

Supplemental Digital Content is available in the text.
Keywords: cardiac hypertrophy, epigenetics, tissue engineering
Abstract
Background:
DNA methylation acts as a mechanism of gene transcription regulation. It has recently gained attention as a possible therapeutic target in cardiac hypertrophy and heart failure. However, its exact role in cardiomyocytes remains controversial. Thus, we knocked out the main de novo DNA methyltransferase in cardiomyocytes, DNMT3A, in human induced pluripotent stem cells. Functional consequences of DNA methylation-deficiency under control and stress conditions were then assessed in human engineered heart tissue from knockout human induced pluripotent stem cell–derived cardiomyocytes.
Methods:
DNMT3A was knocked out in human induced pluripotent stem cells by CRISPR/Cas9gene editing. Fibrin-based engineered heart tissue was generated from knockout and control human induced pluripotent stem cell–derived cardiomyocytes. Development and baseline contractility were analyzed by video-optical recording. Engineered heart tissue was subjected to different stress protocols, including serum starvation, serum variation, and restrictive feeding. Molecular, histological, and ultrastructural analyses were performed afterward.
Results:
Knockout of DNMT3A in human cardiomyocytes had three main consequences for cardiomyocyte morphology and function: (1) Gene expression changes of contractile proteins such as higher atrial gene expression and lower MYH7/MYH6 ratio correlated with different contraction kinetics in knockout versus wild-type; (2) Aberrant activation of the glucose/lipid metabolism regulator peroxisome proliferator-activated receptor gamma was associated with accumulation of lipid vacuoles within knockout cardiomyocytes; (3) Hypoxia-inducible factor 1α protein instability was associated with impaired glucose metabolism and lower glycolytic enzyme expression, rendering knockout-engineered heart tissue sensitive to metabolic stress such as serum withdrawal and restrictive feeding.
Conclusion:
The results suggest an important role of DNA methylation in the normal homeostasis of cardiomyocytes and during cardiac stress, which could make it an interesting target for cardiac therapy.
Clinical Perspective.
What Is New?
DNA methylation by DNA methyltransferase DNMT3A is involved in the homeostasis of human cardiomyocytes.
Aberrant DNA methylation in cardiomyocytes affects contractility and inflicts mitochondrial damage and lipid and glucose metabolism deficits.
Loss of DNMT3A-dependent DNA methylation can be compensated partially by growth factors.
What Are the Clinical Implications?
Processes affecting DNA methylation, such as nutrition and DNA methylation aging, could contribute to the pathogenesis of heart failure.
Addressing DNA methylation could represent a new therapeutic concept for heart disease.
Heart failure represents one of the leading causes of death worldwide, with a steadily increasing incidence.1 Development of heart failure and cardiac hypertrophy is not only associated with structural changes of the heart but also with distinct changes in gene expression patterns known as reactivation of the fetal gene program.2–4 In this context, epigenetic mechanisms such as DNA methylation have gained increasing attention over recent years as potential regulators of these gene expression changes.
DNA methylation, in mammals typically referring to the addition of a methyl group to a DNA cytosine of a CpG dinucleotide residue, is catalyzed by the de novo DNA methyltransferases (DNMTs) DNMT3A and DNMT3B, as well as by the maintenance isoform DNMT1.5–7 Methylation of cytosines in the context of so-called CpG islands, regions of high cytosine-guanidine dinucleotide density often found in promoter regions of genes, is typically associated with a change in expression of genes in close proximity, for example by interference with transcription factor binding or recruitment of repressor complexes.8 However, whereas a contribution of other epigenetic marks such as histone modifications to cardiac disease progression has already been firmly established,9–13 the role of DNA methylation remains controversial. DNA methylation analysis in patients experiencing end-stage heart failure or dilated cardiomyopathy showed distinct methylation patterns in diseased individuals compared with healthy controls.14,15 Moreover, whole-genome DNA methylation analysis by bisulfite sequencing in an in vivo model of cardiac hypertrophy in mice revealed a DNA methylation signature similar to fetal mice,16 arguing for an involvement of DNA methylation in these pathologies. However, further studies using pharmacological inhibition of DNMTs and studies using genetic knockout models of single DNMTs have reached diverging conclusions regarding the importance of DNA methylation in cardiac hypertrophy. Several studies using nucleosidic or nonnucleosidic DNMT inhibitors in both in vitro and in vivo models of cardiac hypertrophy have consistently reported a protective effect of DNMT inhibition on disease progression,17–20 whereas the results from 2 genetic mouse models are conflicting. One study using a cardiomyocyte-specific knockout of Dnmt3b in mice reported an impairment of contractility, exaggerated fibrosis, and cardiac dilation even under nonhypertrophic conditions,21 suggesting an important function of de novo DNA methylation in the heart. In contrast, cardiomyocyte-specific knockout of both de novo DNMTs in mice was devoid of apparent effects on cardiac function or disease progression in transverse aortic constriction–induced cardiac hypertrophy. Even though significant differences in both DNA methylation and gene expression were observed,22 these results suggest a negligible role of DNA methylation for cardiomyocyte function.
Analysis of DNA methylation in human cardiomyocytes under reproducible in vitro conditions could contribute to our understanding and resolve this conflict. Engineered heart tissue (EHT) enables culture of human cardiomyocytes in a 3-dimensional format, which helps to overcome possible interspecies differences that could influence the results.23,24 Moreover, the model opens up the opportunity for a high variety of interventions, some of which are extreme in nature and therefore impossible to perform in vivo.25–27 This might allow for the uncovering of those functions of DNA methylation which (1) are not sufficiently triggered in animal models of disease, (2) develop in a time frame not analyzed in animal models, or (3) are restricted to humans.
The present study therefore aimed to elucidate the role of de novo DNA methylation in human cardiomyocytes by knockout of the main cardiac DNMT isoform, DNMT3A, in human induced pluripotent stem cells (hiPSC) by CRISPR/Cas9 gene editing. The effect of the knockout on cardiomyocyte function under normal and stress conditions was analyzed in the controlled environment of human EHT generated from knockout hiPSC-derived cardiomyocytes.
Methods
A detailed description of the methods can be found in the Data Supplement. All data of high-throughput experiments have been made publicly available through the BioProject database and can be accessed at http://www.ncbi.nlm.nih.gov/bioproject/646856.
CRISPR/Cas9-Mediated DNMT3A Knockout
Culture and genetic engineering of a hiPSC line (UKEi003-C) were approved by the local ethics committee in Hamburg, Germany (approval numbers PV3501 and PV4798). CRISPR/Cas9-mediated knockout of DNMT3A was performed as published.28,29
EHT Generation and Culture
Generation of fibrin-based 24-well format EHT from hiPSC-derived cardiomyocytes and analysis of EHT contractility by video-optical recording were done as described before.23,24,30 Short-term electric stimulation using carbon electrodes of EHTs was performed as published.31
Medium Restrictions and Pharmacological Treatments
EHT substrate preference was analyzed in serum-free medium containing either 1 mmol/L lactate or 5 mmol/L d-glucose as the only energy substrate. In addition to standard horse serum, lipoprotein-deficient serum32 and dialyzed serum were used in EHT experiments. Glucose consumption and lactate production were analyzed with the Radiometer ABL90 Flex. The following substances were used in EHT experiments: 10 µmol/L GW9662 (peroxisome proliferator-activated receptor gamma [PPARγ] inhibitor; Cayman Chemical, #70785); 10 ng/mL basic fibroblast growth factor (PeproTech, #100-18B); 20 ng/mL insulin-like growth factor 1 (PeproTech, #100-11); 10 ng/mL endothelial growth factor (Sino Biological, #10-605-HNAE5); 20 ng/mL transforming growth factor β1 (PeproTech, #100-21C).
Histological Stainings
Antibodies used for immunohistochemistry were MLC2V (Synaptic Systems #SY310111, 1:3000) and Ki-67 (Abcam #Ab15580, 1:100). Lipid-staining of 30 day-old EHTs with Oil Red O (Sigma) was performed as previously described for zebrafish embryos.33,34
Transmission Electron Microscopy
For electron microscopy, ultrathin sections (60 nm) from 30-day-old fixed EHTs were mounted on copper grids and stained with uranyl acetate and lead citrate. Sections were examined and photographed using an EM902 (Zeiss) electron microscope equipped with a TRS 2K digital camera (A. Tröndle, Moorenweis, Germany).
Metabolic Profiling
Cardiomyocyte mitochondrial and glucose metabolism was analyzed with the Seahorse XF96 extracellular flux analyzer using the Glycolysis and Mito Stress Test kits (Agilent) as previously described.35
Gene Expression Analysis
RNA for gene expression analysis was isolated from hiPSC and 30-day-old human EHT with the RNeasy Plus Mini kit (QIAGEN) or TRIzol reagent (Life Technologies). For quantitative polymerase chain reaction (qPCR) analysis, the High-Capacity cDNA Reverse Transcription kit and an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) were used. Expression analysis on the nCounter SPRINT Profiler (NanoString) was performed as described before with 50 ng of total RNA and a customized expression CodeSet.36 RNA sequencing was performed as described by Stenzig et al20 from 500 ng RNA per sample on the Illumina HiSeq 4000 sequencing system. Pathway mapping was performed with the DAVID Functional Annotation tool.37,38
Protein Analysis by Western Blot
Primary antibodies used were DNMT3A (CST #2160, 1:500), p44/42 mitogen-activated protein kinase (ERK1/2; CST #9102, 1:1000), and hypoxia-inducible factor 1α (HIF-1α; Invitrogen #PA1-16601, 1:1000). Secondary antibodies used were α-mouse IgG peroxidase-conjugate (Sigma A3682, 1:10,000) and α-rabbit IgG peroxidase-conjugate (Sigma, A9044, 1:10,000).
DNA Methylation Analysis
Genome-wide DNA methylation of single EHTs was analyzed by reduced representation bisulfite sequencing (RRBS) from 70 ng DNA on the Illumina HiSeq 2500 sequencer as published.20 Targeted DNA methylation analysis was performed by bisulfite conversion and Sanger sequencing of individual clones.
Statistical Methods
If not stated otherwise, data were analyzed using GraphPad Prism software and are plotted as individual data points with their mean (horizontal line) and SEM (vertical line) in a dot plot. For statistical analysis of differences between groups, an unpaired t test (2 groups), a 1-way ANOVA (more than 2 groups), or a 2-way ANOVA (more than 2 groups, repeated measurements) were used as appropriate. Bonferroni posttest was used for correction for multiple testing. Values of P<0.05 were considered to be statistically significant.
Results
CRISPR/Cas9-Mediated Knockout of DNMT3A
To identify the most important DNA methyltransferase in both hiPSC and hiPSC-derived cardiomyocytes, DNMT isoform mRNA abundance was analyzed in both cell types. DNMT3B was the most prominent isoform in undifferentiated hiPSC but was virtually absent from differentiated cells (Figure IA in the Data Supplement). Similar to what had been described for adult murine cardiomyocytes,22 DNMT3A was the DNA methyltransferase isoform with the highest transcript abundance in hiPSC-derived cardiomyocytes. CRISPR/Cas9-mediated knockout of DNMT3A as published by Liao et al29 (Figure IB in the Data Supplement) yielded 3 successfully mutated cell lines carrying either a heterozygous insertion of an adenine nucleotide (DNMT3A+/−), a compound heterozygous insertion of an adenine nucleotide/20-bp deletion (DNMT3A−/− #1), or a homozygous insertion of an adenine nucleotide at the cutting site (DNMT3A−/− #2; Figure II in the Data Supplement). Screening of the 10 most-likely off-targets revealed no unwanted mutations in either of the 3 cell lines. Moreover, analysis of the genomic DNMT3A locus by long-range PCR as described by Kosicki et al39 showed no indications of large insertions or deletions around the cutting site (Figure IC in the Data Supplement). All mutant cell lines yielded beating cardiomyocytes with cardiomyocyte fractions and differentiation efficiencies similar to wild-type cells (Figure III in the Data Supplement). In silico analysis of the effect of the mutations on mRNA sequence predicted induction of a frameshift and a premature stop codon for both the insertion of adenine and the 20-bp deletion (Figure IVA and IVB in the Data Supplement). In line with these results, DNMT3A protein was reduced by ≈40% in the DNMT3A+/− mutant and not detectable in the DNMT3A−/− mutant (Figure ID and IE in the Data Supplement). Similar results were obtained on the mRNA level (Figure IVC in the Data Supplement). Global DNA methylation was analyzed by RRBS, revealing a slightly but significantly lower global DNA methylation state in DNMT3A−/− than in wild-type cardiomyocytes (Figure IF in the Data Supplement), compatible with a loss of DNMT3A mediated de novo methylation.
Knockout of DNMT3A Affects EHT Contractility
DNMT3A-deficiency did not influence EHT development under standard, serum-containing culture conditions. Analysis of average contraction peaks under electric stimulation at 1.5 Hz after 3 weeks of culture, however, revealed significantly faster relaxation in DNMT3A+/− EHTs, which was even more pronounced in the DNMT3A−/− EHTs (Figure 1A and 1B). Contraction and relaxation kinetics are among others determined by the expression ratio of the alpha and beta myosin heavy chain (MHC) isoforms, because the α isoform (encoded by MYH6) has faster kinetic properties than the β isoform (encoded by MYH7). Expression analysis indeed revealed a significantly lower MYH7/MYH6 ratio in DNMT3A−/− EHTs (Figure 1C), which could in part be responsible for the observed faster relaxation. In addition, knockout EHTs showed a significantly higher ratio of atrial myosin light chain (MLC2A) to ventricular myosin light chain (MLC2V, Figure 1D), as well as higher transcript abundance of other typical atrial cardiomyocyte markers, including PITX2 (Figure 1E). In line with this, the PITX2 promoter was found to be significantly hypomethylated in the knockout compared with wild-type (Figure 1F). In accordance with the observed faster contractility, all 3 knockout lines were able to follow pacing impulses to the highest tested frequency, whereas wild-type EHTs gradually lost pacing capture at frequencies >3 Hz (Figure 1G).
Figure 1.

Contraction kinetics of DNA methyltransferase(DNMT) 3A knockoutengineered heart tissue (EHT).
A, Normalized average contraction peaks of EHT from wild-type and knockout lines at a stimulation frequency of 1.5 Hz. Mean±SEM. B, Relaxation times from peak to 80% relaxation at 1.5 Hz. n=7-12 EHTs per group. C, Ratio of myosin heavy chain (MHC) isoforms in wild-type and DNMT3A−/− EHTs. MYH7 and MYH6 abundance was measured by NanoString analysis, n=3. D, Ratio of atrial (MYL7, MLC2A) and ventricular myosin light chain (MYL2, MLC2V) isoforms, measured by RNA sequencing. n=3. E, Relative expression of atrial-specific marker genes. n=3, False discovery rate- (FDR-) adjusted P value. F, PITX2 promoter methylation, measured by reduced representation bisulfite sequencing. n=3–4. G, Pacing capture of wild-type and knockout EHT at frequencies from 0.7 Hz to 6 Hz, beating frequency of each EHT line plotted against the stimulation frequency. Each dot represents a single EHT measurement. ***P<0.001.
Unfortunately, all 3 mutant cell lines were created from a cell line carrying a trisomy of chromosome 1 (Figure II in the Data Supplement; uncovered only after creation of the mutant lines), a common unwanted side effect of prolonged hiPSC culture.40,41 To explore whether phenotype differences in the knockout lines might be related to the trisomy, most experiments were not only performed using the karyotypically normal parent cell line as control (wild-type) but also repeated with the parent control cell line carrying the same trisomy (isogenic control; Figure V in the Data Supplement). Wild-type and isogenic control EHTs were indistinguishable in almost all experiments. The only exception was a significantly faster relaxation and altered MHC isoform ratio in the trisomic isogenic control, which, however, was still much more similar to wild-type than knockout EHTs. Overall, the data support the interpretation that the effects observed in the knockout lines were attributable to the deletion of DNMT3A.
Functional Degeneration Under Serum-Free Conditions
Although force development of knockout EHTs did not differ from the wild-type under standard culture conditions containing 10% serum, striking differences were observed when the EHTs were cultured under serum-free conditions. Wild-type EHTs displayed a steady increase in force in serum-free medium, as usually observed in this model, and retained a regular beating pattern over the whole culture period (Figure 2A, 2B, and 2E). In contrast, knockout EHTs showed a steadily declining force and increasing occurrence of beating arrhythmias (Figure 2C, 2D, and 2F; Figure VI in the Data Supplement). It is important to note that the functional degeneration did not seem to be caused by loss of cardiomyocytes, because the effect was fully reversible by subsequently culturing the EHTs in serum-containing medium again (Figure VIIA through VIID in the Data Supplement). The observed functional decline of knockout EHTs under serum-free conditions suggested that serum contains either an energy source (eg, lipids) or a signaling molecule essential for normal cardiomyocyte function specifically in DNMT3A knockout. To evaluate these possibilities, we cultured knockout EHT with different depleted sera. Lipoprotein-deficient and small molecule-depleted serum (lipoprotein-deficient serum; removal of all lipoproteins plus subsequent dialysis with 30-kDa cut-off) and small molecule-depleted dialysis-only serum (30-kDa cut-off) had a similar negative effect on EHT contractility as serum-free medium (Figure VIIE through VIIG in the Data Supplement). This observation suggested that the absence of 1 or several low-molecular serum components contributed to the decline of knockout EHT contractility in serum-free culture.
Figure 2.

Contraction behavior in serum-free culture conditions.
Force development (A) and RR scatter (B) as a surrogate parameter for beating arrhythmias, with R being the interval between 2 neighboring force peaks, of wild-type and (C–D) knockout engineered heart tissue (EHT) over culture time under both standard serum-containing (green plus) and serum-free culture conditions (red minus). The dotted line marks beginning of serum withdrawal. Mean±SEM, n=51–58 EHTs from 3 batches. Representative contraction peaks of wild-type (E) and knockout EHTs (F) under serum-free conditions.
DNA Methylation and Gene Expression Changes
To analyze the effect of de novo DNMT deficiency on DNA methylation in cardiomyocytes, whole-genome DNA methylation analysis of 30-day-old EHTs was performed by RRBS. Cluster analysis of differentially methylated cytosines revealed closer clustering of all samples to their respective replicates than to those from the other genotype (Figure VIIIA in the Data Supplement). As expected for de novo DNA methylation-deficient EHTs, most of the differentially methylated cytosines showed hypomethylation in knockout EHTs compared with wild-type EHTs (Figure VIIIB in the Data Supplement). Pathway analysis of genes associated with differentially methylated cytosines using the Ingenuity Pathway Analysis tox list analysis revealed an association of DNA methylation differences between knockout versus wild-type EHTs with the cardiac hypertrophy and increases cardiac proliferation pathways, as well as the signaling pathways p53 signaling, transforming growth factor β signaling, NRF2-mediated oxidative stress response, and retinoic acid receptor activation (Figure IX in the Data Supplement).
In line with the concept of DNA methylation-mediated gene silencing, semitargeted gene expression analysis with a custom-designed NanoString panel revealed significantly higher expression of most of the analyzed low-abundance genes in the knockout EHTs compared with wild-type (Figure 3A). In contrast with the low-abundance genes, genes with high transcript abundance in cardiomyocytes did not consistently show increased expression in knockout compared with wild-type, except for MYH6 and ATP2A2 (Figure 3B). It is important to note that genes not expressed at all in wild-type EHTs remained silenced in DNMT3A−/− EHTs (data not shown). Cluster analysis of whole-transcriptome RNA sequencing data moreover revealed close clustering of samples with their respective genotype replicates, arguing for rather specific gene expression changes (Figure VIIIC in the Data Supplement). In line with this, pathway mapping of significantly differentially expressed genes in the knockout versus wild-type EHTs showed association of the gene expression changes with many cardiac-, signaling-, and metabolism-related pathways (Figure 3C). Looking at the differentially expressed genes, only a small number of genes were also associated with differentially methylated cytosines, suggesting rather limited direct gene regulation by DNA methylation in the EHTs (Figure VIIID in the Data Supplement). Instead, the observed DNA methylation changes seemed to have an effect on the activity of upstream regulators, which, in turn, brought about the observed gene expression changes. In line with this, upstream regulator analysis of the RRBS data using Ingenuity Pathway Analysis software identified a number of regulators affected by the observed DNA methylation changes (Table I in the Data Supplement).
Figure 3.
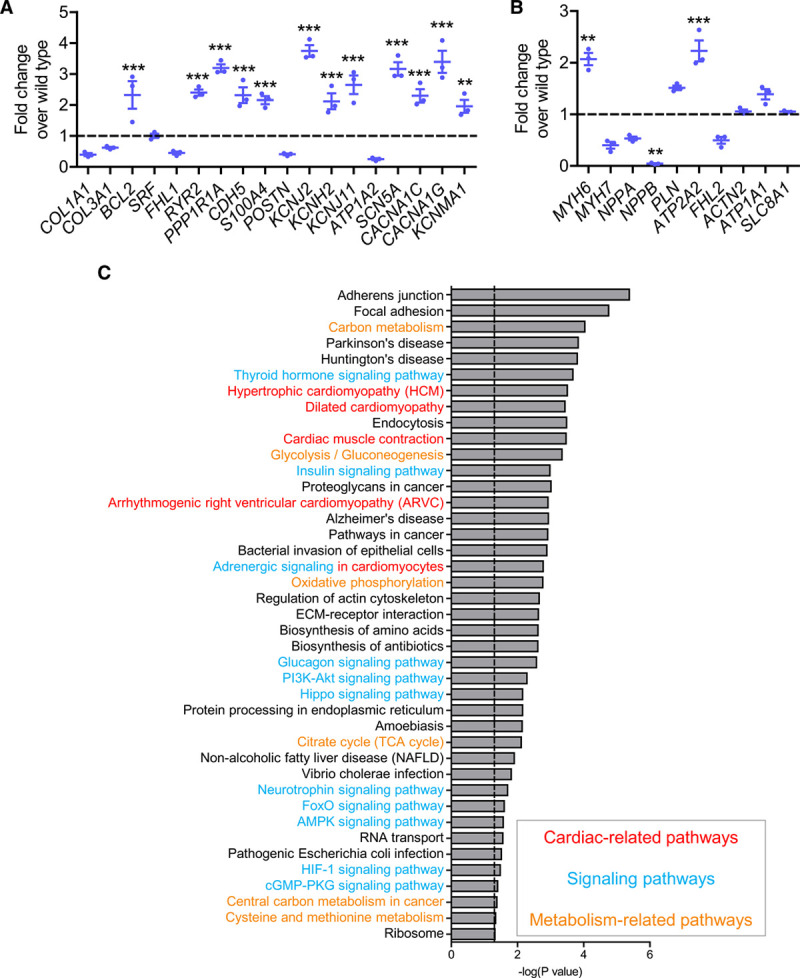
Gene expression changes in homozygous DNA methyltransferase(DNMT) 3A knockout engineered heart tissue(EHT).
Transcript abundance of (A) low-expression genes and (B) high-expression genes in DNMT3A−/− EHTs. All values normalized to wild-type (dashed line); n=3 per group, **P<0.001, ***P<0.001. C, Pathway mapping of significantly differentially expressed genes in DNMT3A−/− vs wild-type. Dotted line represents significance threshold of P<0.05. Red, cardiac-related; blue, signaling-related; yellow, metabolism-related pathways.
Dysregulation of PPARγ Induces Lipid Accumulations
Although no overt differences in EHT development could be observed at first sight, knockout EHTs developed distinct morphological features compared with wild-type EHTs. Knockout EHTs retained a significantly higher overall diameter than wild-type and isogenic control EHTs over the whole culture period starting on day 3 of culture, indicating reduced remodeling (1.3–1.4 mm versus 1.0 mm after 3 weeks of culture; Figure XA through XF in the Data Supplement). The larger EHT diameter observed in knockout EHT, however, did not seem to result from a higher content of extracellular matrix proteins but rather from a higher total cell volume inside the EHTs (Figure XG through XI in the Data Supplement), which was not paralleled by higher proliferation of cells (Figure XI in the Data Supplement). Histological analysis by hematoxylin and eosin staining revealed large vacuolic structures inside the cells of all knockout EHTs, which were absent in wild-type EHTs (Figure 4A and 4B, left panels). Staining of the cardiomyocyte marker MLC2V displayed a clear signal surrounding the vacuoles, suggesting that the vacuoles were indeed situated within the cardiomyocytes (Figure 4A and 4B, middle panels). Lipid staining with Oil Red O on fixed intact EHTs revealed a distinct dotted staining pattern in the knockout EHTs, however seemingly less abundant than the total number of vacuolic cells in each EHT, whereas no staining was detected in wild-type EHTs (Figure 4A and 4B, right panels). Ultrastructural analysis by transmission electron microscopy revealed no difference between knockout and wild-type EHTs with regard to sarcomeric structure alignment (Figure 4C and 4D; Figure XIIA through XIID in the Data Supplement, yellow arrowheads), whereas the mitochondria in knockout cardiomyocytes were swollen and displayed an impaired cristae structure compared with wild-type (black arrowheads), suggesting impairment of mitochondrial metabolic function. Accumulation of lipid droplets was mainly found in (pre)apoptotic cells lacking sarcomeric structures in DNMT3A knockout EHTs but not in wild-type EHTs (Figure XIIE in the Data Supplement). In line with the electron microscopy results, analysis of mitochondrial function by Seahorse technology revealed significantly lower basal and maximal respiration, lower nonmitochondrial respiration, as well as lower ATP production in knockout compared with wild-type cells (Figure XIII in the Data Supplement), clearly supporting an impairment of mitochondrial metabolism in the knockout.
Figure 4.
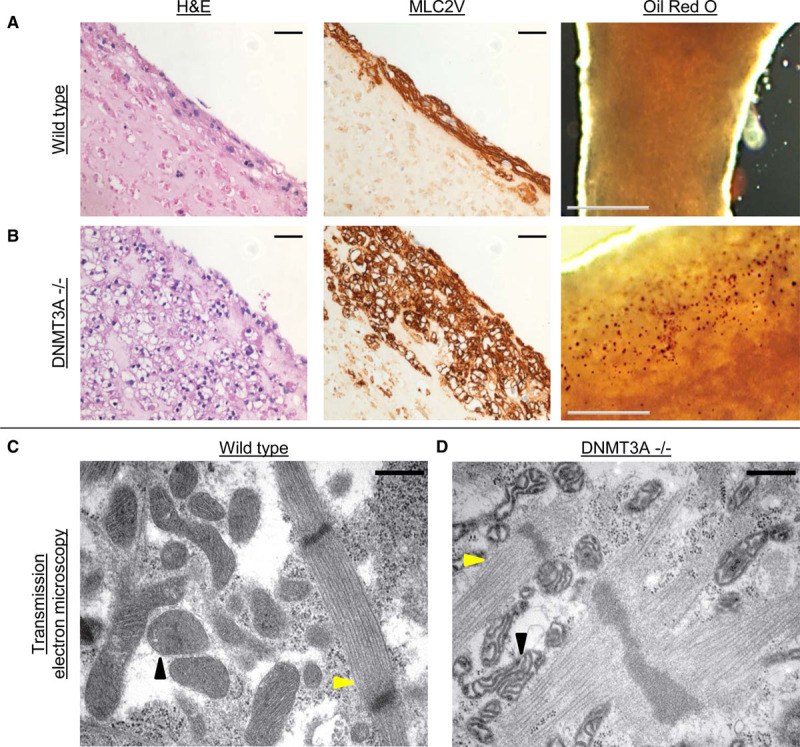
Morphology of DNA methyltransferase(DNMT) 3A knockout engineered heart tissue(EHT).
Histological analysis of (A) wild-type and (B) DNMT3A−/− EHTs. Left, Hematoxylin and eosin staining (H&E); scale bar, 50 µm. Middle, MLC2V staining; scale bar, 50 µm. Right, Lipid staining with Oil Red O; scale bar, 500 µm, red dots mark lipid accumulations. Transmission electron microscopy of wild-type (C) and knockout (D) EHTs showing intact sarcomeric structures but degenerated mitochondria in knockout EHT. Yellow arrowheads, sarcomeres; black arrowheads, mitochondria. Scale bar, 500 nm.
An important regulator of cell lipid metabolism is the PPARγ. Expression analysis revealed a significantly higher PPARγ transcript abundance in the knockout EHTs than in wild-type EHT (Figure 5A). DNA methylation analysis of the PPARγ promoter region revealed lower methylation in 6 of the 7 analyzed CpG sites (Figure 5B), suggesting a direct link between loss of DNMT3A-mediated methylation and PPARγ upregulation. Treatment of knockout EHTs with the PPARγ inhibitor GW9662 starting on day 14 of culture revealed no differences between inhibitor-treated and vehicle-treated EHTs regarding beating frequency, force generation, or occurrence of arrhythmias under both serum-containing and serum-free conditions (Figure XIV in the Data Supplement). However, histological analysis revealed a strong reduction of the vacuolic structures in inhibitor-treated EHTs (Figure 5C). These data suggest that increased PPARγ in the absence of DNMT3A indeed drives cardiomyocyte lipid accumulation but that lipid accumulation and functional decline are two independent phenomena in DNMT3A knockout EHTs. This concept was supported by histological analysis of knockout EHTs cultured in serum-containing medium for the whole culture period, which showed accumulation of lipids despite the absence of any functional impairment (Figure 5D).
Figure 5.

Upregulation ofperoxisome proliferator-activated receptor gamma (PPARγ) is responsible for lipid accumulation inengineered heart tissue (EHT).
A, Normalized PPARγ transcripts in wild-type and DNA methyltransferase (DNMT) 3A−/− EHTs. n=3 EHTs per group, ***P<0.001. B, PPARγ promoter methylation in wild-type and DNMT3A−/− EHTs measured by bisulfite sequencing. Black dot, methylated cytosine; white dot, unmethylated cytosine. C, Hematoxylin and eosin staining (H&E) staining of vehicle (left) and PPARγ-inhibitor GW9662–treated DNMT3A−/− EHTs (right); scale bar, 50 µm. D, H&E staining of DNMT3A−/− EHTs cultured for 4 weeks in serum-containing conditions; scale bar, 50 µm.
DNMT3A-Deficiency Leads to Impairment of Glycolysis by HIF-1α Destabilization
Even small changes in cell metabolism can have an impact on cardiomyocyte contractility because of high energy demand. To analyze the integrity of glucose metabolism of wild-type and knockout cardiomyocytes, glucose and lactate concentrations were measured in EHT medium 24 h after medium change. It is interesting that knockout EHTs consumed less glucose and produced less lactate in serum-free medium, dialysis-only serum, and lipoprotein-deficient serum than in serum-containing standard conditions (Figure XVA and XVB in the Data Supplement). For isogenic control EHTs no difference was observed between the serum conditions. Moreover, knockout EHTs cultured with glucose as the only energy source in serum-free medium showed the same progressive functional decline as EHTs cultured under serum-free conditions, whereas EHTs cultured with lactate as the only energy source retained stable function. Wild-type EHTs, on the other hand, retained stable contractility in both conditions (Figure 6A through 6C), indicating a major difference in glucose handling between wild-type and DNMT3A knockout.
Figure 6.

Effect of DNA methyltransferase(DNMT) 3A knockout on engineered heart tissue(EHT) glucose metabolism.
Effect of restrictive feeding with lactate or glucose-only medium on frequency (A), force (B), and RR scatter, with R being the interval between 2 neighboring force peaks, (C) of wild-type and knockout EHTs. n=8, *P<0.05, ***P<0.001. D, Relative transcript abundance of glycolytic genes in DNMT3A−/− #2 EHTs after 1 week of serum-free culture. n=3, values normalized to wild-type (dashed line). E and F, Quantification of hypoxia-inducible factor 1α (HIF-1α) protein by Western blot of DNMT3A−/− #1 and isogenic control EHTs cultured either with lipoprotein-deficient serum (LDS), control serum, or dialyzed serum. Loading control = ERK1/2; n=2 to 3 EHTs per group.
RNA sequencing of knockout EHTs after 1 week of serum-free culture showed significantly lower transcript abundance of almost all enzymes involved in the glycolysis pathway (Figure 6D). In accordance with the downregulation of glycolytic enzymes in knockout EHTs, the regulator of most glycolytic enzymes, HIF-1α, showed lower protein abundance in knockout compared with isogenic control EHTs (Figure 6E and 6F). In contrast, glycolytic gene expression of knockout EHTs permanently cultured under serum-containing conditions did not differ from wild-type EHTs (Figure XVC in the Data Supplement). In line with this observation, no difference in glycolytic capacity was found between wild-type and knockout cells in the Seahorse Glycolysis Stress Test of cells cultured with the serum replacement B27 for 1 week (Figure XVD and XVE in the Data Supplement). Taken together, these data suggest a destabilization of HIF-1α protein in DNMT3A knockout EHTs, which, under serum-free conditions, leads to an impaired glucose metabolism by deregulation of glycolytic enzymes, which, in turn, causes the observed functional decline of the EHTs.
Serum Growth Factors Counteract Glycolytic Impairment in the Absence of DNMT3A
The observation that small molecule-depleted sera had similar negative effects on EHT contractility as serum depletion suggests an involvement of low-molecular serum components, such as growth factors, in the contractility-preserving effect of serum. In line with this, supplementation of serum-free culture medium with a mixture of growth factors (basic fibroblast growth factor, insulin like-growth factor 1, endothelial growth factor, and transforming growth factor β1) prevented the functional decline of knockout EHTs (Figure 7A). The intervention moreover stabilized HIF-1α protein (Figure 7B and 7C), increased expression of HIF target genes (Figure 7D), and normalized glucose consumption and lactate generation of knockout EHTs (Figure 7E and 7F).
Figure 7.

Effect of growth factor treatment of engineered heart tissue(EHT) under serum-free conditions.
A, Effect of growth factor treatment (GFs; 20 ng/mL transforming growth factor [TGF]-β1, 10 ng/mL basic fibroblast growth factor [bFGF], 20 ng/mL insulin-like growth factor 1 [IGF-1], 10 ng/mL endothelial growth factor [EGF]) on force under serum-free conditions. n=10–15 EHTs from 2 batches per group. B and C, Quantification of hypoxia-inducible factor 1α (HIF-1α) protein by Western blot of wild-type and DNA methyltransferase (DNMT) 3A−/− EHTs cultured either with serum-free medium or with serum-free medium containing GFs. Loading control = ERK1/2; n=3 EHTs per group. D, Relative transcript abundance of glycolytic genes in DNMT3A−/− EHTs after 1 week of serum-free culture ± GFs. n=3, values normalized to wild-type (dashed line). Effect of GF treatment on glucose consumption (E) and lactate generation (F) of wild-type and DNMT3A−/− EHTs. n=4, values normalized to wild-type (dashed line). ns indicates nonsignificant, *P<0.05, **P<0.01, ***P<0.001.
Discussion
The role of DNA methylation in heart disease is a matter of debate. It has been suggested that de novo DNA methylation in terminally differentiated cell types such as cardiomyocytes is of little to no functional importance.22 In contrast with this, the present study revealed clear indications for important functions of DNA methylation in overall cardiomyocyte homeostasis and function. To begin with, the lower global DNA methylation we observed in our DNMT3A knockout EHTs strongly argues for continuous DNA methylation turnover in cardiomyocytes. Our RRBS Ingenuity Pathway Analysis tox list analysis additionally pointed to a role of DNA methylation in cardiomyocytes because it identified differences centered on cardiac-related pathways instead of methylation changes in random, unrelated pathways or none at all. Moreover, these differences in DNA methylation correlated with substantial differences of cardiomyocyte function. In summary, our model revealed 3 main effects of de novo DNA methylation-deficiency in EHT beyond aberrant methylation itself (Figure 8): (1) altered contraction kinetics correlating with different expression of contractile proteins, (2) deregulation of PPARγ and differences in overall EHT morphology comprising a higher diameter and intracellular lipid accumulation, and (3) protein instability of HIF-1α, leading to disturbed glycolysis and functional impairment of EHT under metabolic stress.
Figure 8.

Overview key experiments and findings.
Dashed boxes indicate key experimental findings; colored boxes indicate conclusions drawn from experimental findings.
The first striking difference we observed in our knockout EHTs compared with wild-type controls was a significant acceleration of contraction kinetics. The EHTs displayed a shorter relaxation time, rendering them able to adapt to higher stimulation frequencies than their wild-type counterparts. Gene expression analysis of contractile proteins revealed, among others, an unusual expression ratio of the two myosin heavy chain isoforms MYH7 (encoding the slow twitch β-MHC) and MYH6 (encoding α-MHC). This preferential expression of the faster α-MHC could contribute to the faster relaxation kinetics observed in the knockouts and could be directly attributed to the absence of DNMT3A in those cells: the switch from MYH6 to MYH7 during human cardiomyocyte development has been previously shown to be dependent on DNA methylation-mediated silencing of MYH6.16,42 Moreover, methylation changes in enhancers involved in MYH7 gene regulation43 could contribute to the altered expression. We additionally probed the MYH6 and MYH7 loci for the presence of the repressive histone mark H3K27me3 by ChIP-qPCR (Figure XVI in the Data Supplement). Mutual exclusivity of DNA methylation and this histone mark has been described.44,45 Because we found no differences between wild-type and knockout EHTs regarding H3K27me3, MYH6 could thus have been derepressed by demethylation in knockout. Going beyond direct gene regulation by DNA methylation, DNMTs additionally influence gene expression indirectly, for example by interaction with histone modifying enzymes and chromatin rearrangement8. Moreover, regulation of transcription factor expression by DNA methylation indirectly affects expression of the respective downstream genes. In this context, we performed mapping analysis of the RRBS data for upstream regulator identification. We identified EZH2 as an expression regulator of a set of differentially methylated genes. EZH2 interestingly forms part of the epigenetic repression complex PRC2 and has previously been implied in regulation of MYH6 expression by modulation of GATA446 and could therefore also have been involved in the altered MHC ratio. In addition to the MHC isoforms, we also observed higher expression of atrial-specific genes such as PITX2 and a higher MLC2A/MLC2V ratio in our knockout cardiomyocytes. In particular, the higher PITX2 expression could be involved in the observed faster relaxation kinetics. PITX2 is a key regulator of atrial cell fate decision,47 and atrial cells typically show significantly faster kinetics than their ventricular counterparts.48,49 The lower PITX2 promoter methylation moreover hints at a direct regulation of PITX2 by DNA methylation, as has been previously described in prostate cancer.50 These observations can additionally be read as an indicator that the knockout hiPSC cardiomyocytes are not less cardiomyocyte-like than their wild-type counterpart but instead have a different cell homeostasis status. Moreover, these findings also highlight the importance of a human model in addition to the established mouse knockout models. In contrast with human cardiomyocytes, the predominant myosin heavy chain isoform in adult mouse cardiomyocytes is the faster α-MHC, necessitating no DNA methylation-mediated switch from Myh6 to Myh7. This important species difference could thus explain the lack of a similar effect of Dnmt3a/3b double knockout on mouse cardiomyocyte contractility.22
Histological analyses of the EHTs after 30 days of culture revealed a second striking difference between knockout and wild-type EHTs. Cardiomyocytes lacking DNMT3A accumulated lipid vacuoles in their cytoplasm, similar to what has been previously observed in patients experiencing metabolic syndrome.51 Cardiomyocytes of patients in this study displayed significantly higher expression of PPARγ, which is strongly expressed in adipose tissue and, under physiological conditions, comparatively lowly abundant in cardiomyocytes and is involved in the homeostasis of lipid and glucose metabolism.52,53 In line with these findings, gene expression analysis in our knockout EHTs revealed significantly higher PPARγ transcript abundance compared with wild-type. Treatment with a PPARγ inhibitor was able to greatly reduce the lipid accumulations in knockout cardiomyocytes, arguing for a direct causal relationship of high PPARγ activity and morphological abnormalities, even though the exact downstream pathways remain elusive. This conclusion is further supported by observations from a study in mice, where cardiomyocyte-specific overexpression of PPARγ induced a similar formation of lipid vacuoles in the cytoplasm of cardiomyocytes, providing a direct link between PPARγ and lipid accumulation.54 In line with previous reports,55 we found lower PPARγ promoter methylation accompanying the higher PPARγ transcript abundance, suggesting that the observed higher transcript abundance of PPARγ in the knockout EHTs was a direct cause of the absence of DNA methylation–mediated repression by DNMT3A. The fact that transmission electron microscopy mainly showed accumulation of lipid droplets in apoptotic knockout cells, which had lost their sarcomeric structure, suggests a toxic effect of the accumulated lipids leading to cardiomyocyte remodeling and apoptosis. However, this needs to be further evaluated in the future. Besides lipid accumulation, transmission electron microscopy also revealed severely degenerated and swollen mitochondria in the knockout, which was accompanied by significantly impaired mitochondrial metabolism in the Seahorse experiments. Because DNMT3A has previously been described to be involved in regulating the methylome of mitochondrial DNA,56 the observed mitochondrial impairment could in part be a direct effect of DNMT3A knockout, although further research will have to be conducted on this. It is interesting that Li et al57 have observed lipid accumulations similar to what we saw in our EHTs in mice harboring a conditional cardiomyocyte-specific knockout of Pitx2. Although at a first glance this seems to be contradictory to the higher PITX2 expression we observed, Li et al found the fat-accumulating cells in their knockout model to be of noncardiomyocyte origin, whereas in the EHTs lipid droplets were located inside cardiomyocytes. Moreover, the same study also observed lipid accumulations in mice heterozygous for a knockout of Cox7c, a key mitochondrial PITX2 target gene, even in the presence of endogenous PITX2. This suggests accumulation of lipids to be secondary to an impairment of mitochondrial metabolism rather than a direct effect of PITX2 deletion, which is in line with our observations in the DNMT3A knockout EHTs, showing both mitochondrial metabolism deficits and accumulation of lipids.
The third and most momentous effect of DNMT3A-deficiency in EHT was a severe disturbance of the glycolysis pathway attributable to instability of the master regulator of glycolytic enzymes, HIF-1α, leading to functional deterioration of knockout EHTs under metabolic stress. Our observation was in line with results from a study using siRNA-mediated knockdown of Dnmt3a in mouse cardiomyocytes cultured under low-serum conditions, resulting in lower force and arrhythmic beating.58 In contrast with PPARγ, HIF-1α does not seem to be subject to direct regulation by DNA methylation, because there was no difference in DNA methylation or mRNA abundance of HIF-1α in knockout EHTs compared with wild-type (data not shown). HIF-1α is mainly regulated at the posttranslational level: under normoxic conditions, HIF-1α is hydroxylated by prolyl hydroxylase domain–containing proteins, followed by poly-ubiquitination by the von Hippel–Lindau E3 ubiquitin ligase complex and subsequent proteasomal degradation.59–61 Hypoxia prevents HIF-1α degradation leading to stabilization of the protein which can then act as a transcription factor.62 In addition, oxygen-independent mechanisms including growth factors, nutrient concentrations, and posttranslational modifications such as phosphorylation can influence HIF-1α protein stability.63–65 Thus, the lower protein abundance of HIF-1α which we detected by Western blot likely indicates insufficient stabilization of HIF-1α protein in the knockout cardiomyocytes owing to mechanisms only indirectly governed by DNA methylation. This protein instability resulted in downregulation of many HIF-1α target genes, which, in turn, impaired glucose metabolism and led to decreased contractility under metabolic stress conditions. In the situation of overall perturbed cell homeostasis attributable to DNMT3A knockout, the high abundance of growth factors in serum might have been indispensable for knockout cardiomyocytes to trigger sufficient metabolic activity to sustain contractility and overcome insufficient HIF-1α stability. This hypothesis is supported by the results of the growth factor treatment experiment, showing not only sustained contractility of growth factor–treated knockout EHTs under serum-free conditions but also partially restored HIF-1α protein and glycolytic gene expression, as well as normalization of glucose consumption. Although deregulation of HIF-1α and its target genes did not result in an obvious phenotype under physiological conditions, it nevertheless affected overall cell homeostasis and cardiomyocyte function, which only became apparent under stress conditions. This observation could provide another explanation for the discrepancy between our results and the absence of an effect of the cardiomyocyte-specific knockout which had been described in mice, under both physiological and less harsh pathological conditions.22 Although in our study harsh serum manipulation interventions where necessary to reveal the phenotype, which raises the question whether these findings are relevant for any physiological setting, we believe that they hint at a general importance of DNA methylation in humans. Especially DNA methylation ageing and its counteraction by DNMTs, viewed together with the rising incidence of heart disease in higher age, points toward a possible role of DNMTs in cardiomyocytes that could not possibly have been observed in mice. Taken together, these results suggest an important fine-tuning function of DNMT3A for physiological cardiomyocyte metabolism and function, which needs extreme conditions or possibly long-term exposure to less extreme stimuli during the lifespan of a human being to become apparent.
Taken together, the findings of this study are in line with a more general concept of the role of DNA methylation in gene regulation: Fully silenced genes carry additional repressive chromatin marks on top of transcription-blocking DNA methylation and are therefore not affected by knockout of DNMT3A. In contrast, partial repression of lowly expressed genes seems to be more dependent on promoter DNA methylation state, leading to a leaky expression of the corresponding gene.16 In the absence of repressing DNA methylation marks, expression of leaky genes can therefore be considerably increased, whereas the silenced genes stay repressed by their additional chromatin features. Thus, de novo DNA methylation-deficiency would not be expected to lead to drastic changes of overall transcriptional regulation but rather to influence the fine-tuning of cell homeostasis, as supported by pathway mapping of differentially expressed genes, resulting in many cardiac-, signaling-, and metabolism-related pathways. Because cardiomyocytes are cells with an exceptionally high energy turnover, required for contractile work, they highly rely on a strict regulation of metabolism, and it seems straightforward that metabolism was among the first functions to be disrupted in DNMT3A knockout EHT. This observation is also in line with other studies reporting alterations in metabolic pathways as one of the earliest phenotypic changes in epigenetically modified cells,13,16,66 suggesting epigenetic mechanisms as a means of metabolic fine-tuning in cardiomyocytes.
Limitations
The present study used CRISPR/Cas9 to knock out DNMT3A in hiPSCs to elucidate the role of de novo DNA methylation in hiPSC-derived cardiomyocytes in 3-dimensional EHT. Although this approach has the limitation that DNMT3A was already knocked out in undifferentiated hiPSC and could have therefore influenced the results by impeding cardiac differentiation,67,68 as the lower expression of some highly expressed cardiac genes in the knockout (eg, NPPB) could suggest, there are several arguments for the validity and importance of our results. Our own gene expression analysis in the hiPSC and hiPSC-derived cardiomyocytes, as well as published expression analyses in human embryonic stem cells and cell types derived from these, suggest a much greater importance of the second de novo DNMT isoform, DNMT3B, in undifferentiated cells.29 Moreover, looking at the abundance of the 3 DNMT isoforms over the course of our hiPSC differentiation protocol (Figure XVII in the Data Supplement), we observed no changes in the expression of DNMT1 and DNMT3A, whereas DNMT3B was expressed dynamically, from very high expression in undifferentiated cells toward a steady decline over time. It is interesting that the start of embryoid body contraction coincided with the disappearance of DNMT3B transcripts to background level, suggesting a high importance of DNMT3B for mesodermal induction and early cardiac differentiation but its dispensability in later stages. This finding is also supported by a study on the DNMT isoform activity pattern during mouse development, proposing a sequential activation of Dnmt3b during early development and Dnmt3a during later stages, as well as the fact that a global knockout of Dnmt3b was shown to be embryonically lethal, whereas Dnmt3a knockout mice developed to term and became runted and died only at 3 weeks of age.69 Moreover, looking at the expression pattern of PPARγ (Figure XVIII in the Data Supplement), we observed low levels of PPARγ directly after dissociation but continuously increasing abundance over EHT culture duration, arguing for a later time point of DNMT3A dependent repression. Together, these observations point to a more important function of DNMT3A during later developmental stages instead of early cell type differentiation. In addition, quality controls of the hiPSC-derived cardiomyocytes in the present study revealed no differences between knockout and wild-type cells regarding both the proportion of cardiac troponin T–positive cells and the differentiation efficiency, arguing strongly against a negative impact of the knockout on the outcome of cardiomyocyte differentiation.
Related to this first limitation, it has to be kept in mind that knockout of DNMT3A in undifferentiated hiPSCs will also affect noncardiomyocytes present in the cell mixture after differentiation. This could influence the cardiomyocyte phenotype, for example, by altered paracrine signaling of those cells. However, because the observed phenotype was still present in EHTs from 99% cardiomyocytes (data not shown), we think this effect to be negligible for our experiments.
The karyotypic abnormality which was present in the wild-type line used for CRISPR/Cas9 gene knockout can be considered as another limitation, because the trisomy in itself might already influence EHT morphology and function. However, EHT experiments performed with the isogenic control wild-type cell line, carrying the same trisomy as the knockout lines, indicated that the phenotype observed in the knockout was mostly attributable to lack of DNMT3A and not the trisomy itself. Nonetheless, we cannot fully exclude that some experiments and results might have been biased by the trisomy.
Last, some experiments, such as RNA sequencing and DNA methylation analysis by RRBS, used a rather small (3–4) number of replicates, making statistical assumptions difficult. Because the EHT model overall shows rather small data scattering of replicates and key functional experiments were done at n>50 from different batches of cells, we believe our results to be nonetheless valid.
Conclusions
We observed direct and indirect effects of DNMT3A knockout in hiPSC-derived cardiomyocytes on 3 distinct aspects of cardiomyocyte biology and function—contractility, cell morphology, and cardiomyocyte metabolism—emphasizing the importance of de novo methylation for physiological cardiomyocyte homeostasis. Even though compensatory mechanisms in vivo seem to be able to counterbalance the absence of DNMT3A on the short term and even in a disease situation,22 we believe our findings to be relevant for future therapeutic approaches. In the present study, serum withdrawal as a harsh intervention was able to uncover the severe functional implications of deregulated DNA methylation in the very short timeframe of only a few days. It would therefore be conceivable that long-term deregulation of DNA methylation under less harsh conditions, such as in the setting of aging, could also have a currently underestimated impact on cardiac disease progression in human. Examples for this can be seen in studies on clonal hematopoiesis with indeterminate potential, showing association of DNMT3A mutations in peripheral blood cells with development of cardiovascular diseases over time.70 Viewed together with the results from DNA methylation inhibition in models of cardiac hypertrophy suggesting a positive therapeutic effect of DNA methylation modulation,15,18–20 our results point to a potential relevance of DNA methylation in the setting of cardiac disease, which should be explored in more detail in the future.
Acknowledgments
The authors thank Birgit Klampe, Thomas Schulze, Elisabeth Krämer, Zenia Tiang, Tim Hartmann, Bangfen Pan, and Emanuela Szpotowicz for excellent technical assistance, Bärbel M. Ulmer, Aya Shibamiya, Ingke Braren, and Arne Hansen for hiPSC line generation, Anika E. Knaust and Antonia T. L. Zech for support during CRISPR experiments and differentiation, Eleanor Wong for support with RRBS, Jörg Heeren and Sandra Ehret for providing lipoprotein-deficient serum, and the group of Petra Arck for granting us access to their BGA setup. The authors also thank Kristin Hartmann from the mouse pathology core facility at the University Medical Center Hamburg-Eppendorf (UKE) for histological stainings, Sigrid Fuchs and Jennifer Kaiser at the UKE Institute of Human Genetics for karyotyping, the FACS core facility at the UKE for support with FACS and flow cytometry, and the sequencing core facility at the Genome Institute of Singapore for sequencing of RRBS and RNA samples. J.S., A.M., R.S.Y.F., and T.E. designed the experiments. J.K. and M.N.H. contributed additional conceptual work. A.M., J.S., and T.E. wrote the manuscript. A.M. and S.D.L. performed CRISPR/Cas9 experiments. A.M. performed cardiac differentiation, EHT experiments, blood gas analysis, histology analysis, Oil Red O staining, bisulfite sequencing together with G.H., and analysis and pathway mapping of gene expression and RRBS data. G.H. performed qPCR experiments together with A.M., Western blotting, and RNA/DNA extraction. M.S. performed electron microscopy. D.M. performed Seahorse experiments. C.G.A.-N.performed chromatin immunoprecipitation- (ChIP-)-qPCR. J.S. performed RRBS. Sequencing raw data processing, analysis, and quality control, as well as clustering and Venn diagram preparation was performed by W.L.W.T. and I.L. Prof Hansen is head of the stem cell method development and disease modeling group in the department in which the greater part of the work was carried out and with which both first and last author are affiliated. Prof Hansen and his group were instrumental in establishing all necessary stem cell related procedures, from differentiation, to iPS-CM based tissue engineering and to the establishment of the Crispr technology. He provided protocols, infrastructure, funding and management necessary for the current work, and also personally provided scientific input with regard to problems arising during the course of the work in numerous meetings and personal conversations, as well as in emails exchanged with the first and the last author and several other authors.
Sources of Funding
This work was supported by the German Research Foundation (DFG, STE2596), the Singapore National Medical Research Council (R-172-000-335-511), the National Center for the Replacement, Refinement, and Reduction of Animals in Research (NC3Rs: NC/S001808/1), and the European Research Council (IndivuHeart, NCT02417311).
Disclosures
Drs Eschenhagen and Hirt are cofounders of EHT Technologies GmbH, Hamburg. The other authors report no conflicts.
Supplemental Materials
Expanded Methods and Materials
Data Supplement Figures I–XIX
Data Supplement Tables I–IV
Supplementary Material
Footnotes
Sources of Funding, see page 1577
This manuscript was sent to Prof. Mauro Giacca, Guest Editor, for review by expert referees, editorial decision, and final disposition.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/circulationaha.119.044444.
References
- 1.Ponikowski P, Anker SD, Alhabib KF, Cowie MR, Force TL, Hu S, Joorsma T, Krum H, Rastogi V, Rohde LE, et al. Heart failure: Preventing disease and death worldwide. ESC Heart Fail. 2014; 373:941–955. doi: 10.1002/ehf2.12005 [DOI] [PubMed] [Google Scholar]
- 2.Takahashi T, Allen PD, Izumo S. Expression of A-, B-, and C-type natriuretic peptide genes in failing and developing human ventricles: correlation with expression of the Ca(2+)-ATPase gene. Circ Res. 1992; 71:9–17. doi: 10.1161/01.res.71.1.9 [DOI] [PubMed] [Google Scholar]
- 3.Arai M, Alpert NR, MacLennan DH, Barton P, Periasamy M. Alterations in sarcoplasmic reticulum gene expression in human heart failure: a possible mechanism for alterations in systolic and diastolic properties of the failing myocardium. Circ Res. 1993; 72:463–469. doi: 10.1161/01.res.72.2.463 [DOI] [PubMed] [Google Scholar]
- 4.Locher MR, Razumova MV, Stelzer JE, Norman HS, Moss RL. Effects of low-level & α-myosin heavy chain expression on contractile kinetics in porcine myocardium. Am J Physiol Heart Circ Physiol. 2011; 300:H869–H878. doi: 10.1152/ajpheart.00452.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008; 454:766–770. doi: 10.1038/nature07107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bashtrykov P, Jankevicius G, Jurkowska RZ, Ragozin S, Jeltsch A. The UHRF1 protein stimulates the activity and specificity of the maintenance DNA methyltransferase DNMT1 by an allosteric mechanism. J Biol Chem. 2014; 289:4106–4115. doi: 10.1074/jbc.M113.528893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998; 19:219–220. doi: 10.1038/890 [DOI] [PubMed] [Google Scholar]
- 8.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006; 31:89–97. doi: 10.1016/j.tibs.2005.12.008 [DOI] [PubMed] [Google Scholar]
- 9.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002; 110:479–488. doi: 10.1016/s0092-8674(02)00861-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007; 13:324–331. doi: 10.1038/nm1552 [DOI] [PubMed] [Google Scholar]
- 11.Wei JQ, Shehadeh LA, Mitrani JM, Pessanha M, Slepak TI, Webster KA, Bishopric NH. Quantitative control of adaptive cardiac hypertrophy by acetyltransferase p300. Circulation. 2008; 118:934–946. doi: 10.1161/CIRCULATIONAHA.107.760488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang QJ, Chen HZ, Wang L, Liu DP, Hill JA, Liu ZP. The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J Clin Invest. 2011; 121:2447–2456. doi: 10.1172/JCI46277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kronlage M, Dewenter M, Grosso J, Fleming T, Oehl U, Lehmann LH, Falcão-Pires I, Leite-Moreira AF, Volk N, Gröne HJ, et al. O-GlcNacylation of histone deacetylase 4 protects the diabetic heart from failure. Circulation. 2019; 140:580–594. doi: 10.1161/CIRCULATIONAHA.117.031942 [DOI] [PubMed] [Google Scholar]
- 14.Movassagh M, Choy MK, Knowles DA, Cordeddu L, Haider S, Down T, Siggens L, Vujic A, Simeoni I, Penkett C, et al. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011; 124:2411–2422. doi: 10.1161/CIRCULATIONAHA.111.040071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haas J, Frese KS, Park YJ, Keller A, Vogel B, Lindroth AM, Weichenhan D, Franke J, Fischer S, Bauer A, et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol Med. 2013; 5:413–429. doi: 10.1002/emmm.201201553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilsbach R, Preissl S, Grüning BA, Schnick T, Burger L, Benes V, Würch A, Bönisch U, Günther S, Backofen R, et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun. 2014; 5:5288. doi: 10.1038/ncomms6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao D, Dasgupta C, Chen M, Zhang K, Buchholz J, Xu Z, Zhang L. Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats. Cardiovasc Res. 2014; 101:373–382. doi: 10.1093/cvr/cvt264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watson CJ, Horgan S, Neary R, Glezeva N, Tea I, Corrigan N, McDonald K, Ledwidge M, Baugh J. Epigenetic therapy for the treatment of hypertension-induced cardiac hypertrophy and fibrosis. J Cardiovasc Pharmacol Ther. 2016; 21:127–137. doi: 10.1177/1074248415591698 [DOI] [PubMed] [Google Scholar]
- 19.Stenzig J, Hirt MN, Löser A, Bartholdt LM, Hensel JT, Werner TR, Riemenschneider M, Indenbirken D, Guenther T, Müller C, et al. DNA methylation in an engineered heart tissue model of cardiac hypertrophy: common signatures and effects of DNA methylation inhibitors. Basic Res Cardiol. 2016; 111:9. doi: 10.1007/s00395-015-0528-z [DOI] [PubMed] [Google Scholar]
- 20.Stenzig J, Schneeberger Y, Löser A, Peters BS, Schaefer A, Zhao RR, Ng SL, Höppner G, Geertz B, Hirt MN, et al. Pharmacological inhibition of DNA methylation attenuates pressure overload-induced cardiac hypertrophy in rats. J Mol Cell Cardiol. 2018; 120:53–63. doi: 10.1016/j.yjmcc.2018.05.012 [DOI] [PubMed] [Google Scholar]
- 21.Vujic A, Robinson EL, Ito M, Haider S, Ackers-Johnson M, See K, Methner C, Figg N, Brien P, Roderick HL, et al. Experimental heart failure modelled by the cardiomyocyte-specific loss of an epigenome modifier, DNMT3B. J Mol Cell Cardiol. 2015; 82:174–183. doi: 10.1016/j.yjmcc.2015.03.007 [DOI] [PubMed] [Google Scholar]
- 22.Nührenberg TG, Hammann N, Schnick T, Preißl S, Witten A, Stoll M, Gilsbach R, Neumann FJ, Hein L. Cardiac myocyte de novo DNA methyltransferases 3a/3b are dispensable for cardiac function and remodeling after chronic pressure overload in mice. PLoS One. 2015; 10:e0131019. doi: 10.1371/journal.pone.0131019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mannhardt I, Breckwoldt K, Letuffe-Brenière D, Schaaf S, Schulz H, Neuber C, Benzin A, Werner T, Eder A, Schulze T, et al. Human engineered heart tissue: analysis of contractile force. Stem Cell Reports. 2016; 7:29–42. doi: 10.1016/j.stemcr.2016.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Breckwoldt K, Letuffe-Brenière D, Mannhardt I, Schulze T, Ulmer B, Werner T, Benzin A, Klampe B, Reinsch MC, Laufer S, et al. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat Protoc. 2017; 12:1177–1197. doi: 10.1038/nprot.2017.033 [DOI] [PubMed] [Google Scholar]
- 25.Hirt MN, Sörensen NA, Bartholdt LM, Boeddinghaus J, Schaaf S, Eder A, Vollert I, Stöhr A, Schulze T, Witten A, et al. Increased afterload induces pathological cardiac hypertrophy: a new in vitro model. Basic Res Cardiol. 2012; 107:307. doi: 10.1007/s00395-012-0307-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eder A, Hansen A, Uebeler J, Schulze T, Neuber C, Schaaf S, Yuan L, Christ T, Vos MA, Eschenhagen T. Effects of proarrhythmic drugs on relaxation time and beating pattern in rat engineered heart tissue. Basic Res Cardiol. 2014; 109:436. doi: 10.1007/s00395-014-0436-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eder A, Vollert I, Hansen A, Eschenhagen T. Human engineered heart tissue as a model system for drug testing. Adv Drug Deliv Rev. 2016; 96:214–224. doi: 10.1016/j.addr.2015.05.010 [DOI] [PubMed] [Google Scholar]
- 28.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013; 8:2281–2308. doi: 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao J, Karnik R, Gu H, Ziller MJ, Clement K, Tsankov AM, Akopian V, Gifford CA, Donaghey J, Galonska C, et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet. 2015; 47:469–478. doi: 10.1038/ng.3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen A, Eder A, Bönstrup M, Flato M, Mewe M, Schaaf S, Aksehirlioglu B, Schwoerer AP, Schwörer A, Uebeler J, et al. Development of a drug screening platform based on engineered heart tissue. Circ Res. 2010; 107:35–44. doi: 10.1161/CIRCRESAHA.109.211458 [DOI] [PubMed] [Google Scholar]
- 31.Hirt MN, Boeddinghaus J, Mitchell A, Schaaf S, Börnchen C, Müller C, Schulz H, Hubner N, Stenzig J, Stoehr A, et al. Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. J Mol Cell Cardiol. 2014; 74:151–161. doi: 10.1016/j.yjmcc.2014.05.009 [DOI] [PubMed] [Google Scholar]
- 32.Goldstein JL, Basu SK, Brown MS. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983; 98:241–260. doi: 10.1016/0076-6879(83)98152-1 [DOI] [PubMed] [Google Scholar]
- 33.Schlegel A, Stainier DY. Microsomal triglyceride transfer protein is required for yolk lipid utilization and absorption of dietary lipids in zebrafish larvae. Biochemistry. 2006; 45:15179–15187. doi: 10.1021/bi0619268 [DOI] [PubMed] [Google Scholar]
- 34.Fraher D, Ellis MK, Morrison S, McGee SL, Ward AC, Walder K, Gibert Y. Lipid abundance in zebrafish embryos is regulated by complementary actions of the endocannabinoid system and retinoic acid pathway. Endocrinology. 2015; 156:3596–3609. doi: 10.1210/EN.2015-1315 [DOI] [PubMed] [Google Scholar]
- 35.Mosqueira D, Lis-Slimak K, Denning C. High-throughput phenotyping toolkit for characterizing cellular models of hypertrophic cardiomyopathy in vitro. Methods Protoc. 2019; 2:83. doi: 10.3390/mps2040083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prondzynski M, Krämer E, Laufer SD, Shibamiya A, Pless O, Flenner F, Müller OJ, Münch J, Redwood C, Hansen A, et al. Evaluation of MYBPC3 trans-splicing and gene replacement as therapeutic options in human iPSC-derived cardiomyocytes. Mol Ther Nucleic Acids. 2017; 7:475–486. doi: 10.1016/j.omtn.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 38.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13. doi: 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018; 36:765–771. doi: 10.1038/nbt.4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Närvä E, Autio R, Rahkonen N, Kong L, Harrison N, Kitsberg D, Borghese L, Itskovitz-Eldor J, Rasool O, Dvorak P, et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat Biotechnol. 2010; 28:371–377. doi: 10.1038/nbt.1615 [DOI] [PubMed] [Google Scholar]
- 41.Taapken SM, Nisler BS, Newton MA, Sampsell-Barron TL, Leonhard KA, McIntire EM, Montgomery KD. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat Biotechnol. 2011; 29:313–314. doi: 10.1038/nbt.1835 [DOI] [PubMed] [Google Scholar]
- 42.Han P, Li W, Yang J, Shang C, Lin C-H, Cheng W, Hang CT, Cheng H-L, Chen C-H, Wong J, et al. Epigenetic response to environmental stress: assembly of BRG1–G9a/GLP–DNMT3 repressive chromatin complex on Myh6 promoter in pathologically stressed hearts. Biochim Biophys Acta. 20161–10. doi: 10.1016/j.bbamcr.2016.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934–947. doi: 10.1016/j.cell.2013.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jermann P, Hoerner L, Burger L, Schübeler D. Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc Natl Acad Sci U S A. 2014; 111:E3415–E3421. doi: 10.1073/pnas.1400672111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019; 20:590–607. doi: 10.1038/s41580-019-0159-6 [DOI] [PubMed] [Google Scholar]
- 46.He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, Wang G, Marquez VE, Orkin SH, Pu WT. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012; 26:37–42. doi: 10.1101/gad.173930.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tessari A, Pietrobon M, Notte A, Cifelli G, Gage PJ, Schneider MD, Lembo G, Campione M. Myocardial Pitx2 differentially regulates the left atrial identity and ventricular asymmetric remodeling programs. Circ Res. 2008; 102:813–822. doi: 10.1161/CIRCRESAHA.107.163188 [DOI] [PubMed] [Google Scholar]
- 48.Lemme M, Ulmer BM, Lemoine MD, Zech ATL, Flenner F, Ravens U, Reichenspurner H, Rol-Garcia M, Smith G, Hansen A, et al. Atrial-like engineered heart tissue: an in vitro model of the human atrium. Stem Cell Reports. 2018; 11:1378–1390. doi: 10.1016/j.stemcr.2018.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krause J, Löser A, Lemoine MD, Christ T, Scherschel K, Meyer C, Blankenberg S, Zeller T, Eschenhagen T, Stenzig J. Rat atrial engineered heart tissue: a new in vitro model to study atrial biology. Basic Res Cardiol. 2018; 113:41. doi: 10.1007/s00395-018-0701-2 [DOI] [PubMed] [Google Scholar]
- 50.Vinarskaja A, Schulz WA, Ingenwerth M, Hader C, Arsov C. Association of PITX2 mRNA down-regulation in prostate cancer with promoter hypermethylation and poor prognosis. Urol Oncol. 2013; 31:622–627. doi: 10.1016/j.urolonc.2011.04.010 [DOI] [PubMed] [Google Scholar]
- 51.Marfella R, Di Filippo C, Portoghese M, Barbieri M, Ferraraccio F, Siniscalchi M, Cacciapuoti F, Rossi F, D’Amico M, Paolisso G. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J Lipid Res. 2009; 50:2314–2323. doi: 10.1194/jlr.P900032-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol. 2008; 44:968–975. doi: 10.1016/j.yjmcc.2008.03.021 [DOI] [PubMed] [Google Scholar]
- 53.Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR, van der Vusse GJ, Staels B, van Bilsen M. Peroxisome proliferator-activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res. 2003; 92:518–524. doi: 10.1161/01.RES.0000060700.55247.7C [DOI] [PubMed] [Google Scholar]
- 54.Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, Homma S, Szabolcs MJ, Huang LS, Goldberg IJ. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 2007; 117:2791–2801. doi: 10.1172/JCI30335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rinaldi L, Avgustinova A, Martín M, Datta D, Solanas G, Neus P, Benitah SA. Loss of Dnmt3a and Dnmt3b does not affect epidermal homeostasis but promotes squamous transformation through PPARg. ELife. 2017; 6:1–25. doi: 10.7554/eLife.21697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dou X, Boyd-Kirkup JD, McDermott J, Zhang X, Li F, Rong B, Zhang R, Miao B, Chen P, Cheng H, et al. The strand-biased mitochondrial DNA methylome and its regulation by DNMT3A. Genome Res. 2019; 29:1622–1634. doi: 10.1101/gr.234021.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li L, Tao G, Hill MC, Zhang M, Morikawa Y, Martin JF. Pitx2 maintains mitochondrial function during regeneration to prevent myocardial fat deposition. Development. 2018; 145:dev168609. doi: 10.1242/dev.168609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fang X, Poulsen RR, Wang-Hu J, Shi O, Calvo NS, Simmons CS, Rivkees SA, Wendler CC. Knockdown of DNA methyltransferase 3a alters gene expression and inhibits function of embryonic cardiomyocytes. FASEB J. 2016; 30:3238–3255. doi: 10.1096/fj.201600346R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000; 2:423–427. doi: 10.1038/35017054 [DOI] [PubMed] [Google Scholar]
- 60.Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004; 279:38458–38465. doi: 10.1074/jbc.M406026200 [DOI] [PubMed] [Google Scholar]
- 61.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1 is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998; 95:7987–7992. doi: 10.1073/pnas.95.14.7987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010; 40:294–309. doi: 10.1016/j.molcel.2010.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001; 21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem. 2004; 279:40337–40344. doi: 10.1074/jbc.M403057200 [DOI] [PubMed] [Google Scholar]
- 65.Richard DE, Berra E, Gothié E, Roux D, Pouysségur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1α (HIF-1α) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999; 274:32631–32637. doi: 10.1074/jbc.274.46.32631 [DOI] [PubMed] [Google Scholar]
- 66.Lehmann LH, Jebessa ZH, Kreusser MM, Horsch A, He T, Kronlage M, Dewenter M, Sramek V, Oehl U, Krebs-Haupenthal J, et al. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat Med. 2018; 24:62–72. doi: 10.1038/nm.4452 [DOI] [PubMed] [Google Scholar]
- 67.Ehrlich M. Expression of various genes is controlled by DNA methylation during mammalian development. J Cell Biochem. 2003; 88:899–910. doi: 10.1002/jcb.10464 [DOI] [PubMed] [Google Scholar]
- 68.Kou CY-C, Lau SL-Y, Au K-W, Leung P-Y, Chim SS-C, Fung K-P, Waye MM-Y, Tsui SK-W. Epigenetic regulation of neonatal cardiomyocytes differentiation. Biochem Biophys Res Commun. 2010; 400:278–283. doi: 10.1016/j.bbrc.2010.08.064 [DOI] [PubMed] [Google Scholar]
- 69.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999; 99:247–257. doi: 10.1016/s0092-8674(00)81656-6 [DOI] [PubMed] [Google Scholar]
- 70.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017; 377:111–121. doi: 10.1056/NEJMoa1701719 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.