

Abstract
Background
Powassan virus (POWV) is an emerging cause of severe encephalitis; very little is known about human pathogenicity due to challenges in diagnosis and viral RNA recovery. We present 3 patients with fatal encephalitis due to POWV lineage II (deer tick virus).
Methods
We obtained 27 unique samples, including from brain biopsy and autopsy, and used metagenomic sequencing, quantitative reverse transcriptase polymerase chain reaction, and a newly developed CRISPR-based diagnostic assay to perform the first detailed characterization of POWV compartmentalization and genomics between and within human subjects.
Results
In all 3 patients, imaging and histopathology findings were notable for profound cerebellar involvement. All patients were initially diagnosed with POWV by metagenomic sequencing, and 2 of the 3 had negative clinical testing by serology. We detected POWV RNA in 13 clinical samples; levels were highest in the cerebellum, and there was very little involvement of peripheral tissue. We assembled complete POWV genomes from 8 samples, providing unique information about the strains of POWV lineage II (deer tick virus) that infect humans.
Conclusions
We demonstrate the utility of molecular assays for detecting POWV infection, including in seronegative patients, and nominate viral genomic features that may relate to human infection and neuropathogenicity. The cerebellum was identified as a key target POWV in fatal infection, by radiological and histopathological findings as well as molecular testing.
Keywords: flavivirus, molecular diagnostics, neuropathology, viral genomics
In three patients with fatal encephalitis, Powassan virus was diagnosed by metagenomic sequencing. Imaging, histopathology, and molecular testing of 27 unique samples showed profound cerebellar involvement, and little peripherally. Powassan virus genome sequencing offered insight into strains that infect humans.
Powassan virus (POWV), a tick-borne flavivirus, is an underappreciated cause of encephalitis, with 50% long-term morbidity and 10% mortality [1–3]. Similar to other tick-borne pathogens, POWV detection is increasing [4–6], and seroprevalence studies suggest that additional infections go undetected [7, 8]. Magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) studies are nonspecific, and POWV is diagnosed by serology, a time-consuming test performed at reference centers that is generally restricted to patients with severe encephalitis. The lack of a widely available molecular test limits POWV detection early in its course (eg, before neurological symptoms) or in immunocompromised patients who may not mount an antibody response [9].
The limited number of diagnosed cases and the challenges in recovering POWV RNA from clinical samples have restricted viral genomic and evolutionary analyses. Before this study, only 5 complete POWV genomes from humans had been published, all of which were from the canonical POWV lineage I, which is transmitted by Ixodes cookei ticks and thought to rarely infect humans. By contrast, there have been no human-derived complete genomes and only 4 partial genomes from POWV lineage II (deer tick virus), which is thought to be more frequently responsible for human disease [1, 3] and is present in 1%–4% of Ixodes scapularis in endemic areas [10, 11]. No studies have described the dynamics and compartmentalization of POWV in humans. Here, enhanced genomic methods were used to investigate the within-host diversity of POWV lineage II in correlation with histopathologic findings.
METHODS
Samples
Details are available in the Supplementary Data. Briefly, total nucleic acid was extracted from each fluid sample using the QIAmp Viral RNA Mini kit (Qiagen, Hilden, Germany) [12] and from each formalin-fixed paraffin-embedded (FFPE) sample using Quick-DNA/RNA FFPE (Zymo Research, Irvine, California, USA). Water or CSF from an uninfected patient was included as a negative control. Samples were treated with HL-dsDNase (ArcticZymes, Tromso, Norway) per the manufacturer’s instructions.
Quantitative Reverse Transcription Polymerase Chain Reaction
RNA was screened for POWV using a nonclinical quantitative reverse transcription polymerase chain reaction (RT-qPCR) assay targeting the NS5 gene [13]. Diluted samples (1:4) were tested in triplicate alongside a standard curve and negative controls using Power SYBR Green RNA-to-CT (Applied Biosystems, Foster City, California, USA). The previously published limit of detection for this assay was 10 copies/µL [13], so samples with >10 copies/µL were considered positive, samples with 1–10 copies/µL equivocal, and samples with <1 copy/µL negative.
Specific High-Sensitivity Enzymatic Reporter Unlocking
Specific high-sensitivity enzymatic reporter unlocking (SHERLOCK) is a CRISPR/Cas13-based method that enables sensitive, specific, and multiplexable detection of viral nucleic acid [14–16]. A SHERLOCK assay was designed to target a region of POWV NS5, flanking the RT-qPCR assay target (Supplementary Data). RNA and cDNA from each sample were tested alongside biological negative controls, synthetic RNA-positive controls, and an in-assay water control. RPA was performed using the Twist-Dx RT-RPA kit (Twist Bioscience, San Francisco, California, USA) [15]. For experiments with RNA templates, murine RNase inhibitor (NEB, Ipswich, Massachusetts, USA) was added. Detection was performed in triplicate [14, 15]. For each SHERLOCK experiment, the assay positive threshold was defined as 3 standard deviations above the mean fluorescent signal for the in-assay water control, as described previously [14, 15]. Samples whose fluorescent signals (mean and 1 SD) were above this threshold were considered positive. Samples whose mean fluorescent signal was above this threshold but whose standard deviation crossed the threshold were considered equivocal.
Library Preparation, Hybrid Capture, and Sequencing
FFPE samples underwent RNase-H-based ribosomal RNA depletion [17]. For all samples, metagenomic sequencing libraries were constructed using random hexamer cDNA synthesis and Nextera XT (Illumina, San Diego, California, USA) [13, 17]. Samples were sequenced on an Illumina platform to obtain ~1 million (0.2–1.5 million) 2×100–150-bp paired-end reads per sample. For samples with POWV reads detected, an independent library was constructed from RNA, and both libraries underwent deeper sequencing for POWV genomic analyses. Libraries were enriched for viral material using hybrid capture probes designed to recognize all human pathogenic viruses (VALL; Roche, Basel, Switzerland) as previously described [18] and detailed in the Supplementary Data.
POWV Genome Assembly and Analyses
For Subjects A and C, who did not have a confirmed clinical diagnosis, reads first underwent metagenomic classification by Kraken [19]. For each POWV-positive sample, assembly of a consensus POWV sequence was performed using viral-ngs [20]. Reads were merged across sequencing runs, depleted of human and contaminant reads, and positively filtered against all available POWV genomes (GenBank, June 2019). Contigs were de novo assembled, scaffolded to a POWV reference sequence (HM440559.1), and refined by remapping raw reads to correct for reference bias. Intrasample single nucleotide variants (iSNVs) were identified using vphaser-2 [21], implemented in viral-ngs [20], and required iSNVs to be present in ≥1 forward and ≥1 reverse read in each of 2 independent libraries.
The coding region of POWV sequences obtained from brain samples from patients in this study was aligned with 2 sets of reference sequences from GenBank: 1) all POWV genomes with >90% coverage, and 2) partial lineage II genomes, including additional genomes from humans. Genomes were aligned using MUSCLE [22, 23]. Maximum likelihood phylogenetic trees were constructed using IQ-TREE [24] using a GTR+G substitution model with 1000 ultrafast bootstraps [25].
RESULTS
Molecular Testing Identified POWV in 3 Patients With Fatal Encephalitis Better Than Testing With Serology
We identified 3 patients with fatal encephalitis whose clinical presentations and radiological imaging findings were notable for profound cerebellar involvement. In all 3 patients, POWV was first identified by molecular testing, rather than standard clinical testing by serology.
Subject A was a 79-year-old male physician and gardener from Massachusetts who had prostate cancer treated with hormone therapy. He presented with 1 day of headache and dizziness, then developed altered mental status and fever of 102.2°F. CSF analysis showed glucose = 71 mg/dL, protein = 102 mg/dL, and white blood cell count (WBC) = 284 cells/mL (56% monocytes). MRI showed leptomeningeal enhancement and significant edema in the cerebellum and brainstem, consistent with rhombencephalitis (Figure 1). Routine clinical testing was unrevealing, including CSF multiplex PCR (BioFire FilmArray, Biomerieux). The patient enrolled in our study of pathogen detection by metagenomic sequencing, and POWV lineage II RNA was detected in CSF within 72 hours (Figure 3; Supplementary Table 1). This result was confirmed with a positive POWV IgM capture enzyme-linked immunosorbent assay and plaque reduction neutralization test (PRNT; 1:2560) from serum, though these results were not obtained until ~1 month later. The patient was treated with IVIG but had extremely limited neurological recovery and passed away 3 months after presentation due to complications from prolonged hospitalization. No autopsy was performed.
Figure 1.

Brain magnetic resonance imaging findings. T2-fluid-attenuated inversion recovery sequences show hyperintensity in the cerebellum (top row) and brainstem (bottom row), consistent with rhombencephalitis, in Subject A after 4 days of symptoms, Subject B after 5 days of symptoms, and Subject C after 7 days of symptoms. While highly suspicious for POWV infection, other infectious etiologies as well as autoimmune diseases and paraneoplastic syndromes remain in the differential diagnosis, and confirmation by serology or viral RNA detection is required. Abbreviation: POWV, Powassan virus.
Figure 3.

Comparison of screening results from metagenomic sequencing, RT-qPCR, and SHERLOCK. Rows indicate the type of tissue tested from each of the 3 subjects. For routine metagenomic sequencing, a positive result (+, pink) is defined as at least 1 in 1 million reads mapping to POWV. For RT-qPCR, a positive result (+, pink) is defined as >10 copies/µL, and an equivocal result (+/-, light pink) is defined as 1–10 copies/µL. For SHERLOCK, samples whose fluorescent signal (mean and 1 SD) was above the assay positive threshold were defined as positive (+, pink). Samples whose mean fluorescent signal was above this threshold but whose standard deviation crossed the threshold were defined as equivocal (+/-, light pink). SHERLOCK experimental results are further detailed in Supplementary Figure 1. Quantitative results for metagenomic sequencing, RT-qPCR, and SHERLOCK are in Supplementary Table 1. Abbreviations: CSF, cerebrospinal fluid; POWV, Powassan virus; RT-qPCR, quantitative reverse transcription polymerase chain reaction; SHERLOCK, specific high-sensitivity enzymatic reporter unlocking.
Detailed clinical findings and diagnostic metagenomic sequencing results from Subject B were reported previously [9]. Briefly, he was a 63-year-old man from Massachusetts with prostate adenocarcinoma and follicular lymphoma treated with rituximab. He initially presented with testicular pain and fever, was found to have orchiepididymitis, and then developed meningismus and altered mental status. CSF analysis showed glucose = 62 mg/dL, protein = 83 mg/dL, and WBC = 10 cells/mL (60% lymphocytes). MRI showed cerebellar edema and T2 abnormalities in the periventricular, thalamo-mesencephalic, and basal ganglia regions (Figure 1). Cerebellar biopsy revealed a T-cell inflammatory infiltrate of unclear etiology primarily involving the leptomeninges, but also the cerebellar cortex, with some Purkinje cell loss and Bergmann gliosis. The patient passed away 19 days after presentation, and autopsy showed widespread brain abnormalities consistent with meningoencephalitis that was most severe in the cerebellum, but also involving the brainstem, hippocampus, thalamus, basal ganglia, deep white matter, and cerebral cortex (Figure 2A and B). Foci of chronic inflammation were identified in the left testicle, while the right testicle, heart, lungs, liver, spleen, prostate, kidneys, and bone marrow showed minimal nonspecific abnormalities without histological evidence of infection or recurrent lymphoma. As previously reported, the patient was diagnosed with POWV by RNA detection from premortem serum and CSF; results were reported after death and later confirmed by immunohistochemistry and targeted PCR and sequencing from postmortem brain tissue [9]. POWV serology was negative in the setting of rituximab therapy.
Figure 2.

Correlation of autopsy histopathological findings with POWV detection in Subjects B and C. A, Autopsy tissue collected from Subject B included lung (a), heart (b), liver (c), spleen (d), prostate (e), kidney (f), right testicle (g), left testicle (h), and bone marrow (i). Foci of chronic inflammation were identified in the left testicle, while the remaining specimens showed minimal, nonspecific abnormalities without histological evidence of infection or recurrent lymphoma. B, Brain sections from Subject B included basal ganglia (a), frontal cortex (b), periventricular white matter (c), midbrain (d), thalamus (e), pons (f), and cerebellum (g). Findings of meningoencephalitis, including lymphocytic predominant leptomeningeal, perivascular, and parenchymal inflammation, microgliosis with microglial nodules and neuronophagia, and severe neuronal loss, were present throughout the brain and most predominant in the cerebellum. C, Brain sections from Subject C included frontal cortex (a), thalamus (b), medulla (c), and cerebellum (d). Histological abnormalities were similar to Subject B and most severe in the brainstem and cerebellum. All histological images are of hematoxylin and eosin–stained slides using a 20× objective. The quantity of virus detected by metagenomic sequencing (POWV reads/million) is indicated by the thickness of the boxes surrounding the histological images. Anatomy figures were obtained from Wikimedia Commons under Creative Commons CC0 1.0 Public Domain Dedication. Abbreviation: POWV, Powassan virus.
Subject C was a 75-year-old woman from Massachusetts with remote Hodgkin lymphoma. She presented with tinnitus, headache, and low-grade fever, and reported a recent tick bite. Over the course of 1 week, she exhibited progressive difficulty with ambulation, inability to follow commands, and decorticate posturing. Computed tomography and MRI showed cerebellar edema with leptomeningeal enhancement and cortical/basal ganglia involvement (Figure 1). A cerebellar biopsy on hospital day (HD) 2 (6 days after symptom onset) showed loss of Purkinje cells with no definitive evidence of infection. CSF obtained from an extraventricular drain (EVD) on HD 3 showed glucose = 129 mg/dL, protein = 176 mg/dL, and WBC = 1 cell/mL (39% neutrophils, 61% lymphocytes). Sampling from an EVD may have led to lower than expected pleocytosis [26, 27]. Otherwise, the lack of pleocytosis is remarkable; this has been reported in a patient with POWV encephalitis and chronic lymphocytic leukemia [28]; however, Subject C was not overtly immunocompromised. Premortem infectious workup including CSF serology for POWV was negative. The patient continued to decline due to upward cerebellar herniation and passed away on HD 6 (10 days after presentation). Autopsy revealed severe rhombencephalitis with perivascular, parenchymal, and leptomeningeal lymphocytosis and scattered microglial nodules predominantly affecting the cerebellum and brainstem (Figure 2C), with Duret hemorrhages due to increased intracranial pressure and cerebellar herniation. The heart, lungs, liver, spleen, kidneys, thyroid, and hilar lymph nodes showed minimal nonspecific abnormalities without histological evidence of infection or recurrent lymphoma. POWV was first identified by molecular techniques in this study, and subsequently confirmed by flavivirus NS5 gene RT-qPCR/sequencing and POWV immunohistochemistry of postmortem brain tissue performed by the Centers for Disease Control and Prevention Infectious Diseases Pathology Branch.
An Extensive Set of POWV Clinical Samples Enabled Characterization of Anatomic distribution and comparison of Detection Methods
We assembled a unique and extensive set of 27 clinical samples from these 3 subjects, which allowed us to both define the anatomical distribution of infection and evaluate multiple molecular detection methods. From Subject A, CSF was obtained 2 days after symptom onset, as well as urine, whole blood, and serum from ~1 week after symptom onset. From Subject B, brain biopsy tissue (11 days before death) and autopsy tissues from 7 brain regions and 9 peripheral organs were obtained. From Subject C, CSF (6 days before death), brain biopsy tissue (4 days before death), and autopsy tissues from 4 brain regions were obtained.
Anatomic Distribution of POWV
Extensive tissue screening provided insight into the anatomic distribution of POWV during natural infection. Within the brains of Subjects B and C, levels of POWV RNA were highest in the cerebellum, consistent with the pathological observation of marked Purkinje cell loss and consistent with MRI findings (Figures 1 and 2). POWV RNA was detected in the CSF and/or brain tissue from all 3 subjects and in the testicles of Subject B, but not in other peripheral tissues. The absence of POWV RNA from the blood of Subject A (1 week after symptom onset) is compatible with the hypothesis that POWV confers a short period of viremia, similar to other neuroinvasive flaviviruses [29–31]. The absence of POWV RNA in the urine of Subject A and the kidney of Subject B is notable because other flaviviruses including Zika virus and tick-borne encephalitis virus are often detectable in urine [32, 33].
Assessment of 3 Molecular Detection Methods
In order to advance multiple technologies for the detection of POWV RNA in clinical samples, previously described metagenomic sequencing and POWV qRT-PCR methods were employed [13], along with a novel POWV lineage-specific CRISPR/Cas13-based assay (SHERLOCK) (Supplementary Figures 1 and 2) [14, 15]. POWV RNA was identified in 13 clinical samples (Figure 3; Supplementary Table 1). The methods were concordant when applied to samples with moderate to high levels of POWV, such as brain tissue. However, metagenomic sequencing was the most sensitive for samples with a low level of POWV, including CSF (Figure 3; Supplementary Table 1). Although metagenomic sequencing is a robust and sensitive method, it is also time- and resource-intensive. Therefore, an appealing strategy for improving molecular diagnosis of POWV may be to implement RT-qPCR or SHERLOCK as a rapid screening test and metagenomic sequencing for negative samples with high clinical suspicion.
Sequencing Complete POWV Lineage II Genomes Allowed Comparison Within and Between Individuals
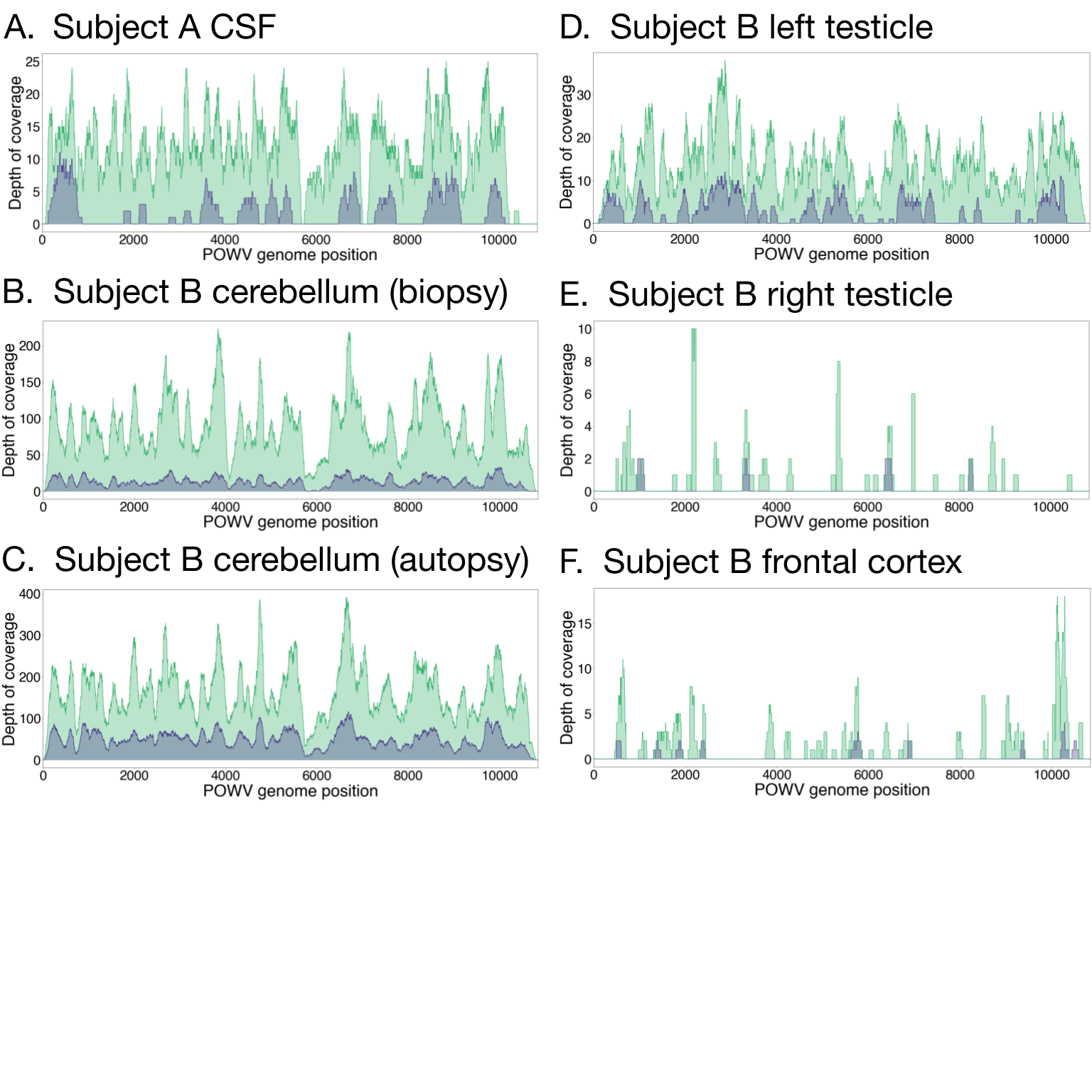
Very little is known about the strains of POWV that infect humans; at the time of this study, only 2 partial POWV lineage II genomes were available from human samples. Here, complete POWV genomes were assembled directly from 8 clinical samples using a combination of metagenomic sequencing [17] and hybrid capture [18] (Supplementary Figure 3), allowing the first insights into POWV diversity within and between infected patients. These samples included CSF from Subject A; cerebellum (from both biopsy and autopsy) and left testicle from Subject B; and cerebellum (from both biopsy and autopsy), thalamus, and medulla from Subject C (Supplementary Table 2). In addition to assembling consensus genomes, we identified intrasample single nucleotide variants (iSNVs) in 5 samples that had moderate to high depth of sequencing (Supplementary Data).
POWV Variation Within the Brain and Between Brain and Periphery
POWV viral genomes were compared between brain and peripheral tissue (Subject B) and within the brain itself (Subject C) to understand POWV compartmentalization. Between the cerebellum and left testicle from Subject B, 5 consensus-level polymorphisms were present (Figure 4A; Supplementary Table 3). At 4 of these 5 sites, the variant in the testicle matched reference genomes, while the variant in the brain was different. Although it is likely that these variants arose by chance, interestingly 1 was a nonsynonymous polymorphism in the NS4b gene, which is thought to play a role in immune evasion in other flavivirus infections [29]. At only 1 site was the testicle variant detected as an iSNV in the cerebellum, suggesting that there was little to no circulation between these compartments by the time of death. In Subject C, consensus sequences were the same between the cerebellum and the thalamus (Figure 4B). Seven iSNVs were present, but their significance is uncertain (Supplementary Material, Supplementary Figure 4B, and Supplementary Table 4).
Figure 4.

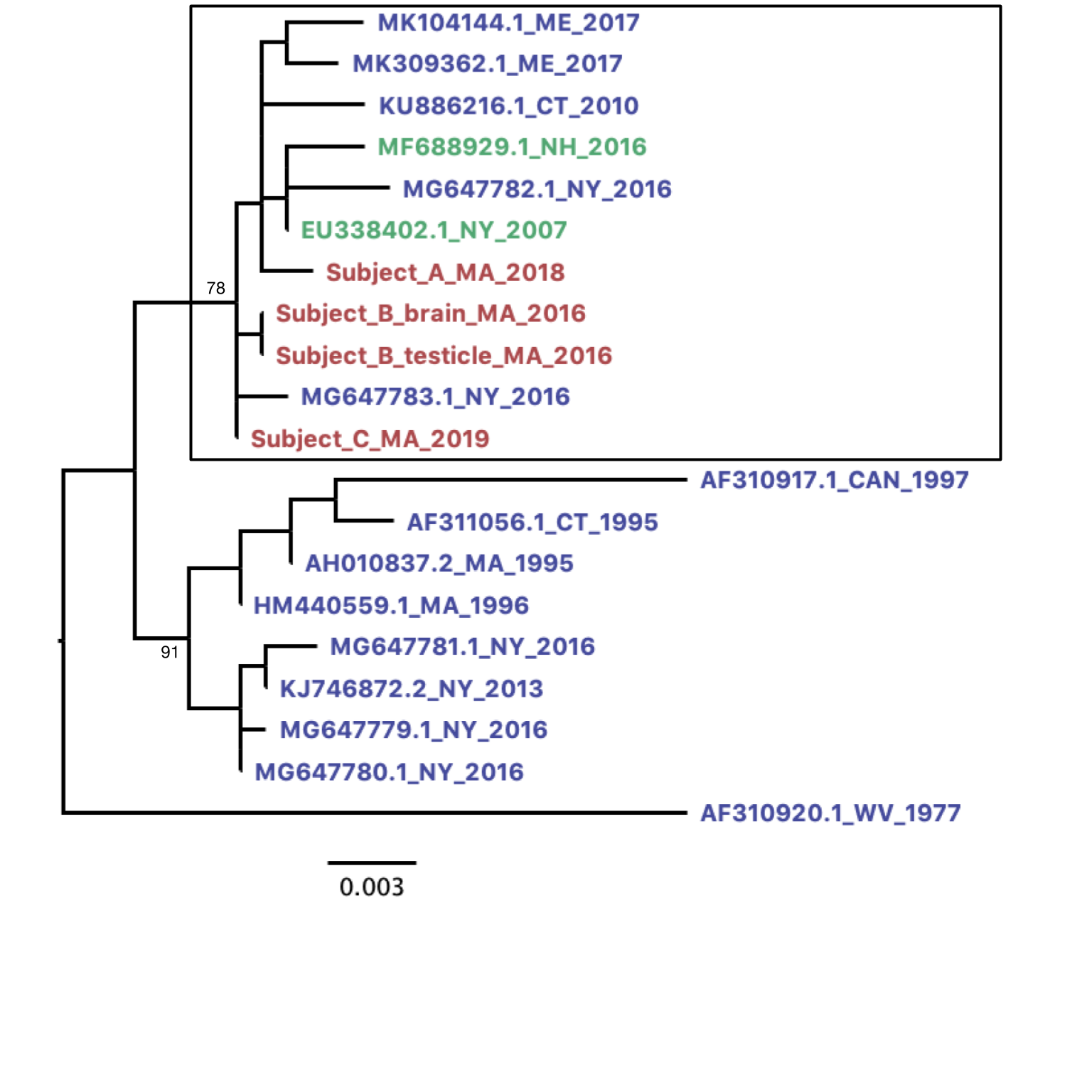
Within-subject POWV variation in tissue samples from Subject B (A) and Subject C (B) and between-subject phylogenetic analysis of consensus POWV genomes (C). The frequency of intrahost variants found in each sample is plotted against the POWV genome position for Subject B (A) and Subject C (B). Variants were defined as differing from all published lineage II POWV genomes from the Northeast, and the genome position is defined relative to POWV reference sequence NC_003687.1. Markers are filled for variants that resulted in an amino acid change. The frequency at which a variant would be detected with 80% probability is shown in Supplementary Figure 4, and specific variant frequencies are shown in Supplementary Tables 4, 5, and 7. C, Unique consensus sequences from this study were compared with all published POWV sequences covering at least 90% of the viral genome. Sequence names reflect GenBank accession number, state, and year. Names are colored by host source (blue indicates Ixodes ticks, green indicates published sequences from humans, and pink indicates novel sequences from humans). Bootstrap (ultrafast [25]) support is shown for major nodes, including those differentiating the subclusters observed in the lineage II Northeast clade. The subcluster containing all POWV genomes from humans is highlighted with a box. Further analysis that includes the only 2 additional published partial POWV genomes from the Northeast is presented in Supplementary Figure 5. Abbreviation: POWV, Powassan virus.
POWV Variation in the Brain Over Time
This study provided the unprecedented opportunity to examine POWV changes over time during human infection by comparing POWV sequences in the cerebella of Subjects B and C at biopsy and autopsy (11 and 4 days apart, respectively). Within each subject, the consensus POWV genomes were the same, indicating no major changes in the virus population over these relatively brief periods. In cerebellum from Subject B, 10 iSNVs were observed (Figure 4A; Supplementary Table 5), 2 of which were nonsynonymous iSNVs in the envelope protein that were detected at relatively high frequency (11% and 6%) at biopsy but were not detected at autopsy. We had a moderate probability (83% and 68%, respectively) of detecting these iSNVs at the same frequency at autopsy (Figure 4A), so it is possible that the apparent loss of these variants is due to chance. However, given that these iSNVs occurred in the envelope protein, which mediates cell entry and is immunogenic [29], it is also possible that they were selected against. In cerebellum from Subject C, 12 iSNVs were observed (Figure 4B; Supplementary Table 4), the most notable of which was a nonsynonymous mutation in the NS3 gene that rose to 10% frequency at autopsy from undetectable at biopsy (if it had also been present at 10% frequency at biopsy, we would have had a 99% probability of detecting it, given the sequencing depth). Caveats to these analyses are that the differences observed may represent fine-scale differences in space rather than time since the biopsy and autopsy tissues were necessarily sampled from different portions of the cerebellum and that the detection of low-frequency variants depends upon sequencing depth (Supplementary Data).
Phylogenetic Analysis Revealsed a POWV Subcluster That Contains all Sequences Derived From Humans
We compared the consensus viral genomes from subjects in this study to published POWV genomes from ticks. As expected, phylogenetic analysis revealed that all patients were infected with POWV lineage II and genomes clustered within the established Northeast geographical clade (Figure 4C). Within the Northeast lineage II clade, there are 2 subclusters that do not appear to be separated by time or geography [11]. Interestingly, all of the human POWV genomes presented in this study belonged to 1 of these subclusters (Figure 4C), as did 2 additional POWV genomes that were previously published, both of which were partial genomes (Supplementary Figure 5). Further work could elucidate whether genomes in this subcluster share genetic features that influence human infection or pathogenicity.
DISCUSSION
POWV is an understudied and likely underdiagnosed cause of severe encephalitis. From 3 patients with fatal encephalitis due to POWV lineage II (deer tick virus), we describe several important findings relevant to clinicians and researchers: the commonality of profound cerebellar involvement along with the lack of peripheral disease, the utility of molecular tests for detecting POWV, and the first complete POWV lineage II genomes derived from humans.
First, cerebellar involvement was notable upon histopathological evaluation of tissue from the 2 subjects who underwent brain biopsy and autopsy, with severe loss of Purkinje cells, diffuse leptomeningeal and parenchymal lymphohistiocytic inflammation, and microgliosis with scattered microglial nodules. MRI findings also indicated cerebellar involvement in all 3 subjects. While not pathognomonic for POWV, this may represent a hallmark of severe disease as it has been described in several additional cases of fatal POWV infection [2, 28, 34], whereas patients with POWV radiographically limited to the basal ganglia and thalamus have recovered [13]. Peripheral tissue was largely uninvolved, with the exception of testicular inflammation in Subject B, consistent with his symptoms [9]; this may indicate immune privilege of the site, and testicular involvement has been observed with other flaviviruses [35].
Second, our results highlight the value of nucleic acid–based diagnostics for POWV, which have been described previously [13] but are not yet routine. All 3 subjects in this study were first diagnosed with POWV by metagenomic sequencing, and 2 ultimately had negative serology. For Subject A, POWV was first identified in CSF by metagenomic sequencing performed in this study; results were returned 3 days after receiving the sample in the research laboratory and 8 days after sample collection, while confirmatory serology results were returned 41 days after sample collection. For Subject B, POWV was first detected by clinical metagenomic sequencing from CSF, while serology was negative in serum and CSF, attributed to rituximab therapy [9]. For Subject C, POWV was first diagnosed by metagenomic sequencing in this study, while serology was negative, which may have been due to early presentation (CSF was collected 7 days after symptom onset) and/or occult immunocompromise. Overall, we found that detection results from targeted POWV RT-qPCR and SHERLOCK assays were concordant with metagenomic sequencing but less sensitive. However, SHERLOCK offers the potential advantages of use with user-friendly and field-deployable lateral-flow assays [15], highly specific polymorphism detection [15], and highly multiplexed assays [16] for detection of numerous different viruses [36].
Finally, RNA sequencing enabled assembly of complete POWV genomes directly from human samples, offering initial insight into the strains of POWV lineage II (deer tick virus) that infect humans. We also provide one of the few descriptions of intrahost viral diversity during human infection for any neuropathogenic flavivirus. The degree of viral quasispecies diversity observed was similar to that seen in POWV in naturally infected ticks [37], POWV in experimentally infected ticks and mice [38], and WNV in 1 case of fatal human encephalitis [39]; however, it is difficult to make exact comparisons because of differences in setting and techniques (Supplementary Data). Our results suggest avenues for further investigation by identifying several polymorphisms of interest: the NS4b consensus-level polymorphism between brain and testes in Subject B (nucleotide position 7463), the 2 nonsynonymous envelope iSNVs in Subject B (nucleotide positions 1441 and 2099), and a nonsynonymous iSNV (nucleotide position 4689) in Subject C, located in a gene (NS3) that has been associated with pathogenicity of related flaviviruses in mouse models [29]. Although it is possible that these polymorphisms arose due to genetic drift, they nevertheless represent a small set of potential adaptations to test for functional relevance.
Overall, this study represents the most comprehensive screening and sequencing of POWV during human infection to date. We demonstrate the feasibility of using molecular detection assays to identify POWV in clinical samples, including from patients with negative serology. Additionally, metagenomic sequencing, in combination with hybrid capture, is a viable technique for assembling viral genomes directly from human tissue. For the first time, complete POWV genomes have been generated, intrahost variation in human tissues examined, and polymorphisms that may affect pathogenicity identified. Not only is characterization of viral localization and genomics in vivo a crucial first step in understanding POWV pathogenicity, but it is hoped that the molecular techniques described here will facilitate the generation of additional POWV genomic data from human subjects. Such work is critically needed to expand the understanding of POWV encephalitis and to enable the development of effective clinical interventions.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are very grateful to the patients, their families, and the many clinicians involved in their care. We would like to thank Sam Telford III for expert assistance with attempts at virus isolation and for helpful discussions on the manuscript. We would like to thank Steve Schaffner for expert assistance with statistical analyses and for helpful discussions on the manuscript.
Financial support. This work was supported by National Institutes of Health (NIH) grants R01AI137424 and K08AI139348, by the Howard Hughes Medical Institute (HHMI), and by Defense Advanced Research Projects Agency (DARPA) grant DC18AC00006.
Disclaimer. The views, opinions, and/or findings expressed should not be interpreted as representing the official views or policies of the Department of Defense or the US government.
Potential conflicts of interest. P.C.S. is a co-founder and consultant for SHERLOCK Biosciences, is a board member of Danaher Corporation, and has equity in both. P.C.S. and C.A.F. are inventors of CRISPR-based diagnostic intellectual property that the Broad Institute has licensed to SHERLOCK for commercialization in the developed world. The authors declare no other competing interests. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Author contributions. Erica Normandin – study design, data collection, data analyses, writing. Isaac Solomon – study design, data collection, data analyses, writing. Siavash Zamirpour – data collection, data analyses, writing. Jacob Lemieux – data collection. Catherine Freije – data collection, data analyses. Shibani Mukerji – data collection, writing. Christopher Tomkins-Tinch – data analyses. Daniel Park – data analyses. Pardis Sabeti – study design, writing. Anne Piantadosi – study design, data collection, data analyses, writing.
Patient consent. This work complies with all relevant ethical regulations and was approved by the Partners Institutional Review Board (IRB). Subject A provided written consent under protocol 2015P001388. Samples were obtained from Subjects B and C with a waiver of consent under protocol 2015P002215. The Broad Institute of MIT and Harvard has relied on Partners IRB review through a standing reliance agreement.
Prior presentations. American Society for Tropical Medicine and Hygiene (ASTMH) Annual Meeting; November 2019; National Harbor, MD. American Society for Microbiology Regional Meeting; October 2018; Albany, NY.
Data availability. All metagenomic sequencing reads (cleaned of human reads) are available on NCBI under BioProject PRJNA659662, and all consensus POWV genomes are available under GenBank accession numbers MT996002, MW001304, MW001305, MW001306. Other raw data (including those for Supplementary Figures 1–4) are available upon request.
Code availability. Source code for the viral-ngs toolkit and WDL pipelines used for genome assembly are freely available on GitHub at https://github.com/broadinstitute/viral-ngs under the terms of a BSD-derived license viewable at https://github.com/broadinstitute/viral-ngs/blob/master/LICENSE. Documentation is available at https://viral-ngs.readthedocs.io/. For this work, we used version 1.25.0-rc1 (doi: /10.5281/zenodo.3509008) and executed the pipelines via DNAnexus using builds available at https://platform.dnanexus.com/projects/F8PQ6380xf5bK0Qk0YPjB17P/data/build/quay.io/broadinstitute/viral-ngs/1.25.0-rc1. All other code is available upon request. This includes code used to generate coverage plots for Supplementary Figure 3, the code used to calculate probabilities reported in Supplementary Tables 4 and 5 and to calculate frequencies plotted in Supplementary Figure 4, and the code used to quantify chimeric reads reported in Supplementary Table 6.
References
- 1. Ebel GD. Update on Powassan virus: emergence of a North American tick-borne flavivirus. Annu Rev Entomol 2010; 55:95–110. [DOI] [PubMed] [Google Scholar]
- 2. Piantadosi A, Rubin DB, McQuillen DP, et al. Emerging cases of Powassan virus encephalitis in New England: clinical presentation, imaging, and review of the literature. Clin Infect Dis 2016; 62:707–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hermance ME, Thangamani S. Powassan virus: an emerging arbovirus of public health concern in North America. Vector Borne Zoonotic Dis 2017; 17:453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nofchissey RA, Deardorff ER, Blevins TM, et al. Seroprevalence of Powassan virus in New England deer, 1979–2010. Am J Trop Med Hyg 2013; 88:1159–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krow-Lucal ER, Lindsey NP, Fischer M, Hills SL. Powassan virus disease in the United States, 2006–2016. Vector Borne Zoonotic Dis 2018; 18:286–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kemenesi G, Bányai K. Tick-borne flaviviruses, with a focus on Powassan virus. Clin Microbiol Rev. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith RP Jr, Elias SP, Cavanaugh CE, et al. Seroprevalence of Borrelia burgdorferi, B. miyamotoi, and Powassan virus in residents bitten by Ixodes ticks, Maine, USA. Emerg Infect Dis 2019; 25:804–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frost HM, Schotthoefer AM, Thomm AM, et al. Serologic evidence of Powassan virus infection in patients with suspected lyme disease. Emerg Infect Dis 2017; 23:1384–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Solomon IH, Spera KM, Ryan SL, et al. Fatal Powassan encephalitis (deer tick virus, lineage II) in a patient with fever and orchitis receiving rituximab. JAMA Neurol 2018; 75:746–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson JF, Armstrong PM. Prevalence and genetic characterization of Powassan virus strains infecting Ixodes scapularis in Connecticut. Am J Trop Med Hyg 2012; 87:754–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robich RM, Cosenza DS, Elias SP, et al. Prevalence and genetic characterization of deer tick virus (Powassan virus, lineage II) in Ixodes scapularis ticks collected in Maine. Am J Trop Med Hyg 2019; 101:467–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gire SK, Goba A, Andersen KG, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014; 345:1369–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Piantadosi A, Kanjilal S, Ganesh V, et al. Rapid detection of Powassan virus in a patient with encephalitis by metagenomic sequencing. Clin Infect Dis 2018; 66:789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gootenberg JS, Abudayyeh OO, Lee JW, et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017; 356:438–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Myhrvold C, Freije CA, Gootenberg JS, et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science 2018; 360:444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gootenberg JS, Abudayyeh OO, Kellner MJ, et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018; 360:439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matranga CB, Andersen KG, Winnicki S, et al. Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol 2014; 15:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Metsky HC, Siddle KJ, Gladden-Young A, et al. ; Viral Hemorrhagic Fever Consortium Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat Biotechnol 2019; 37:160–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 2014; 15:R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tomkins-Tinch C, Park DJ, Ye S, et al. broadinstitute/viral-ngs: v1.19.3 Available at: https://github.com/broadinstitute/viral-ngs/tree/v1.19.310.5281/zenodo.1210274;v1.25.0. https://github.com/broadinstitute/viral-ngs/tree/v1.25.010.5281/zenodo.3509008.
- 21. Yang X, Charlebois P, Macalalad A, et al. V-Phaser 2: variant inference for viral populations. BMC Genomics 2013; 14:674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004; 32:1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004; 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 2015; 32:268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Minh BQ, Nguyen MA, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 2013; 30:1188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gerber J, Tumani H, Kolenda H, Nau R. Lumbar and ventricular CSF protein, leukocytes, and lactate in suspected bacterial CNS infections. Neurology 1998; 51:1710–4. [DOI] [PubMed] [Google Scholar]
- 27. Kakadia B, Zaher M, Badger C, Kavi T. Comparison of lumbar and ventricular cerebrospinal fluid for diagnosis and monitoring of meningitis (P5.9–012). Neurology. 2019; 92(15 Supplement):P5.9–012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tavakoli NP, Wang H, Dupuis M, et al. Fatal case of deer tick virus encephalitis. N Engl J Med 2009; 360:2099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kellman EM, Offerdahl DK, Melik W, Bloom ME. Viral determinants of virulence in tick-borne flaviviruses. Viruses 2018; 10:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holzmann H. Diagnosis of tick-borne encephalitis. Vaccine 2003; 21(Suppl 1):S36–40. [DOI] [PubMed] [Google Scholar]
- 31. Lyons JL, Schaefer PW, Cho TA, Azar MM. Case 34-2017. A 76-year-old man with fever, weight loss, and weakness. N Engl J Med 2017; 377:1878–86. [DOI] [PubMed] [Google Scholar]
- 32. Veje M, Studahl M, Norberg P, et al. Detection of tick-borne encephalitis virus RNA in urine. J Clin Microbiol 2014; 52:4111–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gourinat AC, O’Connor O, Calvez E, et al. Detection of Zika virus in urine. Emerg Infect Dis 2015; 21:84–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cavanaugh CE, Muscat PL, Telford SR 3rd, et al. Fatal deer tick virus infection in Maine. Clin Infect Dis 2017; 65:1043–6. [DOI] [PubMed] [Google Scholar]
- 35. Matusali G, Houzet L, Satie AP, et al. Zika virus infects human testicular tissue and germ cells. J Clin Invest 2018; 128:4697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ackerman CM, Myhrvold C, Thakku SG, et al. Massively multiplexed nucleic acid detection with Cas13. Nature 2020; 582:277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brackney DE, Armstrong PM. Transmission and evolution of tick-borne viruses. Curr Opin Virol 2016; 21:67–74. [DOI] [PubMed] [Google Scholar]
- 38. Grubaugh ND, Rückert C, Armstrong PM, et al. Transmission bottlenecks and RNAi collectively influence tick-borne flavivirus evolution. Virus Evol 2016; 2:vew033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grubaugh ND, Massey A, Shives KD, et al. West Nile virus population structure, injury, and interferon-stimulated gene expression in the brain from a fatal case of encephalitis. Open Forum Infect Dis 2016; 3:XXX–XX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H, Handsaker B, Wysoker A, et al. ; 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011; 27:2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Metsky HC, Matranga CB, Wohl S, et al. Zika virus evolution and spread in the Americas. Nature 2017; 546:411–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Duchêne S, Ho SYW, Holmes EC. Declining transition/transversion ratios through time reveal limitations to the accuracy of nucleotide substitution models. BMC Evol Biol 2015; 15:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haile S, Corbett RD, Bilobram S, et al. Sources of erroneous sequences and artifact chimeric reads in next generation sequencing of genomic DNA from formalin-fixed paraffin-embedded samples. Nucleic Acids Res 2019; 47:e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.