

Abstract
The role of Discoidin Domain Receptors (DDRs) is poorly understood in neurodegeneration. DDRs are upregulated in Alzheimer’s and Parkinson’s disease (PD), and DDRs knockdown reduces neurotoxic protein levels. Here we show that potent and preferential DDR1 inhibitors reduce neurotoxic protein levels in vitro and in vivo. Partial or complete deletion or inhibition of DDR1 in a mouse model challenged with α-synuclein increases autophagy and reduces inflammation and neurotoxic proteins. Significant changes of cerebrospinal fluid microRNAs that control inflammation, neuronal injury, autophagy and vesicular transport genes are observed in PD with and without dementia and Lewy body dementia, but these changes are attenuated or reversed after treatment with the DDR1 inhibitor, nilotinib. Collectively, these data demonstrate that DDR1 regulates autophagy and reduces neurotoxic proteins and inflammation and is a therapeutic target in neurodegeneration.
Introduction
Discoidin Domain Receptor 1 (DDR1) is one of two members of the unique DDR family of receptor tyrosine kinases known to be activated via autophosphorylation predominantly by collagen (1). DDRs (1 and 2) are involved in extracellular matrix remolding, cell adhesion, proliferation, migration and differentiation (2–5). DDRs are implicated in several diseases, including fibrotic disorders, atherosclerosis and cancer (2,5,6). DDR1 is expressed in the central nervous system (CNS) and has been implicated in modulation of microglial activity and matrix metalloproteases (MMPs), leading to blood–brain barrier (BBB) degradation (7). We demonstrated that DDRs are overexpressed postmortem in the nigrostriatum and hippocampus of individuals diagnosed with Parkinson’s disease (PD) and Alzheimer’s disease (AD), respectively (8).
Nilotinib is a tyrosine kinase inhibitor that potently inhibits DDR1 (IC50 = 1–8 nM) (9). We demonstrated that DDR knockdown with shRNA in vivo and in vitro (8) and pharmacological inhibitors of DDR1, including nilotinib and LCB-03-0110, reduce CNS amyloid-β (Aβ), hyperphosphorylated-tau (p-tau) and α-synuclein and increase dopamine levels (10–19). Nilotinib also inhibits Abelson (Abl) tyrosine kinase (IC50 = 20 nM) and is Food and Drug Administration-approved for chronic myeloid leukemia (20). Clinical studies indicate that nilotinib enters the CNS and achieves a pharmacologically relevant cerebrospinal fluid (CSF) concentration (Cmax = 2–4.7 nM) that did not inhibit Abl (11,13,15,21) but sufficient to inhibit DDR1.
Collectively, these data suggest that DDR1 inhibition may be a therapeutic target in neurodegeneration. Therefore, we investigated existing DDR1 inhibitors (nilotinib and LCB-03-0110) and the effects of novel more specific small molecule DDR1 inhibitors in several models of neurodegeneration and DDR1 knockout mice. We discovered that DDR1 regulates autophagy and its inhibition increases clearance of neurotoxic proteins and reduces neuroinflammation. We validated these data with microRNA (miRNA) sequencing (miRNAseq) in CSF from PD, PD with dementia (PDD) and Lewy body dementia (LBD) patients treated with nilotinib.
Results
DDR inhibition via LCB-03-0110
We previously demonstrated that intraperitoneal (I.P) injection of 1.25 and 2.5 mg/kg LCB-03-0110 (LCB) in mice achieves CNS concentrations to inhibit DDRs and Src (10). At these low doses, p-tau and Aβ levels were reduced in a mouse harboring triple mutant amyloid precursor protein (TgAPP), a model of AD and vascular dementia presenting with age-related accumulation of Aβ, p-tau and neuroinflammation (16). A range of 10–100 μM of LCB robustly dephosphorylates DDR1 (Fig. 1A) and DDR2 (Fig. 1B) in B35 rat neuroblastoma cells that have DDRs activated (via phosphorylation) by collagen. Transgenic 3–6 months old C57BL/6J mice were stereotaxically injected unilaterally into the right side of the substantia nigra (SN) with a lentivirus (LV) to express human wild-type α-synuclein. After 21 days of α-synuclein expression, mice were treated with I.P injection of 2.5 mg/kg of LCB or dimethyl sulfoxide (DMSO) once daily for an additional 21 days (Fig. 1C). In the ipsilateral injected midbrains, DDR1 was deactivated by 36% and DDR2 by 50% (Fig. 1D). LV expression resulted in 82% increase of α-synuclein level compared with DMSO-injected controls (Fig. 1E). Importantly, LCB reduced the amount of α-synuclein by 50%—down to the level of mice that did not receive α-synuclein (Fig. 1E). Expression of LV α-synuclein in the ipsilateral SN significantly reduced the number tyrosine hydroxylase (TH)-positive neurons (Fig. 1G and I) compared with the contralateral side (Fig. 1F and I) and LCB treatment partially protected against the loss of TH-positive neurons (Fig. 1H and I).
Figure 1.
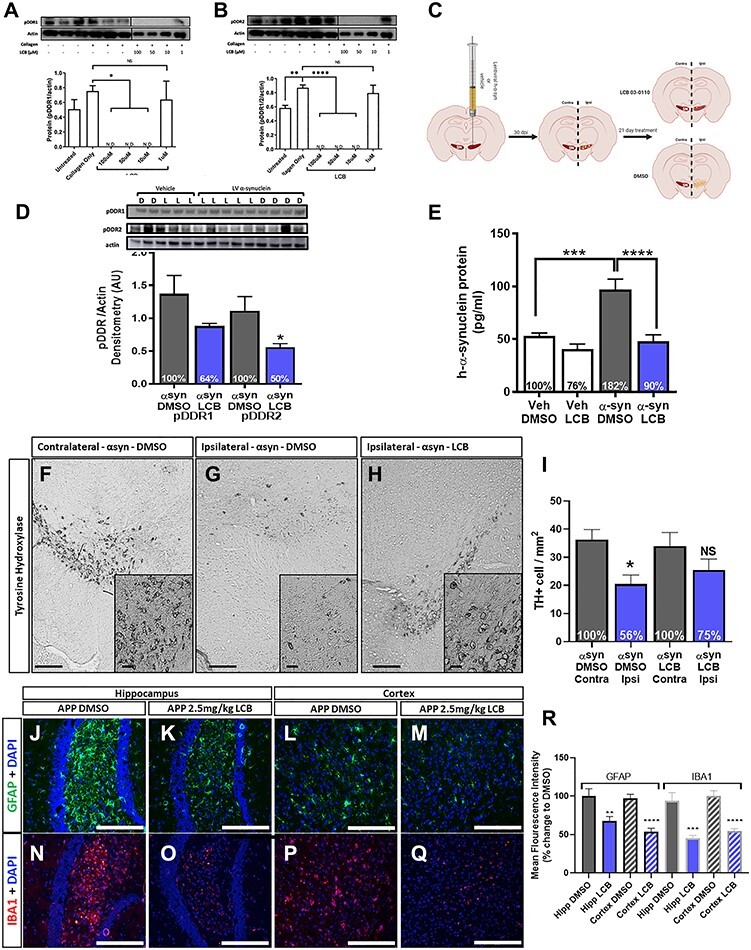
DDR inhibition via LCB-03-0110. (A and B) Representative WB and quantification of pDDR1 and pDDR2, each normalized to actin from B35 rat neuroblastoma stimulated with type-4 collagen for 2 h and treated with LCB-03-0110 in DMSO for 5 h. *P < 0.05, **P < 0.01 and ****P < 0.0001, normal one-way ANOVA; n = 3 per group and three combined into a single sample for treated groups. (C) Human α-synuclein LV gene transfer mouse model study design. LV α-synuclein (αsyn) or vehicle (veh) is unilaterally, stereotaxically injected into the right (ipsilateral) SN of 3–6 months old C57BL/6J mice. α-Synuclein is expressed for 21 days followed by treatment with LCB-03-0110 for 21 days. (D) WB and quantification of pDDR1 and pDDR2, each normalized to actin from right midbrain of αsyn mice treated with DMSO (D) and 2.5 mg/kg LCB-03-0110 (L) for 21 days. *P < 0.05, two-tailed Student’s t test, n = 4 DMSO and six LCB groups. (E) ELISA for human α-synuclein (pg/ml) from right midbrain of αsyn and veh mice treated with DMSO or LCB. ***P < 0.001 and ****P < 0.0001; normal one-way ANOVA; n = 4 DMSO and six LCB groups. (F) Quantification of dopaminergic cells in the SN. (F) Representative image of TH-positive cells in the contralateral non-αsyn injected SN of αsyn mice treated with DMSO. (G) Representative image of TH-positive cells in the ipsilateral αsyn injected SN of αsyn mice treated with DMSO. (H) Representative image of TH-positive cells in the ipsilateral αsyn injected SN of αsyn mice treated with 2.5 mg/kg of LCB. Scale bars, 500 and 20 μm inlay. (I) Quantification of TH-positive cells in the SN. *P < 0.05; normal one-way ANOVA; n = 3 per group, n = 3 sections per mouse and 3 images per section for quantification. Staining for gliosis in TgAPP mice treated with DMSO or 2.5 mg/kg of LCB-03-0110 for 21 days. Hippocampal GFAP and 4', 6-diamidino-2-phenylindole (DAPI) co-staining in (J) DMSO and (K) LCB treated mice. Cortical GFAP and DAPI co-staining in (L) DMSO and (M) LCB-treated mice. Hippocampal IBA1 and DAPI co-staining in (N) DMSO and (O) LCB-treated mice. Cortical IBA1 and DAPI co-staining in (P) DMSO and (Q) LCB-treated mice. Scale bars, 200 μm. (R) Quantification of GFAP and IBA1 MFI as percent change from DMSO-treated animals. **P < 0.01, ***P < 0.001 and ****P < 0.0001; normal one-way ANOVA; n = 2 (DMSO) and 5 (LCB) treated mice; n = 4 coronal sections and 3 images per section for quantification.
We previously demonstrated that LCB reduces neurotoxic Aβ and p-tau in TgAPP mice, and here we show the effects of LCB on neuroinflammation in these mice. LCB significantly reduced glial fibrillary acidic protein (GFAP) staining in the hippocampus (Fig. 1K and R) and cortex (Fig. 1O and R) compared with DMSO (Fig. 1J, N and R). LCB also significantly reduced ionized calcium-binding adapter molecule-1 (IBA-1) staining in the hippocampus (Fig. 1M and R) and cortex (Fig. 1Q and R) compared with DMSO (Fig. 1L, P and R).
Synthesis, effects and potency of novel DDR1 inhibitors in vitro
On the basis of structural analysis of LCB and of metabolic derivative ‘daughter molecules’ identified in the brain extracts of LCB treated wild-type C57BL/6J mice, a small library of selectively substituted thienopyridines including (BK40143 and BK40197) was generated (Fig. 2A, Supplementary Material, Figs S1–S4). Palladium catalyzed Suzuki cross-coupling of commercially available 7-chloro-2-iodothieno[3,2-b]pyridine and m-tolyboronic acid gave the common precursor 7-chloro-2-(m-tolyl)thieno[3,2-b]pyridine in 72% yield. Nucleophilic aromatic substitution at the aryl chloride site with aniline and morpholine then produced BK40143 and BK40197 in 94 and 98% yield, respectively.
Figure 2.
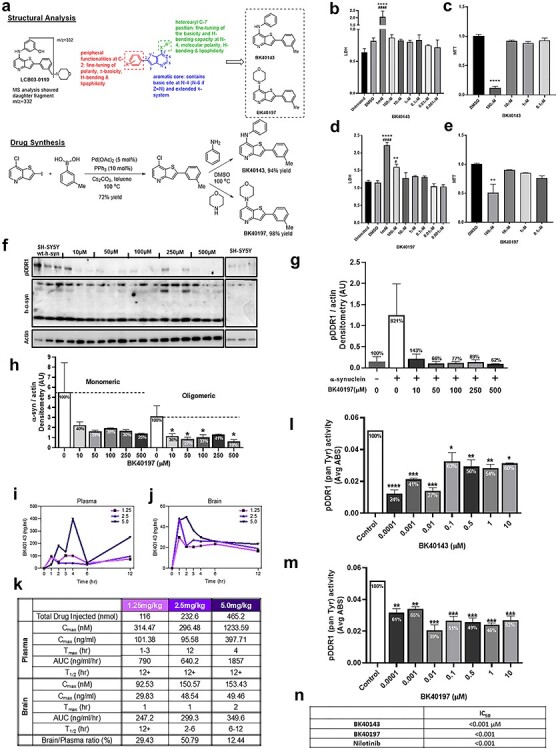
Synthesis, effects and potency of novel DDR1 inhibitors. (A) Schematic of structural analysis of LCB-03-0110, structure activity relationship and synthesis of BK40143 and BK40197. Cell viability of BK40143 and BK40197 via (B) LDH and (C) MTT of DMSO, 1 mM, 100, 10, 1, 0.1, 0.01 and 0.001 μM BK40143-treated and untreated B35 rat neuroblastoma cells. (D) LDH and (E) MTT cell viability assays of DMSO, 1 mM, 100, 10, 1, 0.1, 0.01 and 0.001 μM BK40197-treated and untreated B35 rat neuroblastoma cells. **P < 0.01, ****P < 0.0001 to DMSO group, ####P < 0.0001 to untreated group; normal one-way ANOVA; n = 3 per treatment group. (F) Western blot of pDDR1, α-synuclein and actin SHSY-5Y stably transfected with human α-synuclein treated with 1, 50, 100, 250 and 500 μM of BK40197 for 5 h and untreated. (H) Quantification of monomeric and oligomeric human α-synuclein and (G) pDDR1 with percentage change from untreated group, each normalized to actin. *P < 0.05; normal one-way ANOVA; n = 3 per treatment group. (I) Plasma concentration (nM) and (J) brain concentration (nM) of 1.25, 2.5 and 5 mg/kg doses of BK40143 at 0, 1, 2, 3, 4, 6 and 12 h after I.P injection in C57BL/6 mice determined by liquid chromatography–mass spectrometry. (K) Table of pharmacokinetic properties including Cmax (nM) and (ng/ml), Tmax, area under the curve (AUC) (ng/ml/h), t1/2 (h) and the ratio of brain to plasma concentrations on the basis of 1.25, 2.5 and 5 mg/kg doses of BK40143. ELISA for human pDDR1 (mean absorbance (ABS)) from human wild-type α-synuclein stably transfected SHSY-5Y cells treated with 0.0001, 0.001, 0.01, 0.1, 1 and 10 μM of (L) BK40143 and (M) BK40197 for 5 h and untreated groups. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; normal one-way ANOVA; n = 3 per group. (N) Table of IC50 for BK40143, BK40197, nilotinib for pDDR1.
Cell viability using lactate dehydrogenase (LDH) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays in B35 rat neuroblastoma cells indicated that BK40143 (Fig. 2B and C) and BK40197 (Fig. 2D and E) decreased cell viability near and above 100 μM compared with DMSO-treated controls. We observed that DDR1 is activated by human α-synuclein expression. DDR1 phosphorylation was increased (721%) in human neuroblastoma SH-SY5Y cells stably transfected with human wild-type α-synuclein alone compared with non-transfected cells (Fig. 2F and H) consistent with DDRs upregulation in postmortem brains (8). BK40197 was able to deactivate DDR1 (>80%) and significantly reduce both monomeric and oligomeric human α-synuclein levels (Fig. 2F and G).
Pharmacokinetics and pharmacodynamics
Male and female, 5–6 months old wild-type C57BL/6 mice were I.P injected with DMSO or 1.25, 2.5 and 5 mg/kg 13C-labelled BK40143 to determine the pharmacokinetics of this compound using liquid chromatography–mass spectrometry. BK40143 was detected in the plasma (Fig. 2I) and the brain (Fig. 2J) over 12 h. A maximum dose-dependent concentration was detected in the brain (Cmax = 93–153 nM) at 1 h (1.25 and 2.5 mg/kg) and 2 h (5 mg/kg), whereas the plasma concentration (Cmax = 314–1233 nM) reached maximum at 1–3 h (1.25 mg/kg), 12 h (2.5 mg/kg) and 4 h (5 mg/kg) (Fig. 2K). The half-life (t1/2) was >12 h in the plasma and was varied in the brain to reach 12 h at 1.25 mg/kg dose, 2–6 h at 2.5 mg/kg dose and 6–12 h at 5 mg/kg dose. Overnight treatment of human neuroblastoma SH-SY5Y cells with BK40143 and BK40197 (dissolved in DMSO) inhibits 50% of DDR1 activity (IC50) near or equals 0.001 μM (Fig. 2I, M and N), similar to nilotinib (9).
BK40143 specifically inhibits DDR1 and lowers toxic proteins in ad models
To determine whether BK40143 targets other tyrosine kinases (Src, Fyn, Abl,) that are inhibited by LCB and nilotinib in vivo, rTg4510 mice that express human mutant P301L-tau, were I.P injected with 1.25, 2.5 and 5 mg/kg BK40143 or DMSO once daily for 1 week. Both 1.25 and 2.5 mg/kg BK40143 resulted in a significant ~50% inhibition of DDR1 in total brain extracts, whereas the highest dose did not affect DDR1 phosphorylation level (Fig. 3A and C). There was no inhibition (dephosphorylation) of Src (Fig. 3A and B), Abl tyrosine 412 (Fig. 3A and D), Abl tyrosine 245 (Fig. 3A and E) and Fyn (Fig. 3A and F). Importantly, DDR1 inhibition coincided with a 50% reduction in AT8 p-tau (Ser202/Thr305) levels via immunoblot (Fig. 3A and G) and a significant reduction in p-tau (Thr181) with 2.5 mg/kg via enzyme-linked immunosorbent assay (ELISA) (Fig. 3H).
Figure 3.
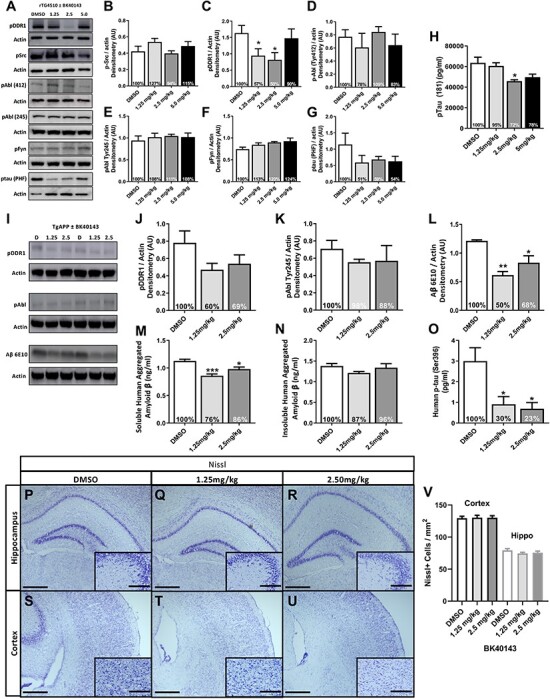
BK40143 specifically inhibits DDR1 and lowers toxic proteins in AD models. (A) Representative western blots of pDDR1, pSrc, pAbl (Tyr412 and Tyr 245), pFyn and pTau paired-helical filament (PHF) from 3 to 4 months old rTG4510 mice treated with 1.25, 2.5 and 5 mg/kg BK40143 or DMSO for 7 days and quantification of (B) pDDR1, (C) pSrc, (D) pAbl (Tyr412), (E) pAbl (Tyr245), (F) pFyn and (G) p-tau (PHF), each normalized to actin with percent change from DMSO-treated animals. (H) ELISA for human pTau (Thr181) (pg/ml) from whole brain. *P < 0.05; normal one-way ANOVA; n = 4 per treatment group. (I–V) Western blot, ELISA and Nissl staining from 7 to 8 months old TgAPP mice treated with 1.25 and 2.5 mg/kg of BK40143 or DMSO for 21 days. (I) Representative western blots of pDDR1, pAbl and Aβ (6E10) with corresponding actin. Quantification of (J) pDDR1 (Tyr796), (K) pAbl (Tyr254) and (L) Aβ (6E10), each normalized to actin with percent change from DMSO-treated group. *P < 0.05, **P < 0.01; normal one-way ANOVA or Student’s t-test. n = 5 per treatment group. ELISA for (M) intracellular soluble human aggregated Aβ (ng/ml), (N) extracellular insoluble human aggregated Aβ (ng/ml) and (O) human p-tau (Ser396) from whole brain. *P < 0.05 and ***P < 0.001; normal one-way ANOVA; n = 5 per treatment group. (P–V) Nissl staining and quantification of the hippocampus and cortex. (P–R) Representative images of hippocampal Nissl staining in (P) DMSO, (Q) 1.25 and (R) 2.5 mg/kg BK40143-treated TgAPP mice. Scale bars, 500 and 20 μm inlay. (S–U) Representative images of cortical Nissl staining in (S) DMSO, (T) 1.25 and (U) 2.5 mg/kg BK40143-treated TgAPP mice. (V) Quantification of Nissl positive cells in the cortex and hippocampus, n = 5 per group, n = 4 coronal sections per mouse and three images per section for quantification.
TgAPP mice were I.P injected with 1.25 and 2.5 mg/kg BK40143 or DMSO once daily for 3 weeks to evaluate long-term exposure of BK40143. BK40143 reduced (40%) DDR1 (Fig. 3I and J) but not Abl (Fig. 3I and K) in total brain lysates via immunoblot. DDR1 inhibition via BK40143 was associated with a significant reduction in soluble Aβ via (6E10) immunoblot (Fig. 3I and L). Soluble synaptic Aβ (Fig. 3M), but not insoluble fractions (Fig. 3N), were reduced via ELISA. BK40143 also significantly reduced p-tau (Ser396) levels in brain lysates (Fig. 3O). Nissl staining in the hippocampus and cortex show no differences in cell number in DMSO-treated mice (Fig. 3P, S and V) versus mice treated with 1.25 mg/kg (Fig. 3Q, T and V) and 2.5 mg/kg (Fig. 3R, U and V) BK40143 for 3 weeks.
BK40143 lowers toxic proteins and improves dopamine utilization
Male and female 5–6 months old transgenic mice harboring human Alanine53Threonin (A53T) α-synuclein mutation associated with familial PD were I.P injected with 2.5 mg/kg of BK40143 once daily for 3 weeks. Human α-synuclein was significantly reduced (39%) in total brain lysates (Fig. 4A) and this reduction (40%) was confirmed (albeit non-significant) via immunoblot (Fig. 4B and C). A53T mice do not exhibit loss of dopaminergic neurons, but an increase in dopamine (30%) and homovanillic acid (HVA) (70%) were observed after BK40143 treatment compared with DMSO (Fig. 4D). Behavioral assessment via open field shows significantly increased velocity of A53T mice (Fig. 4E) with no impact on total distance travelled (Fig. 4F) or time spent moving (Fig. 4G). DDR1 inhibition via BK40143 has no effect on a number of inflammatory chemokines and cytokines except interleukin-9 (IL-9), which trended toward a decrease (20%) associated with a significant reduction in IL-13 and interferon (IFN)-γ in A53T mice compared with DMSO (Supplementary Material, Table S1). IL-9 was significantly increased in TgAPP (Supplementary Material, Table S2), although its level in DMSO-treated TgAPP mice was several folds lower that other transgenic mice (A53T and rTg4510). The levels of the pro-inflammatory cytokine macrophage inflammatory protein (MIP-1a) was significantly reduced in TgAPP treated with BK40143 versus DMSO. Lower concentrations of BK40143 also significantly reduced IL-9 levels in rTg4510 (Supplementary Material, Table S3), but higher doses significantly increased lipopolysaccharide-induced CXC chemokine LIX, or CXCL5 that maintains hematopoietic stem cells compared with DMSO.
Figure 4.
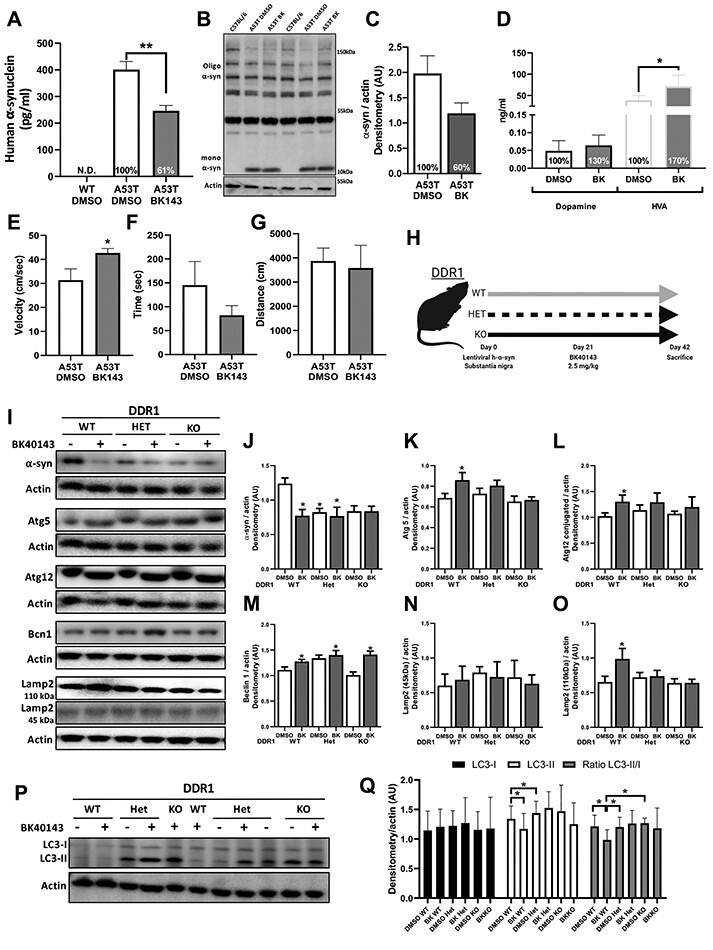
BK40143 lowers toxic proteins, improves dopamine utilization and DDR1 inhibition or deletion boosts autophagy. (A–G) In total, 7–10 months old TgA53T mice treated with 2.5 mg/kg of BK40143 (BK143) and DMSO for 21 days; n = 5 per group. (A) ELISA for human α-synuclein (pg/ml) in TgA53T mice treated with DMSO and BK143 and wild-type C57BL/6J (WT) mice. **P < 0.01; normal one-way ANOVA; n = 5 per group. (B) Representative western blot of human α-synuclein in TgA53T mice and WT control mice. (C) Quantification of monomeric (mono) α-synuclein and percent change from DMSO-treated group in TgA53T mice treated with DMSO and BK143. (D) ELISA for dopamine (ng/ml) and mouse HVA (ng/ml) in TgA53T mice treated with DMSO and BK143. *P < 0.05; two-tailed Student’s t test; n = 5 per group. (E–G) Quantification of open-field behavioral assay. (E) Velocity (cm/s), (F) total ambulation time (s), total distance travelled (cm). *P < 0.05; two-tailed Student’s t test, n = 5 per group. (H–Q) DDR1 WT, DDR1 Het and DDR1 KO human α-synuclein LV gene transfer mice treated with 2.5 mg/kg of BK40143 and DMSO for 21 days. (H) DDR1 human α-synuclein LV gene transfer mouse model study design. LV expressing human α-synuclein vector is unilaterally, stereotaxically injected into the right (ipsilateral) SN of DDR1 WT, Het and KO mice. α-Synuclein is expressed for 21 days followed by treatment with 2.5 mg/kg of BK40143 for 21 days. (I) Representative western blots of human α-synuclein (α-syn), autophagy-related protein 5 (Atg5), conjugated autophagy-related protein 12 (Atg12), Beclin 1 (becn1), Lamp 2 (mature 110 and immature 45 kDA) and corresponding actin from DDR1 WT, Het and KO mice treated with DMSO and 2.5 mg/kg of BK40143 for 21 days. Quantification of (J) α-synuclein, (K) Atg 5, (L) Atg 12, (M) Beclin 1, (N) Lamp 2 (45 kDa) and (O) Lamp 2 (110 kDa), each normalized to actin, from right midbrain of mice treated with DMSO and 2.5 mg/kg BK for 21 days. *P < 0.05; ANOVA; n = 4–8 per group and *P < 0.05; one-tailed Student’s t test for Beclin 1 DMSO versus BK in DDR1 WT group. (P–Q) Western blot and quantification of microtubule-associated protein 1 LC3 in DDR1 WT, Het and KO mice treated with DMSO and 2.5 mg/kg of BK40143. (P) Representative western blot of LC3-I and LC3-II and actin. (Q) Quantification of LC3-I, LC3-II and the ratio of LC3-II/LC3-I, each normalized to actin. *P < 0.05; ANOVA; n = 4–8 per group.
DDR1 inhibition or deletion boosts autophagy
To ascertain the mechanistic effects of DDR1 on protein clearance, LV α-synuclein was injected unilaterally into the SN (right side) of 3–4 months old, male and female DDR1 knockout mice for 30 days (Fig. 4H), followed by I.P injection of 2.5 mg/kg BK40143 once daily for 3 weeks. Human α-synuclein was significantly reduced (37%) in wild-type DDR1 (WT) mice treated with BK40143 (Fig. 4I and J) compared with DMSO, and this significant reduction was observed in heterozygous DDR1 (Het) and homozygous DDR1 null (KO) (albeit non-significant) mice with and without BK40143. The levels of autophagy proteins Atg 5 (Fig. 4I and K) and conjugated Atg 12 (Fig. 4I and L) were significantly increased in DDR1 WT treated with BK40143 compared with DMSO, and these effects were not observed in DDR and DDR1 KO mice. Beclin-1 levels were significantly increased in all DDR1 genotypes (Fig. 4I and M) treated with BK40143 compared with DMSO, indicating autophagosome formation (22). Immature lysosomal-associated membrane protein (LAMP)-2 was not changed (Fig. 4I and N) but mature LAMP-2 was significantly increased (Fig. 4I and O) in DDR1 WT mice treated with BK40143 compared with DMSO. LAMP2 levels did not change in DDR1 Het and DDR1 KO mice. The level of lipidated light chain proteins—LC3-II/LC3-I—was significantly reduced in DDR WT treated with BK40143 compared with DDR WT, DDR1 Het and DDR1 KO treated with DMSO (Fig. 4P and Q), indicating more autophagosome clearance in WT mice treated with BK40143 (17,19). BK40143 had no effects on autophagy proteins, including LC3-II/LC3-I when DDR1 levels were reduced in DDR1 Het and DDR1 KO mice. BK40143 and partial or complete deletion of DDR1 significantly reduced the level of the pro-inflammatory cytokines IL-1a and the IFN-γ-induced protein (IP-10) as well as the level of IL-9 that was significantly lower in Het and KO DDR1 mutants compared with WT (Table 1).
Table 1.
Mouse cytokine and chemokine in DDR1 mice expressing LV α-synuclein and treated with BK40143 once daily for 3 weeks
| DDR1 knockout mice | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | WT | Het | KO | ||||||||
| Treatment | DMSO | 2.5 mg/kg BK40143 | WT drug P-value | DMSO | 2.5 mg/kg BK40143 | Het drug P-value | Het versus WT gene P-value | DMSO | 2.5 mg/kg BK40143 | KO drug P-value | KO versus WT gene P-value |
| n | 5 | 6 | 7 | 6 | 4 | 5 | |||||
| Marker (pg/ml) | Mean ± SD7 (% to DMSO) | Mean ± SD (% to DMSO) | Mean ± SD (% to DMSO) | Mean ± SD (% to DMSO) | Mean ± SD (% to DMSO) | Mean ± SD (% to DMSO) | |||||
| G-CSF | 3.43 ± 0.37 (100%) | 2.66 ± 0.904 (77%) | NS | 3.15 ± 2.06 (91%) | 3.25 ± 0.92 (94%) | NS | NS | 2.56 ± 0.72 (74% | 3.01 ± 1.19 (87%) | NS | NS |
| Eotaxin | 17.62 ± 1.69 (100%) | 18.48 ± 3.93 (104%) | NS | 19.95 ± 6.67 (113%) | 18.64 ± 1.59 (105%) | NS | NS | 16.72 ± 1.32 (94%) | 15.09 ± 2.12 (85%) | NS | NS |
| GM-CSF | 15.68 ± 4.28 (100%) | 12.61 ± 4.09 (80%) | NS | 14.85 ± 5.03 (94%) | 14.91 ± 4.77 (95%) | NS | NS | 12.95 ± 3.15 (82%) | 12 ± 4.30 (76%) | NS | NS |
| IFN-γ | 29.08 ± 7.60 (100%) | 26.99 ± 2.30 (92%) | NS | 26.91 ± 5.53 (92%) | 23.09 ± 1.51 (79%) | NS | NS | 22.03 ± 2.17 (75%) | 20.67 ± 4.13 (71%) | NS | NS |
| IL-1a | 153. 37 ± 24.96 (100%) | 143.32 ± 9.89 (93%) | NS | 147.68 ± 12.36 (96%) | 130.66 ± 6.42 (85%) | 0.011487 | NS | 138.39 ± 22.37 (90%) | 134.38 ± 13.74 (87%) | NS | NS |
| IL-1b | 8.45 ± 1.98 (100%) | 8.10 ± 1.57 (95%) | NS | 9.72 ± 1.57 (115%) | 9.03 ± 3.74 (106%) | NS | NS | 8.66 ± 5.19 (102%) | 8.21 ± 2.87 (97%) | NS | NS |
| IL-2 | 167.38 ± 53.77 (100%) | 170.06 ± 8.37 (101%) | NS | 161.95 ± 19.36 (96%) | 145.16 ± 10.92 (86%) | NS | NS | 154.05 ± 19.97 (92%) | 145.93 ± 11.87 (87%) | NS | NS |
| IL-3 | 0.84 ± 0.12 (100%) | 0.92 ± 0.14 (110%) | NS | 0.91 ± 0.15 (107%) | 0.85 ± 0.14 (101%) | NS | NS | 0.81 ± 0.09 (96%) | 0.75 ± 0.10 (89%) | NS | NS |
| IL-6 | 18 ± 15.99 (100%) | 15.87 ± 4.35 (88%) | NS | 12.2 ± 5.29 (67%) | 8.98 ± 1.28 (49%) | NS | NS | 9.26 ± 4.18 (51%) | 7.57 ± 3.18 (42%) | NS | NS |
| IL-7 | 5.41 ± 0.53 (100%) | 4.78 ± 0.62 (88%) | NS | 4.89 ± 0.28 (90%) | 5.59 ± 0.97 (103%) | NS | NS | 4.61 ± 0.38 (85%) | 4.38 ± 0.39 (80%) | NS | NS |
| IL-9 | 6491.8 ± 1095.36 (100%) | 5762.83 ± 1670.93 (88%) | NS | 5268.71 ± 1010.51 (81%) | 4965.33 ± 518.51 (76%) | NS | <0.0001 | 5667 ± 516.38 (87%) | 4957.8 ± 496.39 (76%) | NS | <0.0001 |
| IL-10 | 18.53 ± 2.58 (100%) | 20.17 ± 2.45 (108%) | NS | 19.77 ± 3.49 (106%) | 18.28 ± 0.53 (98%) | NS | NS | 17.3 ± 1.72 (93%) | 16.49 ± 2.50 (89%) | NS | NS |
| P-40 | 9.24 ± 0.63 (100%) | 9.15 ± 0.93 (98%) | NS | 9.16 ± 1.27 (99%) | 8.77 ± 1.83 (94%) | NS | NS | 7.58 ± 0.32 (82%) | 7.61 ± 2.1 (82%) | NS | NS |
| P-70 | 18.30 ± 2.72 (100%) | 19.15 ± 3.82 (104%) | NS | 17.90 ± 1.96 (97%) | 20.82 ± 4.66 (113%) | NS | NS | 16.9725 ± 4.89 (92%) | 15.35 ± 3.22 (83%) | NS | NS |
| LIF | 2.13 ± 0.25 (100%) | 1.84 ± 0.13 (86%) | 0.034751 | 2.18 ± 0.46 (102%) | 1.99 ± 0.45 (93%) | NS | NS | 1.76 ± 0.22 (82%) | 1.64 ± 0.23 (76%) | NS | NS |
| IL-13 | 217.11 ± 57 (100%) | 79.77 ± 86.38 (36%) | 0.01416 | 82.78 ± 56.03 (38%) | 52.49 ± 32. 42 (24%) | NS | NS | 109.06 ± 47.68 (50%) | 53.99 ± 27. 63 (24%) | NS | NS |
| IL-15 | 35.13 ± 3.12 (100%) | 35.79 ± 3.32 (101%) | NS | 37.51 ± 3.46 (106%) | 35.91 ± 7.20 (102%) | NS | NS | 34.12 ± 4.10 (97%) | 32.33 ± 5.31 (92%) | NS | NS |
| IL-17 | 56.18 ± 8.01 (100%) | 47.31 ± 10.06 (84%) | NS | 40.24 ± 3.68 (71%) | 36.79 ± 2.49 (65%) | NS | NS | 48.21 ± 7.90 (85%) | 41.35 ± 5.87 (73%) | NS | NS |
| IP-10 | 69. 13 ± 15.03 (100%) | 68.63 ± 11.67 (99%) | NS | 63.78 ± 4.16 (92%) | 59.09 ± 2.45 (85%) | 0.034207 | NS | 57.09 ± 2.19 (82%) | 51.68 ± 9.62 (74%) | NS | NS |
| KC | 74.37 ± 11.03 (100%) | 66.34 ± 16.94 (89%) | NS | 61.13 ± 8.62 (82%) | 52.91 ± 4.55 (71%) | NS | NS | 46.73 ± 5.12 (62%) | 43.41 ± 5.70 (58%) | NS | NS |
| MCP-1 | 286.04 ± 67.21 (100%) | 190.21 ± 78.05 (66%) | NS | 175.89 ± 77.85 (61%) | 138.33 ± 52.17 (48%) | NS | NS | 163.67 ± 28.54 (57%) | 134.02 ± 33.6 (46%) | NS | NS |
| MIP-1a | 1018.35 ± 148.98 (100%) | 909.49 ± 166.85 (89%) | NS | 809.13 ± 129.56 (79%) | 705.91 ± 36.49 (69%) | NS | NS | 845.9 ± 85.12 (83%) | 740.25 ± 82.92 (72%) | NS | NS |
| M-CSF | 20.1 ± 1.99 (100%) | 18.04 ± 2.15 (89%) | NS | 21.52 ± 3.44 (107%) | 21.17 ± 3.73 (105%) | NS | NS | 16.26 ± 1.17 (80%) | 15.62 ± 2.92 (77%) | NS | NS |
| MIP-2 | 82.82 ± 6.89 (100%) | 75.92 ± 11.88 (91%) | NS | 80.64 ± 13.74 (97%) | 84.81 ± 14.96 (102%) | NS | NS | 78.28 ± 4.19 (94%) | 76.04 ± 9.06 (91%) | NS | NS |
| MIG | 113.83 ± 13.24(100%) | 97.33 ± 19.39 (85%) | NS | 109.91 ± 36.15 (96%) | 98.055 ± 17.32 (86%) | NS | NS | 92.49 ± 9.59 (81%) | 90.42 ± 8.97 (70%) | NS | NS |
| VEGF | 6.94 ± 1.15 (100%) | 5.59 ± 1.43 (80%) | NS | 5.35 ± 0.82 (81%) | 4.83 ± 0.38 (78%) | NS | NS | 4.93 ± 0.44 (71%) | 4.414 ± 0.86 (67%) | NS | NS |
| TNF-α | 6.23 ± 1.15 (100%) | 4.99 ± 1.07 (80%) | NS | 5.06 ± 0.71 (81%) | 4.90 ± 0.99 (78%) | NS | NS | 4.47 ± 0.85 (71%) | 4.192 ± 0.66 (67%) | NS | NS |
BK40143 significantly reduces pro-inflammatory IL-1a in total brain lysates. IL-9 is significantly reduced in HET and KO mice compared with WT, suggesting that DDR1 deletion lowers IL-9 and the level of the pro-inflammatory downstream cytokine IP-10. GM-CSF: granulocyte–macrophage colony-stimulating factor, Eotaxin: CCL11 chemokine, P40: subunit, of IL-12 that drives polarization of Th17, a subset of T cell suspected to have a key role in the pathophysiology of MS and EAE, P70: IL-12p40 is thought to be a natural antagonist of IL-12p70, acting, at least in part, by competitive binding to the IL-12 receptor, LIF: leukemia inhibitory factor-interleukin 6 family cytokine that are pro-inflammatory, IP10: C-X-C motif chemokine 10 (CXCL10) also known as IFN-γ-IP that plays an pro-inflammatory role in autoimmune diseases, KC: pro-inflammatory chemokine (C-X-C motif) ligand 1 (CXCL1), MIP-1a cytokine also known as CCL3, MIP-2 is a major CXCL2 chemokine, M-CSF: macrophage colony-stimulating factor, MIG: monokine induced by γ-MIG), or CXCL9, is a chemokine induced by IFN-γ, RANTES: known as chemokine (C-C motif) ligand 5 (also CCL5) is a protein produced by T and B Lymphocytes, VEGF: vascular endothelial growth factor, TNF-α: tumor necrosis factor that is a cytokine mainly produced by macrophages. NS: not significant.
Nilotinib alters CSF miRNAs that regulate autophagy genes in PD, PDD and LBD
To verify the effects of DDR1 inhibition, we conducted miRNAseq on CSF from severely advanced PDD and LBD patients (Hoehn and Yahr 3–5) who were treated with 300 mg nilotinib for 6 months (15) and moderately severe PD patients (Hoehn and Yahr 2.5–3) who received 150 or 300 mg nilotinib versus placebo for 1 year (11). Gene target analysis of the statistically significant miRNAs were predicted using human-specific miRTarbase and miRBase mature miRNA databases analysis (Fig. 5A) and changes of miRNAseq were validated by quantitative reverse transcription polymerase chain reaction (RT-qPCR). A total of ~2800 miRNAs were detected in the CSF (Fig. 5B), and 31 miRNAs were significantly changed between baseline and 6 months in PDD and LBD treated with 300 mg nilotinib (Fig. 5B), but only 18 miRNAs were confirmed to significantly (false discovery rate, FDR < 0.05) change (>2-fold) between baseline and end of treatment (6 and 12 months) in the nilotinib-treated PD. The expression levels of three miRNAs (miRNA-4661-5p, miRNA-641 and miRNA-9-5p) were significantly increased in the placebo group, but the expression of these miRNAs was attenuated between baseline and end of treatment in nilotinib-treated group (Fig. 5C). Gene target analysis shows that miRNA-4661-5p controls the BCLAF1 gene. miRNA-641 controls synaptosomal nerve-associated protein (SNAP)-25 gene. Mir-9-5p controls the expression of MMP2, 9 and 13 as well as IL-5 and IL-6 and the nuclear transcription factor Nuclear Factor Kappa B (NF-kB). Two other miRNAs (miRNA-155-5p and miRNA-21-5p) that control the IL-9 gene were significantly increased in the placebo-treated PD group but 150 mg nilotinib further increased miRNA-155-5p but 300 mg increased miRNA-21-5p. These changes in miRNA-155-5p and miRNA-21-5p were not observed in PDD and LBD.
Figure 5.
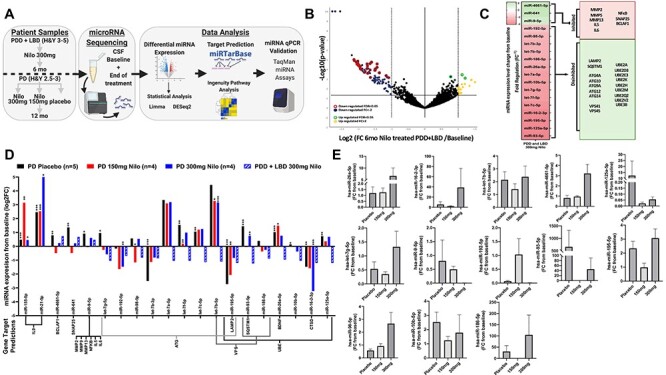
Nilotinib alters miRNAs that regulate autophagy genes in the CSF of PDD, LBD and PD patients. (A) miRNAseq study design; (B) volcano plot of miRNA upregulated and downregulated from baseline in the CSF of five individuals with advanced PD with dementia and LBD (Hoehn and Yahr = 3–5) after 6 months Nilotinib treatment. miRNAs were analyzed using Qiagen Data Analysis Center and DESeq2 and Limma pipelines normalized and plotted as −log10 (P-value) (y-axis) versus log2 (fold-change 6 months Nilo-treated/baseline) (x-axis). Yellow and blue indicate fold change ≥ ± 2, red and green FDR; P < 0.05; Benjamini−Hochberg FDR multiple comparisons. (C) Heatmap (left) of average miRNA changes from baseline (Fig. 5B) as FR (equal to negative inverse of Fold-change (FC)). Predicted gene targets (right) of the significantly differentially expressed miRNA. Predicted target genes determined via human miRTarbase curated database and ingenuity pathway analysis filtered for highly predicted and experimentally observed. miRNAs in red indicate decreased expression and green indicate increased expression. Target genes in red indicate hypothesized inhibition of target mRNA by miRNA and green indicate hypothesized disinhibition of target mRNA. (C) miRNA changes from baseline in individuals with moderate PD (Hoehn and Yahr = 2.5–3) who received 150 (n = 3) or 300 mg (n = 4) nilotinib versus placebo (n = 6) for 1 year and PDD and LBD individuals. Quantification of miRNA expression plotted as log2 FC (y-axis) for each candidate miRNA (x-axis) including PDD and LBD (Fig. 5B and C). Predicted target genes are included below x-axis. *P < 0.05, **P < 0.01, ***P < 0.001; within-group change was analyzed using normalized paired differential genes expression testing via DESeq2 and Limma-based pipelines. (E) RT-qPCR of candidate miRNA in individuals with moderate PD (Hoehn and Yahr = 2.5–3) who received 150 (n = 3) or 300 mg (n = 4) nilotinib or placebo (n = 6) for 1 year using TaqMan advanced miRNA assays. For RT-qPCR, statistically significant change was obtained via ordinary one-way ANOVA with Tukey post hoc test.
Fifteen miRNAs that predominantly control autophagy, vesicular transport and ubiquitination genes were significantly changed in the placebo group between baseline and 1 year. These miRNAs include let-7g-5p, let-7b-3p, let-7a-5p, let-7f-5p, let-7c-5p, let-7b-5p, miRNA-192-5p, which control autophagy-related genes (ATGs) and miRNA-195-5p, which controls ATGs and LAMP-2. Changes were observed in miRNA-93-5p that controls SQSTM1 and miRNA-186-5p, miRNA-26a-5p, miRNA-10b-5p, miRNA-16-2-3p, miRNA-125a-5p that control the E3 ubiquitin ligase UBE gene, the lysosomal enzyme cathepsin-D (CTSD) gene (miRNA-16-2-5p) and vacuolar protein sorting-associated (VPS) genes (let-7c-5p, let-7b-5p, miRNA192-5p, miRNA-93-5p, miRNA186-5p) that control the endosome, autophagy–lysosomal and vesicular transport pathways. The changes of expression levels of these miRNAs were significantly attenuated in the nilotinib-treated groups (Fig. 5C) and these changes in miRNA expression levels were verified using RT-qPCR (Fig. 5D).
Discussion
To our knowledge, this is the first demonstration of the specific role of DDR1 as a therapeutic target in neurodegenerative diseases. The data demonstrate that pharmacological inhibition of DDR1 results in clearance of neurotoxic proteins, including α-synuclein, p-tau and Aβ. Novel DDR1 inhibitors as well as nilotinib enter the brain and achieve pharmacologically relevant concentrations to inhibit DDR1 and activate the autophagy–lysosomal pathway and degrade neurotoxic proteins consistent with our previous data (10,11,13–15,17–19). Partial or complete deletion of DDR1 prevents accumulation of α-synuclein, whereas BK40143 reduces α-synuclein in wild-type control mice, but it fails to further reduce α-synuclein levels in heterozygous or homozygous littermates. These data suggest that deletion or reduction of DDR1 has an optimal effect on basal autophagy, so α-synuclein accumulation maybe quickly degraded when the neuron is challenged with α-synuclein, and perhaps other proteins. The effects of BK40143 on α-synuclein levels were similar to DDR1 deletion in both heterozygous and homozygous mice, and BK40143 has no additive effect on α-synuclein clearance, further suggesting that 50% inhibition of DDR1 is sufficient to boost autophagy (23) and protein clearance mechanisms. miRNAs negatively regulate gene expression via RNA silencing and posttranscriptional regulation of gene expression (24). The increase in miRNAs that control ATG, UBE, SQSTM1 and VPS genes suggests reduced expression of proteins that mediate ubiquitination and the endosomal and autophagy–lysosomal pathways, consistent with our data that show impairment of autophagy in postmortem AD and PD brains (25–27). However, nilotinib attenuates or reverses the level of miRNA expression suggesting an increase in autophagy in agreement with the animal data (14,17–19,28). Furthermore, the increase of miRNAs that control LAMP-2 and CTSD genes suggest reduction or inhibition of lysosomes, but nilotinib reverses these effects. These data are in agreement with the transgenic DDR1 mice and transgenic models and cell culture treated with DDR1 inhibitors (nilotinib, BK40143 and BK40197) that show an increase in autophagy and clearance of neurotoxic proteins, including p-tau, Aβ and α-synuclein (14,17–19,28). Nilotinib effects on the autophagy–lysosomal pathways are also consistent with its effects on reduction of toxic proteins, including oligomeric α-synuclein, p-tau and Aβ in human (11,13,15,21).
Nilotinib effects on SNAP25 and VPS genes that regulate Snap Receptor (SNARE) protein complex assembly to mediate the transport of synaptic vacuoles, including the endosome, lysosome and neurotransmitter-containing vesicles (29,30) are consistent with our data showing increased level of dopamine metabolism in animals (19), PD (11,13,15) and AD patients (21) treated with DDR1 inhibitors, including nilotinib. It was demonstrated that accumulation of oligomeric α-synuclein results in age-dependent impairment of dopamine release, but reduction of oligomeric α-synuclein increases dopamine turnover (31). SNAP-25 gene expresses the trans-SNARE complex that mediates the fusion of synaptic vesicles with the plasma membrane, leading to neurotransmitter release (32). An increase in dopamine level was observed in transgenic animals and PD and AD patients treated with nilotinib (11,13,15,17,19,21,28). Furthermore, nilotinib significantly reduces the level of CSF p-tau in PD patients, consistent with previous data that show nilotinib lowers the level of p-tau in animal models of neurodegeneration (10,11,17,19,33,34). We demonstrated that a reduction of p-tau in nilotinib-treated models of tauopathies enhances astrocyte activity and improves neurotransmitter balance, including glutamate (14). Therefore, autophagy clearance of α-synuclein and p-tau and regulation of SNARE are concurrent with improved astrocytic activity and balance of neurotransmitters (14,16,19,34,35). Taken together, these data suggest that the effect of DDR1 inhibition on neurodegenerative pathology, including PD, may be mediated by multiple changes, representing a ‘biomarker mix’ that includes reduction of CSF oligomeric α-synuclein and p-tau, which affect the brain motor and non-motor systems, and concurrent improvement of dopamine metabolism (11,13,15,21,36). These changes may represent a potential biomarker system—similar to the A/T/N system in AD (37)–that would be investigated in correlation with potential clinical outcomes to study the disease-modifying effects of DDR1 inhibitors.
There was no inflammation or cell death associated with DDR1 deletion or inhibition in animal models, consistent with nilotinib effects. We previously demonstrated that a single dose of nilotinib increases triggered receptor on myeloid (TREM)-2 cells expression in the CSF of PD patients compared with placebo (13), suggesting regulation of microglial activity. However, as p-tau and oligomeric α-synuclein were reduced over 1 year of nilotinib treatment, TREM2 levels were not different in nilotinib-treated versus placebo groups (11). Here we observe that nilotinib reverses the changes of IL-5 and IL-6 and the nuclear transcription factor NF-kB in PD. TREM2 inhibits inflammatory responses in microglia via suppression of NF-kB pathways and is strongly implicated in microglia innate immunity (38). MMPs as well as DDR1 regulate BBB integrity; therefore, the changes observed in MMP gene expression levels between the placebo and nilotinib-treated patients suggest that DDR1 inhibition may affect MMP regulation of both neuroinflammation and the BBB (7,39). In the CSF of PD patients, the increase in BCLAF1 gene that encodes a transcriptional repressor, which interacts with pro-apoptotic members of the Bcl-2 family of proteins, was reversed by nilotinib, consistent with nilotinib effects on cell death and inflammation in animal models (16,34). Most importantly, the decrease in IL-9 is consistent with DDR1 inhibition and deletion in transgenic models, suggesting that DDR1 regulates IL-9 level. CSF miRNA-155-5p and miRNA-21-5p were significantly increased in placebo-treated PD patients, but 150 and 300 mg nilotinib further increased these miRNA levels, suggesting suppression of IL-9 in the nilotinib-treated PD groups. The lack of change of these miRNAs in PDD and LBD after nilotinib treatment may be because of stage of disease. IL-9 is produced predominantly by CD4+ lymphocytes, helper T and regulatory T cells and mast cells and plays a critical role in adaptive immunity and the pathogenesis of inflammatory diseases and cancer (40). The CSF level of IL-9 is increased in AD (41), and IL-9 mRNA is increased in the plasma of PD patients (42). Mitigation of IL-9 effects protects the BBB in stroke (43) and experimental autoimmune encephalomyelitis (EAE) (44). Autophagy is essential for T cell activation, but CD4+ T cells lacking Atg3 or Atg5 show increased IL-9 expression upon differentiation (45), indicating that impairment of autophagy may alter the adaptive immune response. IL-9 expression affects many other cytokines including IL-5 and IL-6 and INF-γ and modulation of its levels profoundly regulates the resolution of inflammation (46,47). Taken together, these data suggest an interplay between autophagy and inflammation because reduction of neurotoxic proteins attenuates the CNS inflammatory response, and conversely, activation of microglial activity leads to phagocytosis of misfolded proteins and subsequently reduces the burden of neuronal autophagy, hence the concurrent changes of inflammatory and autophagy markers.
Lower doses of DDR1 inhibitors appear to be more efficacious at reducing neurotoxic proteins. Although these effects are still poorly understood, several studies showed that low doses of nilotinib enter the brain and degrade α-synuclein, Aβ and p-tau via autophagy in animal models of neurodegeneration (10,17,19,33,34), similar to the effects of BK40143 in vivo. It is likely that at higher doses these molecules have some off-target effects to abrogate the role of DDR1 inhibition or maybe reduce the efficiency of the autophagy–lysosome system. Our data demonstrate a dose-dependent increase in brain concentration of BK40143 in the plasma but maximum concentrations occur between 1 and 2 h or perhaps earlier in the brain and plateau at 15 nM in 2.5 and 5 mg/kg doses. Importantly, brain penetration only increases from 29.4 to 50.7% with 1.25 and 2.5 mg/kg doses, respectively, yet drops to 12.4% with the highest dose of 5 mg/kg. This phenomenon may be because of efflux mechanisms at higher doses via the g-protein systems (48), so a maximum concentration of the drug may be reached in the CNS, independent of plasma concentration. The bioavailability (t1/2) and brain concentration of BK40143 in 5 mg/kg dose may overwhelm the autophagy–lysosome pathway and perhaps reduce its efficiency. Therefore, the selectivity and potency of DDR1 inhibitors allow lower brain concentrations to boost the autophagy–lysosome pathway and avoid off-target side effects. Furthermore, the effects of BK40143 and BK40197 should also be investigated in DDR2−/− models because our previous data showed that shRNA knockdown of either DDR1 or DDR2 reduces neurotoxic protein levels and attenuates microglial and inflammatory signaling (8).
DDR1 inhibition is a therapeutic target to clear neurotoxic proteins in neurodegenerative diseases. DDR1 regulates ubiquitination, the autophagy–lysosome pathway and vesicular transport and control the utilization of brain neurotransmitters. DDR1 effects on MMPs and IL-9 may play a critical role in BBB integrity and neuroinflammation. However, CSF miRNAs that predominantly control autophagy, vesicular transport and ubiquitination genes were significantly changed in the placebo-treated PD patients between baseline and 1 year and this effect was not observed in the nilotinib-treated groups, suggesting that nilotinib attenuates miRNA changes. The changes in CSF miRNAs in PD versus healthy controls are unknown and bigger sample sizes are needed to determine whether nilotinib effects on CSF miRNAs can lead to statistical differences between the groups and whether these changes represent actual reversal in miRNA levels back to healthy individuals. The concurrent changes of vesicular transport and autophagy with neuroinflammation suggest that neurotoxic proteins may be cleared via simultaneous neuronal and glial (microglia and astrocytes) activities.
Materials and Methods
Study design
The primary research objective of this study was to assess, in mouse models of neurodegeneration (including TgA53T, TgAPP, rTG4510 and LV gene transfer synuclein mouse models) and CSF from individuals with severely advanced PDD and LBD and moderate PD, whether specific inhibition of DDR1 could ameliorate end points of disease progression via activation of autophagic mechanisms. In animal experiments, a combination of pharmacological and genetic inhibition of DDR1 was utilized. All animal treatment groups were matched for age, sex and littermate controls. All animal studies were approved by the Georgetown University Institutional Animal Care and Use Committee. Human samples were procured with the Ethics Committee approval and written informed consent. The clinical study was conducted in accordance with Good Clinical Practice guidelines and was approved by the Institutional Review Board (IRB# 2016-0380) at Georgetown University Medical Center (GUAMC).
BK40143 and BK40197 synthesis
Commercially available 7-chloro-2-iodothieno[3,2-b]pyridine, m-tolylboronic acid, aniline, morpholine, reagents, catalysts and solvents were used as purchased without further purification. Nuclear magnetic resonance (NMR) spectra were obtained at 400 MHz (1H NMR) and 100 MHz (13C NMR) in deuterated chloroform (Supplementary Material, Figs S1–S4). Reaction products were purified by column chromatography on silica gel (particle size 40–63 μm). For the synthetic methods and characterization, a mixture of 7-chloro-2-iodothieno[3,2-b]pyridine (500 mg, 1.69 mmol), 3-methylphenylboronic acid (230 mg, 1.69 mmol), palladium (II) acetate (19 mg, 0.084 mmol), triphenylphosphine (44 mg, 0.169 mmol) and cesium carbonate (1.101 g, 3.38 mmol) in 15 ml of toluene was heated at reflux for 24 h (Supplementary Material, Fig. S1A). The reaction mixture was cooled to room temperature and partitioned between water and dichloromethane. The organic layer was washed with saturated aqueous sodium chloride, dried over sodium sulfate, filtered and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel using hexanes–ethyl acetate (EtOAc) (4:1) as mobile phase. The product was obtained as a colorless solid in 72% yield (315 mg, 1.21 mmol). Rf = 0.2 (hexanes/EtOAc, 4:1).
For the nucleophilic aromatic substitution reactions (SI Fig. 1B and C), an 8 ml pressure vessel was charged with 7-chloro-2-(m-tolyl)thieno[3,2-b]pyridine (0.3 mmol), the amine (0.6 mmol) and DMSO (1.0 ml). The pressure vessel was then placed in a 100°C oil bath and stirred until full conversion was achieved on the basis of 1H NMR analysis. The reaction mixture was cooled to room temperature, extracted with EtOAc and washed with water. The combined organic layers were dried over sodium sulfate and the solvent was removed in vacuo. The crude product was purified by flash chromatography on silica gel using hexanes–EtOAc as mobile phase as described later.
N-Phenyl-2-(m-tolyl)thieno[3,2-b]pyridin-7-amine (Supplementary Material, Fig. S1B) was named BK40143 and obtained as a colorless solid in 94% yield (89 mg, 0.282 mmol) from 7-chloro-2-(m-tolyl)thieno[3,2-b]pyridine (78 mg, 0.3 mmol) and aniline (56 mg, 0.6 mmol) in 1 ml of DMSO after 16 h at 100°C by following the general procedure described previously. The crude product was purified using flash chromatography on silica gel using hexanes/EtOAc (1:1). Rf = 0.2 (hexanes/EtOAc, 1:1). Additionally, 4-(2-(m-tolyl)thieno[3,2-b]pyridin-7-yl)morpholine (Supplementary Material, Fig. S1C) was named BK40197 and was obtained as a colorless solid in 98% yield (91 mg, 0.294 mmol) from 7-chloro-2-(m-tolyl)thieno[3,2-b]pyridine (78 mg, 0.3 mmol) and morpholine (52 mg, 0.6 mmol) in 1 ml of DMSO after 4 days at 100°C by following the general procedure described previously. The crude product was purified using flash chromatography on silica gel using hexanes/EtOAc (1:1). Rf = 0.2 (hexanes/EtOAc, 1:1).
Cell lines, transfection and treatment
Human SHSY5Y neuroblastoma cells, were procured from the Tissue Culture and Biobanking Shared Resource at Georgetown University Lombardi Comprehensive Cancer Center. Human embryonic kidney (HEK) 293FT cells were purchased from ThermoFisher Scientific (R70007). Stably transfected SHSY-5Y cells expressing human wild-type α-synuclein or human α-synuclein with A53T mutation were developed via selective resistance to G418 disulfate salt (Sigma-Aldrich, A1720). Cell identity was not further authenticated. B35 and HEK293FT cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (ThermoFisher, 11 965 118) with 10% fetal bovine serum (FBS) (ThermoFisher, 16 000 044) and 1% penicillin–streptomycin (PenStrep, ThermoFisher, 15 140-122) and incubated at 37°C under atmospheric oxygen concentrations (21%) with 5% CO2. SHSY-5Y cells were cultured in DMEM with Ham’s F12 (1:1) (ThermoFisher, 11 765 054) with 20% FBS, 1% PenStrep. Cells were plated at a density tailored to reach 70–80% confluence at the beginning of every experiment. B35 cells were transiently transfected for 24 h with human-tau or human wild-type α-synuclein using Fugene HD transfection reagent (Promega, E2311) according to manufacturer’s protocol before fresh culture media and drug were added. HEK293FT cells were stimulated via 10 μg/ml of rat tail type-1 collagen (Sigma-Aldrich, C7661) for 2 h before fresh culture media and drug were added. Cells were treated with 1 mM, 100, 10, 1, 0.1, 0.01 and 0.001 μM of BK40143 and BK40197 or nilotinib dissolved in DMSO or an equivalent volume of DMSO for 5 h. Cell viability was determined via LDH assay on culture media (ThermoFisher, 88 954) and MTT assay on plated cells (ThermoFisher, V13154). Using separate samples with the same treatment conditions, cells were harvested on ice by removing culture medium and adding 0.2 ml 1× sodium-Tris, Ethylenediamenetetraacetic acid (EDTA), NP-40 (STEN) buffer (50 mM Tris (pH 7.6), 150 mM NaCl, 2 mM EDTA, 0.2% NP-40, 0.2% with Halt protease and phosphatase inhibitor solution (ThermoFisher, 78 446). Cells were detached with a cell scraper and collected into centrifuge tubes and incubated at 4°C for 30 min with agitation. Samples were stored at −80°C and used for additional analyses.
Mice, treatment and stereotaxic surgery
Mice experiments were conducted on (a) TgAPP mice that express neuronally derived human APP gene, 770 isoform, containing the Swedish (K670N/M671L, Dutch E693Q and Iowa D694N mutations under the control of the mouse thymus cell antigen 1, theta, Thy1, promotor (49), (b) rTg4510 mice that express human P301L-tau and have the tet-responsive element (TRE or tetO) and mouse prion protein promotor sequences (PrP or Prnp) directing expression of the P301L mutant variant of human four-repeat microtubule-associated protein-tau (4R0NTau P301L) (50) or (c) TgA53T mice that express mutant arginine to threonine (A53T) human α-synuclein under the control of the prion promotor (51). DDR1 knockout (DDR1KO) mice were generated via homologous recombination by Lexicon Pharmaceuticals Inc. (Woodlands, TX, USA) and procured from Infrafrontier EMMA mouse repository (EM:02343) Wellcome Trust Knockout Mouse Resource. Coding exons 1–3 of the DDR1 gene were replaced with the LacZ/Neo3a cassette resulting in the targeted silencing of the DDR1 gene. DDR1KO, Het and WT littermates were generated at expected Mendelian frequency. DDR1 genotype was confirmed via PCR using DDR1 WT forward primer 5′-GTT GCG TTA CTC CCG AGA TG-3′, DDR1 knockout forward primer 5′-GCA GCG CAT CGC CTT CTA TC-3′ and a common reverse primer 5′-AGA CAA TCT CGA GAT GCT GG-3′. LV gene transfer mice were generated as previously described (17,28,52,53). Briefly, LV constructs encoding α-synuclein were generated in HEK293FT cells, and the supernatants were collected, concentrated by centrifugation, resuspended in culture medium, aliquoted in autoclaved tubes and stored at −80°C. Stereotaxic surgeries were performed to inject 1 × 109 moi LV α-synuclein into the right SN of wild-type C57BL/6J and DDR1 WT, Het and KO mice. α-Synuclein is expressed for 21 days before treatment is started. To verify transgene expression, we routinely PCR with the primers that were used to clone α-synuclein into LV vectors. Treatment groups for all experiments were balanced for age, sex and littermate controls. Mice received treatments of daily I.P injections of 1.25, 2.5, or 5.0 mg/kg of LCB-03-0110 dihydrochloride (Tocris Bioscience, 5592), BK40143 and BK40197 (Medicinal Chemistry Program, Georgetown University) and Nilotinib (Selleckchem Inc., S1033) dissolved in DMSO (Fisher scientific, D128-500,) or an equivalent dose of DMSO only. Treatment periods were either 7 consecutive days or 21 consecutive days as designated in the figure legends.
Pharmacokinetics studies
C57BL/6J mice were injected once with I.P injection of 13C-labeled BK40143. Brain and serum were collected at 2, 4, 6 or 12 h (n = 18 per drug, n = 3 per dose and time point). Animals injected with vehicle (DMSO) were used for background subtraction. Stock solutions of drug (∼1 mg/ml each) were prepared in methanol/dichloromethane (50:50). The serial dilutions for each of the standards were produced for the study separately in methanol/high-performance liquid chromatography grade water (50:50). Preparation of the calibration curve standards and quality samples (QC) was performed by mixing the stock solutions in blank samples. Serum and brain samples were stored at −80°C, then thawed to room temperature prior to preparation. The thawed serum samples (20 μl) were transfused to a tube containing 100 μl of water. The 500 μl extraction solvent, acetonitrile/methanol (50:50) was added to the sample. The mixture was vortexed and incubated on ice for 20 min to accelerate protein precipitation. After incubation, the samples were vortexed before being centrifgued at 1300 rpm for 20 min at 4°C. The supernatant was then collected and transferred to a new tube, dried using speed vacuum and reconstituted in 200 μl of methanol/water (50:50). The mixture was then spun again at 1300 rpm for 20 minutes at 200 at 4°C. The supernatant was then collected into a mass spec sample tube cap and run in the mass spectrometer. For the brain, a small section of the thawed brain sample from each animal was transferred to a flat bottom tube. In total, 200 μl of methanol/water (90:10) was added, and the tissue was homogenized. Acetonitrile was then added to the mixture facilitating protein precipitation. The mixture was then incubated on ice for 10 min. After incubation, the samples were vortexed and centrifuged at 13000 rpm for 20 min at 210 4°C. The supernatant was then collected and transferred to a new tube, dried using speed vacuum and reconstituted in 200 μl of methanol/water (50:50). The mixture was centrifuged at 13000 rpm for 20 min at 4°C. The supernatant was collected into a mass spectrometer sample tube cap and run in the mass spectrometer. The samples were resolved on an Acquity UPLC BEH C18 1.7 μm, 2.1 × 50 mm column online with a triple quadrupole mass spectrometer (Xevo-TQ-S, Waters Corporation) operating in the multiple reaction monitoring (MRM) mode. The sample cone voltage and collision energies were optimized for both analytes to obtain maximum ion intensity for parent and daughter ions using ‘IntelliStart’ feature of MassLynx software (Waters Corporation). The instrument parameters were optimized to gain maximum specificity and sensitivity of ionization for the parent (m/z = 438.25) and daughter ions (m/z = 357.33). Signal intensities from all MRM Q1/Q3 ion pairs for analytes were ranked to ensure selection of the most intense precursor and fragment ion pair for MRM-based quantitation. This approach resulted in the selection of cone voltages and collision energies that maximized the generation of each fragment ion species. An analysis was performed with a six- to eight-point calibration curve; the sample queue was randomized and solvent blanks were injected to assess sample carryover. MRM data were processed using TargetLynx 4.1. The relative quantification values of analytes were determined by calculating the ratio of peak areas of transitions of samples normalized to the peak area of the internal standard.
Tissue collection and preparation
Animals were deeply anesthetized with a mixture of xylazine and ketamine (1:8), and 500 μl of whole blood was collected via cardiac puncture, centrifuged at 2000 × g to precipitate blood cells and the serum was collected. Mice were transcardially exsanguinated with 25 ml of 1× phosphate-buffered saline (PBS). Brains were collected and homogenized in 1.0 ml 1× STEN buffer. Homogenized samples were centrifuged at 12000 × g for 20 min at 4°C and supernatant (soluble protein fraction) was collected and stored at −80°C. Insoluble protein was extracted after removing the supernatant. The tissue pellet was washed with 1× STEN buffer. The pellet was resuspended in 750 μl of 70% formic acid and incubated for 30 min at room temperature followed by a centrifugation at 28000 × g at 4°C for 1 h. The supernatant was collected as the ‘insoluble fraction’. Samples from the 70% formic acid fraction were stored at −80°C and neutralized with 1 M Tris–base (1:20) immediately before use. Protein levels were quantified using Pierce BCA protein assay (ThermoFisher, 23 225) via manufacturer’s instructions. Where immunohistochemistry experiments were performed, the brain of PBS-perfused animals was extracted and separated into two hemispheres. The right hemisphere was used for protein as previously described. The left hemisphere was postfixed in 4% paraformaldehyde in PBS for 48 h at 4°C, followed by cryoprotection in 30% sucrose in PBS for 48 h and sectioned coronally at 20 μm using a cryostat microtome and stored at −20°C for further analysis.
Western blotting
In total, 20 μg of soluble and insoluble proteins extracted from mouse brain lysates were separated on sodium dodecyl sulfate Bis–Tris gels (Invitrogen; Bio-Rad). Proteins were transferred to nitrocellulose before being incubated with primary antibodies (Supplementary Material, Table S7) diluted in 5% non-fat dry milk in TBST overnight at 4°C. Blots were washed then incubated in species-specific secondary antibodies (anti-mouse, Invitrogen, PA1–74421; Anti-rabbit, Cell Signaling, 7074S) were visualized using Super Signal™ West Dura Extended Duration Substrate (Cat. #37071, ThermoFisher) on the Amersham™ Imager 600 (GE Healthcare Life Sciences). Western blots were quantified by densitometry using Image J software.
Immunohistology
In total, 20 μm sections were rinsed in water then PBS and blocked with 10% normal horse serum in PBS for 30 min at room temperature before overnight incubation at 4°C with primary antibodies diluted in 1% normal horse serum. Sections were then washed then incubated in species-specific secondary antibodies or AlexaFluor ® 488-conjugated goat anti-mouse and AlexaFluor ® 594-conjugated goat-anti-rabbit (LifeTechnologies, A11001 and A11012, respectively) diluted at 1:1000 in PBS. Slides were washed and coverslipped with EverBrite hardset mounting medium with DAPI (Biotium, 23 004). Fluorescent quantification was performed in ImageJ, where images were converted to grayscale and intensity was measured and reported as mean fluorescent intensity (MFI). Quantification of TH and Nissl positive cells via crystal violet staining was performed by three counters blinded to treatment groups and averaged together.
Enzyme-linked immunosorbent assay
ELISAs for total tau, Aβ40 and Aβ42 and mouse cytokine/chemokine (Millipore, HNABTMAG 60K and MCYTOMAG-70K, respectively) were conducted using Milliplexed ELISA. Xmap technology uses magnetic microspheres that are internally coded with two fluorescent dyes. Through precise combinations of these two dyes, multiple proteins are simultaneously measured within a sample. Each of these spheres is coated with a specific capture antibody. The capture antibody binds to the detection antibody and a reporter molecule, completing the reaction on the surface of the bead. All samples including placebo and resveratrol at baseline and 52 weeks were analyzed in parallel using the same reagents. A total of 25 μl soluble protein was incubated overnight at 4°C with 25 μl of a mixed bead solution. After washing, samples were incubated with 25 μl detection antibody solution for 1.5 h at room temperature. Streptavidin–phycoerythrin (25 μl) was added to each well containing the 25 μl of detection antibody solution. Samples were then washed and suspended in 100 μl of sheath fluid. Samples were then run on MAGPIX with Xponent software. The median fluorescent intensity data were analyzed using a five-parameter logistic or spline curve-fitting method for calculating analyte concentrations in samples. Specific p-Tau ser396 (Invitrogen, KHB7031), human Tau thr181 (Invitrogen, KHO0631), general dopamine (MyBioSource, 2 884 081), Mouse HVA (MyBioSource, 776 374), aggregated Aβ (ThermoFisher, KHB3491), PathScan phosphorylated DDR1 panTyrosine (Cell Signaling, 7863) and Aβ42 (Invitrogen, KHB3442) were performed according to manufacturer’s protocol on tissue soluble extracts from midbrain lysates in 1XSTEN buffer (see above).
Behavior
Open-field
Mice were placed in the open-field arena apparatus for 60 min. Animals were tracked by photocell beams along the arena floor. Data were collected and analyzed for total distance traveled (cm), total time spent moving (s) and velocity (distance/time) during the 60 min trial. A center zone was digitally defined in the software in the center of the apparatus, and center zone entries, center zone distance travelled (cm) and time spent in center zone (s) during the 60 min trial were recorded.
Human miRNA analysis
The protocol was approved by the Institutional Review Board (IRB #2014-0006) at GUAMC as well as by Georgetown–Howard Universities Center for Clinical and Translational Science (GHUCCTS) scientific review board as previously indicated. All participants were recruited from the Movement Disorders Clinic at MedStar Georgetown University Hospital and were consented at the GHUCCTS Clinical Research Unit. Eligible participants were computer randomized to either 150 or 300 mg groups and enrolled in the study. Patients were deidentified and a Pad-Id was included (e.g. NIL) in the patient’s clinical chart. An independent data safety monitoring board (DSMB) that included a cardiologist, hematologist, clinical pharmacologist, neurologist and biostatistician monitored safety signals in patients in the study.
CSF collection
We previously showed that nilotinib was detected in the CSF for 4 h after administration (15). Lumbar puncture ∼15 ml CSF were performed on all patients ∼2 h after the last levodopa dose and at 1, 2, 3 or 4 h after nilotinib administration. CSF was aliquoted and stored at −80°C. Freeze/thaw cycles were avoided. To avoid CSF contamination with blood, the first 1 ml of CSF collection was discarded and all samples were centrifuged at 1000 × g for 15 min. Samples that contained >25 ng/ml hemoglobin were eliminated and were not tested for biomarkers (11,13,15). The hemoglobin levels in CSF samples were measured using human hemoglobin ELISA Quantitation (Cat # E80-136) Kit (Bethyl Lab Inc, Montgomery, TX, USA) according to the manufacturer’s instructions. All patients demographic information were previously published (11,13,15).
RNA extraction and miRNAseq
Cell-free total RNA was isolated from 200 μl of CSF using the Qiagen miRNAeasy serum/plasma extraction kit (Qiagen, 217 184). Quality control analysis to confirm RNA was performed on each sample using UV–VIS spectroscopy on a Nanodrop ND-1000 (ThermoFisher). As per manufacturer’s instructions, samples were normalized to an input volume of 5 μl RNA eluate to prepare miRNAseq libraries using Qiagen QiaSeq miRNA-seq library preparation kit (Qiagen, 331 502). Next-generation miRNA sequencing was performed on a NextSeq 550 Sequencing System (Illumina) using single-end 1 × 75 bp sequencing to a depth of 25 million raw reads per sample. FASTQ files are uploaded to the online Qiagen Data Analysis Center for miRNA quantification. In the primary QIAseq quantification step, the unique molecular index (UMI) counts are calculated and primary miRNA mapping was performed using a human-specific miRBase mature database. At this point, adaptor sequences from the library preparation process and any low-quality bases are removed. In a secondary QIAseq analysis step, UMI counts are analyzed to calculate changes in miRNA expression generating fold-change values. miRNAseq expressions data (n = 2538) were used in the differential analysis and a total of 43 genes were identified statistically significant when the absolute fold regulation (FR) value ≥2 and FDR < 0.05.
miRNA target predictions
miRNA gene targets were predicted in silico using data mining of MiRtarbase (mirtarbase.mbc.nctu.edu.tw) and miRbase human-specific curated databases of mature miRNA that included experimental support (NGS, quantitative PCR, reporter assay or western blot) through surveying of >8000 scientific articles (54). The gene targets were filtered to include only highly predicted and strong experimental support predictions. Gene targets were illustrated using Venny 2.1 (http://bioinfogp.cnb.csic.es/tools/venny/index.html).
Reverse transcription-quantitative polymerase chain reaction
After miRNAseq, remaining RNA was used for RT-qPCR of miRNAs using TaqMan advanced miRNA assays (ThermoFisher, A25576) according to manufacturer’s instructions. Briefly, miRNA cDNA was reverse transcribed using the TaqMan advanced miRNA cDNA synthesis kit (ThermoFisher, A28007). Pre-formulated primer probe sets, designed to analyze mIRNA expression levels, were purchased from ThermoFisher and prepared with TaqMan Fast Advanced Master Mix (ThermoFisher, 4 444 557). qPCR was performed using a StepOnePlus real-time PCR system (ThermoFisher). All samples were run in duplicate and quantified using a standard curve method and normalized to baseline.
Statistics
For cell culture and animal work, all data are presented as mean ± SEM. All statistical analysis was performed using GraphPad Prism, version 8.0 (GraphPad software Inc.). When comparing averages in two groups, two-tailed Student’s t test or Welch’s t test was performed. When comparing the means of multiple groups, one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison post hoc test was performed. Asterisks denote actual P-value significances (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001) between groups or within groups and is noted in the individual figure legends.
For human CSF miRNA, quantified data are normalized, and paired differential gene expression testing were performed using ‘DESeq2’ and ‘limma’-based pipelines (55,56). Because the fold-change values are positive values (>0) whether they are miRNA that are downregulated (<1) or upregulated (>1) the negative inverse of the fold-change values are reported as FR where now downregulated miRNA are reported as less than zero and upregulated miRNA are reported as greater than zero, which represents a more biologically meaningful way of reporting the quantified sequencing results. Multiple hypothesis testing adjusted P-values or the FDRs were computed using the Benjamini–Hochberg procedure. Differentially expressed miRNAs are determined to be statistically significant when the absolute FR value ≥2 and FDR < 0.05. For RT-qPCR, statistically significant change was obtained via ordinary one-way ANOVA with Tukey post hoc test.
Supplementary Material
Acknowledgements
The authors are thankful to the nurses and staff of the Clinical Research Unit at Georgetown University Medical Center, Center for Translational and Clinical Science, and all staff of the research pharmacy at MedStar Georgetown University Hospital, the Genomics and Epigenomics Shared Resource and the Proteomics and Metabolomics Shared Resource at GUMC throughout this study.
Conflict of Interest statement. CM, CW and BK are listed as inventors on several US and international Georgetown University patents to use nilotinib and other tyrosine kinase inhibitors as a treatment for neurodegenerative diseases. FP and CM are shareholding founders of KeifeRX LLC (a GU spin-out) and CM receives consulting fee from KeifeRX LLC. No other authors declare any conflict of interests with this study.
Contributor Information
Alan J Fowler, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA; Interdisciplinary Program in Neuroscience, Georgetown University Medical Center, Washington, DC 20057, USA; Georgetown Howard Universities Center for Clinical and Translational Sciences, Translational Biomedical Sciences Program, Georgetown University Medical Center, Washington, DC 20057, USA.
Michaeline Hebron, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA.
Kaluvu Balaraman, Department of Chemistry, Georgetown University and Medicinal Chemistry Shared Resource, Georgetown University Medical Center, Washington, DC 20057, USA.
Wangke Shi, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA.
Alexander A Missner, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA.
Jonathan D Greenzaid, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA.
Timothy L Chiu, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA.
Clementina Ullman, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA.
Ethan Weatherdon, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA.
Val Duka, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA.
Yasar Torres-Yaghi, MedStar Georgetown University Hospital, Movement Disorders Clinic, Department of Neurology, Washington, DC 20057, USA.
Fernando L Pagan, MedStar Georgetown University Hospital, Movement Disorders Clinic, Department of Neurology, Washington, DC 20057, USA.
Xiaoguang Liu, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA.
Habtom Ressom, Department of Oncology, Lombardi Comprehensive Cancer Center, Georgetown University Medical Center, Washington, DC 20057, USA.
Jaeil Ahn, Department of Bioinformatics, Biostatistics, and Biomathematics, Georgetown University Medical Center, Washington, DC 20057, USA.
Christian Wolf, Department of Chemistry, Georgetown University and Medicinal Chemistry Shared Resource, Georgetown University Medical Center, Washington, DC 20057, USA.
Charbel Moussa, Department of Neurology, Translational Neurotherapeutics Program, Laboratory for Dementia and Parkinsonism, Lewy Body Dementia Association, Research Center of Excellence, Georgetown University Medical Center, Washington, DC 20057, USA; Interdisciplinary Program in Neuroscience, Georgetown University Medical Center, Washington, DC 20057, USA.
Funding
This work was supported by the Georgetown University Funding to CM, and philanthropies through the Lasky and Barajas Family Fund and other private philanthropies to conduct clinical studies. Nilotinib was provided to all participants by Novartis Pharmaceuticals Corporation. The National Center for Advancing Translational Sciences of the National Institutes of Health (Award Number TL1R001431 to AJF) and young investigator support from MedStar YT-Y.
Authors’ Contributions
AJF conducted the experiments, interpreted the data and contributed to manuscript. MH performed experiments, managed the data and performed CSF analysis. CW and BK designed and provided BK40143 and BK40197. HR oversaw the next-generation sequencing and miRNA analysis. FLP was principal investigator on the clinical trials, and YT-Y contributed to CSF collection and miRNA analysis. WS, AAM, CU, EW, VD, JDG, TLC and XL performed experiments and analyzed the data. JA performed and oversaw statistical analyses. CM conceived the study, designed the experiments, interpreted the results, discussed the findings and wrote the manuscript. All authors read and approved the final manuscript.
References
- 1. Vogel, W., Brakebusch, C., Fassler, R., Alves, F., Ruggiero, F. and Pawson, T. (2000) Discoidin domain receptor 1 is activated independently of beta (1) integrin. J. Biol. Chem., 275, 5779–5784. [DOI] [PubMed] [Google Scholar]
- 2. Vogel, W.F., Abdulhussein, R. and Ford, C.E. (2006) Sensing extracellular matrix: an update on discoidin domain receptor function. Cell. Signal., 18, 1108–1116. [DOI] [PubMed] [Google Scholar]
- 3. Canning, P., Tan, L., Chu, K., Lee, S.W., Gray, N.S. and Bullock, A.N. (2014) Structural mechanisms determining inhibition of the collagen receptor DDR1 by selective and multi-targeted type II kinase inhibitors. J. Mol. Biol., 426, 2457–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim, H.G., Tan, L., Weisberg, E.L., Liu, F., Canning, P., Choi, H.G., Ezell, S.A., Wu, H., Zhao, Z., Wang, J.et al. (2013) Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem. Biol., 8, 2145–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Valiathan, R.R., Marco, M., Leitinger, B., Kleer, C.G. and Fridman, R. (2012) Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev., 31, 295–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Franco, C., Hou, G., Ahmad, P.J., Fu, E.Y., Koh, L., Vogel, W.F. and Bendeck, M.P. (2008) Discoidin domain receptor 1 (ddr1) deletion decreases atherosclerosis by accelerating matrix accumulation and reducing inflammation in low-density lipoprotein receptor-deficient mice. Circ. Res., 102, 1202–1211. [DOI] [PubMed] [Google Scholar]
- 7. Zhu, M., Xing, D., Lu, Z., Fan, Y., Hou, W., Dong, H., Xiong, L. and Dong, H. (2015) DDR1 may play a key role in destruction of the blood-brain barrier after cerebral ischemia-reperfusion. Neurosci. Res., 96, 14–19. [DOI] [PubMed] [Google Scholar]
- 8. Hebron, M., Peyton, M., Liu, X., Gao, X., Wang, R., Lonskaya, I. and Moussa, C.E. (2017) Discoidin domain receptor inhibition reduces neuropathology and attenuates inflammation in neurodegeneration models. J. Neuroimmunol., 311(1–9). [DOI] [PubMed] [Google Scholar]
- 9. Jeitany, M., Leroy, C., Tosti, P., Lafitte, M., Le Guet, J., Simon, V., Bonenfant, D., Robert, B., Grillet, F., Mollevi, C.et al. (2018) Inhibition of DDR1-BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer. EMBO Mol. Med., 10, e7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumar, M., Salem, K., Michel, C.J., Jeffery, J.J., Yan, Y. and Fowler, A.M. (2019) (18)F-Fluoroestradiol positron emission tomography imaging of activating estrogen receptor alpha mutations in breast cancer. J. Nucl. Med., 60, 1247–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pagan, F.L., Hebron, M.L., Wilmarth, B., Torres-Yaghi, Y., Lawler, A., Mundel, E.E., Yusuf, N., Starr, N.J., Anjum, M., Arellano, J.et al. (2019) Nilotinib effects on safety, tolerability, and potential biomarkers in Parkinson disease: a phase 2 randomized clinical trial. JAMA Neurol., 77, 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Robba, V., Fowler, A., Karantana, A., Grindlay, D. and Lindau, T. (2019) Open versus arthroscopic repair of 1B ulnar-sided triangular fibrocartilage complex tears: a systematic review. Hand (N Y) in press, 1558944718815244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pagan, F.L., Hebron, M.L., Wilmarth, B., Torres-Yaghi, Y., Lawler, A., Mundel, E.E., Yusuf, N., Starr, N.J., Arellano, J., Howard, H.H.et al. (2019) Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson's disease. Pharmacol. Res. Perspect., 7, e00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hebron, M.L., Javidnia, M. and Moussa, C.E. (2018) Tau clearance improves astrocytic function and brain glutamate-glutamine cycle. J. Neurol. Sci., 391, 90–99. [DOI] [PubMed] [Google Scholar]
- 15. Pagan, F., Hebron, M., Valadez, E.H., Torres-Yaghi, Y., Huang, X., Mills, R.R., Wilmarth, B.M., Howard, H., Dunn, C., Carlson, A.et al. (2016) Nilotinib effects in Parkinson's disease and dementia with Lewy bodies. J. Parkinsons Dis., 6, 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lonskaya, I., Hebron, M.L., Selby, S.T., Turner, R.S. and Moussa, C.E. (2015) Nilotinib and bosutinib modulate pre-plaque alterations of blood immune markers and neuro-inflammation in Alzheimer's disease models. Neuroscience, 304, 316–327. [DOI] [PubMed] [Google Scholar]
- 17. Lonskaya, I., Hebron, M.L., Desforges, N.M., Schachter, J.B. and Moussa, C.E. (2014) Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. (Berl.), 92, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lonskaya, I., Desforges, N.M., Hebron, M.L. and Moussa, C.E. (2013) Ubiquitination increases parkin activity to promote autophagic alpha-synuclein clearance. PLoS One, 8, e83914. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Hebron, M.L., Lonskaya, I. and Moussa, C.E. (2013) Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson's disease models. Hum. Mol. Genet., 22, 3315–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hijiya, N. and Suttorp, M. (2019) How I treat chronic myeloid leukemia in children and adolescents. Blood, 133, 2374–2384. [DOI] [PubMed] [Google Scholar]
- 21. Turner, R.S., Hebron, M.L., Lawler, A., Mundel, E.E., Yusuf, N., Starr, J.N., Anjum, M., Pagan, F., Torres-Yaghi, Y., Shi, W.et al. (2020) Nilotinib effects on safety, tolerability, and biomarkers in Alzheimer's disease. Ann. Neurol., 88, 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suzuki, K., Akioka, M., Kondo-Kakuta, C., Yamamoto, H. and Ohsumi, Y. (2013) Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J. Cell Sci., 126, 2534–2544. [DOI] [PubMed] [Google Scholar]
- 23. Vehlow, A., Klapproth, E., Jin, S., Hannen, R., Hauswald, M., Bartsch, J.W., Nimsky, C., Temme, A., Leitinger, B. and Cordes, N. (2019) Interaction of discoidin domain receptor 1 with a 14-3-3-Beclin-1-Akt1 complex modulates glioblastoma therapy sensitivity. Cell Rep., 26, 3672–3683 e3677. [DOI] [PubMed] [Google Scholar]
- 24. Bartel, D.P. (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, 116, 281–297. [DOI] [PubMed] [Google Scholar]
- 25. Agha, R.A., Borrelli, M.R., Farwana, R., Koshy, K., Fowler, A.J., Orgill, D.P. and Group, P (2018) The PROCESS 2018 statement: updating consensus preferred reporting of CasE series in surgery (PROCESS) guidelines. Int. J. Surg., 60, 279–282. [DOI] [PubMed] [Google Scholar]
- 26. Lonskaya, I., Hebron, M.L., Algarzae, N.K., Desforges, N. and Moussa, C.E. (2013) Decreased parkin solubility is associated with impairment of autophagy in the nigrostriatum of sporadic Parkinson's disease. Neuroscience, 232, 90–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lonskaya, I., Shekoyan, A.R., Hebron, M.L., Desforges, N., Algarzae, N.K. and Moussa, C.E. (2013) Diminished parkin solubility and co-localization with intraneuronal amyloid-beta are associated with autophagic defects in Alzheimer's disease. J. Alzheimers Dis., 33, 231–247. [DOI] [PubMed] [Google Scholar]
- 28. Lonskaya, I., Hebron, M., Desforges, N.M., Franjie, A. and Moussa, C.E. (2013) Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol. Med., 5, 1247–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shi, Y., Zhang, Y. and Lou, J. (2017) The influence of cell membrane and SNAP25 linker loop on the dynamics and unzipping of SNARE complex. PLoS One, 12, e0176235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nickerson, D.P., Brett, C.L. and Merz, A.J. (2009) Vps-C complexes: gatekeepers of endolysosomal traffic. Curr. Opin. Cell Biol., 21, 543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wegrzynowicz, M., Bar-On, D., Calo, L., Anichtchik, O., Iovino, M., Xia, J., Ryazanov, S., Leonov, A., Giese, A., Dalley, J.W.et al. (2019) Depopulation of dense alpha-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson's disease model. Acta Neuropathol., 138, 575–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rizo, J. (2018) Mechanism of neurotransmitter release coming into focus. Protein Sci., 27, 1364–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hebron, M.L., Lonskaya, I., Sharpe, K., Weerasinghe, P.P., Algarzae, N.K., Shekoyan, A.R. and Moussa, C.E. (2013) Parkin ubiquitinates tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J. Biol. Chem., 288, 4103–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hebron, M.L., Lonskaya, I., Olopade, P., Selby, S.T., Pagan, F. and Moussa, C.E. (2014) Tyrosine kinase inhibition regulates early systemic immune changes and modulates the neuroimmune response in alpha-synucleinopathy. J. Clin. Cell Immunol., 5, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heyburn, L., Hebron, M.L., Smith, J., Winston, C., Bechara, J., Li, Z., Lonskaya, I., Burns, M.P., Harris, B.T. and Moussa, C.E. (2016) Tyrosine kinase inhibition reverses TDP-43 effects on synaptic protein expression, astrocytic function and amino acid dis-homeostasis. J. Neurochem., 139, 610–623. [DOI] [PubMed] [Google Scholar]
- 36. Moussa, C. (2020) Dopamine metabolite biomarkers and testing for disease modification in Parkinson disease-reply. JAMA Neurol., 77, 1039–1040. [DOI] [PubMed] [Google Scholar]
- 37. Jack, C.R., Jr., Bennett, D.A., Blennow, K., Carrillo, M.C., Feldman, H.H., Frisoni, G.B., Hampel, H., Jagust, W.J., Johnson, K.A., Knopman, D.S.et al. (2016) A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology, 87, 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sims, R., van der Lee, S.J., Naj, A.C., Bellenguez, C., Badarinarayan, N., Jakobsdottir, J., Kunkle, B.W., Boland, A., Raybould, R., Bis, J.C.et al. (2017) Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer's disease. Nat. Genet., 49, 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seo, M.C., Kim, S., Kim, S.H., Zheng, L.T., Park, E.K., Lee, W.H. and Suk, K. (2008) Discoidin domain receptor 1 mediates collagen-induced inflammatory activation of microglia in culture. J. Neurosci. Res., 86, 1087–1095. [DOI] [PubMed] [Google Scholar]
- 40. Lee, J.E., Zhu, Z., Bai, Q., Brady, T.J., Xiao, H., Wakefield, M.R. and Fang, Y. (2019) The role of interleukin-9 in cancer. Pathol. Oncol. Res., in press. [DOI] [PubMed] [Google Scholar]
- 41. Wharton, W., Kollhoff, A.L., Gangishetti, U., Verble, D.D., Upadhya, S., Zetterberg, H., Kumar, V., Watts, K.D., Kippels, A.J., Gearing, M.et al. (2019) Interleukin 9 alterations linked to Alzheimer disease in African Americans. Ann. Neurol., 86, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saunders, J.A., Estes, K.A., Kosloski, L.M., Allen, H.E., Dempsey, K.M., Torres-Russotto, D.R., Meza, J.L., Santamaria, P.M., Bertoni, J.M., Murman, D.L.et al. (2012) CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson's disease. J. NeuroImmune Pharmacol., 7, 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tan, S., Shan, Y., Lin, Y., Liao, S., Zhang, B., Zeng, Q., Wang, Y., Deng, Z., Chen, C., Hu, X.et al. (2019) Neutralization of interleukin-9 ameliorates experimental stroke by repairing the blood-brain barrier via down-regulation of astrocyte-derived vascular endothelial growth factor-a. FASEB J., 33, 4376–4387. [DOI] [PubMed] [Google Scholar]
- 44. Rojas-Zuleta, W.G. and Sanchez, E. (2017) IL-9: function, sources, and detection. Methods Mol. Biol., 1585, 21–35. [DOI] [PubMed] [Google Scholar]
- 45. Rivera Vargas, T., Cai, Z., Shen, Y., Dosset, M., Benoit-Lizon, I., Martin, T., Roussey, A., Flavell, R.A., Ghiringhelli, F. and Apetoh, L. (2017) Selective degradation of PU.1 during autophagy represses the differentiation and antitumour activity of TH9 cells. Nat. Commun., 8, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cully, M. (2017) Inflammatory diseases: an IL-9 solution to inflammation resolution. Nat. Rev. Drug Discov., 16, 600–601. [DOI] [PubMed] [Google Scholar]
- 47. Roediger, B. and Weninger, W. (2017) Resolving a chronic inflammation mystery. Nat. Med., 23, 914–916. [DOI] [PubMed] [Google Scholar]
- 48. Hira, D. and Terada, T. (2018) BCRP/ABCG2 and high-alert medications: biochemical, pharmacokinetic, pharmacogenetic, and clinical implications. Biochem. Pharmacol., 147, 201–210. [DOI] [PubMed] [Google Scholar]
- 49. Davis, J., Xu, F., Deane, R., Romanov, G., Previti, M.L., Zeigler, K., Zlokovic, B.V. and Van Nostrand, W.E. (2004) Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem., 279, 20296–20306. [DOI] [PubMed] [Google Scholar]
- 50. Santacruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., Guimaraes, A., DeTure, M., Ramsden, M., McGowan, E.et al. (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science, 309, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Giasson, B.I., Duda, J.E., Quinn, S.M., Zhang, B., Trojanowski, J.Q. and Lee, V.M. (2002) Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron, 34, 521–533. [DOI] [PubMed] [Google Scholar]
- 52. Khandelwal, P.J., Herman, A.M., Hoe, H.S., Rebeck, G.W. and Moussa, C.E. (2011) Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum. Mol. Genet., 20, 2091–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu, X., Hebron, M., Shi, W., Lonskaya, I. and Moussa, C.E. (2019) Ubiquitin specific protease-13 independently regulates parkin ubiquitination and alpha-synuclein clearance in alpha-synucleinopathies. Hum. Mol. Genet., 28, 548–560. [DOI] [PubMed] [Google Scholar]
- 54. Chou, C.H., Shrestha, S., Yang, C.D., Chang, N.W., Lin, Y.L., Liao, K.W., Huang, W.C., Sun, T.H., Tu, S.J., Lee, W.H.et al. (2018) miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res., 46, D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ritchie, M.E., Phipson, B., Wu, D., Hu, Y., Law, C.W., Shi, W. and Smyth, G.K. (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res., 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.