

Abstract
Background
Both genetic and methylation analysis have been shown to provide insight into the diagnosis and prognosis of many brain tumors. However, the implication of methylation profiling and its interaction with genetic alterations in pediatric low-grade gliomas (PLGGs) are unclear.
Methods
We performed a comprehensive analysis of PLGG with long-term clinical follow-up. In total 152 PLGGs were analyzed from a range of pathological subtypes, including 40 gangliogliomas. Complete molecular analysis was compared with genome-wide methylation data and outcome in all patients. For further analysis of specific PLGG groups, including BRAF p.V600E mutant gliomas, we compiled an additional cohort of clinically and genetically defined tumors from 3 large centers.
Results
Unsupervised hierarchical clustering revealed 5 novel subgroups of PLGG. These were dominated by nonneoplastic factors such as tumor location and lymphocytic infiltration. Midline PLGG clustered together while deep hemispheric lesions differed from lesions in the periphery. Mutations were distributed throughout these location-driven clusters of PLGG. A novel methylation cluster suggesting high lymphocyte infiltration was confirmed pathologically and exhibited worse progression-free survival compared with PLGG harboring similar molecular alterations (P = 0.008; multivariate analysis: P = 0.035). Although the current methylation classifier revealed low confidence in 44% of cases and failed to add information in most PLGG, it was helpful in reclassifying rare cases. The addition of histopathological and molecular information to specific methylation subgroups such as pleomorphic xanthoastrocytoma–like tumors could stratify these tumors into low and high risk (P = 0.0014).
Conclusion
The PLGG methylome is affected by multiple nonneoplastic factors. Combined molecular and pathological analysis is key to provide additional information when methylation classification is used for PLGG in the clinical setting.
Keywords: lymphocytic infiltration, methylation profile, pediatric low grade glioma, pleomorphic xanthoastrocytoma
Key Points.
1. Epigenetic subgroups in PLGG are driven by nongenetic factors. Unsupervised clustering reveals that methylation-based subgroups of PLGG are stratified predominantly by tumor location and nonneoplastic cell composition.
2. Epigenetic classification is not sufficient in PLGG. In contrast to other childhood cancers, methylation arrays may lead to unclear or erroneous results in PLGG. In contrast, combined genetic and epigenetic analysis provides invaluable insights into these cancers.
3. Combined genetic and epigenetic analysis of PLGG can be clinically used to predict outcome and for therapeutic decisions. When combining pathological grade, genetic events, and methylation analysis, one can uncover rare groups of PLGG, stratify tumors for outcome, and target the high-risk tumors for precision medicine.
Importance of the Study.
We performed methylation array analysis of PLGGs with comprehensive molecular data and clinical follow-up spanning three decades. This study enabled us to uncover several key insights, including the prognostic importance of lymphocytic infiltration in BRAF p.V600E mutant PLGGs, as well as highlight the pitfalls and benefits of genetic and epigenetic analysis of PLGG.
Pediatric low-grade gliomas (PLGGs) are the most common central nervous system tumors in children.1,2 They arise throughout the neuroaxis and cause significant morbidity and late mortality which is largely dependent on the tumor location, among other clinical variables.1,2 PLGGs encompass a range of pathological entities which overall share similar favorable clinical outcome but do not reliably predict those most likely to recur or transform.
Long-term survival of patients with PLGGs is excellent if gross total resection can be achieved.3 However, management of incompletely resected PLGGs is complex. Until recently, medical therapy for unresected PLGGs consisted of radiation and chemotherapy. These therapies carry significant long-term morbidity,4 and their efficacy is variable. Importantly, since the management of PLGG does not depend on pathological subtypes, some tumors were not biopsied and therapy was empirically based on history and imaging findings. Overall there is a need for more scientific decision making in determining the right therapeutic approach for these patients.
Based on a series of extensive molecular analyses,5 several genetic alterations have been identified in PLGG which converge on the Ras/mitogen-activated protein kinase pathway.5–10 The most common alterations observed in PLGG are KIAA1549-BRAF fusion,9 followed by germline and somatic mutations in neurofibromin 1, and the BRAF p.V600E mutation.11 A variety of relatively rare alterations of fibroblast growth factor receptor 1 (FGFR1), NTRK,8FGFR2,7RAF1,8MYB/MYBL1,5,6,12 and others are also found in these tumors. The impact of these new molecular subgroups on survival and response to conventional and target therapies is still under investigation.
Epigenetic analysis by DNA methylation arrays has had an enormous impact on our understanding, diagnosis, and risk stratification of pediatric brain tumors.13,14 Analysis of the epigenome has uncovered novel molecular entities and allowed more accurate classification of tumors such as embryonal brain tumors16,17 as well as enabling subclassification of tumors such as medulloblastoma and ependymoma.18 This subclassification is also able to identify prognostic risk groups in these tumors and help in tailoring therapy in some cases. Although several papers have tried to stratify PLGG using methylation arrays,19–22 the interaction of molecular genetic events, location, and morphology with methylation status in these tumors is not clear. Furthermore, since genetic signatures are emerging to have impact on long-term outcome of PLGG, the relative contribution of methylation arrays in classifying these tumors remains relatively unknown.
In order to shed light on the above issues we performed a comprehensive analysis of a large carefully defined cohort of PLGG with full genetic, morphologic, and methylation data aligned with clinical and outcome data. Our data underscore the limitations and potential roles of epigenetic analysis with current molecular classification of PLGG.
Materials and Methods
Tumor Materials
We performed full molecular analysis combined with morphological and clinical outcome analysis on 152 PLGGs which were wildtype for H3F3A, including 136 from the Hospital for Sick Children (SickKids) and 16 BRAF p.V600E mutant PLGGs from St Jude Children’s Research Hospital (St Jude) (Table 1).
Table 1.
Clinical and molecular information of cases for the genome-wide methylation analysis
| Total Number of Enrolled Cases, 152 | ||
|---|---|---|
| Location | Hemisphere | 83 |
| Thalamus/hypothalamus | 35 | |
| Cerebellum | 22 | |
| Brainstem/spine | 12 | |
| Pathological diagnosis | Pilocytic/pilomyxoid astrocytoma | 48 |
| Ganglioglioma | 40 | |
| Diffuse astrocytoma | 22 | |
| Pleomorphic xanthoastrocytoma (PXA) | 16 | |
| Other low grade gliomas | 26 | |
| Molecular status | BRAF V600E mutation | 79 |
| BRAF fusions | 39 | |
| FGFR1-TACC1 fusion | 6 | |
| FGFR2 fusions | 4 | |
| MYBL1 amplification | 2 | |
| ALK fusions | 2 | |
| Others | 20 | |
| Observation period, median (range) | 5.4 y (0‒28.0) |
To assess the confidence of the Heidelberg classifier for PLGG, in addition to these 152, a further 70 cases from St Jude’s were included (total 222 cases).
For analysis of degree of lymphocyte infiltration on pathological slides, 70 BRAF p.V600E mutant cases from SickKids were assessed, including 45 cases with either high or low Leukocytes Unmethylated for Purity (LUMP) score based on methylation profiling (see below), as well as an additional 25 BRAF p.V600E cases were used.
To assess the role of methylation in the context of tumor grade and molecular data, we also submitted a cohort of 14 BRAF p.V600E mutant high-grade gliomas (including anaplastic pleomorphic xanthoastrocytomas [PXA]) for methylation analysis from SickKids and NYU. A schematic of patient samples and workflow is shown in Supplementary Figure 1. This study was approved by the institutional research ethics board from each respective institution.
Molecular Genetic Analysis
BRAF p.V600E mutations were detected by droplet digital (DD)-PCR (Bio-Rad) and/or by an immunohistochemistry test approved by the Clinical Laboratory Improvement Amendments.11,23 Multiple fusions including KIAA1549-BRAF fusions, BRAF duplication, and FGFR-transforming acidic coiled-coil protein 1 (TACC1) fusions were evaluated using NanoString and/or fluorescent in situ hybridization.24,25 Status of cyclin-dependent kinase inhibitor 2A (CDKN2A) was analyzed using copy number analysis with signal data of methylation array,13 or DD-PCR.11 All methods have been described previously. Anaplastic lymphoma kinase (ALK) fusions and FGFR2 fusions were found by RNA sequencing as described elsewhere.26,27
Genome-Wide DNA Methylation Analysis
We performed comprehensive methylation analysis of all tumors using the Illumina Infinium HumanMethylationEPIC (EPIC array) or HumanMethylation450k (450k array) BeadChip array, which includes 866 238 and 485 512 cytosine-phosphate-guanine (CpG) sites, respectively, for analysis. Further details on the specifics of analysis and additional information are provided in the Supplementary data.
LUMP score was discovered by Aran et al to estimate tumor purity.29 The authors selected 44 CpG sites that are involved in both unmethylated probes of methylation data of normal immune cells and methylated probes of those of tumor samples from The Cancer Genome Atlas database. LUMP score can be calculated by average methylation levels of the probes divided by 0.85.
Histopathology
Pathological diagnoses were assigned according to the 2016 World Health Organization classification of the central nervous system.28 Cases with insufficient material to assign a specific category but which were clearly low grade were assigned a “low grade astrocytoma” diagnosis in our cohort. Anaplastic PXA was included in the pediatric high-grade glioma cohort.
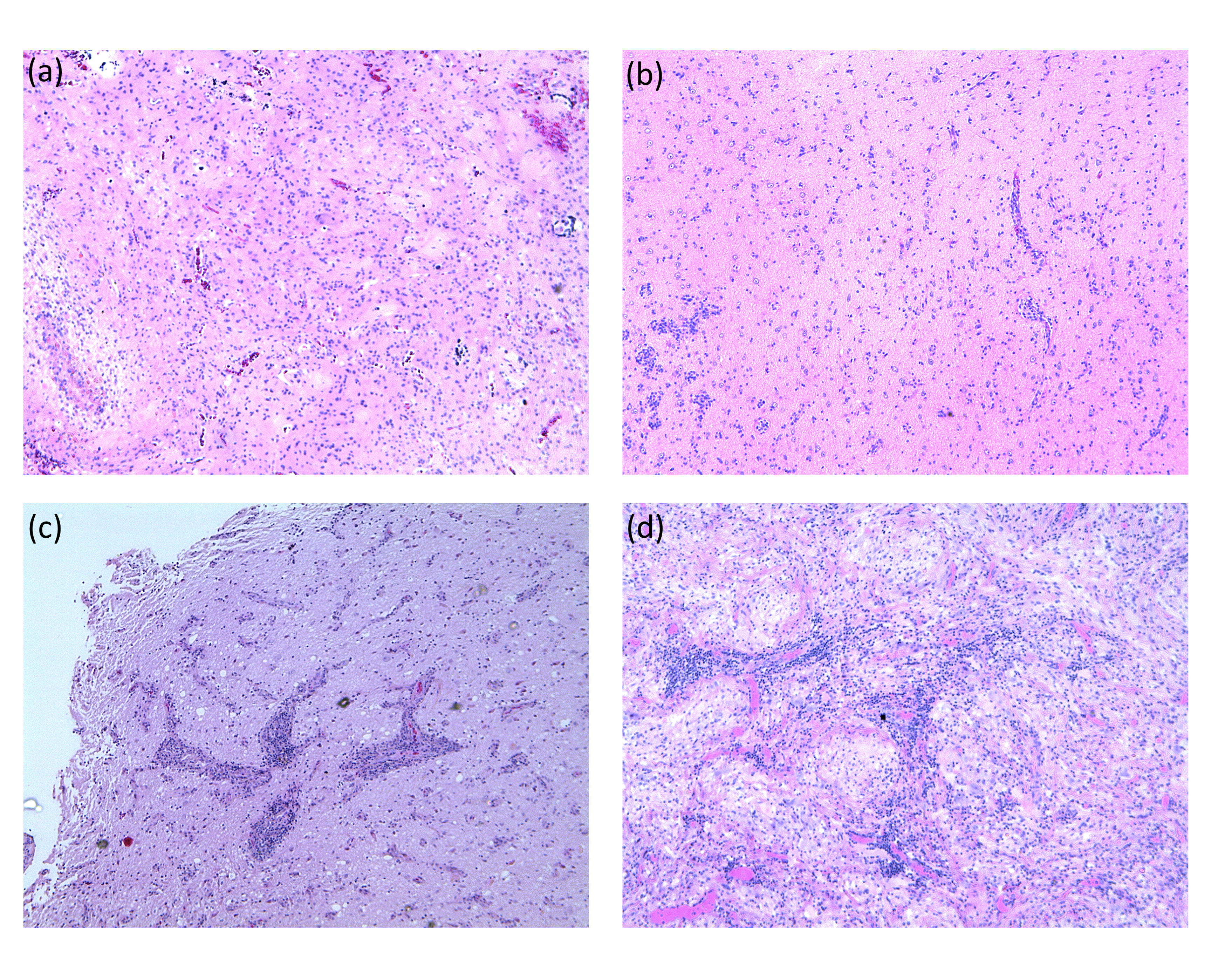
Hematoxylin and eosin–stained slides corresponding to the block from which tissue was sent for methylation analysis were reviewed for degree of lymphocytic infiltration. Cases were assigned a “high” score if prominent (>2 cells thick) perivascular lymphocytic infiltrates were identified within the tumor. All other cases were assigned a “low” score. Representative slides are shown in Supplementary Figure 2.
Statistical Analysis
For statistical analysis, subgroup comparisons were performed by t-test, Pearson’s chi-square test, Fisher’s exact test, or Wilcoxon rank-sum test. Overall survival (OS) was defined as the time from initial diagnosis until death. Progression-free survival (PFS) was defined as the time from initial diagnosis until relapse. Survival curves were plotted using the Kaplan–Meier method. The log-rank test and Cox proportional hazards model were used to detect differences in survival between different groups of patients. Two-sided tests were used for all analyses, and P-values <0.05 were considered significant. JMP 9 (SAS Institute) was used for all analyses.
Results
The PLGG Methylome Is Affected by Key Cellular and Molecular Factors
For PLGG clustering, 152 tumors were analyzed. These included a variety of pathological diagnoses, ages, tumor locations and molecular alterations. Detailed data is provided in Table 1. As an initial step, we performed unsupervised hierarchical clustering of genome-wide methylation data from all 152 PLGG (Fig. 1A). PLGG could be divided into 3 major clusters, with cluster 2 and 3 each having an additional 2 subclusters. These reflected tumor location and additional cellular and molecular events. Consensus clustering and t-distributed stochastic neighbor embedding plotting supported the hierarchical clustering results (Supplementary Figure 3). Cluster 1 consisted mostly of midline PLGG from both the cerebellum and hypothalamus/optic pathway. Cluster 3 consisted of hemispheric tumors which could be further subdivided into 2 groups, clusters 3A and 3B. Detailed imaging analysis of these tumors revealed that tumors attached to the midline were enriched in cluster 3A, while cluster 3B was enriched for more peripherally located hemispheric tumors (P = 0.001; Fig. 1B). Overall, tumor location was a more important determinant of cluster than any molecular or morphological feature. Interestingly, cluster 2 encompassed 2 distinct subgroups where molecular and non-tumor-related factors played a key role. Cluster 2A uniformly comprised hemispheric tumors with both BRAF p.V600E mutation and CDKN2A homozygous deletion. These cluster 2A PLGGs were all Heidelberg classifier PXA-like tumors; however, not all Heidelberg classifier PXA-like tumors were included in cluster 2A. Cluster 2B was enriched for tumors with lower LUMP scores.29 LUMP score is a surrogate marker of leukocyte/lymphocyte infiltration in the specimen calculated from the methylation data, suggesting that tumors in cluster 2B are affected by high leukocyte/lymphocyte infiltration (Fig. 1A).
Fig. 1.

Stratification of PLGG into clusters based on epigenetic profiling. (A) Unsupervised hierarchical clustering of methylation data of 152 PLGGs with 5000 probes showing highest median absolute deviation. The tumors are divided into 3 clusters termed cluster 1, 2, and 3. The following information is indicated below the heatmap: tumor location, molecular status, pathology, CDKN2A status, and the Heidelberg classifier result. (B) Prevalence of tumors attaching to the midline among hemispheric tumors in cluster 3. (C–G) Representative MRI of hemispheric PLGGs in cluster 3A and (H –I) those in cluster 3B.
Biological and Clinical Impact of Cluster 2 PLGG
Since cluster 2B suggested immune infiltration of these PLGGs, we further studied the specific tumors within this group. Progression-free survival was significantly worse for BRAF p.V600E mutant tumors with low LUMP score (P = 0.008; Fig. 2A, B). Low LUMP score was independent of other clinical and molecular variables in these tumors (hazard ratio [HR] 2.41; 95% CI for HR, 1.06–5.42, P = 0.035; Table 2a). Interestingly, the LUMP score was not prognostic in BRAF wildtype tumors (P = 0.37, data not shown). To verify that LUMP score accurately reflected leukocyte/lymphocyte infiltration, we reviewed the pathological slides of the BRAF p.V600E mutant PLGGs with LUMP score data (n =45). To further test the role of lymphocytic infiltration in BRAF p.V600E mutant PLGG we assessed this in a further series of 25 cases. Lymphocyte infiltration was confirmed in tumors with low LUMP scores (Supplementary Figure 2). Further, BRAF p.V600E mutant PLGG with high lymphocytic infiltration, as defined by pathologic assessment, had worse PFS than those without infiltration (P = 0.01; Fig. 2C) on univariate analysis and approached significance on multivariate analysis (P = 0.053; Table 2b). These data indicate that high lymphocytic infiltration may be an important prognostic factor in BRAF p.V600E mutant PLGGs.
Fig. 2.

Molecular and clinical analysis of PLGG in cluster 2. (A) Bar chart of the distribution of LUMP score in PLGGs depending on BRAF V600E status. (B) PFS of BRAF V600E mutant PLGGs stratified by LUMP score. (C) PFS of BRAF V600E mutant PLGGs depending on degree of lymphocytic infiltration by pathology.
Table 2.
Multivariate analysis of PFS among BRAF V600E mutant PLGGs
| a. Multivariate Analysis of PFS among BRAF V600E Mutant PLGGs | |||
|---|---|---|---|
| Variable | Hazard Ratio (HR) | 95% CI for HR | P-value |
| Low LUMP score | 2.41 | 1.06–5.42 | 0.035 |
| CDKN2A homozygous deletion | 8.95 | 2.97–33.19 | <0.0001 |
| Non–hemispheric/cerebellar location | 4.69 | 1.46–18.98 | 0.008 |
| No gross total resection | 1.05 | 0.37–2.83 | 0.91 |
| Age <3 y | 1.35 | 0.48–4.15 | 0.57 |
| b. Multivariate Analysis of PFS among BRAF V600E Mutant PLGGs | |||
| Variable | Hazard Ratio (HR) | 95% CI for HR | P-value |
| High lymphocytic infiltration | 3.33 | 0.98–12.73 | 0.053 |
| CDKN2A homozygous deletion | 21.75 | 4.75–153.95 | 0.001 |
| Non–hemispheric/cerebellar location | 4.19 | 0.85–32.36 | 0.08 |
| No gross total resection | 1.30 | 0.32–4.76 | 0.68 |
| Age <3 y | 2.15 | 0.41–18.77 | 0.38 |
Limitations of the Methylation-Based Classifier in PLGG
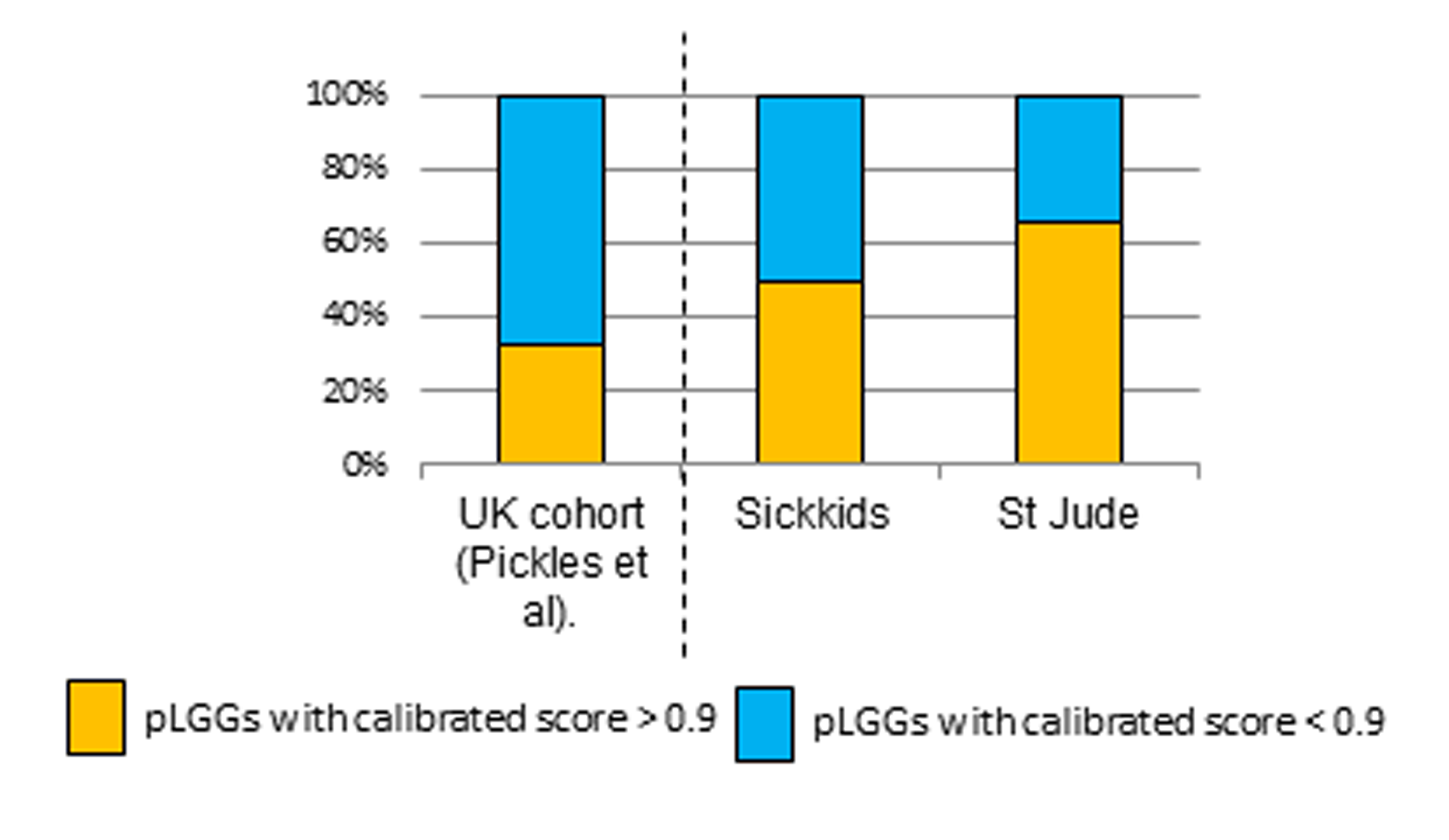
In order to assess the clinical role of methylation-based classification in PLGG, we used the available methylation-based molecular classifier13 and applied it to all tumors from different institutions (Fig. 3). The classifier failed to reach high confidence with a calibrated score of more than 0.9, which reflects the reliability of the diagnosis,13 in 51% of cases (69/136 cases) at SickKids and 34% (29/86 cases) at St Jude. This is comparable to 67% (57/85 cases) of a population-based cohort in the United Kingdom14 (Fig. 3A, Supplementary Figure 4). Importantly, none of the PLGG diagnosed by pathology was changed to high-grade glioma by the classifier. BRAF p.V600E mutation and BRAF fusions, which are critical for clinical risk stratification and molecular targeting therapy, were observed across multiple methylation subclasses (Fig. 3A).
Fig. 3.

Epigenetic, clinical, and molecular features of study cohort. (A) Oncoprint of the cases. The Heidelberg classifier is indicated in the top column. The following information is indicated below the figure: confidence of the classifier showing the calibrated score more than 0.9 or not, Institution, tumor location, pathology, and molecular status, including BRAF V600E mutation, BRAF fusions, FGFR1-TACC1 fusion, FGFR2 fusions, MYBL1 alterations, ALK fusions, and CDKN2A status. (B) OS of the cases diagnosed as “PXA like” by the classifier. Green dot line indicates a survival curve of all of the cases. Red and blue lines are indicated survival curves depending on histological grade. (C) OS of pediatric gliomas with both BRAF-V600E mutation and CDKN2A homozygous deletion stratified by the histological grade. (D) Survival of the same cohort as stratified by the classifier.
We then further examined the additional value of the methylation classifier to the current molecular and pathological scheme. Some rare alterations such as FGFR1-TACC1, FGFR2 fusions, MYBL1 alterations, and ALK fusions clustered together within the classifier except one FGFR1-TACC1 fused case. These still were not called as such but rather clustered with other PLGG. In one case, accounting for 0.6% of our cohort, clinical management would have benefited from using the classifier. The case was a 2.7-year-old patient with localized brainstem low-grade glioma without metastasis at diagnosis (Supplementary Figure 5a). The pathological diagnosis was diffuse astrocytoma. No alterations were detected by NanoString or DD-PCR. This patient experienced disseminated recurrence starting at 2.92 years after diagnosis (Supplementary Figure 5b) and died from the disease at 5.18 years. Methylation data from the primary tumor revealed a diagnosis of diffuse leptomeningeal glioneural tumor (DLGNT) accompanied by 1p loss30(Supplementary Figure 5c).
In contrast, 10.9% of PLGGs (29 out of 222 cases) were termed as nontumor tissue by the classifier, including reactive inflammation and control normal brain. These were mostly low confidence tumors which had clinical, imaging, and pathological diagnosis of PLGG, and all harbored molecular alterations such as BRAF p.V600E or BRAF fusions observed in PLGG. Additionally, 4 PLGGs in which BRAF was altered had nonglial tumor diagnoses (2 cases of meningioma and 2 cases of craniopharyngioma) by the classifier (Supplementary Table).
Combined Methylation, Pathological, and Molecular Analysis Redefines PXA-Like Tumors
One of the possible strengths of methylation analysis is in classifying BRAF p.V600E mutant/CDKN2A deleted tumors as a unique clinical entity termed PXA-like gliomas.31 PXA-like tumors are defined as tumors diagnosed as “PXA” with high confidence (calibrated score >0.9) by the Heidelberg classifier.13,31 In order to shed light onto this group of gliomas we constructed an additional cohort of pediatric tumors termed PXA-like tumors by the Heidelberg classifier from 3 separate institutions (SickKids, St Jude, and NYU). In total, 27 PXA-like tumors had high confidence by the classifier regardless of the underlying pathology, which included 17 low grade (13 PXAs, 2 gangliogliomas, 1 desmoplastic infantile ganglioglioma, 1 low grade astrocytoma not otherwise specified) and 10 high grade histologies (5 anaplastic PXAs, 3 glioblastomas, 1 anaplastic astrocytoma, and 1 anaplastic ganglioglioma). All had both BRAF V600E mutation and CDKN2A homozygous deletion except for 3 cases (1 PXA, 1 BRAF wildtype/CDKN2A deleted anaplastic PXA harboring, and 1 BRAF p.V600E mutant/CDKN2A wildtype PXA). The 5-year OS for the entire cohort was 82%, which is similar to previously described PXA-like cohorts.31 Strikingly, these tumors could be divided into 2 groups based on tumor pathological grade. PXA-like PLGGs had significantly better outcome compared with PXA-like high grade gliomas. At 5 years, all PXA-like PLGGs were alive, while only 34% of high grade tumors survived (P = 0.0014; Fig. 3B). We then tested all BRAF p.V600E mutated/CDKN2A homozygous deleted tumors regardless of their pathology or methylation classification (n =32). These included 20 low grade (14 PXAs, 3 gangliogliomas, 2 low grade astrocytomas, and 1 desmoplastic infantile ganglioglioma) and 12 high grade tumors (5 glioblastomas, 4 anaplastic PXAs, 1 anaplastic astrocytoma, 1 anaplastic ganglioglioma, and 1 gliosarcoma). The majority of these tumors were called “PXA” by the classifier (24 cases with high confidence [PXA-like tumors] and 4 cases with low confidence [calibrated scores were 0.28, 0.51, 0.57, and 0.71, respectively]), while 4 tumors were termed as diverse pathological types. While pathological grading determined survival in this cohort (Fig. 3C), there were no prognostic differences between “PXA-like” and “non PXA-like” in cases with both genetic alterations (Fig. 3D).
Discussion
In this study of the largest PLGG cohort of combined long-term outcome data with morphological, molecular, and epigenetic status, we are able to redefine the role of methylation arrays as a clinical and biological tool in these cancers. Our study provides insight into the role of tumor location and other nonneoplastic variables in PLGG and describes the potential and limitations of genome-wide methylation data in clinical classification and prognostic stratification of PLGG groups.
Unsupervised clustering is usually the first step to provide an overview of emerging subgroups and their biological role in tumor analysis. In PLGGs, tumor location was previously reported to drive differences in methylation status by several groups.20,32 Our analysis of clusters 1 and 3 provides further clinical and biological insight into these data. Although midline optic and cerebellar tumors clustered together, our observation that specific locations within the cerebral hemispheres have a different profile and therefore potentially cell of origin is intriguing. For example, cluster 3B, which is enriched with peripherally located hemispheric tumors, contained, as expected, numerous BRAF p.V600E mutant tumors (Figures 1–2). Since cluster 3A, which represents more centrally located tumors, harbored similar alterations, identification of this subcluster may have clinical and biological value in the future. As resection is extremely important in BRAF p.V600E mutant PLGG,11 and very difficult in cluster 3A tumors, targeting the different cell of origin may be an important part of the management and future approach to these tumors. Additional future research, such as single cell analysis, which is emerging as a tool to dissect the biological vulnerabilities of tumor cell of origin, may be an important aspect of such tumors with similar molecular alterations but different origin.33
Cluster 2B provides a yet underappreciated look at PLGG. Our data suggest that LUMP score correlates with lymphocytic infiltration and may be associated with tumor progression. Lymphocytic infiltration is well known to occur in PLGGs, particularly gangliogliomas, PXA, and pilocytic astrocytomas,28,34 and one study by Dahiya et al showed worse PFS for cases with high chronic inflammatory infiltrates.34 Although validation in a larger, prospective cohort is required, our findings support those of the Dahiya study and suggest that lymphocytic infiltration may be a marker for recurrence of at least BRAF p.V600E mutant PLGG. Interestingly, numerous studies have shown that increased lymphocytic infiltration is a good prognostic factor in most cancers.35 More rarely, lymphocytic infiltration negatively affecting patients’ prognosis has been reported, such as in clear cell renal cell carcinomas (ccRCC), diffuse large B-cell lymphomas, and Hodgkin lymphomas.36–38 Interestingly, the latter tumors also respond dramatically to immune checkpoint inhibition. Giraldo et al described the mechanism of the correlation between lymphocytic infiltration and worse prognosis of patients with ccRCC. They detected a high prevalence of regulatory T cells and polyclonal CD8+ T cells with reduced cytotoxic capacity and low ability to recognize tumor associated antigens.36 It is thus intriguing to hypothesize that these PLGG may be more susceptible to combination therapies with immune checkpoint inhibitors.
Another important clinical aspect of this study is redefining the potential and pitfalls of methylation arrays in PLGG. Based on our data, it is clear that in PLGG, methylation clustering alone has major issues in reaching high confidence calls and is less useful than in other tumors in defining clinically relevant subgroups. This is very different from other pediatric brain tumors where methylation arrays are a robust tool for tumor classification and management. In our opinion these differences are important to discuss as they explain other issues with cancer molecular analysis.
First, in contrast to most childhood embryonal and high grade tumors, the cell population is diverse in a biopsy of PLGG and includes multiple normal and reactive cells which may skew the methylation-based diagnosis. Although normal neurons and glial cells, which are intermixed in some infiltrative biopsies, may complicate the cell of origin analysis, as discussed above, methylation-based LUMP score indicative of high immune cell infiltration may serve as a tool for tumor prognostication and targeting therapies.
Second, while in many tumors, such as posterior fossa ependymoma and other embryonal tumors, specific genetic alterations do not exist to support subgroups, recent data reveal that unique, usually mutually exclusive driver mutation can be found in almost all PLGG and help in both tumor stratification and defining therapy in these tumors. In the era of targeted therapy, this role of genetic stratification will increase and perhaps overshadow the role of epigenetic analysis in PLGG.
Third, the unique ability of methylation arrays to find novel rare subgroups in many tumor types which lead to uncovering genetic alterations in these tumors has also shown benefit in PLGG. For example, the Heidelberg classifier seems to be the best tool to diagnose DLGNT.
In summary, we suggest that in PLGG, a combined histopathological, molecular, and epigenetic approach may be required for management of these tumors. While in the majority of PLGG, histopatholgical and genetic analysis will suffice in initial classification. In rare cases where cheaper molecular tools fail to establish the diagnosis, methylation arrays may provide an important tool for correct stratification. Furthermore, future studies will help in verifying the role of epigenetic analysis in targeting the cell of origin and the microenvironment in PLGG.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was supported by endowed funds from the Government of Canada through Genome Canada and the Ontario Genomics Institute (OGI-121); A Kid’s Brain Tumor Cure; Brain Tumour Research Assistance and Information Network; The Elmaagacli Foundation; Livewise foundation: The Pediatric Low-Grade Astrocytoma Foundation; Meagan’s Walk; B.r.a.i.n.child Canada; Canadian Cancer Society (Grant # 702296); Canadian Institute of Health Research (Grant # 159805); Restracomp Scholarship and Fellowship funds from the Garron Family Chair in Childhood Cancer Research at The Hospital for Sick Children (S.R, M.Z, A.L, and K.F); Canadian Institute of Health Research (CGS-M) scholarship (S.R.); Ontario Graduate Scholarship (S.R.); Tokyo Children’s Cancer Study Group (TCCSG) scholarship of the Gold Ribbons Network of Japan (K.F.); The Marie-Josée and Henry R. Kravis Center for Molecular Oncology; The National Cancer Institute Cancer Center Core Grant No. P30-CA008748; The American Lebanese Syrian Associated Charities (ALSAC) of St Jude Children’s Research Hospital. Sohn Conference Foundation, the Freidberg Charitable Foundation, and the Making Headway Foundation grants (M.S.).
Conflict of interest statement.
Authors disclose no conflict of interest.
Authorship statement.
Experimental design: KF, CH, UT. Data collection: KF, YM, RT, MZ, SR, AGS, JB, AA, BL, JW, KZ, AL, DT, TJP, LH, NL, JD, JTR, PD, AVK, MDT, UB, AH, GZ, KA, VR, EB, MS, DE, CH, UT. Data analysis: KF, YM, AL, VR, CH, UT. Interpretation of the data: KF, YM, MS, DE, CH, UT. Manuscript writing: KF, CH, UT.
References
- 1. Jones DTW, Kieran MW, Bouffet E, et al. Pediatric low-grade gliomas: next biologically driven steps. Neuro Oncol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Packer RJ, Pfister S, Bouffet E, et al. Pediatric low-grade gliomas: implications of the biologic era. Neuro Oncol. 2017;19(6):750–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stokland T, Liu JF, Ironside JW, et al. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro Oncol. 2010;12(12):1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsang DS, Murphy ES, Merchant TE. Radiation therapy for optic pathway and hypothalamic low-grade gliomas in children. Int J Radiat Oncol Biol Phys. 2017;99(3):642–651. [DOI] [PubMed] [Google Scholar]
- 5. Ramkissoon LA, Horowitz PM, Craig JM, et al. Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A. 2013;110(20):8188–8193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bandopadhayay P, Ramkissoon LA, Jain P, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet. 2016;48(3):273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huse JT, Snuderl M, Jones DT, et al. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): an epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol. 2017;133(3):417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones DT, Hutter B, Jäger N, et al. ; International Cancer Genome Consortium PedBrain Tumor Project Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45(8):927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45(6):602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lassaletta A, Zapotocky M, Mistry M, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol. 2017;35(25):2934–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wefers AK, Stichel D, Schrimpf D, et al. Isomorphic diffuse glioma is a morphologically and molecularly distinct tumour entity with recurrent gene fusions of MYBL1 or MYB and a benign disease course. Acta Neuropathol. 2020;139(1):193–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pickles JC, Fairchild AR, Stone TJ, et al. DNA methylation-based profiling for paediatric CNS tumour diagnosis and treatment: a population-based study. Lancet Child Adolesc Health. 2020;4(2):121–130. [DOI] [PubMed] [Google Scholar]
- 15. Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. [DOI] [PubMed] [Google Scholar]
- 16. Hovestadt V, Remke M, Kool M, et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 2013;125(6):913–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mack SC, Witt H, Piro RM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506(7489):445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sturm D, Orr BA, Toprak UH, et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell. 2016;164(5):1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jeyapalan JN, Doctor GT, Jones TA, et al. DNA methylation analysis of paediatric low-grade astrocytomas identifies a tumour-specific hypomethylation signature in pilocytic astrocytomas. Acta Neuropathol Commun. 2016;4(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lambert SR, Witt H, Hovestadt V, et al. Differential expression and methylation of brain developmental genes define location-specific subsets of pilocytic astrocytoma. Acta Neuropathol. 2013;126(2):291–301. [DOI] [PubMed] [Google Scholar]
- 21. Qaddoumi I, Orisme W, Wen J, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016;131(6):833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stone TJ, Keeley A, Virasami A, et al. Comprehensive molecular characterisation of epilepsy-associated glioneuronal tumours. Acta Neuropathol. 2018;135(1):115–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mistry M, Zhukova N, Merico D, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol. 2015;33(9):1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hawkins C, Walker E, Mohamed N, et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res. 2011;17(14):4790–4798. [DOI] [PubMed] [Google Scholar]
- 25. Ryall S, Arnoldo A, Krishnatry R, et al. Multiplex detection of pediatric low-grade glioma signature fusion transcripts and duplications using the NanoString Ncounter system. J Neuropathol Exp Neurol. 2017;76(7):562–570. [DOI] [PubMed] [Google Scholar]
- 26. Guerreiro Stucklin AS, Ryall S, Fukuoka K, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun. 2019;10(1):4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ryall S, Zapotocky M, Fukuoka K, et al. The genetic landscape of pediatric low-grade gliomas: incidence, prognosis, and response to therapy. Neuro Oncol. 2018;20(suppl_2):i114. [Google Scholar]
- 28. Louis DN, et al. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon, France: International Agency for Research on Cancer;2016. [Google Scholar]
- 29. Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun. 2015;6:8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deng MY, Sill M, Chiang J, et al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol. 2018;136(2):239–253. [DOI] [PubMed] [Google Scholar]
- 31. Korshunov A, Ryzhova M, Hovestadt V, et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015;129(5):669–678. [DOI] [PubMed] [Google Scholar]
- 32. Sexton-Oates A, Dodgshun A, Hovestadt V, et al. Methylation profiling of paediatric pilocytic astrocytoma reveals variants specifically associated with tumour location and predictive of recurrence. Mol Oncol. 2018;12(8):1219–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vladoiu MC, El-Hamamy I, Donovan LK, et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature. 2019;572(7767):67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR. BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol. 2013;125(6):901–910. [DOI] [PubMed] [Google Scholar]
- 35. Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–734. [DOI] [PubMed] [Google Scholar]
- 36. Giraldo NA, Becht E, Vano Y, et al. Tumor-infiltrating and peripheral blood T-cell immunophenotypes predict early relapse in localized clear cell renal cell carcinoma. Clin Cancer Res. 2017;23(15): 4416–4428. [DOI] [PubMed] [Google Scholar]
- 37. Muris JJ, Meijer CJ, Cillessen SA, et al. Prognostic significance of activated cytotoxic T-lymphocytes in primary nodal diffuse large B-cell lymphomas. Leukemia. 2004;18(3):589–596. [DOI] [PubMed] [Google Scholar]
- 38. Riemersma SA, Oudejans JJ, Vonk MJ, et al. High numbers of tumour-infiltrating activated cytotoxic T lymphocytes, and frequent loss of HLA class I and II expression, are features of aggressive B cell lymphomas of the brain and testis. J Pathol. 2005;206(3):328–336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.