

Abstract
DOCK3 is a member of the DOCK family of guanine nucleotide exchange factors that regulate cell migration, fusion and viability. Previously, we identified a dysregulated miR-486/DOCK3 signaling cascade in dystrophin-deficient muscle, which resulted in the overexpression of DOCK3; however, little is known about the role of DOCK3 in muscle. Here, we characterize the functional role of DOCK3 in normal and dystrophic skeletal muscle. Utilizing Dock3 global knockout (Dock3 KO) mice, we found that the haploinsufficiency of Dock3 in Duchenne muscular dystrophy mice improved dystrophic muscle pathologies; however, complete loss of Dock3 worsened muscle function. Adult Dock3 KO mice have impaired muscle function and Dock3 KO myoblasts are defective for myogenic differentiation. Transcriptomic analyses of Dock3 KO muscles reveal a decrease in myogenic factors and pathways involved in muscle differentiation. These studies identify DOCK3 as a novel modulator of muscle health and may yield therapeutic targets for treating dystrophic muscle symptoms.
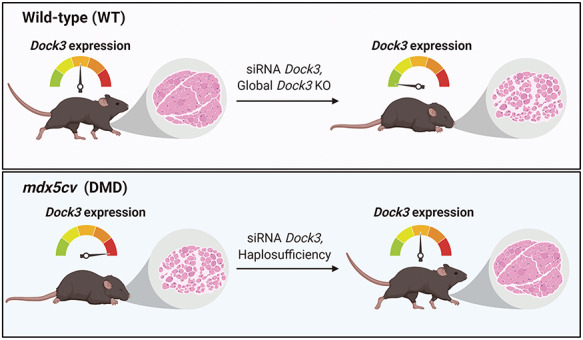
Graphical Abstract
Graphical Abstract.
Introduction
Skeletal muscle formation requires a complex coordination of a myriad of signaling factors and cell populations to efficiently form mature myofibers capable of generating muscle force. Duchenne muscular dystrophy (DMD) is an X-linked neuromuscular disorder caused by pathogenic mutations in the DYSTROPHIN gene resulting in the lack of functional dystrophin protein. This dystrophin-deficiency results in a degradation of the link between filamentous actin and the extracellular matrix predisposing to structural damage. In DMD patients, after the loss of ambulation, their skeletal muscles begin to progressively deteriorate and are replaced with adipocytes, inflammatory cells and necrotic tissue. While the dystrophin protein has been thought of as a classical muscle structural protein, recent evidence suggests that it also plays a significant role in the regulation of various cellular signaling pathways involved in dystrophic pathology (1–5).
The DOCK (dedicator of cytokinesis) family of proteins functions as key signaling factors that regulate cellular migration, fusion and survival in different cell and tissue types (6). DOCK1 and DOCK5 have been shown to play key roles in myoblast fusion through CRK1/MAPK14 protein interactions (7,8). Additional studies have demonstrated that DOCK3 and DOCK4 activity can regulate key downstream pathways such as Sonic hedgehog to regulate axonal guidance and RAC1 activation (9). DOCK3 (previously called MOCA or modifier of cell adhesion) was shown to be essential for normal neuronal function, and Dock3 global knockout (KO) mice develop plaques in the brain (10). Human patients with loss-of-function DOCK3 variants develop intellectual disability, ataxia and developmental delay and have low muscle tone from birth (11–13). There is strong genetic evidence that the disease severity of patients with DOCK3 pathogenic variants is linked to the degree of which DOCK3 can activate RAC1 signaling as a guanine nucleotide exchange factor (GEF) protein (13). Previously, we identified a muscle-enriched microRNA, miR-486, as a regulator of dystrophin-deficient skeletal muscle pathology in mice and human muscle biopsies through its negative regulation of DOCK3 mRNA expression levels (14,15). We hypothesized that DOCK3 may be a direct regulator of muscle function and that its dysregulation in DMD may play significant downstream consequences in the progression of the disease. We assessed the roles of DOCK3 in DMD pathology in both zebrafish and mouse models of the disease. We analyzed Dock3 KO mouse muscles and found significant perturbations in muscle integrity and function. Furthermore, Dock3 KO mouse muscle cells were analyzed for their ability to proliferate, differentiate and viability in culture. Transcriptomic analyses of Dock3 KO tibialis anterior (TA) muscles revealed a significant downregulation of several muscle pathways including the fusogen myomixer (Mymx), also referred to as myomerger or minion (16–18). This work demonstrates the critical role of DOCK3 in normal myogenesis and a gene-dosing effect of DOCK3 expression levels on dystrophin-deficient muscle health.
Results
Dock3 gene expression is upregulated in dystrophic muscles
We first aimed to assess endogenous Dock3 gene expression levels in normal and dystrophin-deficient muscle. Previously, we established DOCK3 as an mRNA target of miR-486, a muscle-enriched miRNA whose expression is reduced in with dystrophic disease progression (14). We evaluated Dock3 gene expression in our DMD mouse model, mdx5cv, and compared with wild-type (WT) (C57BL/6 J strain) aged-matched male mice. The mdx5cv mice have significantly fewer revertant dystrophic fibers, increased muscle weakness and an overall greater dystrophic pathology than the classical mdx strain of mice (19,20). We observed Dock3 mRNA expression to be significantly elevated in the mdx5cv muscles compared with aged-matched WT control muscles across three different muscles types including the TA, soleus and diaphragm starting at 1 month of life (Fig. 1A–C). These findings of elevated Dock3 mRNA were found to be consistent throughout adulthood into aged (12-month-old) mdx5cv muscles (Fig. 1A–C).
Figure 1.
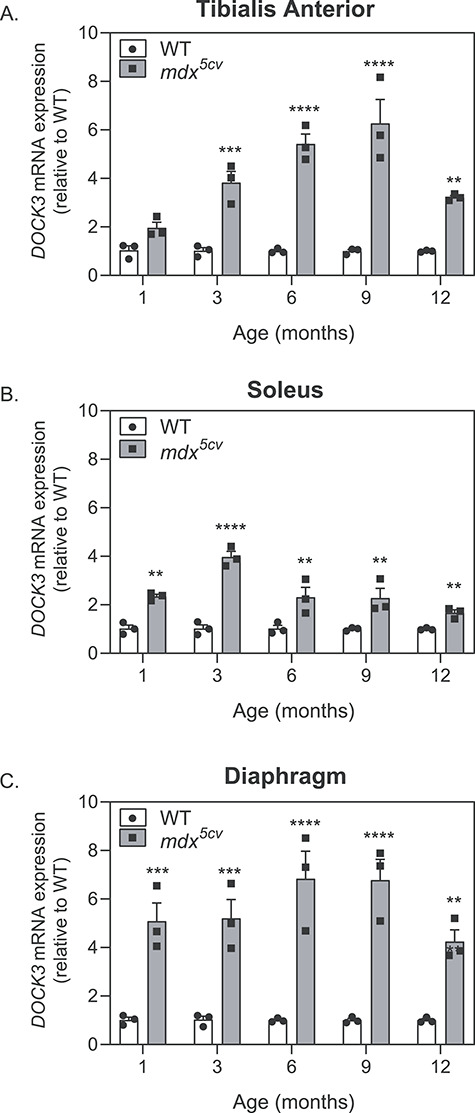
Dock3 mRNA expression levels are increased in adult mdx5cv mouse skeletal muscles. Real-time qPCR data from WT and mdx5cv mice taken from (A) TA, (B) soleus and (C) diaphragm muscles at 1, 3, 6, 9 and 12 months of age demonstrating the significant increase in Dock3 mRNA expression in mdx5cv dystrophic mouse muscle compared with WT muscle (n = 3 per genotype and cohort, performed in triplicate; two-way ANOVA with Tukey’s correction; **P < 0.01, ***P < 0.001).
Knockdown of dock3 mRNA in dystrophic zebrafish larvae improves muscle structure and overall outcomes
Zebrafish are a powerful model system for DMD disease modeling due to their translucent ex vivo development, large offspring sizes, ease of genetic manipulation and ability to uptake compounds in their skin and gills during early developmental stages (21–23). Zebrafish Dock3 protein amino acid sequences showed a strong amount of evolutionary conservation to mouse and human DOCK3 protein sequences (85% homology with mouse and human; Supplementary Material, Fig. S1). We performed in situ hybridization (ISH) for zebrafish dock3 mRNA expression in WT (AB strain) zebrafish embryos at the early stages of development including prior to and after muscle formation. Zebrafish dock3 mRNA was detected at low levels during early somite formation with strong expression in the developing brain, neural tube and dorsal muscle by 4 days post-fertilization (dpf) (Fig. 2A). The sapje zebrafish is a widely used DMD model that has the severe muscle weakness and myofiber degradation comparable to those observed in the later stages of DMD patient skeletal muscles (21,24,25). Unlike mammals, zebrafish lack definitive sex chromosomes and the dystrophin (dmd) gene in zebrafish is located on chromosome 1 (26). Mating of sapje heterozygote fish results in a Mendelian percentage of 25% affected zebrafish that can be easily identified via birefringence assay (24,25). The sapje homozygote mutant larvae showed severe myofiber disruption in affected larvae versus unaffected (sapje heterozygotes and WT) larvae (25,27–29) (Fig. 2B). Given our findings of increased Dock3 expression in mdx5cv muscles, we tested whether the modulation of zebrafish dock3 expression might affect dystrophic outcomes. We observed significantly fewer than expected affected sapje zebrafish in our dock3 morphant (dock3 MO) zebrafish compared with control morphants when using a low dose of dock3 morpholino (1 ng) (Fig. 2C). However, the high dose of dock3 morpholino (6 ng) resulted in a higher number of affected sapje zebrafish, suggesting that there may be an optimal dose for blocking dystrophic pathology. These results were further confirmed using histochemical staining of F-actin in the dorsal muscles of the 1 ng dock3 MO sapje zebrafish, which revealed fewer areas of myofiber detachment and overall more integrity compared with control MO sapje homozygotes, while the converse was observed for the high dose dock3 morphant (Fig. 2D). Altogether, these data suggest that DOCK3 expression levels are strongly linked towards dystrophic pathologies and overall outcomes in DMD zebrafish.
Figure 2.
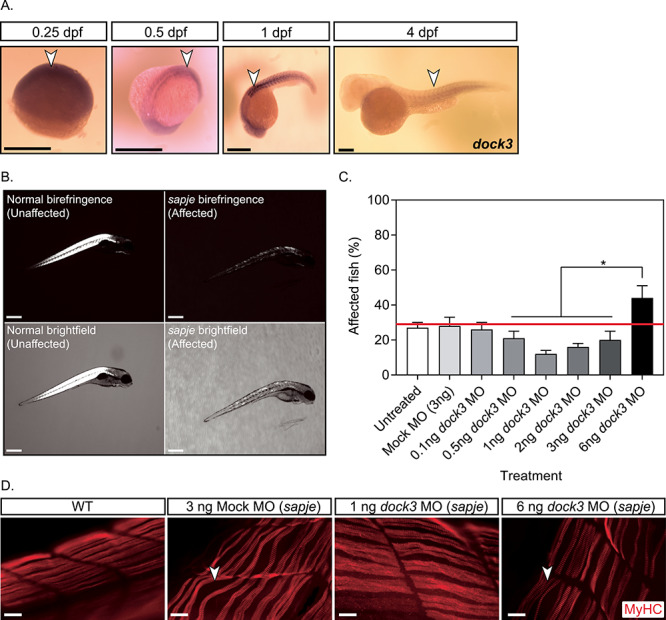
Knockdown of dock3 mRNA in DMD zebrafish reduces dystrophic pathology. (A) ISH of zebrafish dock3 mRNA during early developmental time points. Note the robust expression of zebrafish dock3 mRNA in muscle tissues at stages of early muscle formation and muscle pioneer cell fusion. Not shown sense probes are used as internal controls. Arrowheads demarcate dock3 mRNA ISH signal. Scale bar = 100 μm. (B) Representative images of normal and sapje mutant birefringence morphology. Scale bar = 200 μm. (C) Quantification of muscle birefringence shows that 1 ng of dock3 MO improves muscle birefringence scoring in sapje mutant zebrafish, while 6 ng of dock3 MO worsens muscle birefringence scoring. (n = 3 experimental replicates; one-way ANOVA with Tukey’s correction; *P < 0.05). (D) MyHC (F-59 antibody) immunofluorescent staining of 4 dpf WT (uninjected) or sapje mutant larvae injected with control (mock) morpholino, or dock3 MO (1 or 6 ng). Arrowheads demarcate myofiber tears from the sarcolemmal membrane. Scale bar = 40 μm.
Knockdown of DOCK3 in isolated human DMD myoblasts increases myogenic fusion
To investigate the effects of DOCK3 on myoblast fusion in dystrophin-deficient cells, we infected DMD and normal primary human myoblasts with lentiviral particles of either shRNA DOCK3, shRNA scrambled (i.e. non-targeting) or a mock control (Fig. 3A). DMD myoblasts have poor terminal differentiation into myotubes (30,31). We observed a significantly higher myogenic fusion index in the shRNAi DOCK3 DMD myotubes compared with either shRNA scrambled or mock-treated DMD muscle cells (Fig. 3A and B). To validate the DOCK3 mRNA knock-down, we performed western blot analysis for DOCK3 protein levels and confirmed a marked reduction of DOCK3 protein expression (Fig. 3C). These data reveal that the knockdown of DOCK3 mRNA in human DMD myoblasts improves myogenic fusion percentages despite dystrophin-deficiency.
Figure 3.
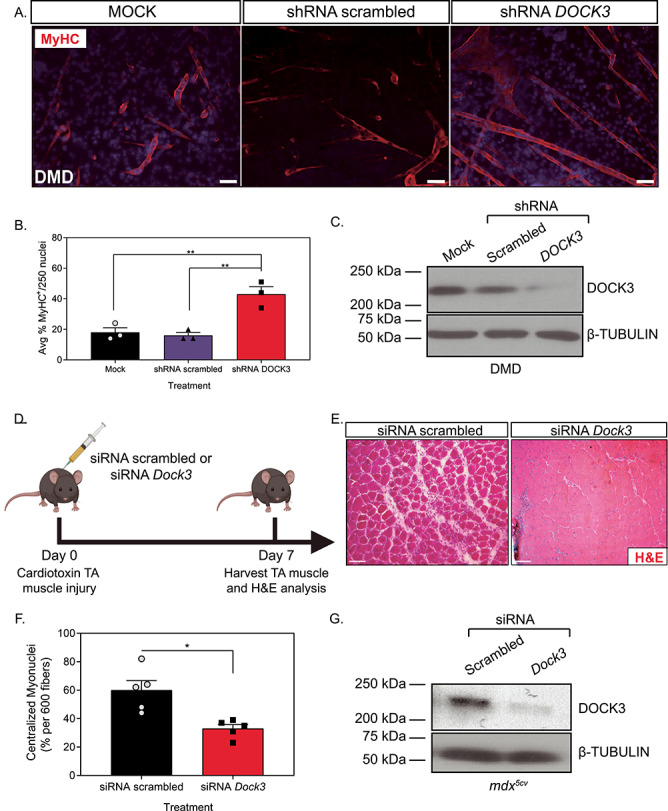
Knockdown of DOCK3 in DMD patient muscle cells and mdx5cv mice increases myogenic differentiation and improves pathology. MyHC staining reveals that the knockdown of DOCK3 protein in human DMD primary myotubes increases myogenic fusion indices. (A) Immunofluorescence of MyHC (MF-20 antibody, DSHB Iowa, red) and nucleus marker (DAPI, blue) of Day 7 differentiated human DMD primary myotubes either transduced with mock (control), shRNA scrambled (negative control) and shRNA DOCK3 lentiviral particles [transduced at a multiplicity of infection (MOI) of 10]. Scale bar = 50 μm. (B) Summary graph of average myogenic fusion indices as calculated by percentage of nuclei inside of MyHC+ cells out of 250 nuclei, as previously described (68). DMD primary myotubes transduced with shRNA DOCK3 demonstrated significantly higher amount of myogenic fusion. (n = 3 experimental replicates, one-way ANOVA with Tukey’s correction, *P < 0.05). (C) Western blot analysis of whole cell lysates taken from Day 7 differentiated DMD myotubes transduced with mock (control), shRNA scrambled (shRNA internal control) or shRNA DOCK3 knockdown lentiviral particles, demonstrating successful knockdown of DOCK3 protein expression. (D) Schematic showing co-injection in mdx5cv mice of cardiotoxin (10 μM ctx) as well as either siRNA double-stranded (ds) oligos to inhibit mouse Dock3 mRNA (siRNA Dock3) or a scrambled ds oligo (siRNA scrambled; control). Experiment was performed in each leg and mice were evaluated 7 days post ctx injury. (E) Representative H&E staining of the siRNA-injected mdx5cv muscles 7 days post ctx. Scale bar = 50 μm. (F) Summary graph showing reduced centralized myonuclei when mdx5cv mice were injected with siRNA Dock3 (n = 5, student’s t-test, two-tailed, *P < 0.05). (G) Western blot analysis of whole muscle lysates from siRNA scrambled and siRNA Dock3 muscle demonstrating successful knockdown of Dock3 protein.
Knockdown of Dock3 in mdx5cv mice improves cardiotoxin-induced muscle injury repair
We next investigated the role of DOCK3 in DMD pathology using the mdx5cv mouse model of DMD. Given our findings of DOCK3 in myoblast fusion, we hypothesized that DOCK3 might play a role in muscle repair following injury. Mdx5cv mice were co-injected with cardiotoxin (ctx) into the TA muscles along with either siRNA scrambled (control) or mouse Dock3 siRNA (Fig. 3D). The mdx5cv mice treated with Dock3 siRNA had a more improved muscle injury repair pathology and muscle architecture compared with siRNA scrambled-treated mice 7 days after injury (Fig. 3E). The siRNA Dock3-treated mdx5cv mice had significantly less centralized myonuclei than the control group (Fig. 3F). To validate Dock3 mRNA knockdown, we performed western blot analysis which confirmed a strong reduction of DOCK3 protein expression levels (Fig. 3G). These results support a potential beneficial effect of reducing DOCK3 expression in the mdx5cv mice during muscle injury-induced repair and regeneration.
Haploinsufficiency of Dock3 improves muscle pathologies in mdx5cv mice
Given our previous findings demonstrating that DOCK3 expression levels are elevated in DMD patients and mdx5cv mice, we used Dock3 global KO mice and evaluated their muscles on dystrophic mdx5cv strain background in three separate cohorts. Muscle architecture was assessed at 6 months of age across all groups by measuring a number of centralized myonuclei and fiber size distribution via hematoxylin and eosin (H&E) histochemistry (Fig. 4A–C), as well as measuring the severity of fibrosis via Masson’s trichrome staining (Fig. 4D and E). Consistent with our initial findings, the global Dock3 KO mice on mdx5cv background [Dock3:mdx5cv double KO (DKO)] mice developed exacerbated muscle weakness and all Dock3:mdx5cv DKO mice expired by 6 months of age due to diaphragm muscle weakness (data not shown). Muscle structural analyses revealed severe myofiber disruption and fibrosis in the Dock3:mdx5cv DKO mice compared with mdx5cv (Fig. 4A–D). Interestingly, the haploinsufficiency of Dock3 in mdx5cv mice (Dock3+/−:mdx5cv) improved muscle fiber architecture, larger myofiber sizes and improved muscle pathology in comparison with the mdx5cv and Dock3:mdx5cv DKO double mutant mice (Fig. 4A–D). Western blot analysis revealed that Dock3+/−:mdx5cv mice had a 75% reduction in DOCK3 protein compared with mdx5cv mice (Fig. 4F and G). These findings suggest that Dock3 dosage levels influence the pathologic outcomes of dystrophin-deficient muscles and that the haploinsufficiency of Dock3 is beneficial in dystrophic muscle pathology.
Figure 4.
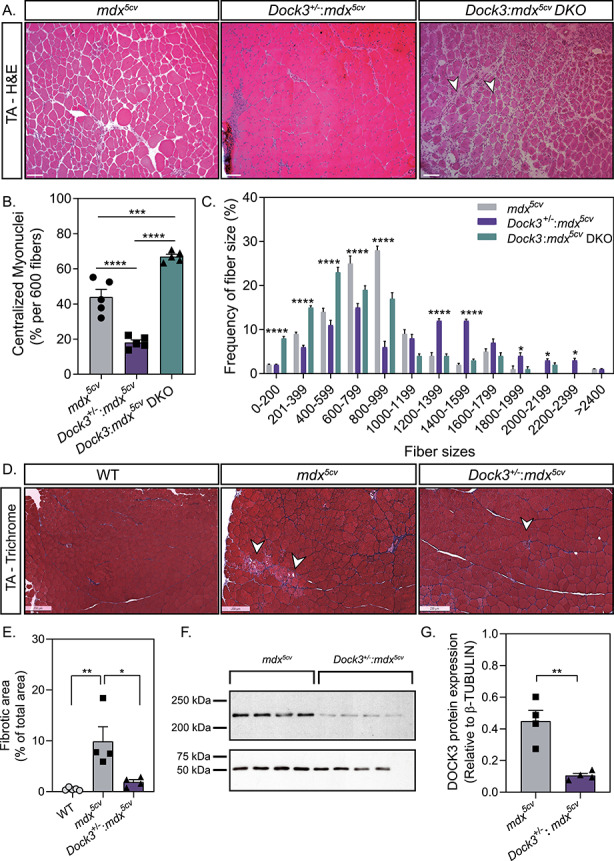
Gene dosage of Dock3 affects muscle architecture and dystrophic pathology in mdx5cv mice. (A) H&E immunohistochemistry of mdx5cv, Dock3+/−:mdx5cv and Dock3:mdx5cv DKO to visualize muscle morphology. Scale bar = 50 μm. Arrowheads indicate necrotic/fibrotic muscle. (B) Percent of centralized myonuclei out of 600 myofibers/TA comparing Dock3 alleles on mdx5cv genetic background. (n = 5, one-way ANOVA with Tukey’s correction; **P < 0.01, ****P < 0.0001). (C) Comparison of fiber size distribution curve based on CSA (μm2) among Dock3 alleles on mdx5cv genetic background. (n = 5; two-way ANOVA with Tukey’s correction; *P < 0.05, ****P < 0.0001). (D) Masson’s Trichrome stain to visualize fibrotic area in WT, mdx5cv and Dock3+/−:mdx5cv. Scale bar = 250 μm. Arrowheads indicate fibrotic muscle. (E) Percent of fibrotic area comparing WT, mdx5cv and Dock3+/−:mdx5cv TA muscle (n = 4, one-way ANOVA with Tukey’s correction; *P < 0.05, **P < 0.01). (F) Western blot analysis of whole muscle lysate from mdx5cv and Dock3+/−:mdx5cv mice for DOCK3 protein expression level quantification. (G) Quantification of DOCK3 protein expression level (n = 4, student’s t-test, two-tailed, **P < 0.01).
Adult Dock3 KO mice have abnormal muscle architecture, muscle mass and metabolism
Given our data demonstrating that the loss of DOCK3 expression in dystrophin-deficient muscle results in differential muscle effects, we further interrogated the role of DOCK3 expression in muscle health and metabolism in WT mice. Global Dock3 KO mice had increased centralized myonuclei and altered fiber size distribution, indicating impaired muscle health compared with control WT mice (Fig. 5A–C). In addition, we observed significant muscle fiber atrophy reflected in smaller myofiber frequencies in the Dock3 KO mouse muscle compared with control WT muscles (Fig. 5C). Additionally, adult Dock3 KO mice had significantly lower body weight compared with WT mice at 6 months of age (Fig. 5D). Quantitative magnetic resonance (QMR) imaging in Dock3 KO mice revealed reduced lean mass (Fig. 5E), while there was no effect on total fat mass (Fig. 5F) compared with WT mice. As muscle is the primary source of glucose and given our previous findings linking DOCK3 to the PTEN/AKT muscle growth pathway, we next measured the ability of Dock3 KO mice to respond to a glucose challenge via a glucose tolerance test (GTT) (32). The GTT revealed that Dock3 KO mice had a significantly impaired response to the glucose bolus, indicating a deficit in glucose metabolic processing (Fig. 5G). We then investigated the role of Dock3 in global locomotor function using open field activity tracking to record basal activity levels in both aged-matched (6-month-old) WT and Dock3 KO male mice. Dock3 KO mice demonstrated significantly decreased distance traveled and average velocity compared with WT mice, indicating a reduction in basal locomotor function (Fig. 5H and I; Supplementary Material, Fig. S2). This demonstrates an decrease in locomotor ability in Dock3 KO mice at 6 months of age similar to that reported for the mdx mouse model and consistent with our findings that Dock3 KO mice have significant muscle weakness and myofiber atrophy (33–40). Additionally, we assessed cardiac function in WT and Dock3 KO mice and found no significant differences in functional parameters (Supplementary Material, Fig. S3). These findings implicate a crucial role for DOCK3 in the maintenance of muscle mass and metabolic function in skeletal muscle.
Figure 5.
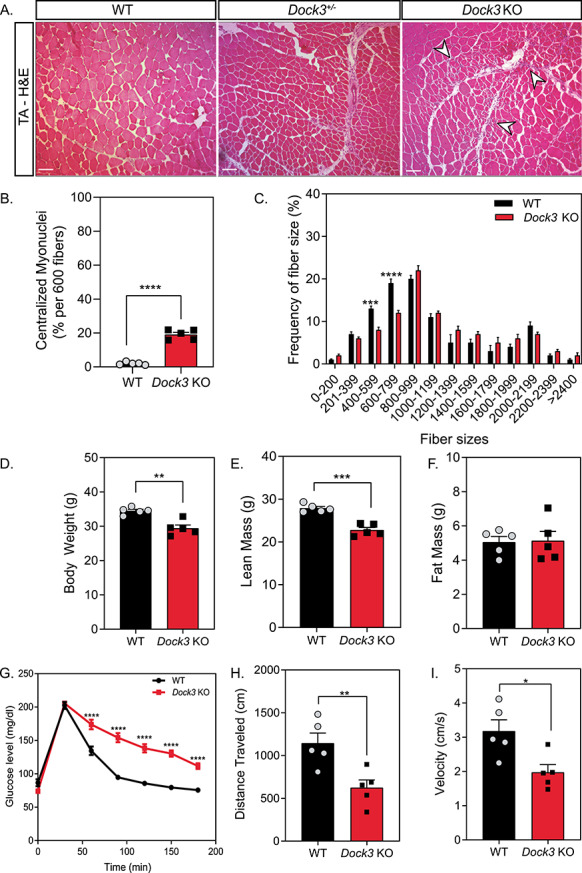
Adult Dock3 KO mice have abnormal muscle architecture, muscle mass and metabolism. (A) H&E immunohistochemistry of Dock3+/+ (WT), Dock3+/− and Dock3 KO TA adult muscle on WT background. Scale bar = 50 μm. Arrowheads indicate necrotic/fibrotic muscle. (B) Percent of centralized myonuclei out of 600 myofibers/TA comparing WT and Dock3 KO mice. (n = 5, student’s t-test, two-tailed, ****P < 0.0001). (C) Comparison of WT and Dock3 KO mice fiber size distribution curve based on CSA (μm2). (n = 5; two-way ANOVA with Tukey’s correction; ***P < 0.001, ****P < 0.0001). (D) Compared with WT mice, Dock3 KO mice were significantly lower in gross body weight, (E) had significantly decreased lean mass and (F) had no difference in fat mass observed. (n = 5; student’s t-test, two-tailed; **P < 0.01, ***P < 0.001). (G) GTT revealed that Dock3 KO mice are glucose intolerant. (n = 5; two-way ANOVA with Tukey’s correction; ****P < 0.0001). (H) Dock3 KO mice had significantly decreased distance traveled (n = 5; student’s t-test, two-tailed; **P < 0.01). (I) Dock3 KO mice had significantly decreased average velocity (n = 5; student’s t-test, two-tailed; *P < 0.05).
RNA-sequencing of Dock3 KO muscles reveals dysregulated muscle transcripts
To determine what pathways may be affected by the loss of DOCK3 protein in muscle, we performed RNA-sequencing (RNA-seq) on adult TA skeletal muscles from WT and Dock3 KO mice. Transcriptome analysis revealed 280 differentially expressed genes (DEGs) as shown by volcano plot (157 downregulated in blue and 123 upregulated in orange) (Fig. 6A). Gene ontology (GO) analyses revealed significant alterations in muscle structural and metabolic signaling pathways in the Dock3 KO mice (Fig. 6B), which is consistent with the severe Dock3 KO phenotype observed. We identified the top 10 up- and down-regulated transcripts, excluding undefined transcripts, of Dock3 KO mice compared with WT by sorting top genes according to the statistical significance (adjusted P-value) and the strength of fold change induced (log2-fold change) (Fig. 6C and D). We confirmed a significant decrease in Dock3 mRNA transcript in the Dock3 KO mouse muscles (Fig. 6D). These findings reveal significant transcriptomic changes in important muscle structural and metabolic signaling pathways in the absence of DOCK3 protein.
Figure 6.
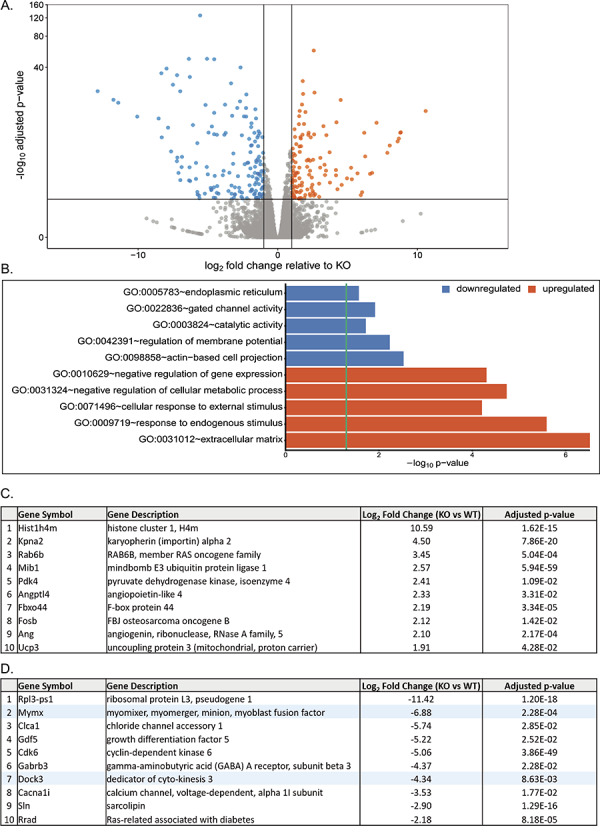
RNA-sequencing analysis of Dock3 KO TA muscles reveals numerous dysregulated mRNA transcripts and signaling pathways. (A) Summary graph of differentially regulated genes, with blue representing downregulated genes and orange representing upregulated genes relative to KO. (B) Top downregulated and upregulated GO terms as determined by GO analyses compared with WT. (C) Top 10 upregulated genes in Dock3 KO mice compared with WT. (D) Top 10 downregulated genes in Dock3 KO mice compared with WT. Highlighted in blue are specific genes of interest.
Dock3 KO myoblasts fail to differentiate and have reduced myogenic fusion capabilities
Previous studies have demonstrated that DOCK1 and DOCK5 are essential for normal myoblast fusion and differentiation via actin nucleation factors (7,8,41). Additionally, previous studies have demonstrated that DOCK3 interacts with the WAVE complex to activate RAC1, modulate actin dynamics and promote cellular outgrowth (42). We postulated that DOCK3’s function in modulating actin dynamics might influence muscle differentiation and fusion capabilities. We tested whether lentiviral shRNA knockdown of DOCK3 mRNA affected myogenic differentiation capabilities in primary human muscle cells. Healthy human myoblasts were treated with either mock, shRNA scrambled (negative control) or shRNA DOCK3 and assessed for myogenic differentiation capability using myosin heavy chain (MyHC) fluorescence (Fig. 7A). Myoblasts treated with shRNA DOCK3 had significantly impaired myogenic differentiation compared with other control groups as evidenced by decreased amounts of MyHC-positive cells (Fig. 7B). Western blot analysis validated the reduction of DOCK3 protein levels by shRNA DOCK3 mRNA knockdown (Fig. 7C). Moreover, we replicated this experiment using primary myoblasts isolated from our Dock3 KO mice and WT mice and measured their myogenic differentiation capacity. We isolated primary myoblasts from adult WT and Dock3 KO mouse TA muscles and evaluated their ability to fuse upon exposure to reduced serum medium. Dock3 KO myoblasts showed poor myogenic differentiation capacity with an overall reduced amount of myotubes compared with aged-matched WT controls (Fig. 7D and E). Taken together, these studies demonstrate a critical role of DOCK3 protein in myogenic differentiation and myotube formation. As we noted a significant decrease in expression levels of the myogenic fusion peptide myomixer (Mymx-Myomerger-Minion) in Dock3 KO TA muscles (Fig. 7D), we postulated that this decrease in myomixer-myomerger-minion expression may be the cause of the limited myogenic differentiation capacity of the Dock3 KO myoblasts. We overexpressed FLAG-tagged Mymx (henceforth referred to as Mymx OE) or GFP in Dock3 KO myoblasts. The Dock3 KO myoblasts transfected with Mymx-Myomerger-Minion overexpression plasmid demonstrated a slightly increased percentage of multi-nucleated cells as observed by increase in the myogenic fusion index compared with the GFP-control (Supplementary Material, Fig. S4). Moreover, we found no significant difference in myogenic differentiation factors between Mymx OE and GFP Dock3 KO myoblasts, indicating that overexpression of myomixer has minimal effects in improving the myogenic differentiation capability in the Dock3 KO myoblasts in Dock3 KO myoblasts (Supplementary Material, Fig. S4).
Figure 7.
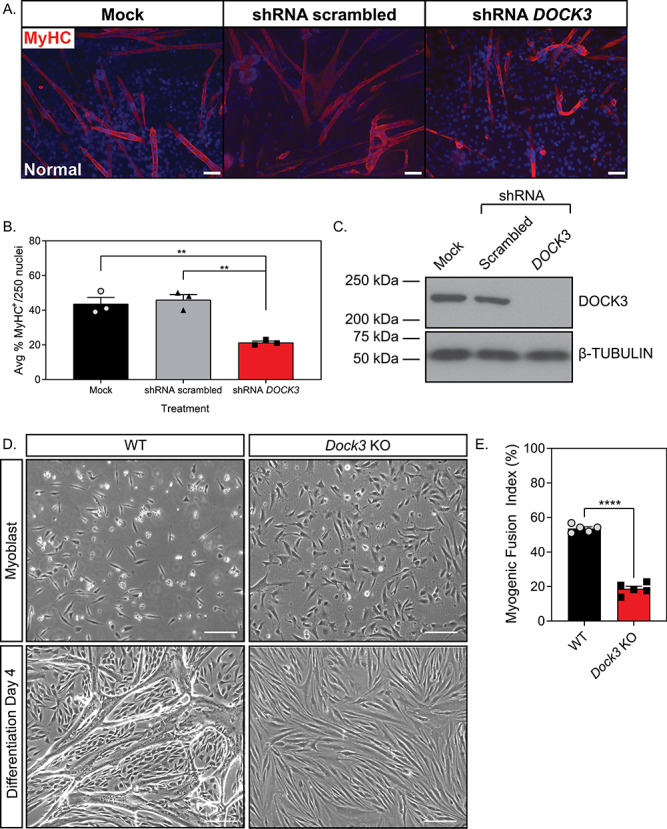
DOCK3 is essential for normal myoblast fusion. (A) MyHC staining reveals that knockdown of DOCK3 protein in normal human primary myotubes decreases myogenic fusion indices. MyHC (MF-20 antibody, DSHB Iowa, red) immunofluorescence of Day 7 differentiated primary myotubes along with DAPI (blue). MOCK, shRNA scrambled and shRNA DOCK3 lentiviral particles were transduced at an MOI of 10. Scale bar = 50 μm. (B) Average myogenic fusion indices as calculated by percentage of nuclei inside of MyHC+ cells out of 250 nuclei as previously described (68). Myotubes with shRNA DOCK3 knockdown have significantly less myogenic fusion than shRNA scrambled control (n = 3 experimental replicates; one-way ANOVA with Tukey’s correction; *P < 0.05). (C) Western blot analysis of whole cell lysates taken from normal human Day 7 differentiated myotubes transduced with mock (control), scrambled (shRNA internal control) or shRNA DOCK3 knockdown inhibitor lentiviral particles showing successful knockdown of DOCK3 protein expression. (D) Representative images of WT and Dock3 KO mouse myoblasts undergoing differentiation. (E) Quantification of myogenic fusion index where Dock3 KO mouse myoblasts has significantly less myogenic fusion than WT mouse myoblasts. Scale bar = 100 μm. (n = 5; student’s t-test, two-tailed; ****P < 0.0001).
Discussion
We have identified DOCK3 as a modifier of normal and dystrophin-deficient skeletal muscle health and function. First, we showed that DOCK3 is strongly elevated in expression levels in dystrophin-deficient mouse skeletal muscles consistent with the DOCK3 expression profile in human patients.8 While complete loss of Dock3 expression in Dock3 KO and Dock3:mdx5cv DKO mice have detrimental effects on skeletal muscles, haploinsufficiency of Dock3+/−:mdx5cv improves myofiber size and skeletal muscle pathology. These findings were further validated in the zebrafish sapje DMD zebrafish. Taken as a whole, these studies demonstrated that the reduction of Dock3 expression in DMD models prevents the severity of dystrophic muscle pathology. These findings potentially represent the discovery of DOCK3 as new therapeutic target for DMD, as previous findings have identified factors that provide protective haploinsufficiency in mdx mice. For example, the haploinsufficiency of the transcription factors Six4 and Six5 in mdx mice similarly resulted in improved muscle grip strength and extended life span when haploinsufficient in mdx mice (43). While the roles of other DOCK proteins in muscle are not well described, it is possible that DOCK3 may have some overlapping and non-overlapping functions with DOCK4 based on similar protein sequence structure and domains. Studies in the Dock4 KO mice revealed an essential role for DOCK4 in social behaviors and that the DOCK4-mediated activation of RAC1 in neurons could be overcome by constitutive RAC1 overexpression (44). Additional studies demonstrating that DOCK3 expression levels are tightly linked to RAC1 activation suggest that RAC1 activation may compensate for the loss of DOCK3 in muscle (45–47). Given our previous findings demonstrating that DOCK3 overexpression activates PTEN/AKT signaling in DMD muscle, the restoration of DOCK3 expression levels to that of WT muscle may restore RAC1 activation and could prevent the activation of cell death pathways that occurs in dystrophic muscles (14,48).
Complete loss of DOCK3 expression in both WT and dystrophin-deficient muscle resulted in worse muscle health, decreased myoblast fusion, glucose intolerance and defective muscle locomotive function. This is consistent with the reporting of clinical muscle hypotonia and muscle weakness in patients with pathogenic DOCK3 variants (11–13). It is also consistent with the essential role of DOCK3 in axonal growth and the formation of protein aggregates leading to neurological deficits (10). Indeed, the compound loss of DOCK3 and the dystrophin protein likely impairs the myofiber membrane integrity and ability of RAC1 to be activated at the myofiber membrane. Thus, resulting in the exacerbated muscle pathologies that we observed in the Dock3−/−:mdx5cv double KO mice. Previous studies have demonstrated an essential role for dystrophin and an intact myofiber membrane to activate RAC1 (49,50). These studies are in compliment with ours and highlight an essential requirement for the dystrophin-associated protein complex as a requisite signaling hub for proper downstream signaling processes in skeletal muscle. The mechanism of action of DOCK3 in myogenesis and its role as a GEF remains of key interest to understand its function in muscle disease. As a GEF, DOCK3 activates RAC1, a Rho-GTPase, which has been shown to be tightly regulated in myogenesis (51). Overexpression of a constitutively active RAC1 in C2C12 mouse skeletal myoblasts inhibits differentiation demonstrating the importance of RAC1 activation status in regulating temporal signals in myogenesis (52). Similar to the glucose impairment, we observed that in the Dock3 KO mice, the muscle Rac1 KO mice show impaired glucose processing (Raun et al., 2018). Interestingly, it has been shown that the overexpression of Def-6 (also called HMGB1), a GEF protein similar to DOCK3, impairs the downregulation of RAC1 activity and results in inhibited myogenic differentiation (53). These studies provide mechanistic insight into the molecular interplay between DOCK3 and RAC1 in dystrophin-deficient muscles and myogenesis.
Additionally, we observed multiple myogenic differentiation factors that were decreased in expression levels in the Dock3 KO muscles via transcriptomic pathway analysis. A key question remains on whether DOCK3 functions to alter the filamentous actin in myoblasts or works through another mechanism to regulate the myogenic differentiation process. Our studies revealed that the overexpression of the myomixer-myomerger-minion peptide in Dock3 KO myoblasts while slightly increasing the myogenic differentiation capability did not fully restore normal myogenic differentiation. It remains likely that DOCK3 has essential interactions with actin and other key molecules that may be the source of the impaired myogenic differentiation observed in the Dock3 KO myoblasts.
Our findings provide evidence that DOCK3 plays a key role in normal muscle fusion and affects dystrophin-deficient muscle pathologies and outcomes. Furthermore, as DOCK3 expression levels are tightly correlated with dystrophic disease progression, DOCK3 may be a potential DMD biomarker that could be used to measure disease severity in muscle. Moreover, our findings also provide strong evidence for DOCK3 as a potential novel therapeutic target for the treatment of DMD. Future experiments aimed at further elucidating the mechanism of DOCK3 in muscle differentiation, signaling and its contribution towards muscle metabolic regulation are warranted.
Materials and Methods
Mice
Dock3 global KO mice (Jax mice stock# 033736) were obtained from the laboratory of Dr David Shubert (Salk Institute) and have been previously described (10). These mice contain a beta-galactosidase cassette integrated in-frame into exon 2 of mouse Dock3 locus resulting in a complete disruption of the gene. WT (C57BL/6 J; stock# 000664) and mdx5cv (stock# 002379) were originally obtained from Jackson Labs (Bar Harbor, ME). All mice were maintained on the C57BL/6 J strain background. All mouse strains were maintained under standard housing and feeding conditions with the University of Alabama at Birmingham Animal Resources Facility under pathogen-free, sterile conditions under the animal protocol number 21393.
Zebrafish morpholino experiments
WT AB and sapje (dmdta222a mutation on the AB strain background) zebrafish were used for all zebrafish morpholino experiments. A dock3 morpholino (5′-GCCTCAGATCAATCAACTCGTTC AT-3′) and a control FITC-labeled (5′-CCTCTTACCTCAGTTACAAT TTATA-3′) non-targeting morpholino (Gene Tools LLC; Philomath, OR) were used for all morpholino injections. Zebrafish morpholinos were injected into one-cell embryos obtained from sapje heterozygote matings at amounts of 0.1, 0.5, 1, 2, 3 and 6 nanograms (ng) in a solution of 1x Danieau buffer, water and phenol red (Sigma Aldrich, St. Louis, MO) as previously described (54). For MyHC zebrafish larvae were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) overnight at 4°C and then following 3x washes in 1xPBS for 5 min were incubated for 1 h at room temperature with F-59 (MyHC; Developmental Studies Hybridoma Bank, Iowa City, IA) in the dark. Larvae were then washed 3x in 1xPBS for five min per wash and then incubated with anti-mouse Alexa Fluor-568 (Invitrogen; Cat# A-11004) for 45 min at room temperature in the dark. After an additional three washes in 1x PBS, the fish were then placed on superfrosted slides (FisherScientific; Waltham, MA) and imaged on a Nikon Eclipse E-1000 microscope. All adult fish were fed a standard diet of Artemia salina (brine shrimp) three times per day under a 14 h on, 10 h off light cycle in 3 L tanks with a density of no more than 20 fish per tank as standard of care guidelines. All zebrafish strains were maintained University of Alabama at Birmingham Animal Resources Aquatics Facility under pathogen-free conditions under the animal protocol number 20320.
Zebrafish muscle birefringence assay
Subsequently following the locomotor assay, birefringence assay was performed on 5 dpf larvae as previously described. Larvae were anesthetized using MS-222 and placed on microscope with polarizing light attachment. In this assay, when the polarized light is shone on the larvae, a white light is refracted back and captured in the camera if the muscle is healthy and has organized fibers. However, if the muscle is broken down and disorganized (worsened muscle phenotype), the polarized light will shine straight through and will appear in the camera as black (absence of white refracted light).
ISH zebrafish experiments
The zebrafish dock3 mRNA (dock3–201 ENSEMBLE transcript; ENSDARG00000063180) was used as a template and an antisense region containing 300 base pairs sequence of the end of dock3 mRNA coding sequence was used for in vitro transcription probe synthesis. In vitro transcription reactions performed using the MegaScript Sp6 (Cat# AM 1330) and MegaScript T7 Transcription (Cat# AM 1334) kits (ThermoFisher Scientific) were used following the manufacturer’s guidelines. Morpholino injected zebrafish embryos were placed in fish water containing 1x PTU (Sigma; Cat# P7629) at 24 h post-fertilization. ISH reactions were performed on 4% paraformaldehyde (Electron Microscopy Sciences; Hatfield, PA; Cat# 15710) zebrafish embryos using the DIG RNA Labeling Kit (MilliporeSigma; Burlington, MA; Cat# 11175025910) following a standardized protocol (55). The DIG-labeled embryos were imaged under the dissection scope (Nikon, SMZ1500; Tokyo, Japan) with EXFO Fluorescence illumination system, X-Cite 120 (Photonic Solutions Inc.; Edinburgh, UK), using a Nikon Eclipse E-1000 microscope attached to a Hamamatsu digital camera, and images were acquired using Openlab software version 3.1.5 (Improvision; PerkinElmer; Waltham, MA).
Human muscle samples
Primary vastus lateralis muscle biopsies from normal and DMD patients were obtained from consented patients under the approved Boston Children’s Hospital protocol 03-12-205 and described elsewhere (14,15). Additional samples were collected under the UAB protocol 300 002 164. All patient samples were de-identified and the secondary confirmation of DMD mutations was performed using MLPA analysis and/or exome sequencing (56).
Mouse activity tracking
Mouse locomotive activity measurements were performed as previously described (57). Twenty-four hours prior to experiment termination and tissue harvest, mice were analyzed for locomotive activity using the Ethovision XT software platform (Noldus; Leesburg, VA) with isolated individual chambers adapted from a previously described protocol (14). Mice were adapted to the room and open-field chambers 1 day prior to activity and were given a 5-min additional adaptation period prior to activity recording. Mouse activity was recorded for 6 min with no external stimulation.
Myofiber and myogenic fusion calculations
The cross-sectional area (CSA) of the skeletal muscle sections was calculated in a manner previously described (14). Briefly, 600 TA myofibers were counted and CSA (μm2) was measured via a series of overlapping H&E microscopy images, and quantified in Fiji software platform (58). Myogenic fusion indices were determined by immunofluorescent staining using the MF-20 (MyHC) as a marker of myogenic differentiation. Fusion of myoblasts was determined by the detection of more than one nuclei within an MyHC+ myotube.
DEXA QMR imaging
To evaluate body composition (fat and lean tissue mass) in vivo, 6-month male WT and Dock3 KO mice (5 mice/genotype) were measured using the EchoMRI™ 3-in-1 composition analyzer (software version 2016, Echo Medical, Houston, TX). Individual fat and lean mass measurements were measured in grams (g) and were analyzed using student’s t-test between WT and Dock3 KO mice.
Glucose tolerance test
Mice were fasted for 16 h prior to afternoon administration of a bolus of D-glucose (MilliporeSigma; Cat# G8270) which was intraperitoneal injected at a concentration of 3 mg/gram of mouse body weight. Blood glucose was measured on a commercially obtained glucometer (Nipro Diagnostics; Ft. Lauderdale, FL) using 10 μL of whole serum from tail bleeds placed on standardized glucose meter test strips.
siRNA knockdown in cardiotoxin-injured muscles
Adult male mdx5cv mice (4–6 months old) were injected in their TA skeletal muscles with cardiotoxin (10 μM) and siRNA pooled oligos (10 μg; GE Healthcare Dharmacon Inc; Lafayette, CO) containing either scrambled siRNAs or siRNAs targeting mouse Dock3 mRNA following a protocol previously established (59). The contralateral TA muscle was used as a sham phosphate-buffered saline (ThermoFisher Scientific; Waltham, MA; Cat#10010049) control. Seven days following injections, mice were euthanized, and their TA skeletal muscles were slow-frozen in TissuePlus O.C.T (FisherScientific; Hampton, NH; Cat#23–730-571), for histological and molecular analysis.
Immunofluorescence and immunohistochemistry
Mouse skeletal muscles were cryo-frozen in a TissuePlus O.C.T (FisherScientific; Hampton, NH; Cat#23–730-571) Isopentane (FisherScientific; Cat# AC397221000) liquid nitrogen bath as unfixed tissues. Mouse hearts were perfusion fixed in 10% neutral buffered formalin (MilliporeSigma) overnight at 4°C, which was performed before embedding the tissue sample into paraffin blocks. Blocks were later cut on a cryostat and 7- to 10-μm-thick sections were placed on SuperFrost Plus Gold slides (ThermoFisher; Cat# FT4981GLPLUS). H&E staining was performed as previously described (15). For immunofluorescent staining, following de-paraffinization, the slides were incubated in eBioscience IHC Antigen Retrieval Solution (ThermoFisher; Cat# 00–4955-58) and washed in 1x PBS three times for 5 min, and then incubated with blocking reagent from the Mouse-on-Mouse (M.O.M) kit (Vector Laboratories; Burlingame, CA; Cat# BMK-2202). Slides were incubated for 1 h at room temperature in primary antibody.
Western blot
Protein lysates were obtained from either pestle-homogenized tissues or cell lysates in M-PER lysis buffer (ThermoFisher; Cat# 78501) supplemented with 1x Complete Mini EDTA-Free protease inhibitor cocktail tablets (Roche Applied Sciences; Cat# 04693159001; Penzburg, Germany). Protein lysates were quantified using a Pierce BCA Protein Assay Kit (ThermoFisher; Cat# 23225). Unless stated otherwise, 50 μg of whole protein lysate was used for all immunoblots, and electrophoretically resolved on 4–20% Novex tris-glycine gels (ThermoFisher: Cat# XPO4205BOX). Protein samples were then transferred to 0.2 μm PVDF membranes (ThermoFisher: Cat# LC2002), blocked in 0.1x TBS-Tween in 5% non-fat milk for 1 h before being incubated in primary antibody overnight at 4°C with gentle rocking. Blots were washed in 0.1% TBS-Tween three times for 5-min intervals before being incubated with secondary antisera (mouse or rabbit) conjugated to horse radish peroxidase (HRP) for 1 h at room temperature with gentle agitation. Membranes were then washed in 0.1% TBS-Tween three times for 5-min intervals before being incubated with secondary antibodies (either mouse or rabbit) conjugated to HRP for 1 h at room temperature with gentle agitation. Following another three washes for 15-min intervals at room temperature, membranes were then treated with RapidStep ECL Reagent (MilliporeSigma; Cat# 345818-100ML) and exposed to X-ray film (Genesee Scientific). Some Western blot images were acquired using a Typhoon Variable Mode Imager (Amersham Pharmacia; Little Chalfont, UK). Some membranes were stripped using Restore Plus Western Blot Stripping Buffer (ThermoScientific; Cat# 46428) and later probed with a different primary antibody.
RNA-sequencing and data analyses
Adult male 6-month-old WT and Dock3 KO mouse TA muscles (n = 3 muscles per genotype cohort) were biopsied, and total RNA was extracted following mechanical homogenization using the GenElute Total RNA Purification Kit (MilliporeSigma; Cat# RNB100-50RXN) following the manufacturer’s guidelines. The total RNA was amplified using the Sure Select Stranded RNA-Seq kit (Agilent Technologies; Santa Clara, CA) using standard protocols. A ribominus kit (ThermoFisher; Cat# K155002) was used to deplete large ribosomal RNAs. The sequencing was done on the Illumina HiSeq2500 in paired-end mode following the manufacturer’s protocols by the UAB Genomics Core Facility.
All biological replicates contained a minimum of 35.7 million reads with an average number of 39.6 million reads across the replicates. The FASTQ files were uploaded to the UAB high-performance computer cluster for bioinformatics analysis with the following custom pipeline built in the Snakemake workflow system (v5.2.2) (60): first, quality and control of the reads were assessed using FastQC, and trimming of the Illumina adapters and bases with quality scores of less than 20 were performed with Trim_Galore! (v0.4.5). Following trimming, the transcripts were quasi-mapped and quantified with Salmon (61) (v0.12.0, with ‘—gencode’ flag for index generation and ‘-l ISR’, ‘—gcBias’ and ‘—validateMappings’ flags for quasi-mapping) to the mm10 mouse transcriptome from Gencode release 21. The average quasi-mapping rate was 70.4% and the logs of reports were summarized and visualized using MultiQC (62) (v1.6). The quantification results were imported into a local RStudio session (R version 3.5.3) and the package ‘tximport’ (63) (v1.10.0) was utilized for gene-level summarization. Differential expression analysis was conducted with DESeq2 (64) package (v1.22.1). Following count normalization, principal component analysis was performed and genes were defined as DEGs if they passed a statistical cutoff containing an adjusted P-value <0.05 (Benjamini–Hochberg false discovery rate method) and if they contained an absolute log2-fold change ≥ 1. Functional annotation enrichment analysis was performed in the NIH Database for Annotation, Visualization and Integrated Discovery (DAVID, v6.8) by separately submitting upregulated and downregulated DEGs. A P-value <0.05 cutoff was applied to identify GO terms.
The FASTQ files of the current study have been uploaded to NCBI’s Gene Expression Omnibus under accession number GSE141621.
Real-time quantitative PCR
Total RNA was extracted using the miRVana (ThermoFisher; Cat# AM1560) kit following the manufacturer’s protocol. One microgram of total RNA was reverse transcribed using the Taqman Reverse Transcription kit following the manufacturer’s protocol (Applied Biosystems; Foster City, CA; Cat# N8080234). Taqman assay probes were all purchased from Applied Biosystems corresponding to each individual gene. Quantitative PCR (qPCR) Taqman reactions were performed using Taqman Universal PCR Master Mix (Applied Biosystems; Cat# 4304437). Samples were run on the Fluidigm Biomark HD platform (Fluidigm Corp.; San Francisco, CA) in 96.96 dynamic array plates. Relative expression values were calculated using the manufacturer’s software and further confirmed using the 2–ΔΔCt method (65).
Muscle myoblast cell cultures
Primary muscle cells were isolated from postnatal 7-day-old WT or Dock3 KO mouse hindlimbs via a pre-plate purification strategy for enrichment of a slow-adhering purification as previously described (66,67). Primary muscle cells were later evaluated for myogenic capacity via immunofluorescent staining of desmin to ensure for a myogenic population greater than 95%. Muscle cells were grown in skeletal muscle growth medium (Promocell; Heidelberg, Germany; Cat# C-23060) supplemented with 1x antibiotic-antimycotic (ThermoFisher Scientific; Cat# 15240062) and 1 ng/10 mL rhFGF (Promega; Madison, WI; Cat# G5071). Cells were seeded at a density of 1.5 × 106 per 10 cm2 plate or 2-chamber slides (Corning Inc.; Corning, NY) coated with 1% rat tail collagen (Millipore Sigma; Cat# 08–115). When cells reached 90% confluency, they were switched to differentiation medium, which is composed of DMEM GlutaMAX™, 1% of Antibiotic-Anti-mycotic (Invitrogen: Cat# 1520) and Promocell Human Skeletal Muscle Differentiation Media (Cat# C-23061). Myogenic fusion assays and immunofluorescent labeling were performed as previously described (68).
Nucleofection of myomixer fusion protein in mouse Dock3 KO primary myoblasts
Myomixer-FLAG was packaged in the pLVX-TetON (Takara Bio; Mountain View, CA) plasmid and have been described in previous literature (69). The plasmid was subcloned into pIRES-1a-hrGFP (Stratagene; San Diego, CA) plasmid. Nucleofection of Dock3 KO primary myoblasts in culture was done according to the optimized protocol for skeletal muscle myoblasts following the manufacturer’s protocol (Lonza; Basel, Switzerland). Briefly, myoblasts were grown in their respective culture media in 100 cm2 plates until they reached 80% confluency, corresponding to approximately 1.5 x 106 cells. The medium was removed from the plate and cells were trypsinized with 1 mL of trypsin–EDTA (Invitrogen) to detach them completely. Subsequently, 4 mL of medium was added to the plate and then the cells and the culture medium were centrifuged at 1000× g for 5 min at room temperature. The supernatant was then removed, and cell pellets were resuspended in 100 μL of Nucleofector solution (Lonza) in which either 2 ug of myomixer plasmid or 2 ug pmaxGFP plasmid were added to respective cohorts. The cells were analyzed 24–48 h after the nucleofection by fluorescence microscopy with an inverted Leica microscope. After 48 h post-nucleofection, proliferation media was removed and washed with 1× PBS (Dulbecco) which was aspirated and replaced with differentiation media.
Myogenic fusion assays
Myoblasts from Dock3 global KO mice were nucleofected with myomixer-FLAG and its GFP control in four-well chamber slides at 100 000 cells/per well, separately (as described above). To assess myogenic fusion, cells were fixed at day 4 of differentiation with 4% paraformaldehyde for 20 min at 4°C followed by permeabilization with 0.1% triton™X-100 (Sigma Aldrich) for 30 min at room temperature and blocking in 1xPBS with 1% fetal bovine serum (Gibco). Cells were incubated with primary antibody for MyHC (MF-20—1:100, Developmental Studies Hybridoma Bank) overnight at 4°C, followed by three washes with 1x PBS and incubation with secondary antibody Alexa Fluor 555 (Invitrogen). Slides were washed and mounted with Vectashield® Mounting Media (Vector) with DAPI. Images were obtained in Zeiss 710 confocal system at 20X. The fusion index was calculated as follows: (MF20-stained myocytes containing ≥ 2 nuclei/total number of nuclei) × 100, as previously described (70).
CLUSTAL Omega alignment
CLUSTAL Omega alignments using freely available software (https://www.ebi.ac.uk/Tools/msa/clustalo/) as described (71). Accession numbers for protein sequences: zebrafish (Danio rerio, XP_009294754.1), mouse (Mus musculus, NP_700462.2) and human (Homo sapiens, NP_004938.1).
Cardiac functional measurements: transthoracic echocardiography
The Vevo 3100 (VisualSonics Inc, Canada) imaging system was used for echocardiography to evaluate individual parameters of cardiac structure and function in vivo. Six-month male WT and Dock3 KO mice (5 mice/genotype) were anesthetized using 1–2% isofluorane continuously suppled from surgivet vaporizer, and both long- and short-axis high-resolution images were acquired for heart function measurements and analysis. Each individual parametric measurement was compared between WT and Dock3 KO mice using a student’s t-test (Supplementary Material, Fig. S3).
Statistical analyses
All graphs were represented as mean ± SEM. Unless otherwise mentioned, a two-tailed student’s t-test was performed for all single comparisons and either a one-way or two-way analysis of variance (ANOVA) with Tukey correction was performed for all multiple comparisons. GraphPad Prism version 8 software (GraphPad Software; San Diego, CA) was used for all statistical analyses. An a priori hypothesis of *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 was used for all data analysis.
Supplementary Material
Acknowledgements
We would like to thank Dr David Schubert (Salk Institute) for the generous gift of the Dock3 KO mice. We would like to thank Dr Michael Crowley and the staff of the UAB Genomics Core Facility for RNA-sequencing processing and initial analyses. We would like to thank Eddie Bradley for the mouse cardiovascular function analyses. We would like to thank Drs Timothy Nagy and Maria Johnson at the UAB Small Animal Phenotyping Core for DEXA measurements. We would like to thank Drs David Schneider (UAB), Glenn Rowe (UAB) and Emanuela Gussoni (Boston Children’s Hospital) for critical reading of our manuscript. We would like to thank Dr Susan Farmer and her staff for care and maintenance of our animals. Lastly, we would like to thank the patients and their families who contributed samples that were used in this study and helped to make this research possible.
Conflict of Interest statement. All authors declare no conflicts of interest.
Contributor Information
Andrea L Reid, Division of Neurology, Department of Pediatrics, The University of Alabama at Birmingham and Children’s of Alabama, Birmingham, AL 35294, USA.
Yimin Wang, Division of Neurology, Department of Pediatrics, The University of Alabama at Birmingham and Children’s of Alabama, Birmingham, AL 35294, USA.
Adrienne Samani, Division of Neurology, Department of Pediatrics, The University of Alabama at Birmingham and Children’s of Alabama, Birmingham, AL 35294, USA.
Rylie M Hightower, Division of Neurology, Department of Pediatrics, The University of Alabama at Birmingham and Children’s of Alabama, Birmingham, AL 35294, USA; UAB Center for Exercise Medicine, Birmingham, AL 35294, USA.
Michael A Lopez, Division of Neurology, Department of Pediatrics, The University of Alabama at Birmingham and Children’s of Alabama, Birmingham, AL 35294, USA; UAB Center for Exercise Medicine, Birmingham, AL 35294, USA.
Shawn R Gilbert, Department of Orthopedic Surgery, The University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Lara Ianov, Civitan International Research Center, The University of Alabama at Birmingham, Birmingham, AL 35294, USA.
David K Crossman, Department of Genetics, The University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Louis J Dell’Italia, Birmingham Veteran Affairs Medical Center, Birmingham, AL 35233, USA; Division of Cardiovascular Disease, Department of Medicine, The University of Alabama at Birmingham, Birmingham, AL 35294, USA; Department of Cell, Developmental, and Integrative Biology, The University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Douglas P Millay, Division of Molecular Cardiovascular Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA.
Thomas van Groen, Department of Cell, Developmental, and Integrative Biology, The University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Ganesh V Halade, Division of Cardiovascular Sciences, Department of Medicine, University of South Florida, Tampa, FL 33602, USA.
Matthew S Alexander, Division of Neurology, Department of Pediatrics, The University of Alabama at Birmingham and Children’s of Alabama, Birmingham, AL 35294, USA; UAB Center for Exercise Medicine, Birmingham, AL 35294, USA; Civitan International Research Center, The University of Alabama at Birmingham, Birmingham, AL 35294, USA; Department of Genetics, The University of Alabama at Birmingham, Birmingham, AL 35294, USA.
Funding
R.M.H. is a member of the NIH-supported RoadMap Scholars Program grant and is also supported by a training fellowship sponsored by the University of Alabama at Birmingham Center for Exercise Medicine grant number T32HD071866. R.M.H. is a funded by an NIH National Institute of Neurological Disorders and Stroke (NINDS) F99/K00 grant (F99NS118718). The UAB Small Animal Phenotyping Core supported by the NIH Nutrition & Obesity Research Center (P30DK056336) and the Mouse Cardiovascular Core Vevo 3100 Mouse Ultrasound Facility for this project. Research reported in this publication was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH and HHS of the National Institutes of Health under award number R01HD095897 awarded to M.S.A. M.S.A. is also a co-investigator on an NIH NIAMS award R21AR074006. M.S.A. is funded by a Muscular Dystrophy Association grant (MDA41854). UAB Small Animal Phenotyping Core supported by the NIH Nutrition & Obesity Research Center P30DK056336, Diabetes Research Center P30DK079626 and the UAB Nathan Shock Center P30AG050886A. T.V.G. is funded by NIH P30NS47466. L.J. D. is funded by NIH grant P01HL051952 and the Department of Veterans Grant 1CX000993-01. G.V.H. is funded by NIH R01 grants HL132989 and HL144788. D.P.M. is funded by NIH grants R01 grants AR068286 and AG059605.
References
- 1. Ballarino M., Morlando M., Fatica A. and Bozzoni I. (2016) Non-coding RNAs in muscle differentiation and musculoskeletal disease. J. Clin. Invest., 126, 2021–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cacchiarelli D., Incitti T., Martone J., Cesana M., Cazzella V., Santini T., Sthandier O. and Bozzoni I. (2011) miR-31 modulates dystrophin expression: new implications for Duchenne muscular dystrophy therapy. EMBO Rep., 12, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waldrop M.A., Gumienny F., El Husayni S., Frank D.E., Weiss R.B. and Flanigan K.M. (2018) Low-level dystrophin expression attenuating the dystrophinopathy phenotype. Neuromuscul Disord, 28, 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peter A.K. and Crosbie R.H. (2006) Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp. Cell Res., 312, 2580–2591. [DOI] [PubMed] [Google Scholar]
- 5. Dumont N.A., Wang Y.X., Maltzahn J., Pasut A., Bentzinger C.F., Brun C.E. and Rudnicki M.A. (2015) Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med., 21, 1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Laurin M. and Côté J.-F. (2014) Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev., 28, 533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moore C.A., Parkin C.A., Bidet Y. and Ingham P.W. (2007) A role for the myoblast city homologues Dock1 and Dock5 and the adaptor proteins Crk and Crk-like in zebrafish myoblast fusion. Development, 134, 3145–3153. [DOI] [PubMed] [Google Scholar]
- 8. Laurin M., Fradet N., Blangy A., Hall A., Vuori K. and Côté J.-F. (2008) The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo. Proc. Natl. Acad. Sci., 105, 15446–15451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Makihara S., Morin S., Ferent J., Côté J.-F., Yam P.T. and Charron F. (2018) Polarized Dock activity drives Shh-mediated axon guidance. Dev. Cell, 46, 410–425.e417. [DOI] [PubMed] [Google Scholar]
- 10. Chen Q., Peto C.A., Shelton G.D., Mizisin A., Sawchenko P.E. and Schubert D. (2009) Loss of modifier of cell adhesion reveals a pathway leading to axonal degeneration. J. Neurosci., 29, 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Helbig K.L., Mroske C., Moorthy D., Sajan S.A. and Velinov M. (2017) Biallelic loss-of-function variants in DOCK3 cause muscle hypotonia, ataxia, and intellectual disability. Clin. Genet., 92, 430–433. doi: 10.1111/cge.12995. [DOI] [PubMed] [Google Scholar]
- 12. Iwata-Otsubo A., Ritter A.L., Weckselbatt B., Ryan N.R., Burgess D., Conlin L.K. and Izumi K. (2018) DOCK3-related neurodevelopmental syndrome: Biallelic intragenic deletion of DOCK3 in a boy with developmental delay and hypotonia. Am. J. Med. Genet. A, 176, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wiltrout K., Ferrer A., Laar I., Namekata K., Harada T., Klee E.W., Zimmerman M.T., Cousin M.A., Kempainen J.L., Babovic-Vuksanovic D. et al. (2019) Variants in DOCK3 cause developmental delay and hypotonia. Eur. J. Hum. Genet., 27, 1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alexander M.S., Casar J.C., Motohashi N., Vieira N.M., Eisenberg I., Marshall J.L., Gasperini M.J., Lek A., Myers J.A., Estrella E.A. et al. (2014) MicroRNA-486–dependent modulation of DOCK3/PTEN/AKT signaling pathways improves muscular dystrophy–associated symptoms. J. Clin. Invest., 124, 2651–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alexander M., Casar J.C., Motohashi N., Myers J., Eisenberg I., Gonzalez R., Estrella E., Kang P., Kawahara G. and Kunkel L. (2011) Regulation of DMD pathology by an ankyrin-encoded miRNA. Skelet. Muscle, 1, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quinn M.E., Goh Q., Kurosaka M., Gamage D.G., Petrany M.J., Prasad V. and Millay D.P. (2017) Myomerger induces fusion of non-fusogenic cells and is required for skeletal muscle development. Nat. Commun., 8, 15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bi P., McAnally J.R., Shelton J.M., Sánchez-Ortiz E., Bassel-Duby R. and Olson E.N. (2018) Fusogenic micropeptide Myomixer is essential for satellite cell fusion and muscle regeneration. Proc. Natl. Acad. Sci., 115, 3864–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Q., Vashisht A.A., O’Rourke J., Corbel S.Y., Moran R., Romero A., Miraglia L., Zhang J., Durrant E., Schmedt C. et al. (2017) The microprotein minion controls cell fusion and muscle formation. Nat. Commun., 8, 15664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Im W.B., Phelps S.F., Copen E.H., Adams E.G., Slightom J.L. and Chamberlain J.S. (1996) Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet., 5, 1149–1153. [DOI] [PubMed] [Google Scholar]
- 20. Beastrom N., Lu H., Macke A., Canan B.D., Johnson E.K., Penton C.M., Kaspar B.K., Rodino-Klapac L.R., Zhou L., Janssen P.M.L. et al. (2011) mdx5cv mice manifest more severe muscle dysfunction and diaphragm force deficits than do mdx mice. Am. J. Pathol., 179, 2464–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berger J., Berger S., Hall T.E., Lieschke G.J. and Currie P.D. (2010) Dystrophin-deficient zebrafish feature aspects of the Duchenne muscular dystrophy pathology. Neuromuscul. Disord., 20, 826–832. [DOI] [PubMed] [Google Scholar]
- 22. Li M., Hromowyk K.J., Amacher S.L. and Currie P.D. (2017) The Zebrafish: Disease Models and Chemical Screens In Detrich H.W., Westerfield M. and Zon L.I. (eds), Methods in Cell Biology. Academic Press, Amersterdam, Netherlands, Vol. 138, pp. 347–380. [DOI] [PubMed] [Google Scholar]
- 23. Berger J. and Currie P.D. (2012) Zebrafish models flex their muscles to shed light on muscular dystrophies. Dis. Models Mech., 5, 726–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bassett D. and Currie P.D. (2004) Identification of a zebrafish model of muscular dystrophy. Clin. Exp. Pharmacol. Physiol., 31, 537–540. [DOI] [PubMed] [Google Scholar]
- 25. Bassett D.I., Bryson-Richardson R.J., Daggett D.F., Gautier P., Keenan D.G. and Currie P.D. (2003) Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development, 130, 5851–5860. [DOI] [PubMed] [Google Scholar]
- 26. Jin H., Tan S., Hermanowski J., Böhm S., Pacheco S., McCauley J.M., Greener M.J., Hinits Y., Hughes S.M., Sharpe P.T. et al. (2007) The dystrotelin, dystrophin and dystrobrevin superfamily: new paralogues and old isoforms. BMC Genomics, 8, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kawahara G., Karpf J.A., Myers J.A., Alexander M.S., Guyon J.R. and Kunkel L.M. (2011) Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci., 108, 5331–5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guyon J.R., Mosley A.N., Zhou Y., O'Brien K.F., Sheng X., Chiang K., Davidson A.J., Volinski J.M., Zon L.I. and Kunkel L.M. (2003) The dystrophin associated protein complex in zebrafish. Hum. Mol. Genet., 12, 601–615. [PubMed] [Google Scholar]
- 29. Berger J., Sztal T. and Currie P.D. (2012) Quantification of birefringence readily measures the level of muscle damage in zebrafish. Biochem. Biophys. Res. Commun., 423, 785–788. [DOI] [PubMed] [Google Scholar]
- 30. Blau H.M., Webster C. and Pavlath G.K. (1983) Defective myoblasts identified in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci., 80, 4856–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blau H.M., Webster C., Chiu C.-P., Guttman S. and Chandler F. (1983) Differentiation properties of pure populations of human dystrophic muscle cells. Exp. Cell Res., 144, 495–503. [DOI] [PubMed] [Google Scholar]
- 32. Chrousos G.P. (2016) Part 5 - Diabetes Mellitus In Jameson J.L., Groot L.J., Kretser D.M., Giudice L.C., Grossman A.B., Melmed S., Potts J.T. and Weir G.C. (eds), Endocrinology: Adult and Pediatric (Seventh Edition). W.B. Saunders, Philadelphia, pp. 1727–1740.e1725. [Google Scholar]
- 33. Dunn J.F. and Zaim-Wadghiri Y. (1999) Quantitative magnetic resonance imaging of the mdx mouse model of Duchenne muscular dystrophy. Muscle Nerve, 22, 1367–1371. [DOI] [PubMed] [Google Scholar]
- 34. Pratt S.J.P., Xu S., Mullins R.J. and Lovering R.M. (2013) Temporal changes in magnetic resonance imaging in the mdx mouse. BMC Res. Notes, 6, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bostock E.L., Edwards B.T., Jacques M.F., Pogson J.T.S., Reeves N.D., Onambele-Pearson G.L. and Morse C.I. (2018) Impaired glucose tolerance in adults with Duchenne and Becker muscular dystrophy. Nutrients, 10, 1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brazeau G.A., Mathew M. and Entrikin R.K. (1992) Serum and organ indices of the mdx dystrophic mouse. Res. Commun. Chem. Pathol. Pharmacol., 77, 179–189. [PubMed] [Google Scholar]
- 37. Stapleton D.I., Lau X., Flores M., Trieu J., Gehrig S.M., Chee A., Naim T., Lynch G.S. and Koopman R. (2014) Dysfunctional muscle and liver glycogen metabolism in mdx dystrophic mice. PLoS One, 9, e91514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ahmad N., Welch I., Grange R., Hadway J., Dhanvantari S., Hill D., Lee T.-Y. and Hoffman L.M. (2011) Use of imaging biomarkers to assess perfusion and glucose metabolism in the skeletal muscle of dystrophic mice. BMC Musculoskelet. Disord., 12, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mokhtarian A. and Even P.C. (1996) Effect of intraperitoneal injection of glucose on glucose oxidation and energy expenditure in the mdx mouse model of Duchenne muscular dystrophy. Pflugers Arch., 432, 379. [DOI] [PubMed] [Google Scholar]
- 40. Even P.C., Decrouy A. and Chinet A. (1994) Defective regulation of energy metabolism in mdx mouse skeletal muscles. Biochem. J., 304, 649–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaipa B.R., Shao H., Schäfer G., Trinkewitz T., Groth V., Liu J., Beck L., Bogdan S., Abmayr S.M. and Önel S.-F. (2013) Dock mediates scar- and WASp-dependent actin polymerization through interaction with cell adhesion molecules in founder cells and fusion-competent myoblasts. J. Cell Sci., 126, 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Namekata K., Harada C., Taya C., Guo X., Kimura H., Parada L.F. and Harada T. (2010) Dock3 induces axonal outgrowth by stimulating membrane recruitment of the WAVE complex. Proc. Natl. Acad. Sci., 107, 7586–7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yajima H. and Kawakami K. (2016) Low Six4 and Six5 gene dosage improves dystrophic phenotype and prolongs life span of mdx mice. Develop. Growth Differ., 58, 546–561. [DOI] [PubMed] [Google Scholar]
- 44. Guo D., Peng Y., Wang L., Sun X., Wang X., Liang C., Yang X., Li S., Xu J., Ye W.-C. et al. (2019) Autism-like social deficit generated by Dock4 deficiency is rescued by restoration of Rac1 activity and NMDA receptor function. Mol. Psychiatry. doi: 10.1038/s41380-019-0472-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Namekata K., Enokido Y., Iwasawa K. and Kimura H. (2004) MOCA induces membrane spreading by activating Rac1. J. Biol. Chem., 279, 14331–14337. [DOI] [PubMed] [Google Scholar]
- 46. Namekata K., Harada C., Guo X., Kimura A., Kittaka D., Watanabe H. and Harada T. (2012) Dock3 stimulates axonal outgrowth via GSK-3β-mediated microtubule assembly. J. Neurosci., 32, 264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang W.-H., Lan H.-Y., Huang C.-H., Tai S.-K., Tzeng C.-H., Kao S.-Y., Wu K.-J., Hung M.-C. and Yang M.-H. (2012) RAC1 activation mediates Twist1-induced cancer cell migration. Nat. Cell Biol., 14, 366–374. [DOI] [PubMed] [Google Scholar]
- 48. Vieira N.M., Spinazzola J.M., Alexander M.S., Moreira Y.B., Kawahara G., Gibbs D.E., Mead L.C., Verjovski-Almeida S., Zatz M. and Kunkel L.M. (2017) Repression of phosphatidylinositol transfer protein α ameliorates the pathology of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci., 114, 6080–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Oak S.A., Zhou Y.W. and Jarrett H.W. (2003) Skeletal muscle signaling pathway through the dystrophin glycoprotein complex and Rac1. J. Biol. Chem., 278, 39287–39295. [DOI] [PubMed] [Google Scholar]
- 50. Chockalingam P.S., Cholera R., Oak S.A., Zheng Y., Jarrett H.W. and Thomason D.B. (2002) Dystrophin-glycoprotein complex and Ras and rho GTPase signaling are altered in muscle atrophy. Am. J. Physiol. Cell Physiol., 283, C500–C511. [DOI] [PubMed] [Google Scholar]
- 51. Heller H., Gredinger E. and Bengal E. (2001) Rac1 inhibits myogenic differentiation by preventing the complete withdrawal of myoblasts from the cell cycle. J. Biol. Chem., 276, 37307–37316. [DOI] [PubMed] [Google Scholar]
- 52. Bai Y., Guo D., Sun X., Tang G., Liao T., Peng Y., Xu J. and Shi L. (2018) Balanced Rac1 activity controls formation and maintenance of neuromuscular acetylcholine receptor clusters. J. Cell Sci., 131, jcs215251. [DOI] [PubMed] [Google Scholar]
- 53. Samson T., Will C., Knoblauch A., Sharek L., Mark K., Burridge K. and Wixler V. (2007) Def-6, a guanine nucleotide exchange factor for Rac1, interacts with the skeletal muscle integrin chain α7A and influences myoblast differentiation. J. Biol. Chem., 282, 15730–15742. [DOI] [PubMed] [Google Scholar]
- 54. Vieira N.M., Elvers I., Alexander M.S., Moreira Y.B., Eran A., Gomes J.P., Marshall J.L., Karlsson E.K., Verjovski-Almeida S., Lindblad-Toh K. et al. (2015) Jagged 1 rescues the Duchenne muscular dystrophy phenotype. Cell, 163, 1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thisse C. and Thisse B. (2008) High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protocols, 3, 59–69. [DOI] [PubMed] [Google Scholar]
- 56. Wildforster V. and Dekomien G. (2009) Detecting copy number variations in autosomal recessive limb-girdle muscular dystrophies using a multiplex ligation-dependent probe amplification (MLPA) assay. Mol. Cell. Probes, 23, 55–59. [DOI] [PubMed] [Google Scholar]
- 57. Hightower R.M., Reid A.L., Gibbs D.E., Wang Y., Widrick J.J., Kunkel L.M., Kastenschmidt J.M., Villalta S.A., Groen T. and Chang H. (2020) The SINE compound KPT-350 blocks dystrophic pathologies in DMD zebrafish and mice. Mol. Ther., 28, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ahrens H.E., Henze H., Schüler S.C., Schmidt M., Hüttner S.S. and Maltzahn J. (2019) Analyzing satellite cell function during skeletal muscle regeneration by cardiotoxin injury and injection of self-delivering siRNA in vivo. J. Vis. Exp. doi: 10.3791/6019. [DOI] [PubMed] [Google Scholar]
- 60. Koster J. and Rahmann S. (2012) Snakemake--a scalable bioinformatics workflow engine. Bioinformatics, 28, 2520–2522. [DOI] [PubMed] [Google Scholar]
- 61. Patro R., Duggal G., Love M.I., Irizarry R.A. and Kingsford C. (2017) Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods, 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ewels P., Magnusson M., Lundin S. and Kaller M. (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Soneson C., Love M.I. and Robinson M.D. (2015) Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res, 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Love M.I., Huber W. and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Livak K.J. and Schmittgen T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-[Delta][Delta]CT method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 66. Cheung T.H., Quach N.L., Charville G.W., Liu L., Park L., Edalati A., Yoo B., Hoang P. and Rando T.A. (2012) Maintenance of muscle stem-cell quiescence by microRNA-489. Nature, 482, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gharaibeh B., Lu A., Tebbets J., Zheng B., Feduska J., Crisan M., Peault B., Cummins J. and Huard J. (2008) Isolation of a slowly adhering cell fraction containing stem cells from murine skeletal muscle by the preplate technique. Nat. Protocols, 3, 1501–1509. [DOI] [PubMed] [Google Scholar]
- 68. Alexander M.S., Kawahara G., Motohashi N., Casar J.C., Eisenberg I., Myers J.A., Gasperini M.J., Estrella E.A., Kho A.T., Mitsuhashi S. et al. (2013) MicroRNA-199a is induced in dystrophic muscle and affects WNT signaling, cell proliferation, and myogenic differentiation. Cell Death Differ., 20, 1194–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Leikina E., Gamage D.G., Prasad V., Goykhberg J., Crowe M., Diao J., Kozlov M.M., Chernomordik L.V. and Millay D.P. (2018) Myomaker and Myomerger work independently to control distinct steps of membrane remodeling during myoblast fusion. Dev. Cell, 46, 767–780.e767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vadivelu S.K., Kurzbauer R., Dieplinger B., Zweyer M., Schafer R., Wernig A., Vietor I. and Huber L.A. (2004) Muscle regeneration and myogenic differentiation defects in mice lacking TIS7. Mol. Cell. Biol., 24, 3514–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sievers F. and Higgins D.G. (2018) Clustal omega for making accurate alignments of many protein sequences. Protein Sci., 27, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.