

Abstract
Mutant huntingtin (mHTT) is associated with mitochondria, but the exact mitochondrial location of mHTT has not been definitively established. Recently, it was reported that mHTT is present in the intermembrane space and inhibits mitochondrial protein import by interacting with TIM23, a major component of mitochondrial protein import machinery, but evidence for functional ramifications were not provided. We assessed mHTT location using synaptic and nonsynaptic mitochondria isolated from brains of YAC128 mice and subjected to alkali treatment or limited trypsin digestion. Mitochondria were purified either with discontinuous Percoll gradient or with anti-TOM22-conjugated iron microbeads. We also used mitochondria isolated from postmortem brain tissues of unaffected individuals and HD patients. Our results demonstrate that mHTT is located on the cytosolic side of the mitochondrial outer membrane (MOM) but does not cross it. This refutes the hypothesis that mHTT may interact with TIM23 and inhibit mitochondrial protein import. The levels of expression of nuclear-encoded, TIM23-transported mitochondrial proteins ACO2, TUFM, IDH3A, CLPP and mitochondrially encoded and synthesized protein mtCO1 were similar in mitochondria from YAC128 mice and their wild-type littermates as well as in mitochondria from postmortem brain tissues of unaffected individuals and HD patients, supporting the lack of deficit in mitochondrial protein import. Regardless of purification technique, mitochondria from YAC128 and WT mice had similar respiratory activities and mitochondrial membrane potentials. Thus, our data argue against mHTT crossing the MOM and entering into the mitochondrial intermembrane space, making it highly unlikely that mHTT interacts with TIM23 and inhibits protein import in intact mitochondria.
Introduction
Huntington’s disease (HD) is an autosomal dominant, hereditary neurodegenerative disorder associated with the abnormal expansion of CAG repeats in the HTT gene, which encodes a polyglutamine tract in huntingtin protein (HTT), a 350 kDa protein ubiquitously expressed in various tissues (1). This single mutation results in the expression of mutant HTT (mHTT) with an elongated polyglutamine stretch near the N-terminus and is linked to a loss of medium spiny neurons in the striatum (2). A clear mechanistic connection between the HTT gene mutation and neuronal loss, resulting in neurological defects in HD, remains unclear. In early studies, association of mHTT with mitochondria was demonstrated (3–5). Consequently, an interaction of mHTT with mitochondria leading to mitochondrial dysfunction has been proposed to be a mechanism contributing to HD pathogenesis (6–11).
The possible mechanism of the proposed mHTT effect on mitochondria remains obscure. It is obvious, however, that if mHTT directly exerts a deleterious effect on mitochondria, its mechanism should strongly depend on the localization of mHTT on or within mitochondria. In 2004, Choo et al. treated mitochondria isolated from SH-SY5Y cells with trypsin and found that wild-type huntingtin (wtHTT) was completely degraded whereas voltage-dependent anion channel (VDAC), cytochrome c oxidase and cytochrome c (Cyt c) were spared (3). This finding indicated that wtHTT resided on the cytosolic side of the mitochondrial outer membrane (MOM). To the best of our knowledge, up until recently, that was the only study specifically addressing mitochondrial location of HTT.
Recently, Yano et al. Reported that mHTT interacts with TIM23 (10), a protein in the mitochondrial inner membrane (MIM) and a major component of mitochondrial protein import machinery (12). According to the authors, this interaction leads to inhibition of mitochondrial protein import that potentially could result in depletion of nuclear-encoded, TIM23-transported mitochondrial proteins and subsequent mitochondrial dysfunction. The proposed mechanism implied mHTT translocation through the MOM and localization in the intermembrane space. However, neither localization of mHTT in the intermembrane space nor alteration in expression of nuclear-encoded, TIM23-transported mitochondrial proteins was demonstrated (10). Very recently, Yablonska et al. (11), using a limited trypsin digestion assay, reported that mHTT resides in the intermembrane space. This finding supported the authors’ previous observation, which argued for mHTT-induced inhibition of TIM23 (10). Surprisingly, Yablonska et al. (11) did not find any significant difference in expression of TIM23-transported mitochondrial proteins. However, in HD mitochondria, the authors found a 6-fold increase in expression of mitochondrially encoded and synthesized protein mtCO1. Consequently, in subsequent experiments measuring expression of TIM23-transported mitochondrial proteins, the authors adjusted protein loading to the gel according to mtCO1 expression level (2.5 μg versus 15 μg protein per lane for HD and WT mitochondria, respectively). As a result of this ‘visual correction’, the authors found much lower signals from mitochondrial proteins in HD compared with WT mitochondria (11), suggesting a decreased expression of TIM23-transported mitochondrial proteins. Yet, the same group previously showed the lack of alterations in respiratory activities of HD compared with WT mitochondria (10). Thus, there is a glaring discrepancy among reported location of mHTT, inhibition of TIM23 and the lack of alterations in respiratory activity of HD mitochondria (10,11).
To resolve this discrepancy, in the present study, we revisited the question regarding mHTT location and functional ramifications of mHTT interaction with mitochondria. To assess the possible impact of the proposed TIM23 inhibition, we compared expression of randomly chosen, nuclear-encoded, TIM23-transported mitochondrial proteins ACO2, TUFM, IDH3A, CLPP between WT and HD mitochondria and compared expression of nuclear-encoded mitochondrial proteins with expression of mitochondrially encoded and synthesized protein mtCO1 in control and HD mitochondria. We used mitochondria purified from YAC128 mice, an established mouse model of HD (13) and their genetic background FVB/NJ mice as well as from postmortem brain tissues of unaffected individuals and HD patients. In addition, we assessed mitochondrial respiration and membrane potential using synaptic and nonsynaptic brain mitochondria purified with Percoll gradient or anti-TOM22 iron microbeads from WT and YAC128 mice. The results of our study unequivocally demonstrate extramitochondrial location of mHTT and similar expression of mitochondrial proteins in WT and HD mitochondria, arguing against mHTT-mediated inhibition of mitochondrial protein import. Consistent with these findings, our data clearly indicate the lack of respiratory and membrane potential deficits in mitochondria from YAC128 mice, which argues against the contribution of mitochondrial dysfunction to HD pathogenesis.
Results
Establishing the precise location of mHTT in or on mitochondria is of utmost importance for our understanding of possible mechanisms of putative mHTT action on mitochondria and development of potential treatments for HD. In our study, we used alkali treatment and limited trypsin digestion assays (3) to determine the location of mHTT on or within mitochondria. Alkali treatment of nonsynaptic mitochondria isolated by anti-TOM22 iron microbeads purification from brains of YAC128 mice, expressing human mHTT with a 128 glutamine stretch at the N-terminus (13), resulted in complete removal of mHTT from mitochondria (Fig. 1). Simultaneously, cytochrome c (Cyt c), a 13 kDa intermembrane space protein, was also lost, whereas Smac, a 27 kDa pro-apoptotic protein from the intermembrane space, was retained in mitochondria. Here and in other experiments, VDAC1 was used as a loading control. Figure 1B shows a statistical summary of these experiments. The removal of Cyt c suggested that the integrity of the MOM was compromised by the alkali treatment and it became permeable for proteins with molecular weight at least ~13 kDa. On the other hand, Smac retention after alkali treatment suggested that MOM permeabilization was limited to lower molecular weight proteins. Consequently, if mHTT, with a molecular weight of 350 kDa, were to be located in the intermembrane space, it would most likely remain in mitochondria following alkali treatment. However, that was not the case, suggesting that mHTT was located on the cytosolic side of the MOM.
Figure 1.
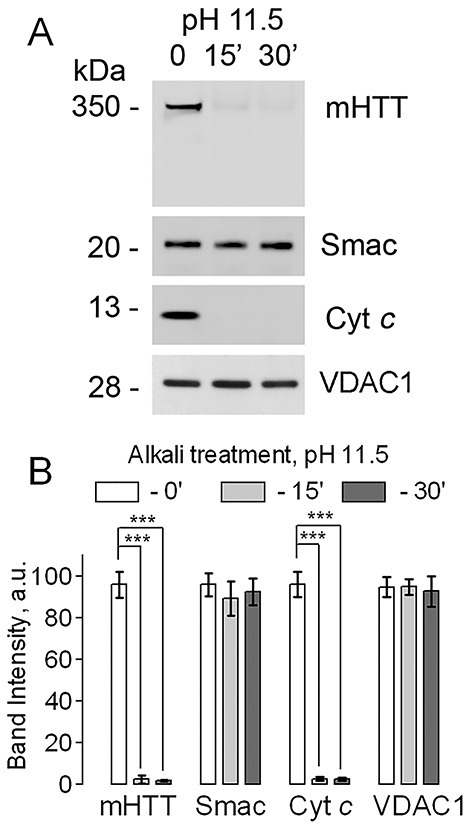
Removal of mHTT from brain mitochondria of YAC128 mice by alkali treatment. In A, mitochondria were purified with anti-TOM22 iron microbeads. Mitochondria (60 μg protein) were incubated on ice at pH 11.5 (in standard incubation medium supplemented with 0.1 M Na2HCO3) for 0, 15, or 30 min. Then, mitochondria were pelleted and used for immunoblotting (20 μg protein per lane). The voltage-dependent anion channel isoform 1 (VDAC1) was used as a loading control. The degradation or lack thereof of cytochrome c (Cyt c) and Smac, respectively, was used to assess the integrity of the mitochondrial outer membrane (MOM). The theoretical molecular weight of Smac is 27 kDa, but according to the manufacturer, on the gel the Smac band appears at about 20 kDa. In B, statistical summary of alkali treatment experiments. Data are mean ± SD, N = 5 experiments with separate independent mitochondrial preparations, ***P < 0.001.
In the following experiments, we used a limited trypsin digestion assay to pinpoint the location of mHTT association with mitochondria (3). We used nonsynaptic and synaptic mitochondria isolated from brains of YAC128 mice. In these experiments, we used two types of mitochondrial preparations: Percoll gradient-purified (14) and anti-TOM22 iron microbeads-purified mitochondria (15). We also used different concentrations of trypsin (5, 25 and 100 μg/ml) and different lengths of exposure to trypsin (30 and 60 min). The trypsin digestion experiments were performed on ice to preserve mitochondrial membrane integrity. Under these conditions, low concentration of trypsin (5 μg/ml) did not effectively digest mHTT in the microbeads-purified nonsynaptic mitochondria and partly, but statistically significantly, degraded mHTT in synaptic mitochondria (Fig. 2). With Percoll-purified mitochondria, low concentration of trypsin (5 μg/ml) degraded mHTT in both nonsynaptic and synaptic mitochondria (Supplementary Material, Fig. S1). Higher concentration of trypsin (25 μg/ml) produced partial to nearly complete mHTT degradation depending on the method of mitochondrial purification, the type of mitochondria and the length of exposure to trypsin (Fig. 2 and Supplementary Material, Fig. S1). The highest tested trypsin concentration (100 μg/ml) completely degraded mHTT within 60 min of trypsin treatment (Fig. 2 and Supplementary Material, Fig. S1). At the same time, Cyt c and endonuclease G (EndoG), both mitochondrial intermembrane space proteins, were left intact in mitochondria, indicating that the barrier properties of the MOM remained preserved and the intermembrane space was not accessible to trypsin. TOM20 was used as a marker of the MOM (Supplementary Material, Fig. S2). TOM20 has a large cytosolic domain (16) that can be degraded by trypsin, leading to disappearance of the TOM20 band. Similar results were obtained by Choo et al. (3) and by Yablonska et al. (11). Our experiments with limited trypsin digestion are consistent with alkali treatment experiments (Fig. 1), supporting the notion that mHTT does not cross the MOM and is located on the cytosolic side of the MOM.
Figure 2.
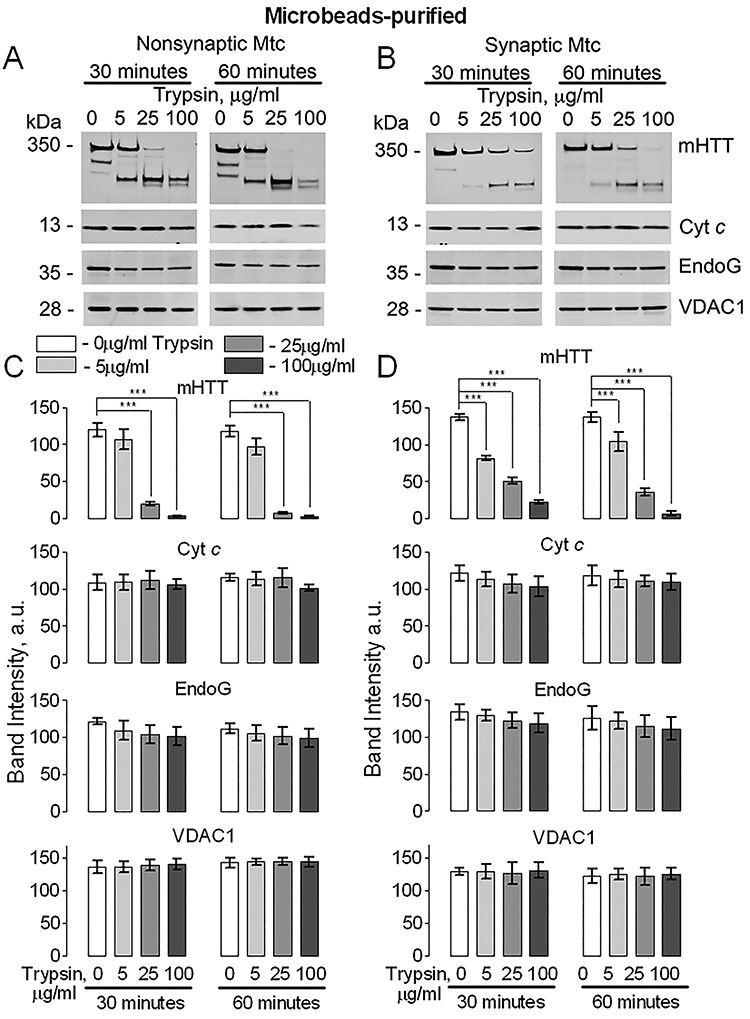
Trypsin digestion of mHTT in mitochondrial fractions purified from brains of YAC128 mice with anti-TOM22 antibody conjugated to iron microbeads. Purified nonsynaptic (A) and synaptic (B) mitochondria (both 80 μg protein) were treated on ice with 0, 5, 25 or 100 μg/ml trypsin for 30 or 60 min. Then, mitochondria were pelleted and used for immunoblotting (20 μg protein per lane). The lack of degradation of cytochrome c (Cyt c) and Endonuclease G (EndoG), proteins of the intermembrane space, was used to assess the integrity of the mitochondrial outer membrane (MOM). In A and B, representative western blots with anti-mHTT 1C2, anti-Cyt c and anti-EndoG antibodies. The VDAC1 immunoblotting was used as a control for protein loading. In C and D, the statistical summary of immunoblotting experiments. Data are mean ± SD, N = 5 experiments with separate independent mitochondrial preparations, ***P < 0.001.
Next, we extended our study to mitochondria isolated from postmortem brain tissues of HD patients. The main characteristics of human brain samples are shown in Supplemental Material, Table S1. The human mitochondria were purified with iron microbeads conjugated to anti-TOM22 following the described protocol (15) with minor modifications. In these experiments, incubation of human brain mitochondria with 25 or 100 μg/ml trypsin for 60 min resulted in complete degradation of mHTT (Fig. 3A and Supplementary Material, Fig. S3). However, in addition to mHTT, EndoG was also completely lost (Fig. 3A and Supplementary Material, Fig. S3), suggesting that the MOM was permeabilized and either intermembrane proteins could escape from mitochondria or trypsin could enter the intermembrane space. TOM20 was used as a marker of the MOM (Fig. 3), but again it was degraded readily by trypsin, leading to disappearance of the TOM20 band, similar to results shown by Choo et al. (3) and Yablonska et al. (11).
Figure 3.
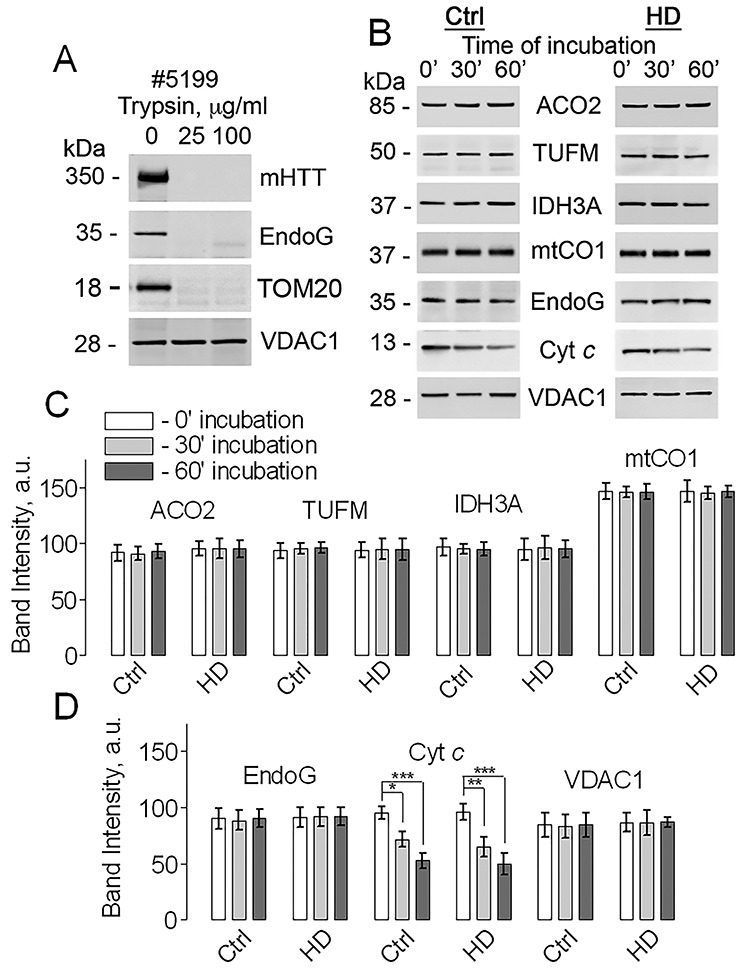
Trypsin digestion of mHTT and evaluation of protein leakage in mitochondria purified from postmortem human brain tissues. In A, trypsin digestion of mHTT in mitochondria from postmortem human brain tissue. Representative western blots with anti-mHTT 1C2 and anti-EndoG antibodies are shown. TOM20 was used as a marker of the MOM. TOM20 has a large cytosolic domain (16) that can be degraded by trypsin, leading to disappearance of the TOM20 band. Similar results were obtained by Choo et al. (3) and by Yablonska et al. (11). The VDAC1 immunoblotting was used as a control for protein loading. Mitochondria were purified with anti-TOM22 iron microbeads. Frozen human brain tissues were placed in ice-cold isolation medium and slowly defrosted on ice. Purified mitochondria (80 μg protein) were treated on ice with 0, 25, or 100 μg/ml trypsin for 60 min. Then, mitochondria were pelleted and used for immunoblotting (20 μg protein per lane). Degradation of Endonuclease G (EndoG), a protein of the intermembrane space, was used to assess the integrity of the mitochondrial outer membrane (MOM). In B, evaluation of protein leakage from brain mitochondria purified from postmortem human brain tissue. Mitochondria (80 μg protein) were incubated on ice for 0, 30 and 60 min and then the levels of different proteins were assessed using immunoblotting. Representative western blots with anti-ACO2, anti-TUFM, anti-IDH3A, anti-mtCO1, anti-EndoG and anti-Cyt c antibodies are shown. The VDAC1 immunoblotting was used as a loading control. Ctrl, samples from unaffected individual (Sample ID #5404); HD, samples from HD patient (Sample ID #5104). In C and D, the statistical summary of densitometry of different protein bands. Data are mean ± SD, N = 5 separate independent mitochondrial preparations, *P < 0.05, **P < 0.01, ***P < 0.001.
The loss of MOM integrity could be attributable to the effect of freezing–thawing of postmortem human brain samples. Previously, it was shown that a freezing–thawing procedure without cryopreserving mitochondria can severely damage the MOM and the mitochondrial inner membrane (MIM) (17). The possible damage to the MOM and the MIM because of freezing–thawing of postmortem human brain samples could lead to leakage of proteins from the intermembrane space and mitochondrial matrix. This leakage may substantially interfere with assessment of mitochondrial protein expression in human brain mitochondria. Consequently, in the following experiments, we evaluated protein leakage from the intermembrane space and mitochondrial matrix of mitochondria isolated from postmortem brain tissues of unaffected individuals and HD patients. Mitochondria were purified using anti-TOM22 iron microbeads and incubated in the incubation medium (80 μg protein) on ice for 30 and 60 min. Then, mitochondria were pelleted and the level of different mitochondrial proteins was assessed using immunoblotting. We assessed the level of intermembrane proteins Cyt c and EndoG, the level of nuclear-encoded TIM23-transported mitochondrial matrix proteins ACO2, TUFM, IDH3A and the level of mitochondrially-encoded and -synthesized mtCO1 (Fig. 3B–D). Figure 3C and D show statistical summaries of these experiments. Incubation of human mitochondria on ice did not result in a detectable leakage of proteins except for Cyt c, suggesting that in human mitochondria from postmortem brain tissues the MOM, for the most part, retained its barrier properties. This also suggested that in the experiments with trypsin treatment of human mitochondria the loss of EndoG and, possibly, mHTT (Fig. 3A and Supplementary Material, Fig. S3) could be attributable to trypsin entry into the intermembrane space and trypsin-mediated EndoG and mHTT degradation. This, however, does not exclude the possibility that mHTT is located on the cytosolic side of the MOM. Thus, the loss of MOM integrity precluded us from definitively establishing mHTT location in mitochondria isolated from postmortem brain tissues of HD patients.
Next, we evaluated in detail the potential effect of mHTT on expression of nuclear-encoded, TIM23-transported proteins ACO2, TUFM, IDH3A and CLPP as well as expression of mitochondrially encoded and synthesized matrix protein mtCO1. We used mitochondria isolated from fresh brains of WT and YAC128 mice (Fig. 4) as well as mitochondria purified from human postmortem brain tissues of unaffected individuals and HD patients (Fig. 5). In both cases, with mouse and human mitochondria, the expression of TIM23-transported proteins was normalized to mitochondrially encoded and synthesized protein mtCO1 and to VDAC1, a protein of the MOM. We did not find a significant difference in expression of the TIM23-transported mitochondrial proteins comparing samples from WT and YAC128 mice as well as from unaffected individuals and HD patients. This indicated the lack of deficit in mitochondrial TIM23-based protein import, which is consistent with extramitochondrial location of mHTT (Figs. 1 and 2) and suggested the lack of mHTT-mediated inhibition of TIM23. Consistent with these data, the lack of altered protein expression in mitochondria from WT and HD mice and striatal cells has been reported by us (18–20) and by other groups (5,21).
Figure 4.
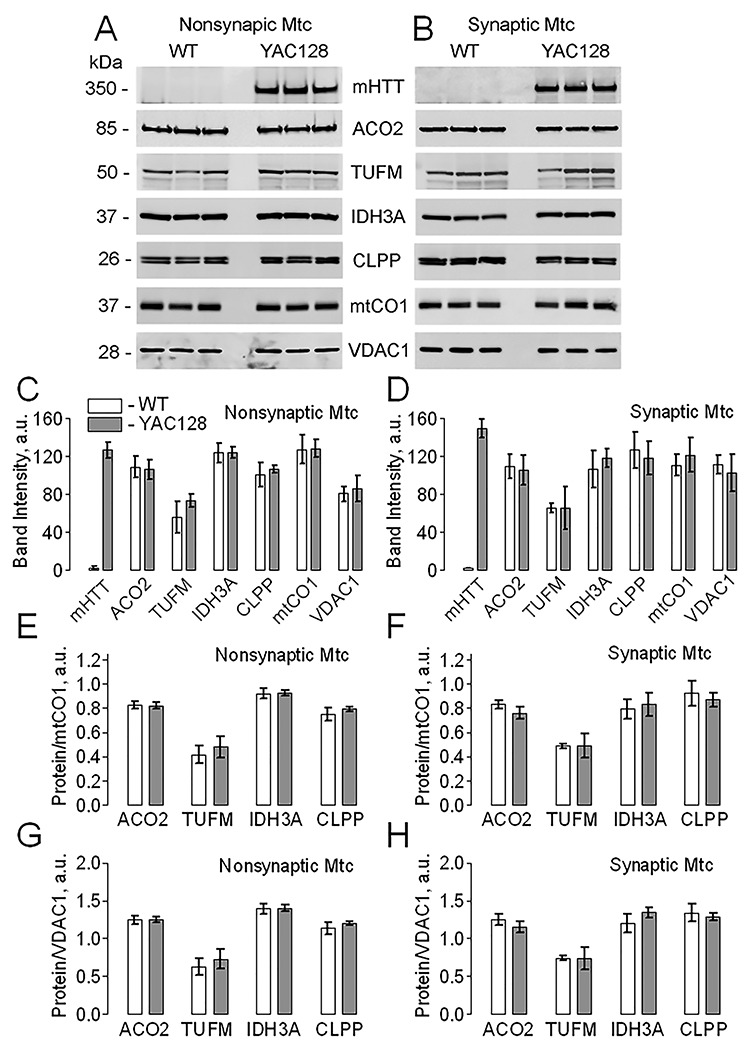
The effect of mHTT on expression level of mitochondrially encoded and mitochondrially synthesized protein mtCO1 and nuclear-encoded, TIM23-transported mitochondrial proteins ACO2, TUFM, IDH3A and CLPP in nonsynaptic and synaptic brain mitochondria from WT and YAC128 mice. Mitochondria were purified with anti-TOM22 iron microbeads. In A and B, representative western blots of nonsynaptic and synaptic mitochondria, respectively, with anti-mHTT 1C2, anti-ACO2, anti-TUFM, anti-IDH3A, anti-CLPP and anti-mtCO1 antibodies. The VDAC1 immunoblotting was used as a loading control. Mitochondria were loaded at 20 μg protein per lane. In C and D, the statistical summary of densitometry of different protein bands for immunoblotting experiments with nonsynaptic and synaptic mitochondria, respectively. In E-H, the statistical summary of densitometry of different protein bands normalized to either mtCO1 (E, F) or VDAC1 (G, H) for immunoblotting experiments with either nonsynaptic (E, G) or synaptic mitochondria (F, H). Data are mean ± SD, N = 3 separate independent mitochondrial preparations.
Figure 5.
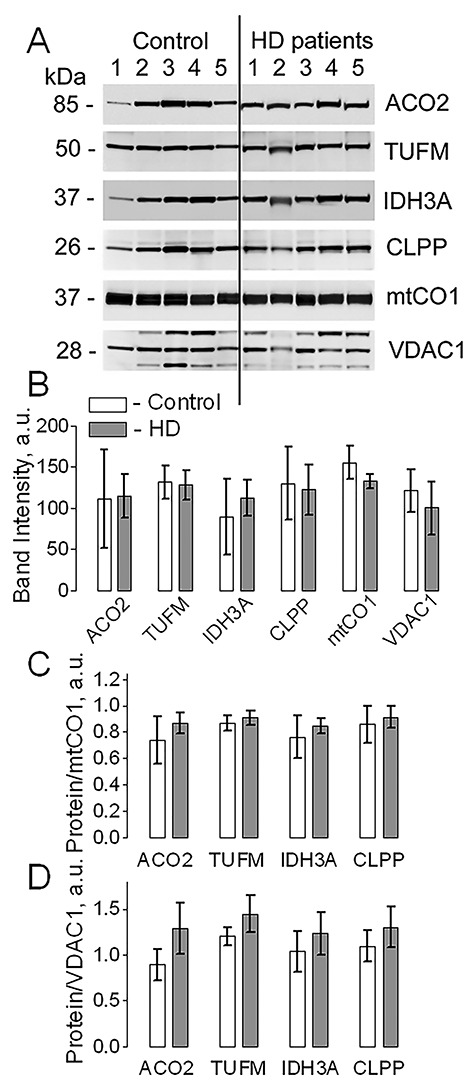
The effect of mHTT on expression level of mitochondrially encoded and mitochondrially synthesized protein mtCO1 and nuclear-encoded, TIM23-transported mitochondrial proteins ACO2, TUFM, IDH3A and CLPP in mitochondria purified from postmortem human brain tissues of unaffected individuals (Control) and HD patients. Mitochondria were purified with anti-TOM22 iron microbeads. In A, representative western blots with anti-ACO2, anti-TUFM, anti-IDH3A, anti-CLPP and anti-mtCO1 antibodies. The VDAC1 immunoblotting was used as a loading control. Mitochondria were loaded at 20 μg protein per lane. In B, the statistical summary of densitometry of different protein bands. In C and D, expression of different proteins normalized either to mtCO1 (C) or to VDAC1 (D). Data are mean ± SD, N = 5 separate independent mitochondrial preparations.
Finally, we assessed the possible effect of mHTT on mitochondrial respiration and membrane potential in nonsynaptic and synaptic mitochondria purified either with Percoll gradient or with iron microbeads. Recently, Hubbard et al. (15) proposed that purification of brain mitochondria with anti-TOM22-conjugated iron microbeads may help to preserve the most vulnerable, damaged mitochondria, thus, unmasking previously undetected deleterious effects of various neuropathologies, including HD. In our experiments, we did not find a significant difference in respiration of mitochondria from brains of WT and YAC128 mice purified by either method (Fig. 6 and Supplementary Material, Fig. S4). Similarly, we did not find a difference in mitochondrial membrane potential between WT and HD mitochondria. On the other hand, mitochondria purified with Percoll gradient demonstrated much higher respiratory activity compared with microbeads-purified mitochondria. Notably, following depletion of added adenosine diphosphate (ADP), the Percoll gradient-purified mitochondria could return to slower V4 respiration, whereas microbeads-purified mitochondria could not, suggesting the presence of mitochondrial debris with ATPase activity in the preparation of microbeads-purified mitochondria (Fig. 6 and Supplementary Material, Fig. S4). In addition, Percoll gradient-purified mitochondria accumulated much larger amounts of tetraphenylphosphonium (TPP+), a probe for mitochondrial membrane potential (22), suggesting either higher membrane potential in Percoll gradient-purified mitochondria compared with microbeads-purified mitochondria or a larger fraction of damaged, depolarized organelles in microbeads-purified mitochondrial preparations (Fig. 6 and Supplementary Material, Fig. S4).
Figure 6.
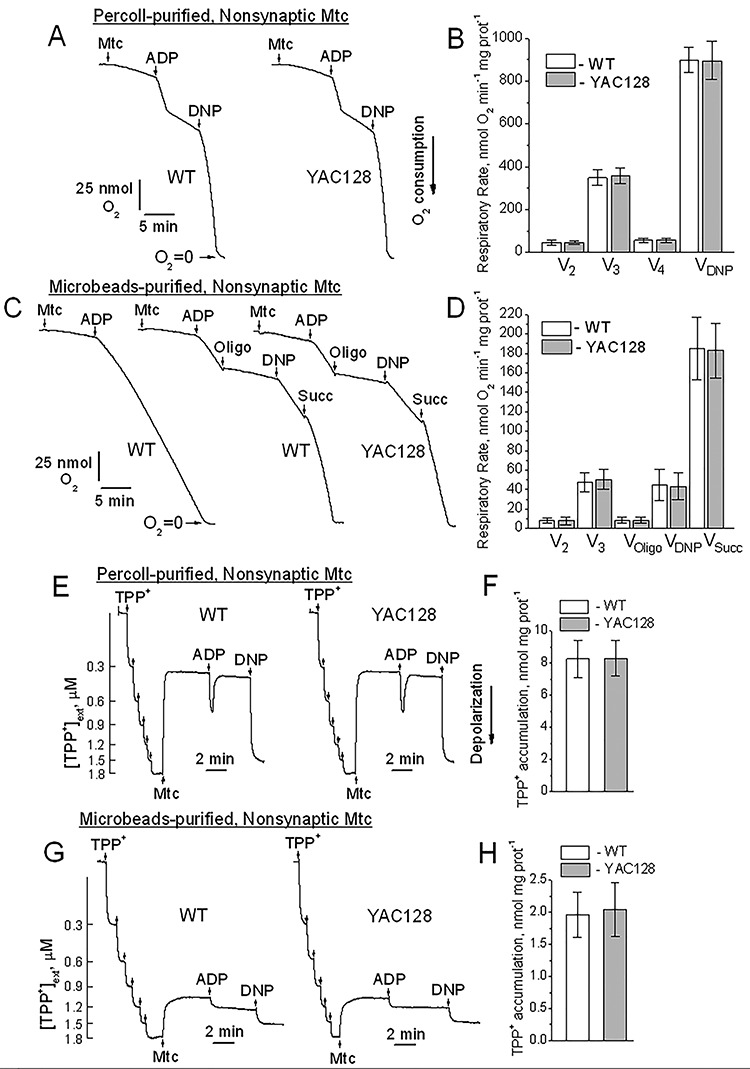
Respiratory activity and membrane potential assessed by tetraphenylphosphonium (TPP+) accumulation in brain nonsynaptic mitochondria purified from WT and YAC128 mice with either discontinuous Percoll gradient or anti-TOM22 iron microbeads. In A and C, representative traces of oxygen consumption by nonsynaptic mitochondria purified with discontinuous Percoll gradient (A) or anti-TOM22 iron microbeads (C). Where indicated, Percoll gradient-purified mitochondria (75 μg protein) or microbeads-purified mitochondria (200 μg protein), ADP (250 μM), oligomycin (Oligo, 1 μM), 2,4-dinitrophenol (2,4-DNP, 60 μM) and succinate (Succ, 3 mM) were added to mitochondria. The incubation medium was supplemented with 3 mM pyruvate and 1 mM malate. In B and D, the statistical summary of respiratory rates of Percoll gradient-purified (B) or anti-TOM22 iron microbeads-purified (D) nonsynaptic mitochondria, respectively. Data are mean ± SD, N = 5 separate independent mitochondrial preparations. In E and G, representative TPP+ traces produced with nonsynaptic mitochondria purified with discontinuous Percoll gradient (E) or anti-TOM22 iron microbeads (G), respectively. Where indicated, Percoll gradient-purified mitochondria (75 μg protein) or microbeads-purified mitochondria (200 μg protein), ADP (250 μM) and 2,4-dinitrophenol (2,4-DNP, 60 μM) were added to mitochondria. The incubation medium was supplemented with 3 mM pyruvate and 1 mM malate. In F and H, the statistical summary of TPP+ accumulation by Percoll gradient-purified (F) or anti-TOM22 iron microbeads-purified (H) nonsynaptic mitochondria, respectively. Data are mean ± SD, N = 5 separate independent mitochondrial preparations.
Discussion
Despite extensive studies, the exact mechanism of pathogenic effect of mHTT remains elusive (23). One of the hypotheses posits that mHTT damages mitochondria, leading to mitochondrial dysfunction (3,6–9,24–26). However, how mHTT can damage mitochondria is not clear. Earlier, Yano et al. (10) reported mHTT-mediated inhibition of TIM23, a key component of the mitochondrial protein import machinery located in the MIM. This finding suggested that mHTT could cross the MOM, enter the intermembrane space and interact with TIM23. This potentially could lead to depletion of nuclear-encoded mitochondrial proteins and subsequent mitochondrial dysfunction (10). However, despite observed inhibition of TIM23, respiration of mitochondria exposed to mHTT remained unchanged (10), suggesting that the level of mitochondrial proteins was not significantly altered. Indeed, in our previous studies, we demonstrated that expression of mitochondrial proteins in brain mitochondria isolated from YAC128 and R6/2 mice and their WT littermates was not different (18–20). Interestingly, in the paper by Yablonska et al. (11), the authors reported that expression of nuclear-encoded, TIM23-transported mitochondrial proteins was similar in mitochondria from immortalized striatal cells ST-Hdh-Q7/Q7 (Control cells) and ST-Hdh-Q111/Q111 (HD cells). This observation was made based on equal protein loading of gels for electrophoresis (15 μg of protein per lane). However, the authors also found that expression of the mitochondrially encoded and synthesized, nonimported protein mtCO1 was about 6 times higher in mitochondria from HD cells (11). Mutant HTT is absent in the mitochondrial matrix and, therefore, it is not clear how mHTT could dramatically increase expression of mitochondrially encoded and synthesized mtCO1. Nevertheless, Yablonska et al. (11) ‘visually corrected’ western blots by loading gels for electrophoresis with 2.5 μg protein per lane of HD mitochondria versus 15 μg of protein per lane of WT mitochondria. As a result, the authors predictably found significantly lower immunoblotting signals from nuclear-encoded, TIM23-transported proteins in HD mitochondria compared with WT mitochondria. In our experiments with mitochondria isolated from postmortem brain tissues of unaffected individuals and HD patients and from fresh brains of WT and YAC128 mice, we did not find a significant difference in expression of mtCO1 as well as in expression of randomly-chosen, nuclear-encoded, TIM23-transported mitochondrial proteins ACO2, TUFM, IDH3A and CLPP. Thus, our data argue against decreased expression of mitochondrial proteins in HD mitochondria. This suggests that mitochondrial protein import machinery in mitochondria from HD patients and HD mice is not inhibited. Furthermore, this also suggests that mHTT does not inhibit TIM23.
In 2004, Choo et al. (3) investigated the location of wtHTT in mitochondria. To determine the location of wtHTT, the authors used limited trypsin digestion applied to isolated mitochondria incubated on ice with increasing trypsin concentrations for 30 min. When the authors used low concentrations of trypsin (1 and 5 μg/ml) to determine huntingtin location on mitochondria, they did not find complete degradation of wtHTT and, as a result, obtained inconclusive results. Treatment of isolated mitochondria with 25 μg/ml trypsin also produced only partial degradation of wtHTT (3). However, treatment with 100 μg/ml trypsin completely degraded wtHTT (3). Under these conditions, Cyt c was retained in mitochondria, indicating well-preserved MOM integrity. Based on these findings, the authors concluded that wtHTT is located on the cytosolic side of the MOM (3). We substantiated these observations in our experiments with freshly isolated total brain mitochondria from YAC128 mice. Incubation of mitochondria with 5, 25 and even 100 μg/ml of trypsin (synaptic mitochondria) for 30 min (Fig. 2) was not sufficient for complete degradation of mHTT. In our experiments, mitochondria were incubated with trypsin on ice to preserve mitochondrial membrane integrity. Consequently, one of the reasons for incomplete degradation of mHTT during 30 min of exposure to trypsin might be the low temperature, at which enzymatic activity of trypsin is significantly reduced.
Yablonska et al. (11) also used a trypsin digestion assay to determine the location of mHTT and wtHTT in mitochondria. Similar to Choo et al. (2004), Yablonska et al. (11) found ~20% of total HTT (mHTT plus wtHTT) remained associated with mitochondria after treatment with 25 μg/ml trypsin. Based on this finding and in contrast to Choo et al. (3), the authors concluded that mHTT can cross the MOM and enter the intermembrane space. Unfortunately, Yablonska et al. (11) did not use higher concentrations of trypsin and, therefore, their conclusion regarding mHTT location seems specious.
Yablonska et al. (11) used mitochondria from immortalized striatal cells ST-Hdh-Q7/Q7 (Control cells) and ST-Hdh-Q111/Q111 (HD cells) purified with iron microbeads conjugated to anti-TOM22. Choo et al. (3) used discontinuous sucrose gradient to purify mitochondria isolated from SH-SY5Y cells. In their study (3,11), it is not clear whether the difference in purification methods or in the use of different types of cells could influence the results produced. In our study, we used synaptic and nonsynaptic mitochondria isolated from brains of YAC128 and WT mice, and purified by two different techniques: with either discontinuous Percoll gradient (14) or anti-TOM22 iron microbeads (15). Regardless of the purification technique used, we found complete degradation of mHTT by trypsin without significant loss of intermembrane space proteins Cyt c and EndoG, indicating that mHTT is located on the cytosolic side of the MOM, consistent with the findings by Choo et al. (3) regarding wtHTT. Our data are also consistent with the previously reported lack of difference in protein expression in WT and HD mitochondria (5,18–21). At the same time, our results strongly argue against the possibility of mHTT interaction with proteins of the intermembrane space and MIM, such as TIM23.
In our study, we used total brain mitochondria, synaptic and nonsynaptic, isolated from fresh brains of YAC128 mice and human mitochondria isolated from frozen striatal tissues of HD patients. The results presented in our manuscript demonstrate an important limitation of the use of mitochondria isolated from frozen brain tissue: the integrity of MOM in mitochondria from frozen brain tissues appeared to be compromised and trypsin degraded not only mHTT, but also EnodG, an intermembrane space protein. This is not a unique finding. Previously, it was clearly shown that freezing brain tissue impairs integrity of the MOM (17). We confirmed this observation in our study. Consequently, a direct comparison of mitochondria isolated from fresh brain tissue and frozen brains in the experiments with trypsin treatment seems unsuitable. Nevertheless, the results of our study clearly indicate the lack of alterations in expression of nuclear-encoded mitochondrial proteins in both mitochondria from fresh YAC128 brains and in mitochondria from frozen brains of HD patients compared with mitochondria from brains of WT mice and brains of unaffected individuals, respectively, arguing against inhibition of mitochondrial protein import by mHTT.
In our previous study, we investigated the effect of mHTT on functions and protein expression in brain mitochondria isolated from R6/2 mice (19). This study unequivocally demonstrated the lack of functional deficits and alterations in protein expression in brain mitochondria from R6/2 mice, similar to brain mitochondria from YAC128 mice (18). We also investigated the effect of mutant huntingtin specifically on striatal mitochondria from YAC128 mice (20). These studies clearly demonstrated the lack of functional defects as well as the lack of alterations in expression of nuclear-encoded mitochondrial proteins in both total brain and striatal mitochondria.
The data presented in this paper, supported by our previously published results with Percoll-purified mitochondria, indicate the lack of mitochondrial dysfunction despite interaction of mHTT with mitochondria (14,18–20,27,28). Recently, however, Hubbard et al. (15) proposed that the use of anti-TOM22-conjugated iron microbeads for mitochondrial purification may improve the yield of purified mitochondria and unmask previously overlooked mitochondrial defects associated with various neuropathologies, including HD. According to the authors, the anti-TOM22 microbeads purification technique may preserve the most vulnerable damaged mitochondria, which could potentially be lost during density gradient purification. The authors convincingly proved their hypothesis using a mild traumatic brain injury (TBI) model by demonstrating decreased State III respiration of brain nonsynaptic mitochondria after isolation with anti-TOM 22 microbeads, but not with Ficoll gradient purification (15). In our study, we compared WT and HD synaptic and nonsynaptic mitochondria purified with Percoll gradient and with anti-TOM22 iron microbeads. We found that Percoll-purified mitochondria had much higher respiratory activity normalized per mg protein and accumulate much larger amounts of TPP+ compared with mitochondria purified with microbeads. In addition, failure of microbeads-purified mitochondria to return to slower V4 respiration following addition of ADP indicates the presence of mitochondrial debris with ATPase activity. This confirms that the anti-TOM22 microbeads technique allows collection of damaged, dysfunctional mitochondria compared with Percoll gradient purification. However, mitochondrial damage most likely may not be attributable to the pathology, but rather could result from the isolation procedure involving tissue homogenization (29). Indeed, despite lower respiratory activity and reduced accumulation of TPP+ compared with Percoll-purified mitochondria, anti-TOM22 microbeads-purified mitochondria from WT and YAC128 mice were functionally indistinguishable, similar to mitochondria purified with Percoll gradient. Thus, although mitochondria purified with anti-TOM22 microbeads unmasked mitochondrial alterations following mild TBI (15), this approach, similar to Percoll gradient purification, failed to demonstrate mHTT-mediated mitochondrial dysfunction, confirming the lack of mitochondrial functional abnormalities in HD, at least, at pre-symptomatic and early symptomatic stages (14,18–20,27,28,30–34). In our recent paper, we demonstrated the lack of mitochondrial bioenergetic deficits in human striatal neurons converted from induced pluripotent stem cells derived from HD patients (28). Our results were recently undeniably confirmed by Sarah Tabrizi’s group, which demonstrated the lack of statistically significant mitochondrial bioenergetic defects in human neural stem cells and neurons expressing mutant exon 1 huntingtin fragments with 71 CAG repeats (35). It has to be mentioned that, in the same study, the authors found some mitochondrial alterations in cells expressing mutant exon 1 huntingtin fragments with 122 CAG repeats (35), but this length of CAG repeats is extremely rare and in the vast majority of HD patients with adult-onset of HD, the length of the CAG repeats varies between 40 and 60 (36,37).
The lack of mHTT-mediated impairment of mitochondrial bioenergetics does not exclude mHTT effect on other mitochondrial functions, e.g. mitochondrial dynamics (38–40). Mutant HTT may also lead to metabolic reprograming in the cell (34). However, these effects most likely take place outside of mitochondria, on the cytosolic side of the MOM, and may involve interaction of mHTT with proteins participating in autophagy/mitophagy (41) as well as in regulation of mitochondrial morphology and motility (42,43).
Materials and Methods
Materials
Pyruvate, malate, ethylene glycol tetraacetic acid (EGTA; Cat# E4378), ADP (Cat# A5285), oligomycin (Cat# 75351), 2,4-dinitrophenol (Cat# D198501), trypsin (Cat# T1426) and trypsin inhibitor (Cat# T9128) were purchased from Sigma (St. Louis, MO, USA). Tetraphenylphosphonium chloride (Cat# 88060) was from Fluka (Buchs, Switzerland). Percoll (Cat# 17089101) was from GE Healthcare Bio-Sciences (Pittsburgh, PA, USA). Bovine serum albumin (BSA), free from free fatty acids (Cat# 152401), was from MP Biomedicals (Irvine, CA, USA). Protease Inhibitor Cocktail (Cat# 04693124001) was from Roche (Indianapolis, IN, USA). Mitochondria MidiMACS Starting Kit, mouse tissue (Cat #130–097-039) and Mitochondria Isolation Kit, human (Cat #130–094532) were from Miltenyi Biotec (Auburn, CA, USA). All materials were purchased no more than 6 months before use.
Human postmortem brain tissues
Human postmortem striatal brain tissues were obtained from University of Maryland Brain and Tissue Bank (UMDBTB) which is a Brain and Tissue Repository of the NIH NeuroBioBank. The length of CAG repeats in the HTT gene of HD patients was determined by Laragen Inc. (Culver City, CA, USA). The samples were stored at −86°C. Before the experiment, samples were placed in ice-cold Isolation Buffer 1 and slowly thawed on ice.
Animals
All procedures with animals were performed in compliance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals as well as in accordance with the Indiana University School of Medicine Institutional Animal Care and Use Committee approved protocol. Wild-type FVB/NJ mice and transgenic HD YAC128 mice of both sexes were used in this study. Breeding pairs of mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and breeding colonies were established in the Laboratory Animals Research Center (LARC) at Indiana University School of Medicine, Indianapolis, IN, USA. Male YAC128 mice were bred with female FVB/NJ mice (background strain). Every mouse was genotyped and weighed 19-25 g at 2–4 months of age. The mice were housed under standard conditions with free access to food and water. All mice were housed in polycarbonate cages, 3 mice per cage. For our experiments, we used 2- to 4-month-old YAC128 mice and their wild-type (WT) littermates. There is evidence that sex hormones may influence mitochondrial functions (44). However, HD affects men and women equally (45,46). Therefore, throughout this study, mice of both sexes were used. YAC128 mice express full-length human mHTT, including upstream and downstream regulatory elements, containing a polyglutamine (polyQ) region of 128 glutamines.
Genotyping
All offspring used for experiments were genotyped using a PCR assay on tail DNA. PCR of tail DNA was performed according to the protocol provided by Jackson Laboratories with oligonucleotide primers oIMR6533 (GGCTGAGGAAGCTGAGGAG) and TmoIMR1594 (CCGCTCAGGTTCTGCTTTTA) (Invitrogen, Carlsbad, CA, USA). The PCR reaction mixture consisted of 1 μl DNA template and 24 μl Platinum PCR SuperMix (Invitrogen; Cat# 12532024) containing 0.39 μM of each primer for a total volume of 25 μl. Cycling conditions were 5 min at 95°C, and then 35 cycles of 30 s at 95°C, 30 s at 56°C, 60 s at 72°C, followed by 10 min at 72°C. Reaction products were visualized on a 1.2% agarose gel run at 100 V for 60 min with Tris-acetate-EDTA running buffer containing 1X GelRed™ Nucleic Acid Gel Stain (Biotium, Hayward, CA, USA; Cat# 41003).
Purification of brain synaptic and nonsynaptic mitochondria with discontinuous Percoll gradient
Percoll gradient-purified total brain nonsynaptic and synaptic mitochondria from 2- to 4-month-old YAC128 and age-matched wild-type FVB/NJ littermates were isolated as we described previously with some modifications (14). Both wild-type and mutant huntingtin are expressed in every brain cell of HD mice and HD patients, and, consequently, the use of total brain mitochondria to study the effect of mutant huntingtin on mitochondria is well justified. To purify nonsynaptic and synaptic mitochondria with Percoll gradient, brains of three FVB/NJ and YAC128 mice each were used for a single isolation procedure. All procedures were performed at 2–4°C. Following brain tissue homogenization in Isolation Buffer 1 in a 15 ml glass Dounce homogenizer (10 strokes with pestle A, 30 strokes with pestle B) on ice, homogenate was diluted with 30 ml of Isolation Buffer 1 and centrifuged for 10 min at 2400 rpm in a Beckman Avanti J-26XP centrifuge, rotor JA-25.50 (700 × g). The composition of Isolation Buffer 1: 225 mM mannitol, 75 mM sucrose, 0.1% BSA free from fatty acid, 10 mM Hepes, pH 7.4 adjusted with KOH, 1 mM EGTA. It was previously suggested that BSA may interfere with mHTT binding to mitochondria (47). However, in our previous study, we have shown that BSA does not replace mHTT from mitochondria (27). Therefore, in the present study with isolated mitochondria, BSA was used to preserve mitochondrial integrity (48). After the 1st centrifugation, supernatant was centrifuged for 10 min at 12500 rpm (18 900 × g) in a Beckman Avanti J-26XP centrifuge, rotor JA-25.50. The pellet was resuspended in 35 ml of Isolation Buffer 2 and centrifuged for 10 min at 12500 rpm (18 900 × g) in a Beckman Avanti J-26XP centrifuge, rotor JA-25.50. The composition of Isolation Buffer 2: 225 mM mannitol, 75 mM sucrose, 10 mM Hepes, pH 7.4 adjusted with KOH, 0.1 mM EGTA. Subsequently, the pellet was resuspended in 5 ml of Isolation Buffer 3. The composition of Isolation Buffer 3: 395 mM sucrose, 0.1 mM EGTA, 10 mM Hepes, pH 7.4. The suspension was layered onto the top of a discontinuous Percoll gradient (26%/40%) in Beckman Ultra-Clear centrifuge tubes and centrifuged for 28 min at 15 500 rpm (41 100 × g) in a Beckman Optima L100K ultracentrifuge, bucket rotor SW41Ti. The 26% and 40% Percoll solutions were prepared in Percoll Buffer. The composition of Percoll Buffer: 320 mM sucrose, 1 mM EGTA, 10 mM Hepes, pH 7.4. Following this centrifugation, nonsynaptic mitochondria were collected for further washes and synaptosomes were collected for isolation of synaptic mitochondria. Nonsynaptic mitochondria were resuspended in Isolation Buffer 3 and centrifuged for 20 min at 15 500 rpm (41 100 × g) in a Beckman Optima L100K ultracentrifuge, bucket rotor SW41Ti. The pellet was resuspended in Isolation Buffer 3 and centrifuged again for 20 min at 15 500 rpm (41 100 × g) in a Beckman Optima L100K ultracentrifuge, bucket rotor SW41Ti. The pellet was collected, resuspended in Isolation Buffer 3 and stored on ice. This was a stock suspension of brain nonsynaptic mitochondria.
Synaptic mitochondria were isolated from synaptosomes by nitrogen cavitation method using an ice-cold nitrogen cell disruption vessel (Parr Instrument Co., Moline, IL; Cat# 4639) as we described previously with some modifications (14). Briefly, the synaptosomes acquired after a discontinuous Percoll gradient purification were transferred to a 10 ml glass beaker on ice and placed into the nitrogen vessel on ice under 1100 psi for 13 min. Then, the ruptured synaptosomes were layered onto a discontinuous Percoll gradient (24/40%) and centrifuged for 28 min at 15 500 rpm (41 100 × g) in a Beckman Optima L100K ultracentrifuge, bucket rotor SW41Ti. Synaptic mitochondria were resuspended in Isolation Buffer 3 and centrifuged twice for 20 min each time at 15 500 rpm (41 100 × g) in a Beckman Optima, L100K ultracentrifuge, bucket rotor SW41Ti as described in this section for the final centrifugations of nonsynaptic mitochondria. The final pellet was collected, resuspended in Isolation Buffer 3 and the stock suspension of synaptic mitochondria was stored on ice.
Purification of brain synaptic and nonsynaptic mitochondria with anti-TOM22 iron microbeads
Mitochondria from brains of WT and YAC128 mice were purified using Mitochondria MidiMACS Starting Kit, mouse tissue (Miltenyi Biotec, Cat #130-097-039) following the protocol described by Hubbard et al. (15) with some modifications. Briefly, after brain tissue from a single mouse was homogenized in Isolation Buffer 1 in a 7 ml glass Dounce homogenizer (10 strokes with pestle A, 30 strokes with pestle B) on ice, homogenate was centrifuged for 10 min at 2400 rpm in a Beckman Avanti J-26XP centrifuge, rotor JA-25.50 (700 × g). All procedures were performed at 2–4°C. After the first centrifugation, supernatant was centrifuged for 10 min at 12 500 rpm (18 900 × g) in a Beckman Avanti J-26XP centrifuge, rotor JA-25.50. The pellet was resuspended in 4 ml of Isolation Buffer 2 and was filtered through a filter with 30 μm pores (Mitochondria Isolation Kit, mouse tissue, Miltenyi Biotec, Cat #130-097-039). One ml of filtrate was diluted with 8 ml of Isolation Buffer 2 and 100 μl of iron microbeads conjugated to anti-TOM22 antibody were added and incubated for 30 min on a nutator Labnet GyroMini S0500 at 4°C. A MACS column was rinsed with 3 ml of Isolation Buffer 2 and then labeled tissue homogenate was passed through a MACS column that had been inserted into a MACS magnetic separator. Then, the column remaining in the MACS magnetic separator was rinsed three times with 3 ml of Isolation Buffer 2. The column was removed from the MACS magnetic separator, rinsed with 1.5 ml of Isolation Buffer 2 and mitochondrial fraction was eluted in the centrifuge tube. Mitochondria were washed once by centrifuging in Isolation Buffer 2. The final stock suspension of purified nonsynaptic mitochondria contained about 6 mg protein/ml and was stored on ice.
To purify synaptic mitochondria with anti-TOM22 iron microbeads, synaptosomes prepared from one mouse brain were ruptured by nitrogen cavitation as described previously. The ruptured synaptosomes were filtered through a filter with 30 μm pores (Mitochondria Isolation Kit, mouse tissue, Miltenyi Biotec, Cat #130-097-039). One milliliter of filtrate was diluted with 8 ml of Isolation Buffer 2 and then mixed with 100 μl of iron microbeads conjugated to anti-TOM22 antibody and incubated for 30 min on a nutator Labnet GyroMini S0500 at 4°C. Then, synaptic mitochondria were purified similarly to nonsynaptic mitochondria as described previously. The final stock suspension of purified synaptic mitochondria contained about 6 mg protein/ml and was stored on ice.
To purify mitochondria from human postmortem brain tissues, 150–200 mg of human brain samples were used. Mitochondria were purified using iron microbeads conjugated to anti-TOM22 antibody (Mitochondria Isolation Kit, human, Miltenyi Biotec, Cat #130-094-532) as described previously for nonsynaptic mouse mitochondria. Briefly, a frozen human brain slice was placed into a 25 ml beaker filled with ice-cold Isolation Buffer 1 and allowed to thaw on ice for 15 min. The brain slice was homogenized in Isolation Buffer 1 in a 7 ml glass Dounce homogenizer (15 strokes with pestle A, 40 strokes with pestle B) on ice, and then homogenate was centrifuged as described previously for the first two centrifugation steps with mouse brain homogenate. Following the second centrifugation, the pellet was resuspended in 2 ml Isolation Medium II and filtered as described previously. Filtrate was diluted with 7 ml Isolation Medium II, incubated with 100 μl of iron microbeads conjugated to anti-TOM22 antibody, passed through a MACS column that had been inserted into a MACS separator and eluted as described previously. Mitochondria were washed once by centrifuging in Isolation Buffer 2. The final stock suspension of purified human mitochondria contained about 2 mg protein/ml and was stored on ice.
Alkali and Trypsin treatment
For alkali treatment experiments, mouse brain mitochondria (60 μg protein) were incubated on ice at pH 11.5 (in the standard incubation medium supplemented with 0.1 M Na2HCO3 as described in (11)) for 0, 15 or 30 min. Then, mitochondria were pelleted and used for immunoblotting (20 μg protein per lane). For trypsin treatment experiments, mouse or human brain mitochondria (80 μg protein) were incubated without or with different concentrations of trypsin (5, 25, 100 μg/ml) for 30 or 60 min on ice. At the end of the experiment, trypsin was inhibited by trypsin inhibitor (0.5 mg/ml), mitochondria were treated with Protease Inhibitor Cocktail (Roche), pelleted and mitochondrial pellets were taken for immunoblotting analysis (20 μg protein per lane).
Mitochondrial respiration and membrane potential
Mitochondrial respiration was measured with a Clark-type oxygen electrode in a tightly sealed 0.4 ml thermostated chamber at 37°C under continuous stirring. Experiments were performed in the standard incubation medium containing 125 mM KCl, 0.5 mM MgCl2, 3 mM KH2PO4, 10 mM Hepes, pH 7.4, 10 μM EGTA, 0.1% BSA free from fatty acids, supplemented with 3 mM pyruvate plus 1 mM malate. The slope of the oxygen electrode trace corresponds to the respiratory rate. Mitochondrial membrane potential was measured in the standard incubation medium at 37°C with a tetraphenylphosphonium (TPP+)-sensitive electrode by following TPP+ distribution between the incubation medium and mitochondria (22). A decrease in external TPP+ concentration corresponds to mitochondrial polarization, whereas an increase in TPP+ in the incubation medium corresponds to depolarization.
Immunoblotting
Brain synaptic and nonsynaptic mitochondria as well as human brain mitochondria pretreated with Protease Inhibitor Cocktail (Roche, Indianapolis, IN, USA; Cat# 04693124001) were incubated with NuPAGE LDS sample buffer (Invitrogen, Carlsbad, CA, USA; Cat# B0007) supplemented with a reducing agent at 70°C for 15 min. For detection of mHTT, Tris-acetate gels (3–8%, Invitrogen; Cat# EA0375) were used for electrophoresis. For detection of all other proteins, Bis-Tris gels (4–12%, Invitrogen; Cat# NP0335) were used. Proteins were transferred to a Hybond-ECL nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA; Cat# RPN78D) following electrophoresis. Blots were incubated at room temperature for 1 h in a blocking solution that consisted of either protein-free blocking buffer (Pierce, Rockford, IL, USA; Cat# 37572) plus 0.15% Triton X-100, 5% milk, phosphate-buffered saline, pH 7.2, plus 0.15% Triton X-100, or 5% BSA, phosphate-buffered saline, pH 7.2, plus 0.15% Triton X-100. After blocking, blots were incubated with either mouse monoclonal anti-polyQ 1C2 (mAb 1574, Millipore, Temecula, CA, USA, 1: 1000), mouse monoclonal anti-aconitase 2 (ACO2) (ab110321, Abcam, 1:1000), rabbit polyclonal anti-CLPP (15698-1-AP, ProteinTech, 1:500), rabbit polyclonal anti-IDH3A (15909-1-AP, ProteinTech, 1:1000), rabbit polyclonal anti-TUFM (A52852, EpiGentek, 1:1000), mouse monoclonal anti-mtCO1 (ab 14 705, Abcam, 1:2000) rabbit polyclonal anti-Endonuclease G (EndoG) (PC684, Calbiochem, 1:1000), mouse monoclonal anti-cytochrome c (Cyt c) (mAb897-SP, R&D Systems, 1:1000), rabbit polyclonal anti-Smac (2411, ProSci, 1:1000), mouse monoclonal anti-TOM20 (612 278, BD Biosciences, 1:5000), or rabbit polyclonal anti-VDAC1 (10866-1-AP, ProteinTech, 1:1000). Blots then were incubated with goat anti-mouse IgG or goat anti-rabbit IgG (both at 1:20000) coupled with horseradish peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and developed with Supersignal West Pico chemiluminescent reagents (Pierce; Cat# 32106). Molecular mass marker HiMark Prestained High Molecular Weight Protein Standard (7 μl, Invitrogen; Cat# LC5699) for 3–8% Tris-acetate gels or Page Ruler Plus Prestained Protein Ladder (5 μl, Thermo Fisher; Cat# 26619) for 4–12% Bis-Tris gels were used to determine molecular masses. Band densities were quantified with NIH ImageJ 1.48v software (http://rsb.info.nih.gov//ij).
Statistics
Data are shown as mean ± SD of indicated number of independent experiments. Statistical analysis of the experimental results consisted of unpaired t-test or one-way analysis of variance followed by Bonferroni’s post hoc test if applicable (GraphPad Prism 4.0, GraphPad Software Inc., San Diego, CA, USA).
Supplementary Material
Acknowledgements
This study was supported by National Institutes of Health grant R01 NS098772 and in part by a grant from Indiana Traumatic Spinal Cord & Brain Injury Research Fund to N.B.
Conflict of Interest statement. The authors declare that they do not have conflict of interest.
Contributor Information
James Hamilton, Department of Pharmacology and Toxicology.
Tatiana Brustovetsky, Department of Pharmacology and Toxicology.
Rajesh Khanna, Department of Pharmacology, University of Arizona, Tucson, AZ 85721, USA.
Nickolay Brustovetsky, Department of Pharmacology and Toxicology; Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
References
- 1. MacDonald M.E., Ambrose C.M., Duyao M.P., Myers R.H., Lin C., Srinidhi L., Barnes G., Taylor S.A., James M., Groot N. et al. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- 2. Vonsattel J.P. and DiFiglia M. (1998) Huntington disease. J. Neuropathol. Exp. Neurol., 57, 369–384. [DOI] [PubMed] [Google Scholar]
- 3. Choo Y.S., Johnson G.V., MacDonald M., Detloff P.J. and Lesort M. (2004) Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet., 13, 1407–1420. [DOI] [PubMed] [Google Scholar]
- 4. Rockabrand E., Slepko N., Pantalone A., Nukala V.N., Kazantsev A., Marsh J.L., Sullivan P.G., Steffan J.S., Sensi S.L. and Thompson L.M. (2007) The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet., 16, 61–77. [DOI] [PubMed] [Google Scholar]
- 5. Orr A.L., Li S., Wang C.E., Li H., Wang J., Rong J., Xu X., Mastroberardino P.G., Greenamyre J.T. and Li X.J. (2008) N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci., 28, 2783–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Panov A.V., Gutekunst C.A., Leavitt B.R., Hayden M.R., Burke J.R., Strittmatter W.J. and Greenamyre J.T. (2002) Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat. Neurosci., 5, 731–736. [DOI] [PubMed] [Google Scholar]
- 7. Tabrizi S.J., Workman J., Hart P.E., Mangiarini L., Mahal A., Bates G., Cooper J.M. and Schapira A.H. (2000) Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann. Neurol., 47, 80–86. [DOI] [PubMed] [Google Scholar]
- 8. Damiano M., Diguet E., Malgorn C., D'Aurelio M., Galvan L., Petit F., Benhaim L., Guillermier M., Houitte D., Dufour N. et al. (2013) A role of mitochondrial complex II defects in genetic models of Huntington's disease expressing N-terminal fragments of mutant huntingtin. Hum. Mol. Genet., 22, 3869–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aidt F.H., Nielsen S.M., Kanters J., Pesta D., Nielsen T.T., Norremolle A., Hasholt L., Christiansen M. and Hagen C.M. (2013) Dysfunctional mitochondrial respiration in the striatum of the Huntington's disease transgenic R6/2 mouse model. PLoS. Curr., 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yano H., Baranov S.V., Baranova O.V., Kim J., Pan Y., Yablonska S., Carlisle D.L., Ferrante R.J., Kim A.H. and Friedlander R.M. (2014) Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci., 17, 822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yablonska S., Ganesan V., Ferrando L.M., Kim J., Pyzel A., Baranova O.V., Khattar N.K., Larkin T.M., Baranov S.V., Chen N. et al. (2019) Mutant huntingtin disrupts mitochondrial proteostasis by interacting with TIM23. Proc. Natl. Acad. Sci. U. S. A., 116, 16593–16602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mokranjac D. and Neupert W. (2010) The many faces of the mitochondrial TIM23 complex. Biochim. Biophys. Acta, 1797, 1045–1054. [DOI] [PubMed] [Google Scholar]
- 13. Slow E.J., van R.J., Rogers D., Coleman S.H., Graham R.K., Deng Y., Oh R., Bissada N., Hossain S.M., Yang Y.Z. et al. (2003) Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet., 12, 1555–1567. [DOI] [PubMed] [Google Scholar]
- 14. Hamilton J., Brustovetsky T. and Brustovetsky N. (2019) Mutant huntingtin fails to directly impair brain mitochondria. J. Neurochem., 151, 716–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hubbard W.B., Harwood C.L., Prajapati P., Springer J.E., Saatman K.E. and Sullivan P.G. (2019) Fractionated mitochondrial magnetic separation for isolation of synaptic mitochondria from brain tissue. Sci. Rep., 9, 9656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schleiff E. and Turnbull J.L. (1998) Functional and structural properties of the mitochondrial outer membrane receptor tom 20. Biochemistry, 37, 13043–13051. [DOI] [PubMed] [Google Scholar]
- 17. Valenti D., de B.L., De F.B., Ricceri L. and Vacca R.A. (2014) Preservation of mitochondrial functional integrity in mitochondria isolated from small cryopreserved mouse brain areas. Anal. Biochem., 444, 25–31. [DOI] [PubMed] [Google Scholar]
- 18. Hamilton J., Pellman J.J., Brustovetsky T., Harris R.A. and Brustovetsky N. (2015) Oxidative metabolism in YAC128 mouse model of Huntington's disease. Hum. Mol. Genet., 24, 4862–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hamilton J., Pellman J.J., Brustovetsky T., Harris R.A. and Brustovetsky N. (2016) Oxidative metabolism and Ca2+ handling in isolated brain mitochondria and striatal neurons from R6/2 mice, a model of Huntington's disease. Hum. Mol. Genet., 25, 2762–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hamilton J., Brustovetsky T. and Brustovetsky N. (2017) Oxidative metabolism and Ca2+ handling in striatal mitochondria from YAC128 mice, a model of Huntington's disease. Neurochem. Int., 109, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Milakovic T. and Johnson G.V. (2005) Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem., 280, 30773–30782. [DOI] [PubMed] [Google Scholar]
- 22. Kamo N., Muratsugu M., Hongoh R. and Kobatake Y. (1979) Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J. Membr. Biol., 49, 105–121. [DOI] [PubMed] [Google Scholar]
- 23. Brustovetsky N. (2016) Mutant Huntingtin and elusive defects in oxidative metabolism and mitochondrial calcium handling. Mol. Neurobiol., 53, 2944–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fernandes H.B., Baimbridge K.G., Church J., Hayden M.R. and Raymond L.A. (2007) Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA-induced apoptosis in YAC128 model of Huntington's disease. J. Neurosci., 27, 13614–13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lim D., Fedrizzi L., Tartari M., Zuccato C., Cattaneo E., Brini M. and Carafoli E. (2008) Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J. Biol. Chem., 283, 5780–5789. [DOI] [PubMed] [Google Scholar]
- 26. Kim S.H., Thomas C.A., Andre V.M., Cummings D.M., Cepeda C., Levine M.S. and Ehrlich M.E. (2011) Forebrain striatal-specific expression of mutant huntingtin protein in vivo induces cell-autonomous age-dependent alterations in sensitivity to excitotoxicity and mitochondrial function. ASN. Neuro, 3, e00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pellman J.J., Hamilton J., Brustovetsky T. and Brustovetsky N. (2015) Ca (2+) handling in isolated brain mitochondria and cultured neurons derived from the YAC128 mouse model of Huntington's disease. J. Neurochem., 134, 652–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hamilton J., Brustovetsky T., Sridhar A., Pan Y., Cummins T.R., Meyer J.S. and Brustovetsky N. (2020) Energy metabolism and mitochondrial superoxide anion production in pre-symptomatic striatal neurons derived from human-induced pluripotent stem cells expressing mutant Huntingtin. Mol. Neurobiol., 57, 668–684. [DOI] [PubMed] [Google Scholar]
- 29. Picard M., Taivassalo T., Gouspillou G. and Hepple R.T. (2011) Mitochondria: isolation, structure and function. J. Physiol., 589, 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olah J., Klivenyi P., Gardian G., Vecsei L., Orosz F., Kovacs G.G., Westerhoff H.V. and Ovadi J. (2008) Increased glucose metabolism and ATP level in brain tissue of Huntington's disease transgenic mice. FEBS J., 275, 4740–4755. [DOI] [PubMed] [Google Scholar]
- 31. Guidetti P., Charles V., Chen E.Y., Reddy P.H., Kordower J.H., Whetsell W.O. Jr., Schwarcz R. and Tagle D.A. (2001) Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp. Neurol., 169, 340–350. [DOI] [PubMed] [Google Scholar]
- 32. Oliveira J.M., Jekabsons M.B., Chen S., Lin A., Rego A.C., Goncalves J., Ellerby L.M. and Nicholls D.G. (2007) Mitochondrial dysfunction in Huntington's disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J. Neurochem., 101, 241–249. [DOI] [PubMed] [Google Scholar]
- 33. Boussicault L., Herard A.S., Calingasan N., Petit F., Malgorn C., Merienne N., Jan C., Gaillard M.C., Lerchundi R., Barros L.F. et al. (2014) Impaired brain energy metabolism in the BACHD mouse model of Huntington's disease: critical role of astrocyte-neuron interactions. J. Cereb. Blood Flow Metab., 34, 1500–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Polyzos A.A., Lee D.Y., Datta R., Hauser M., Budworth H., Holt A., Mihalik S., Goldschmidt P., Frankel K., Trego K. et al. (2019) Metabolic reprogramming in astrocytes distinguishes region-specific neuronal susceptibility in Huntington mice. Cell Metab., 29, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ghosh R., Wood-Kaczmar A., Dobson L., Smith E.J., Sirinathsinghji E.C., Kriston-Vizi J., Hargreaves I.P., Heaton R., Herrmann F., Abramov A.Y. et al. (2020) Expression of mutant exon 1 huntingtin fragments in human neural stem cells and neurons causes inclusion formation and mitochondrial dysfunction. FASEB J.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kremer B., Goldberg P., Andrew S.E., Theilmann J., Telenius H., Zeisler J., Squitieri F., Lin B., Bassett A. and Almqvist E. (1994) A worldwide study of the Huntington's disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med., 330, 1401–1406. [DOI] [PubMed] [Google Scholar]
- 37. Myers R.H. (2004) Huntington's disease genetics. Neuro Rx., 1, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Costa V., Giacomello M., Hudec R., Lopreiato R., Ermak G., Lim D., Malorni W., Davies K.J., Carafoli E. and Scorrano L. (2010) Mitochondrial fission and cristae disruption increase the response of cell models of Huntington's disease to apoptotic stimuli. EMBO Mol. Med., 2, 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Song W., Chen J., Petrilli A., Liot G., Klinglmayr E., Zhou Y., Poquiz P., Tjong J., Pouladi M.A., Hayden M.R. et al. (2011) Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med., 17, 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shirendeb U., Reddy A.P., Manczak M., Calkins M.J., Mao P., Tagle D.A. and Reddy P.H. (2011) Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: implications for selective neuronal damage. Hum. Mol. Genet., 20, 1438–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martinez-Vicente M., Talloczy Z., Wong E., Tang G., Koga H., Kaushik S., de V.R., Arias E., Harris S., Sulzer D. and Cuervo A.M. (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat. Neurosci., 13, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shirendeb U.P., Calkins M.J., Manczak M., Anekonda V., Dufour B., McBride J.L., Mao P. and Reddy P.H. (2012) Mutant huntingtin's interaction with mitochondrial protein Drp 1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington's disease. Hum. Mol. Genet., 21, 406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Trushina E., Dyer R.B., Badger J.D., Ure D., Eide L., Tran D.D., Vrieze B.T., Legendre-Guillemin V., McPherson P.S., Mandavilli B.S. et al. (2004) Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol., 24, 8195–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gaignard P., Liere P., Therond P., Schumacher M., Slama A. and Guennoun R. (2017) Role of sex hormones on brain mitochondrial function, with special reference to aging and neurodegenerative diseases. Front. Aging Neurosci., 9, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Novak M.J. and Tabrizi S.J. (2010) Huntington's disease. BMJ, 340, 34–40. [DOI] [PubMed] [Google Scholar]
- 46. World Health Organization (2016) Human Genomics in Global Health: Genes and human disease. www. who.int/genomics/public/geneticdiseases/en/index 2.html#HD.
- 47. Panov A.V., Burke J.R., Strittmatter W.J. and Greenamyre J.T. (2003) In vitro effects of polyglutamine tracts on Ca2+−dependent depolarization of rat and human mitochondria: relevance to Huntington's disease. Arch. Biochem. Biophys., 410, 1–6. [DOI] [PubMed] [Google Scholar]
- 48. Lai J.C.K. and Clark J.B. (1989) In Boulton A.A., Baker G.B. and Butterworth R.F. (eds), Neuromethods. Humana Press, Clifton, NJ, pp. 43–98. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.