol Neurodegener 2016;11:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jancso N, Jancso-Gabor A, Szolcsanyi J. Direct evidence for neurogenic inflammation and its prevention by denervation and by pretreatment with capsaicin. Br J Pharmacol Chemother 1967;31:138–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kerner-Rossi M, Gulinello M, Walkley S, Dobrenis K. Pathobiology of Christianson syndrome: linking disrupted endosomal-lysosomal function with intellectual disability and sensory impairments. Neurobiol Learn Mem 2018;165:106867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li Y, Tatsui CE, Rhines LD, North RY, Harrison DS, Cassidy RM, Johansson CA, Kosturakis AK, Edwards DD, Zhang H, Dougherty PM. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. PAIN 2017;158:417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lin Q, Wu J, Willis WD. Dorsal root reflexes and cutaneous neurogenic inflammation after intradermal injection of capsaicin in rats. J Neurophysiol 1999;82:2602–11. [DOI] [PubMed] [Google Scholar]
- [20].Mellman I, Fuchs R, Helenius A. Acidification of the endocytic and exocytic pathways. Annu Rev Biochem 1986;55:663–700. [DOI] [PubMed] [Google Scholar]
- [21].Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, Hahn CD, Kannu P, Kobayashi J, Minassian BA, Moharir M, Siriwardena K, Weiss SK, Weksberg R, Snead OC., III Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 2015;56:707–16. [DOI] [PubMed] [Google Scholar]
- [22].Mouchbahani-Constance S, Lesperance LS, Petitjean H, Davidova A, Macpherson A, Prescott SA, Sharif-Naeini R. Lionfish venom elicits pain predominantly through the activation of nonpeptidergic nociceptors. PAIN 2018;159:2255–66. [DOI] [PubMed] [Google Scholar]
- [23].Nizon M, Kury S, Pereon Y, Besnard T, Quinquis D, Boisseau P, Marsaud T, Magot A, Mussini JM, Mayrargue E, Barbarot S, Bezieau S, Isidor B. ARL6IP1 mutation causes congenital insensitivity to pain, acromutilation and spastic paraplegia. Clin Genet 2018;93:169–72. [DOI] [PubMed] [Google Scholar]
- [24].Ouyang Q, Lizarraga SB, Schmidt M, Yang U, Gong J, Ellisor D, Kauer JA, Morrow EM. Christianson syndrome protein NHE6 modulates TrkB endosomal signaling required for neuronal circuit development. Neuron 2013;80:97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pescosolido MF, Stein DM, Schmidt M, Moufawad El AC, Sabbagh M, Rogg JM, Tantravahi U, McLean RL, Liu JS, Poduri A, Morrow EM. Genetic and phenotypic diversity of NHE6 mutations in Christianson syndrome. Ann Neurol 2014;76:581–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Petitjean H, FBB, Tsao D, Davidova A, Sotocinal SG, Mogil JS, Kania A, Sharif-Naeini R. Recruitment of spinoparabrachial neurons by dorsal horn calretinin neurons. Cell Rep 2019;28:1429–38 e1424. [DOI] [PubMed] [Google Scholar]
- [27].Petitjean H, Hugel S, Barthas F, Bohren Y, Barrot M, Yalcin I, Schlichter R. Activation of transient receptor potential vanilloid 2-expressing primary afferents stimulates synaptic transmission in the deep dorsal horn of the rat spinal cord and elicits mechanical hyperalgesia. Eur J Neurosci 2014;40:3189–201. [DOI] [PubMed] [Google Scholar]
- [28].Porreca F, Navratilova E. Reward, motivation, and emotion of pain and its relief. PAIN 2017;158(suppl 1):S43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ravichandra KS, Kandregula CR, Koya S, Lakhotia D. Congenital insensitivity to pain and anhydrosis: diagnostic and therapeutic dilemmas revisited. Int J Clin Pediatr Dent 2015;8:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Scherrer G, Low SA, Wang X, Zhang J, Yamanaka H, Urban R, Solorzano C, Harper B, Hnasko TS, Edwards RH, Basbaum AI. VGLUT2 expression in primary afferent neurons is essential for normal acute pain and injury-induced heat hypersensitivity. Proc Natl Acad Sci U S A 2010;107:22296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sharif-Naeini R, Folgering JH, Bichet D, Duprat F, Lauritzen I, Arhatte M, Jodar M, Dedman A, Chatelain FC, Schulte U, Retailleau K, Loufrani L, Patel A, Sachs F, Delmas P, Peters DJ, Honore E. Polycystin-1 and -2 dosage regulates pressure sensing. Cell 2009;139:587–96. [DOI] [PubMed] [Google Scholar]
- [32].Sikora J, Leddy J, Gulinello M, Walkley SU. X-linked Christianson syndrome: heterozygous female Slc9a6 knockout mice develop mosaic neuropathological changes and related behavioral abnormalities. Dis Model Mech 2016;9:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Skeik N, Rooke TW, Davis MD, Davis DM, Kalsi H, Kurth I, Richardson RC. Severe case and literature review of primary erythromelalgia: novel SCN9A gene mutation. Vasc Med 2012;17:44–9. [DOI] [PubMed] [Google Scholar]
- [34].Stromme P, Dobrenis K, Sillitoe RV, Gulinello M, Ali NF, Davidson C, Micsenyi MC, Stephney G, Ellevog L, Klungland A, Walkley SU. X-linked Angelman-like syndrome caused by Slc9a6 knockout in mice exhibits evidence of endosomal-lysosomal dysfunction. Brain 2011;134(pt 11):3369–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tang Z, Chen Z, Tang B, Jiang H. Primary erythromelalgia: a review. Orphanet J Rare Dis 2015;10:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O'Meara S, Latimer C, Dicks E, Menzies A, Stephens P, Blow M, Greenman C, Xue Y, Tyler-Smith C, Thompson D, Gray K, Andrews J, Barthorpe S, Buck G, Cole J, Dunmore R, Jones D, Maddison M, Mironenko T, Turner R, Turrell K, Varian J, West S, Widaa S, Wray P, Teague J, Butler A, Jenkinson A, Jia M, Richardson D, Shepherd R, Wooster R, Tejada MI, Martinez F, Carvill G, Goliath R, de Brouwer AP, van BH, Van EH, Chelly J, Raynaud M, Ropers HH, Abidi FE, Srivastava AK, Cox J, Luo Y, Mallya U, Moon J, Parnau J, Mohammed S, Tolmie JL, Shoubridge C, Corbett M, Gardner A, Haan E, Rujirabanjerd S, Shaw M, Vandeleur L, Fullston T, Easton DF, Boyle J, Partington M, Hackett A, Field M, Skinner C, Stevenson RE, Bobrow M, Turner G, Schwartz CE, Gecz J, Raymond FL, Futreal PA, Stratton MR. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 2009;41:535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Teter K, Chandy G, Quinones B, Pereyra K, Machen T, Moore HP. Cellubrevin-targeted fluorescence uncovers heterogeneity in the recycling endosomes. J Biol Chem 1998;273:19625–33. [DOI] [PubMed] [Google Scholar]
- [38].Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 2010;11:823–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Woods CG, Babiker MO, Horrocks I, Tolmie J, Kurth I. The phenotype of congenital insensitivity to pain due to the NaV1.9 variant p.L811P. Eur J Hum Genet 2015;23:561–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Xinhan L, Matsushita M, Numaza M, Taguchi A, Mitsui K, Kanazawa H. Na+/H+ exchanger isoform 6 (NHE6/SLC9A6) is involved in clathrin-dependent endocytosis of transferrin. Am J Physiol Cell Physiol 2011;301:C1431–44. [DOI] [PubMed] [Google Scholar]
- [41].Yamashiro DJ, Tycko B, Fluss SR, Maxfield FR. Segregation of transferrin to a mildly acidic (pH 6.5) para-Golgi compartment in the recycling pathway. Cell 1984;37:789–800. [DOI] [PubMed] [Google Scholar]
- [42].Zanni G, Barresi S, Cohen R, Specchio N, Basel-Vanagaite L, Valente EM, Shuper A, Vigevano F, Bertini E. A novel mutation in the endosomal Na+/H+ exchanger NHE6 (SLC9A6) causes Christianson syndrome with electrical status epilepticus during slow-wave sleep (ESES). Epilepsy Res 2014;108:811–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B77.
A video abstract associated with this article can be found at http://links.lww.com/PAIN/B126.