

Abstract
The deamination of adenosine to inosine at the wobble position of tRNA is an essential post-transcriptional RNA modification required for wobble decoding in bacteria and eukaryotes. In humans, the wobble inosine modification is catalyzed by the heterodimeric ADAT2/3 complex. Here, we describe novel pathogenic ADAT3 variants impairing adenosine deaminase activity through a distinct mechanism that can be corrected through expression of the heterodimeric ADAT2 subunit. The variants were identified in a family in which all three siblings exhibit intellectual disability linked to biallelic variants in the ADAT3 locus. The biallelic ADAT3 variants result in a missense variant converting alanine to valine at a conserved residue or the introduction of a premature stop codon in the deaminase domain. Fibroblast cells derived from two ID-affected individuals exhibit a reduction in tRNA wobble inosine levels and severely diminished adenosine tRNA deaminase activity. Notably, the ADAT3 variants exhibit impaired interaction with the ADAT2 subunit and alterations in ADAT2-dependent nuclear localization. Based upon these findings, we find that tRNA adenosine deaminase activity and wobble inosine modification can be rescued in patient cells by overexpression of the ADAT2 catalytic subunit. These results uncover a key role for the inactive ADAT3 deaminase domain in proper assembly with ADAT2 and demonstrate that ADAT2/3 nuclear import is required for maintaining proper levels of the wobble inosine modification in tRNA.
Keywords: tRNA modification, inosine, ADAT3, adenosine deaminase, intellectual disability
INTRODUCTION
The human Adenosine Deaminase Acting on tRNA 3 (ADAT3) gene encodes a tRNA modification protein that has been linked to an autosomal-recessive form of ID (Alazami et al. 2013; El-Hattab et al. 2016; Salehi Chaleshtori et al. 2018; Sharkia et al. 2019; Thomas et al. 2019). Human ADAT3 forms an essential enzyme complex with its heterodimeric partner ADAT2 to catalyze the deamination of adenosine to inosine at the wobble position on a subset of tRNAs (Torres et al. 2015; Ramos et al. 2019). The presence of inosine at the wobble position is conserved in bacteria and eukaryotes (Gerber and Keller 1999; Wolf et al. 2002; Tsutsumi et al. 2007; Ragone et al. 2011; Zhou et al. 2014). The wobble inosine modification plays an essential function in organisms by expanding the decoding capacity of a tRNA thereby allowing a single wobble inosine-containing tRNA to decode -C, -A, and -U ending codons (Unger and Takemura 1973).
In E. coli, only tRNA-Arg-ACG is modified at the wobble position with inosine by the homodimeric TadA complex (Wolf et al. 2002). In contrast, eukaryotes contain multiple tRNA isoacceptors that are deaminated by the ADAT2/3 complex to yield inosine, including eight tRNA isoacceptors in human cells (Torres et al. 2015). This expansion of wobble inosine-containing tRNAs in eukaryotes is hypothesized to have occurred with the increased usage of specific codons in eukarya (Rafels-Ybern et al. 2017, 2019). In all cases, the inosine modification is essential for life in order to decode specific -C and -A ending codons as the genome does not encode a cognate tRNA to decode every codon (Rafels-Ybern et al. 2019).
A single base pair variant in the human ADAT3 gene is the most common cause of autosomal recessive ID in Saudi Arabia, with a carrier frequency of 1% (MIM #615302) (Alazami et al. 2013; El-Hattab et al. 2016; Sharkia et al. 2019). The ID-associated ADAT3 variant leads to a missense mutation altering valine at position 144 of ADAT3 to methionine (NM_138422.3, c.430 G > A, p.V144M). We have previously shown that individuals who are homozygous for the ADAT3–V144M variant exhibit a severe decrease in inosine modification on tRNAs (Ramos et al. 2019). Additionally, the ADAT3–V144M variant can still associate with ADAT2 but forms an adenosine deaminase complex that is greatly impaired in enzymatic activity.
Besides the ADAT3 c.430 G > A (p.V144M) variant, only two other ADAT3 variants have been reported in the human population that are associated with intellectual disability. These reports include an Iranian 6-yr-old girl homozygous for the ADAT3 c.99_106dupGAGCCCGG, p.(Glu36Glyfs*44) (Salehi Chaleshtori et al. 2018) and two siblings of European descent carrying the compound heterozygous variants ADAT3 c.587C > T, (p.Ala196Val) and c.586_587delinsTT, (p.Ala196Leu) (Thomas et al. 2019). However, the molecular and cellular effects of these ADAT3 variants have not been characterized to any extent.
Here, we identify two novel compound heterozygous variants in ADAT3 in three siblings presenting with intellectual disability, absent speech, seizures, and strabismus. Similar to patients with the ADAT3–V144M variant, we find that these children exhibit severe deficiency in wobble inosine modification. Notably, we identify a unique form of ADAT3 etiology in which ADAT2–ADAT3 interaction is perturbed leading to alterations in enzymatic activity and subcellular localization. Strikingly, we find that overexpression of the ADAT2 catalytic subunit can recover tRNA adenosine deaminase activity and wobble inosine modification in patient cells. Altogether, these results uncover a distinct form of ADAT3-associated ID that can be rescued through expression of the heterodimeric ADAT2 partner protein, thereby presenting a possible avenue for correcting diminished wobble inosine modification in patient cells.
RESULTS
Identification of compound variants in ADAT3 linked to neurodevelopmental disorder
Through clinical referral for individuals with neurodevelopmental disorders, we identified a family consisting of nonconsanguineous parents and three children, one boy and two girls of ages 13, 9, and 4, respectively (Fig. 1A,B). Exome sequencing performed on the children (II:1, II:2, and II:3) revealed two novel variants of uncertain significance in the Adenosine Deaminase Acting on tRNA 3 (ADAT3) gene (Fig. 1C). Follow-up Sanger sequencing validated the presence of the compound heterozygous variants in all three children (Supplemental Fig. 1). The children were compound heterozygous for a paternally inherited c.587C > T p.[A196V] variant and a maternally inherited c.820C > T p.[Q274*] variant (Fig. 1C, ADAT3 [NM_138422.3]). No individuals have previously been reported to contain both variants. Both the single amino acid substitution and the frameshift variant occur at highly conserved residues in the conserved deaminase domain of ADAT3 (Fig. 1D; Supplemental Fig. 2A). The c.587C > T p.[A196V] missense variant is predicted to have a deleterious effect by multiple computational tools (SIFT, PolyPhen-2, MutationTaster), and there are seven heterozygous carriers in the gnomAD database (v2.1.1; allele frequency: 8.976 × 10−5). The truncating variant, c.820C > T p.[Q274*], has been observed in two heterozygous carriers in gnomAD (allele frequency: 8.933 × 10−6). RT-PCR of ADAT3 mRNA from blood samples revealed expression of both allelic variants in the two older siblings while the third child has not been tested (Supplemental Fig. 2B).
FIGURE 1.

Biallelic heterozygous variants in ADAT3 linked to intellectual disability. (A) Front and side views of each child at ages 12.5, 8.5, and 3.5 yr, respectively. (B) Pedigree of affected family. All three children have intellectual disability. (C) Whole exome sequencing identified two heterozygous variants of ADAT3 in the children. The father is a heterozygous carrier of the c.587C > T variant, the mother is a heterozygous carrier of the c.820C > T variant. (D) Location of changes in the ADAT3 protein caused by biallelic variants identified here or a previously reported founder variant in ADAT3. The cytidine and deoxycytidylate deaminase zinc-binding region is shown in light blue and the zinc chelating residues in dark blue. (E) Immunoblot of the indicated proteins from fibroblast cellular lysates obtained from the parents (I:1, I:2) and children (II:1, II:2) of the ID-affected family shown in Figure 1. (F) Quantification of ADAT3 and ADAT2 levels after normalization to actin. N = 3. Ordinary One-Way Anova—ADAT3/ACTIN: I:2 ns = 0.06, II:1* = 0.04, II:2 * = 0.01. Ordinary One-Way Anova—ADAT2/ACTIN: I:2 ns = 0.11, II:1** = 0.009, II:2 ** = 0.002.
All three siblings present with severe ID, absent speech, absence seizures (onset at 3 yr of age) and sleep disturbances. Moreover, the siblings exhibit short stature, intermittent strabismus and deeply set eyes. The ear lobes are short and superior helix squared. They also have a notched alae nasea, low hanging columella, a short and deep philtrum, prominent teeth and tapering fingers (Fig. 1A). Comparison to previously characterized individuals with ID linked to ADAT3 revealed a common set of features that included both cognitive and dysmorphic characteristics (Supplemental Table 1; Supplemental Information).
To determine the impact of the compound ADAT3 variants, skin fibroblasts were harvested from both parents and the two eldest children. Samples were not collected from the third child due to the young age. Based upon immunoblotting of whole-cell lysates from skin fibroblasts, the children (II:1 and II:2; A196V/Q274*) exhibited a slight, but significant decrease in ADAT3 protein levels compared to the father (I:1-WT/A196V) (Fig. 1E,F, ADAT3). Since we did not observe a truncated ADAT3 protein product in the mother or children, the decrease in ADAT3 protein levels is consistent with reduced levels of full-length ADAT3 caused by the ADAT3–Q274* allele. A decrease in protein stability of the ADAT3–A196V protein and/or global protein synthesis could also contribute to the reduced ADAT3 levels in the children. Interestingly, we also detected a slight decrease in ADAT2 protein levels in the children (Fig. 1E,F, ADAT2). The decrease in ADAT2 levels suggests that ADAT2 stability could be linked to ADAT3 protein levels or a perturbation in cellular protein translation.
Individuals harboring the compound ADAT3 variants exhibit decreased wobble inosine modification in tRNA
We next investigated whether the ADAT3 variants impacted inosine modification status on endogenous tRNA substrates. The levels of inosine tRNA modification between the fibroblast cell lines was compared using liquid chromatography-mass spectrometry (LC-MS) as previously described (Ramos et al. 2019). After normalization to an average tRNA, we found that the fibroblasts from each child exhibited a ∼50% reduction in inosine levels compared to the parental fibroblasts (Fig. 2A, inosine/tRNA). As comparison, no significant change in the ratio of the abundant m1A modification was detected between the four individuals (Fig. 2A, m1A/tRNA).
FIGURE 2.
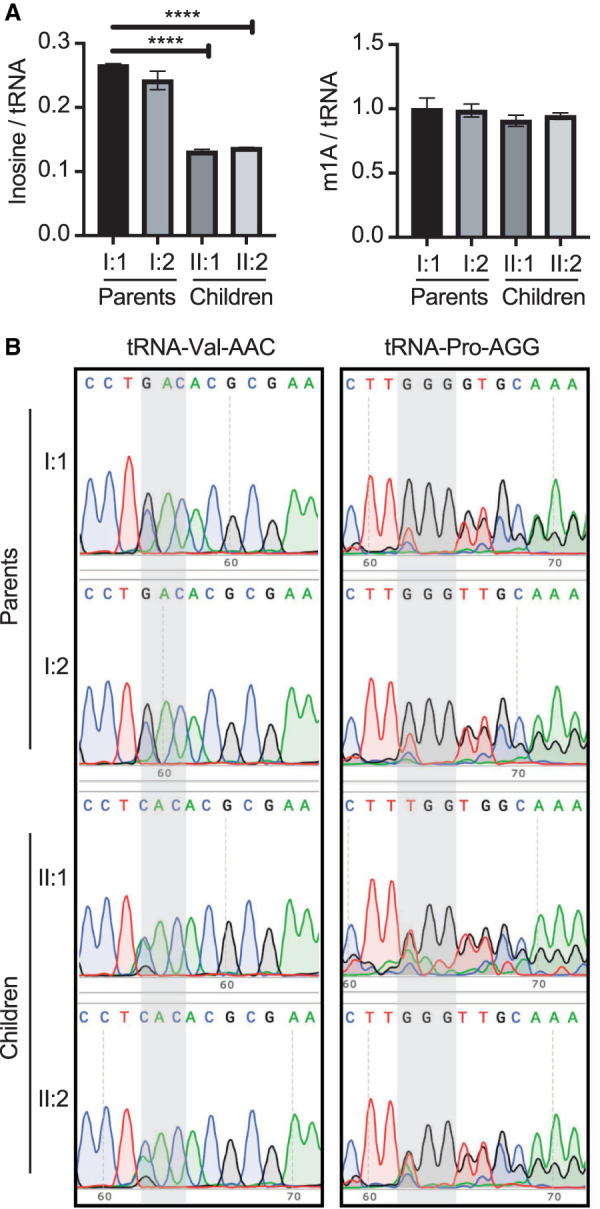
Reduced wobble inosine modification in the affected children harboring biallelic ADAT3 variants. (A) LC-MS analysis of nucleosides prepared from small RNAs from fibroblasts of the indicated individuals. Inosine signal is normalized to an average tRNA molecule. Unpaired t-test (****) P = <0.0001. (B) RT-PCR sequencing analysis of the indicated tRNAs prepared from fibroblast cells harvested from the parents and children. Both children exhibit reduced wobble inosine signal compared to heterozygous parents. Gray highlights the anticodon loop. Inosine is read as G, while a tRNA that is unmodified will contain an A.
As additional verification for reduced wobble inosine modification, we monitored wobble inosine status in specific tRNAs using RT-PCR sequencing. Since inosine is a guanosine (G) analog, the presence of inosine can be detected by sequencing of cDNA products generated by RT-PCR of cellular tRNA (Motorin et al. 2007; Kawahara 2012; Wulff et al. 2017). Using this strategy, we measured the wobble inosine status of tRNA-Val-AAC and tRNA-Pro-AGG in the fibroblast cell lines. We also sequenced additional inosine-containing tRNA isoacceptors but the high sequence similarity between isodecoder variants prevented clear interpretation as previously noted by our laboratory and others (Torres et al. 2018; Ramos et al. 2019).
Fibroblasts prepared from both parental family members exhibited nearly complete adenosine to inosine modification at the wobble position on tRNA-Val-AAC, as shown by the presence of a G peak and the absence of an A peak (Fig. 2B, Valine-AAC, parents I:1 and I:2). In contrast, fibroblasts prepared from the two affected offspring individuals exhibited a reduction in inosine modification as revealed by the A peak signal being greater than the G peak (Fig. 2B, Valine-AAC, children II:1 and II:2). The presence of the C peak is due to tRNA-Val-CAC, which is nearly identical to tRNA-Val-AAC and is recognized by the same oligos used for RT-PCR amplification (Wulff et al. 2017). We also detected a similar decrease in inosine modification when examining the sequencing chromatograms of tRNA-Pro-AGG. While the A peak is absent from the chromatograms of the two parental heterozygous controls, the two affected individuals with ID exhibited a detectable A peak (Fig. 2B, tRNA-Proline-AGG). Altogether, these results demonstrate that fibroblast cells harboring biallelic variants of ADAT3 exhibit reduced inosine modification in at least two different tRNA isoacceptors. Moreover, the reduced inosine modification in the tRNA of patient fibroblast cells suggest that additional wobble inosine-containing tRNAs are likely to be impacted by the biallelic ADAT3 variants.
Reduced tRNA adenosine deaminase activity in patient fibroblasts harboring compound ADAT3 variants
We next measured the levels of tRNA adenosine deaminase activity between the fibroblast cell lines to elucidate the cause for the reduced wobble inosine modification in the children. We performed an adenosine deaminase activity assay using whole cell lysates extracted from fibroblasts and in vitro transcribed tRNA substrates internally radiolabeled at adenosine nucleotides. As model substrates, we used tRNA-Pro-AGG and tRNA-Thr-AGU, which are known to contain the inosine modification at position 34 (Rafels-Ybern et al. 2015; Torres et al. 2015). Moreover, these tRNA substrates are known to contain the m1A modification, which can be used an internal control as noted below. After incubation of radiolabeled tRNA with the cellular lysates, the tRNA was digested to nucleoside monophosphates and separated using thin-layer chromatography (TLC) to monitor inosine formation based upon the detection of inosine monophosphate (IMP) (Grosjean et al. 2004). As an internal control, adenosine deaminase activity was normalized against the formation of 1-methyladenosine (m1A) in the tRNA substrates, which is catalyzed by the separate TRMT6/TRMT61 complex in cellular extracts (Guy and Phizicky 2014).
Using this assay, no detectable formation of inosine or m1A was observed when either tRNA substrate was incubated with buffer alone (Fig. 3A,B, lane 1, Buffer). In lanes where tRNA had been incubated with cell lysates derived from parental fibroblasts, we detected the formation of IMP along with 1-methyladenosine (m1A) (Fig. 3A,B, lanes 2 and 3, I:1 and I:2, parents). In contrast, only a background level of inosine formation was detected in cell extracts derived from the children with the compound ADAT3 variants (Fig. 3A,B, lanes 4 and 5, II:1 and II:2, children). Quantification of IMP formation revealed a significant reduction in adenosine deaminase activity from cellular extracts prepared from the children compared to the parents (Fig. 3C,D, % IMP formation). In contrast, the level of m1A formation on the same tRNA substrates was not significantly different between extracts derived from cells of the parents or children (Fig. 3C,D, % m1A formation). These results indicate that fibroblasts harboring the biallelic ADAT3 variants exhibit defects in catalyzing inosine formation on tRNA substrates, consistent with reduced inosine modification in their tRNAs.
FIGURE 3.

Individuals with the biallelic ADAT3 variants exhibit reduced adenosine deaminase activity. (A) Representative TLC plate of IMP formation after whole-cell extracts were incubated with in vitro transcribed tRNA-Pro-AGG. (B) Representative TLC plate of IMP formation after whole-cell extracts were incubated with in vitro transcribed tRNA-Thr-AGU. (C) Quantification of %IMP formation/total radioactive signal on tRNA-Proline-AGG. Unpaired t-test: ns = 0.53, (**) P = 0.001. Quantification of %m1A formation in the two samples harvested from the affected siblings indicate the extracts are active. Unpaired t-test: I:2 ns = 0.34, II:1 ns = 0.20, II:2 ns = 0.25. (D) Quantification of %IMP formation/total radioactive signal on tRNA-Threonine-AGU. Unpaired t-test: ns = 0.98, (*) P = 0.024. The formation of m1A in the two samples harvested from the affected siblings indicate the extracts are active. Unpaired t-test: I:2 ns = 0.28, II:1 ns = 0.35, II:2 ns = 0.44. Each point is one biological replicate. Three biological replicates were performed. The column on the graph represents the mean and the error bar represents the standard deviation of the three replicates.
ID-associated ADAT3 variants exhibit defects in ADAT2 interaction
Both the A196V missense variant and Q274* truncation lie within the conserved noncatalytic deaminase domain of ADAT3. Moreover, the Q274* variant would truncate nearly half the entire deaminase domain. Previous studies have shown that bacterial TadA forms a homodimer through an interface within the deaminase domain that includes the A196 residue (Kim et al. 2006). The ADAT2 subunit is predicted to heterodimerize with ADAT3 to form an active adenosine deaminase complex using a similar interaction as the bacterial TadA dimer (Elias and Huang 2005; Spears et al. 2011). Thus, we investigated whether the compound ADAT3 variants impact heterodimerization with the ADAT2 subunit. To assay subunit interactions, we expressed ADAT3 variants fused to a Twin-Strep Tag along with His-tagged ADAT2 in 293T human embryonic cells (Fig. 4A, lanes 2 through 4). The Strep-tag allows for purification of proteins under native conditions using streptactin-conjugated resin, allowing for maintenance of protein–protein interactions (Schmidt et al. 2013). To accurately determine the amount of copurifying ADAT2 and ensure the signal was within the linear range of detection, we loaded increasing amounts of the purified ADAT2/3 complexes for quantification (Fig. 4A, lanes 6 through 14). Notably, we detected a reduced amount of ADAT2 that copurified with ADAT3–A196V than with ADAT3–WT (Fig. 4A, compare ADAT2 in lanes 6–8 to lanes 9–11). Quantification of the blots revealed a ∼50% reduction in copurifying ADAT2 with ADAT3–A196V compared to ADAT3–WT (Fig. 4B). The ADAT3–Q274* variant exhibited greatly reduced copurification of His-ADAT2 (Fig. 4A, Q274*, lanes 12–14 and Fig. 4B). These results indicate that the A196V variant impairs the interaction of ADAT3 with ADAT2 while the truncation of the ADAT3 carboxyl terminus greatly diminishes interaction with ADAT2.
FIGURE 4.

ADAT3 variants exhibit decreased ADAT2 interaction without detectable impact on endogenous ADAT2 expression or wobble inosine levels: (A) Representative ADAT3-Strep purifications with copurifying HIS-ADAT2. Input represents 5% of the starting extract while purifications represent threefold dilutions of the final eluted samples. (B) Quantification of ADAT3-STREP copurification. N = 7 purifications. Unpaired t-test (****) P = <0.0001. (C) Immunoblot of cellular extracts from 293T cells transfected with the indicated constructs in triplicate and probed against the STREP tag for ADAT3 expression, ADAT2, and ACTIN. (D) Quantification of ADAT2 signal relative to ACTIN signal. (E) RT-PCR sequencing chromatograms of tRNA-Val-AAC and tRNA-Pro-AGG harvested from cells transfected with the indicated constructs in triplicate.
In addition to monitoring the interaction between ADAT2 and ADAT3, we also tested whether expression of the ADAT3 variants could cause dominant negative effects on ADAT2 levels or wobble inosine modification. The ADAT3 variants were overexpressed in 293T cells using the same procedure as above (Fig. 4C). Based upon three independent replicates of each transfection, we detected no major change in ADAT2 levels between cells transfected with vector alone versus the ADAT3 constructs (Fig. 4D). RT-PCR analysis of wobble inosine modification in tRNA-Val-AAC or Pro-AGG revealed no detectable increase in unmodified wobble adenosine in cells expressing ADAT3–WT, A196V, or Q274* compared to control cells (Fig. 4E). These results suggest that the newly identified ADAT3 variants do not act in a dominant negative fashion to affect ADAT2 protein levels or wobble inosine modification. The lack of a dominant negative effect is also consistent with the lack of unmodified wobble adenosine in cells isolated from either parent who express the variant allele.
ADAT3–A196V and Q274* exhibit alterations in subcellular localization
The interaction of ADAT2 with ADAT3 is required for translocation of ADAT3 into the nucleus (Torres et al. 2015). Since the newly identified ADAT3 variants in this study exhibit weakened or disrupted interaction with ADAT2, we investigated whether the ADAT3 variants also exhibited perturbations in subcellular localization. We used green fluorescent protein (GFP) tagging to visualize ADAT3 localization due to the low levels of endogenous ADAT3, which precludes accurate determination of cellular localization by microscopy (data not shown). We fused the ADAT3–WT, A196V, and Q274* variants to an amino-terminal GFP tag which were expressed in HeLa human cervical cells by transient transfection. We then monitored and binned GFP–ADAT3 localization into three categories: primarily cytoplasmic, ubiquitous distribution between the cytoplasm and nucleus, or primarily nuclear (see Materials and Methods for quantification parameters).
After transient transfection with the ADAT3–WT plasmid, we found that the majority of HeLa cells exhibited cytoplasmic distribution of GFP–ADAT3–WT (Fig. 5A, quantified in 5B, column 1). Similarly, the majority of cells expressing either GFP–ADAT3–A196V or the Q274* variants exhibited primarily cytoplasmic GFP signal (Fig. 5A, quantified in 4B, columns 2 and 3). As shown previously (Torres et al. 2015; Ramos et al. 2019), coexpression of ADAT2 with GFP–ADAT3–WT promotes the translocation of ADAT3 into the nucleus resulting in the majority of cells displaying GFP–ADAT3 localization in the nucleus compared to GFP–ADAT3 expression without ADAT2 (Fig. 5A,B, compare column 1 to column 4). ADAT2 coexpression could also promote the partial localization of GFP–ADAT3–A196V into the nucleus as noted by the increase in percentage of cells with ubiquitous or nuclear GFP localization (Fig. 5A,B, compare columns 2 and 5). However, in contrast to GFP–ADAT3–WT, the majority of cells expressing GFP–ADAT3–A196V with ADAT2 exhibited a ubiquitous distribution rather than being primarily nuclear (Fig. 5A,B, compare columns 4 and 5). The GFP–ADAT3–Q274* variant remained primarily in the cytoplasm without or with ADAT2 coexpression (Fig. 5A,B, compare columns 3 and 6). The decreased nuclear localization of ADAT3–A196V and ADAT3–Q274* in the presence of ADAT2 is consistent with the reduced interaction between ADAT3–A196V with ADAT2 and the near lack of interaction between ADAT3–Q274* with ADAT2 as shown above (Fig. 4A). Moreover, these results suggest that the reduced inosine modification in patients with the compound ADAT3 variants could be due to perturbations in enzymatic activity as well as subcellular localization caused by defects in ADAT2 interaction.
FIGURE 5.

ADAT3 variants exhibit perturbations in ADAT2-dependent subcellular localization which can be rescued through overexpression of ADAT2. (A) Localization of GFP–ADAT3–WT and variants, without and with coexpression of ADAT2 in HeLa human cells. (B) Quantification of ADAT3–WT and ADAT3–A196V without or with coexpression of ADAT2. Localization was binned into three groups, primarily cytoplasmic, ubiquitous, and primarily nuclear. The amount of ADAT2 plasmid that was cotransfected with ADAT3 plasmid is indicated. Error bars represent standard deviation.
Since ADAT2 is limiting for formation of the ADAT2/3 heterodimer and subsequent nuclear translocation, we hypothesized that overexpression of ADAT2 could potentially rescue the localization defect of the ADAT3–A196V variant caused by its reduced interaction with ADAT2. To test this hypothesis, we performed transient transfection of GFP–ADAT3–WT and the GFP–ADAT3 variants along with increasing amounts of plasmid expressing ADAT2. Increasing the amount of ADAT2 plasmid that was cotransfected with ADAT3–WT had no major change in the percentage of cells with nuclear localization, suggesting that the lowest amount of ADAT2 plasmid resulted in the stoichiometric expression of ADAT2 with ADAT3–WT (Fig. 5B, compare column 4 to columns 7 and 10). As noted above, only ∼5% of cells transfected with GFP–ADAT3–A196V and the lowest amount of ADAT2 plasmid exhibited nuclear localization of ADAT3 (Fig. 5B, column 5). Intriguingly, increasing the amount of ADAT2 expression was sufficient to elevate the percentage of cells with primarily nuclear localization of GFP–ADAT3–A196V to ∼23%, and shifted the remaining cytoplasmic portion from 30% to 12% (Fig. 5B, compare column 5 to columns 8 and 11). In contrast, increasing the amount of ADAT2 expression had no effect on ADAT3–Q274* localization (Fig. 5B, compare column 6 to columns 9 and 12). These results suggest that increasing the cellular concentration of ADAT2 can promote interaction with ADAT3–A196V to induce increased nuclear translocation of ADAT3 while ADAT3–Q274* is completely abrogated in interaction with ADAT2.
ADAT2 overexpression in patient fibroblasts restores tRNA adenosine deaminase activity and wobble inosine modification
The heterodimeric ADAT2/3 complex has been shown to catalyze inosine formation on nuclear pre-tRNAs, consistent with the localization of the ADAT2/3 complex in the nucleus (Torres et al. 2015). The rescue of ADAT3–A196V nuclear localization via increased expression of ADAT2 suggests that overexpression of ADAT2 could promote the association between ADAT3–A196V and ADAT2 to increase the levels of ADAT2/3 complexes in the nucleus. Encouraged by the above results, we tested whether overexpression of ADAT2 in patient fibroblasts is sufficient to restore wild-type levels of enzymatic activity and consequently, wobble inosine modification.
For stable recombinant protein expression in fibroblasts, we generated lentiviral expression constructs expressing either GFP as a negative control or untagged ADAT2 (Fig. 6A). After stable integration of the vectors into parental or patient fibroblasts, we monitored protein expression via immunoblot (Fig. 6B). Using this system, recombinant ADAT2 was overexpressed by 5 to 10 times above endogenous levels of ADAT2 in the stable fibroblast cell lines compared to control cell lines expressing only GFP (Fig. 6C).
FIGURE 6.

Overexpression of ADAT2 restores adenosine deaminase activity in fibroblasts harboring biallelic ADAT3 variants. (A) ADAT2 overexpression strategy using lentiviral infection of human fibroblast cell lines. (B) Immunoblot of the indicated fibroblast cell lines infected with either control virus expressing GFP or untagged ADAT2. (C) Quantification of ADAT2 overexpression. N = 3. Error bar represents standard deviation. (D) TLC plate of IMP formation after whole-cell extracts were incubated with internally radiolabeled in vitro transcribed tRNA-Pro-AGG. (E) TLC plate of IMP formation after whole-cell extracts were incubated with internally radiolabeled in vitro transcribed tRNA-Thr-AGU. (F,G) Quantification of IMP product formation in tRNA-Pro-AGG and tRNA-Thr-AGU. The first graph is IMP formation over total radioactive signal. The right graphs are m1A formation over total radioactive signal. Each point is one biological replicate. Three biological replicates were performed. The bar on the graph represents the mean and the error bar represents the standard deviation of the three replicates.
We next determined whether overexpression of ADAT2 could impact the levels of adenosine deaminase activity in cellular extracts. Using the in vitro adenosine deaminase activity assay described above, we monitored IMP formation with tRNA-Pro-AGG and tRNA-Thr-AGU, using whole-cell extracts overexpressing either GFP or ADAT2. As observed above, the fibroblasts harvested from the parental cell lines exhibited both IMP and m1A formation while fibroblasts from the individuals with the biallelic ADAT3 variants displayed no detectable IMP formation at the amount of extract used in the assay (Fig. 6D,E, compare lanes 1 and 2 to lanes 3 and 4). In contrast, extracts prepared from patient fibroblasts overexpressing ADAT2 exhibited readily detectable adenosine deaminase on either tRNA-Pro-AGG or tRNA-Thr-AGU (Fig. 6D,E, compare lanes 3 and 4 to lanes 7 and 8). While the level of adenosine deaminase activity in the patient fibroblasts overexpressing ADAT2 was still reduced relative to extracts prepared from the parental fibroblasts, there was significantly higher activity compared to the control patient fibroblasts expressing just GFP alone (Fig. 6F,G). These results indicate that overexpression of ADAT2 is sufficient to restore adenosine deaminase activity in patient fibroblasts harboring the compound heterozygous ADAT3 variants.
The restoration of adenosine deaminase activity in patient fibroblasts via ADAT2 overexpression suggests the possibility that inosine modification levels could also be rescued. To compare wobble inosine modification levels, we performed LC-MS on nucleosides from digested small RNAs extracted from the fibroblast cells overexpressing GFP or ADAT2. Quantification of inosine showed a significant restoration of inosine in the patient cells overexpressing ADAT2 (Fig. 7A, compare II:1 and II:2 without or with overexpression of ADAT2). In contrast, ADAT2 overexpression had no significant effect on m1A levels in any of the cell lines (Fig. 7B). As further validation for rescue of wobble inosine modification in patient fibroblasts, we monitored wobble inosine formation using RT-PCR sequencing of tRNA-Val-AAC and Pro-AGG. In parental fibroblasts expressing GFP alone, we detected a predominant G peak for the inosine modification at the wobble position of tRNA-Val-AAC and Pro-AGG that was not detectably altered by ADAT2 overexpression (Fig. 7C, Parents I:1 and I:2). In contrast, the patient cells containing the biallelic ADAT3 variants infected with GFP alone exhibited reduced inosine in tRNA-Val-AAC and Pro-AGG as detected by the predominant A peak (Fig. 7C, GFP, Children II:1 and II:2). Strikingly, overexpression of ADAT2 in the patient fibroblasts resulted in the restoration of wobble inosine in both tRNA-Val-AAC and tRNA-Pro-AGG (Fig. 7C, ADAT2, II:1 and II:2, children). These results demonstrate the potential to rescue the essential inosine modification in patient cells with ADAT3 variants through the overexpression of the ADAT2 heterodimeric binding partner.
FIGURE 7.

Overexpression of the ADAT2 subunit can rescue wobble inosine modification in patient fibroblasts from individuals harboring biallelic ADAT3 variants. (A) LC-MS analysis of digested nucleosides from small RNAs of the indicated fibroblast cell lines. Inosine and m1A levels per tRNA molecule were calculated. Unpaired t-test II:1 (*) P = 0.01, II:2 (*) P = 0.03. (B) m1A modification does not change with overexpression of ADAT2. Unpaired t-test II:1 ns = 0.7, II:2 ns = 0.1. (C) Sequencing of substrates tRNA-Val-AAC and tRNA-Pro-AGG. Gray shading highlights anticodon. +GFP are fibroblasts overexpressing GFP and +ADAT2 are fibroblasts overexpressing ADAT2. In fibroblast cells expressing GFP, inosine is reduced on the children, as seen by increased green peak over black peak. Fibroblasts overexpressing ADAT2 restore inosine formation on these two substrates, as seen by lack of green peak in II:1 and II:2 +ADAT2.
DISCUSSION
In this study, we identify and characterize novel compound heterozygous variants in ADAT3 that underscore the key role of wobble inosine modification in proper neurodevelopment. Three affected siblings carry a paternally inherited missense variant (c.587C > T; p.[Ala196Val]) and a maternally inherited truncated variant (c.820C > T; p.[Gln274*]). The three siblings in our study share many of the features previously described for ADAT3 deficiency (Supplemental Table 1). In addition to ID, other commonly reported features include hypotonia, short stature, feeding difficulties, strabismus and microcephaly. In almost all cases absent or limited speech and motor delay were also described. Microcephaly has also been identified in a subset of previously described cases associated with ADAT3 variants and was only observed in the youngest sister of this family, consistent with this sporadic occurrence.
All characterized patient cells with either the previously described ADAT3–V144M variant or the compound heterozygous variants exhibit reduced wobble-inosine tRNA modification. This observation supports the idea that reduced wobble inosine may be the major underlying cause of ID in patients with pathogenic ADAT3 variants. Considering the essential requirement for wobble inosine modification for decoding of certain codons, it is not surprising that reduced inosine modification could have severe impact on neurodevelopment. Interestingly, a recent study has found that human embryonic stem cells optimize translation of codons that are dependent upon tRNAs containing the wobble inosine modifications (Bornelov et al. 2019). Moreover, the study found that inosine levels are highest in human pluripotent embryonic stem cells before differentiation. Future studies using ribosome profiling of patient cells would yield further insight into the mRNAs and the developmental pathways that are impacted by reduced wobble inosine modification.
The A196V and Q274* variants we report here differ from the previously characterized V144M variant in that the amino acid residue alterations lie within the conserved adenosine deaminase domain. The molecular and cellular phenotypes of the ADAT3 variants described in this study also differ from the biochemical defects observed with the previously described ADAT3–V144M variant. In that case, the V144M substitution alters ADAT3 protein folding without any detectable change in ADAT2 interaction or ADAT2-dependent nuclear localization (Ramos et al. 2019). In contrast, we found here that ADAT3–A196V and Q274* exhibit decreased or severely reduced interaction with ADAT2 and perturbations in ADAT2-dependent nuclear localization. While alanine and valine are both hydrophobic, the additional two methyl groups in valine could clash with residues in ADAT2 along the interaction interface. Thus, the alanine-to-valine substitution could alter the proper folding of the ADAT3 interaction interface with ADAT2. In turn, the reduced ADAT2 interaction would result in a decreased population of ADAT3 which is shuttled into the nucleus to catalyze inosine modification in pre-tRNAs destined for the cytoplasm.
Notably, we could suppress the nuclear localization defect exhibited by the ADAT3–A196V variant by increasing the expression of ADAT2. Interestingly, a previous study has identified a point mutation in the fission yeast Schizosaccharomyces pombe (S. pombe) homolog of ADAT3, Tad3p, which also impairs interaction with the ADAT2 homolog, Tad2p (Tsutsumi et al. 2007). Like the ADAT3–A196V variant described here, the Tad3p point mutation converts serine 275 in Tad3p to asparagine (S275N) in the adenosine deaminase domain. Similar to our findings, S. pombe cells expressing only Tad3-S275N exhibit reduced wobble inosine modification that can be rescued by Tad2p overexpression. The rescue of wobble inosine modification levels in S. pombe and in human cells presented here could be due to increased ADAT2 concentrations which shifts the equilibrium toward formation of the heterodimer.
Intellectual disability represents a neurodevelopmental disorder characterized by early onset cognitive impairments and limitations in adaptive behavior that can be caused by environmental or genetic factors (Patel et al. 2018; Bhaumik and Alexander 2020). In addition to ADAT3, whole exome sequencing has identified numerous variants in tRNA modification enzymes as the cause of neurodevelopmental disorders such as ID (Torres et al. 2014; Pereira et al. 2018; Ramos and Fu 2019). These studies highlight the emerging role of tRNA modification in proper gene expression and brain development (Angelova et al. 2018; Tuorto and Parlato 2019).
The findings presented here suggest a potential therapeutic strategy for ADAT3-associated intellectual disability disorders based on the overexpression of ADAT2 to rescue inosine tRNA modifications. Similarly, coexpression of ADAT2 to stoichiometric levels reduced the misfolding properties of the previously characterized ADAT3–V144M variant associated with ID (Ramos et al. 2019). However, many challenges and questions remain before this presents a viable option for therapy. For example, it remains unknown where and when wobble inosine modification is required for proper neurodevelopment. Based upon the increased dependency of stem cells on inosine modification, the requirement for proper wobble inosine modification levels could be very early in development or differentiation (Bornelov et al. 2019). Moreover, the requirement for the wobble inosine modification to decode a subset of codons suggests that certain proteins could be more significantly impacted by reduced wobble inosine levels in humans. However, it is unknown which mRNA transcripts are dependent upon wobble inosine for proper translation in human cells and whether ADAT2 overexpression is sufficient to rescue perturbations in translation. Finally, the overexpression of ADAT2 could cause nonspecific deamination of additional RNA substrates leading to off-target effects. Despite these hurdles, our studies provide the first insight into a potential strategy for modulating inosine levels in cells harboring ADAT3 variants.
MATERIALS AND METHODS
Cell lines
Human primary epithelial skin fibroblasts were cultured in Eagle's Minimum Essential Media (EMEM) (ATCC) containing 15% fetal bovine serum and 1% Penicillin/Streptomycin. Cells were grown at 37°C, 5% Oxygen, and 5% CO2. HeLa S3 human cervical carcinoma and 293T human embryonic kidney cell lines were cultured in Dulbecco's Minimal Essential Medium (DMEM) containing 10% fetal bovine serum, 2 mM L-alanyl-L-glutamine (GlutaMax, Gibco) and 1% Penicillin/Streptomycin. Cells were grown at 37°C, 20% Oxygen, and 5% CO2.
Sequencing of tRNAs
Sequencing of tRNAs was performed identically to Ramos et al. (2019). RNA extraction was performed on 1 × 106 human fibroblast cell lines using TRIzol LS reagent (Thermo Fisher). For RT-PCR, total RNA (∼1.25 µg) was reverse transcribed for tRNA-Val-AAC and Pro-AGG using Superscript IV enzyme followed by the QIAquick PCR purification kit. cDNA was then PCR amplified using Herculase II DNA polymerase (Agilent Genomics). The PCR product was gel extracted and analyzed by Sanger sequencing (ACGT, Inc). The following primers were used:
Val RT primer: TGTTTCCGCCTGGTTTTG
Val PCR primer F: GAACTAAGCTTGTTCAGAGTTCTACAGTCCGGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATGTTTCCGTAGTGTAGTGGTTATCAC
Val PCR primer R: CACT TGTTTCCGCCTGGTTTTGATCCAGGGACC
Pro RT primer: GGGCTCGTCCGGGATTT
Pro PCR primer F: GTTCAGAGTTCTACAGTCCGGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATGGCTCGTTGGTCTAGGGGTA
Pro PCR primer R: GGGCTCGTCCGGGATTT
Mass spectrometry of nucleosides
In the first step, the total RNA was fractionated on an AdvanceBio SEC column (300 A pore size, 2.7 µm particle size, 7.8 by 300 mm; Agilent) at 40°C using isocratic elution with 0.1 M NH4OAc buffer at pH 7 to separate the total tRNA fraction from larger RNA species. The tRNA was collected in 1 mL and lyophilized in the SpeedVac, until 50 µL remained in the vial. By adding 5 µL ammonium acetate (5 M) and 125 µL ice-cold and pure ethanol (100%), the tRNA was incubated overnight and precipitated by centrifugation (12,000g, 4°C, 30 min.). The tRNA pellets were resolved in 30 µL MilliQ water and their concentrations were measured using an IMPLEN nano-photometer (Implen). A maximum of 500 ng tRNA was processed and analyzed by LC-MS using an MRM method as previously described (Heiss et al. 2017; Ramos et al. 2019). The level of each modified nucleoside (I, m1A) was normalized to an average tRNA. The amount of injected tRNA was calculated by the detected amount of C in pmol and the average content of C in an average tRNA.
Transient transfection
Transient transfection and cellular extract production were performed as previously described (Dewe et al. 2017). In brief, 2.5 × 106 293T HEK cells were transiently transfected by calcium phosphate DNA precipitation with 10–20 µg of plasmid DNA followed by preparation of lysate by hypotonic freeze-thaw lysis 48 h post-transfection. For purification, whole cell extract from transiently transfected cells cell lines (1 mg of total protein) was rotated for 2 h at 4°C in lysis buffer (20 mM HEPES at pH 7.9, 2 mM MgCl2, 0.2 mM EGTA, 10% glycerol, 1 mM DTT, 0.1 mM PMSF, 0.1% NP-40) with 200 mM NaCl. Resin was washed three times using the same buffer followed by protein analysis. Strep-tagged proteins were purified using MagSTREP “type3” XT beads, 5% suspension (IBA Lifesciences) and eluted with desthiobiotin.
Western blotting
Western blots were performed as previously described (Ramos et al. 2019). Briefly, cellular extracts and purified protein samples were fractionated on NuPAGE Bis-Tris polyacrylamide gels (Thermo Scientific) followed by transfer to Immobilon FL PVDF membrane (Millipore) for immunoblotting. For analysis of skin fibroblasts, 1 × 106 cells were harvested and proteins were extracted using radioisotope immunoprecipitation assay (RIPA) buffer (50 mM TrisHCl, pH 7.5, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA). Antibodies were against the following proteins : 6xHis tag (MA1-21315, Thermo Fisher), GFP (sc-9996, Santa Cruz Biotechnology), Strep-tag II-tag (NC9261069, Thermo Fisher), ADAT3 (Abcam, ab192987), ADAT3 (H00113179-B01P, Abnova), ADAT2 (ab135429, Abcam), and actin (CST). Primary antibodies were detected using IRDye 800CW Goat anti-Mouse IgG (SA5-35521, Thermofisher) or Rabbit (SA5-35571, Thermofisher) or Rat (925-32219, LI-COR Biosciences), or IRDye 680RD Goat anti-Mouse IgG (926-68070, LI-COR Biosciences) or Rabbit (925-68071). Immunoblots were scanned using direct infrared fluorescence via the Odyssey System (LI-COR Biosciences).
Overexpression of ADAT2
The open reading frame of ADAT2 was PCR amplified with restriction overhangs and cloned into PLJM vector (Addgene: 19219). The vector was then cotransfected with packaging plasmids (psPAX2 and pMD2.G) into 293T cells for lentivirus production. The fibroblast cell lines were subsequently infected with the lentivirus in the presence of polybrene followed by stable clone selection using 100 µg/mL puromycin
Enzymatic assays
Enzymatic assays were performed as previously described (Ramos et al. 2019). Internally radiolabeled tRNA substrates were prepared by T7 in vitro transcription of DNA templates generated by PCR amplification. Oligonucleotides containing the T7 promoter upstream of tRNA sequences were PCR amplified using Herculase II DNA polymerase or Taq DNA Polymerase (New England Biolabs) followed by agarose gel purification of PCR amplification products. In vitro transcription was performed using Optizyme T7 RNA polymerase (Fisher Scientific) with 10 mM each of UTP, CTP, and GTP, 1 mM of ATP, and 250 µCi of [α-32P]-ATP (3000 Ci/mmol, 10 mCi/ml). In vitro transcription reactions were incubated at 37°C for 2 h followed by DNase treatment and purification using RNA Clean and Concentrator columns (Zymo Research). Full-length tRNA transcripts were verified on a 15% Polyacrylamide-urea gel stained with SYBR Gold nucleic acid stain (Thermo Fisher). Before conducting enzymatic assays, all tRNA substrates were refolded by thermal denaturation at 95°C for 2 min in buffer containing a final concentration 5 mM TRIS pH 7.5 and 0.16 mM EDTA, quick chilling on ice for 2 min and refolding at 37°C in the presence of Hepes pH 7.5, MgCl2, and NaCl.
For adenosine deaminase assays, ∼150 ng of refolded tRNA substrate was incubated with either skin fibroblast extract or Strep-purified ADAT3. Reactions were incubated at 37°C for 30min—1 h and RNA was purified using RNA Clean and Concentrator columns. The tRNA was eluted in 10 µL of water and was subjected to nuclease P1 digestion overnight in total volume of 13 µL and 0.125 units of P1 in 250 mM Ammonium Acetate pH 5.35. Half of the P1-nuclease treated samples were spotted on POLYGRAM polyester Cellulose MN 300 plates (Macherey Nagel) run in solvent B (0.1 M sodium phosphate buffer pH 6.8:NH4 sulfate:n-propanol (100∶60∶2 [v∶w∶v]). Phosphorimaging was conducted on a Bio-Rad Personal Molecular Imager followed by analysis using NIH ImageJ software.
Microscopy
Procedure was previously described (Ramos et al. 2019). Hela cells were plated at 2.5 × 105 cells on a six-well plate. Cells were transfected 1 d after plating with a 1.25–5 µg of DNA using Lipofectamine 3000. Cells were imaged 24 h post-transfection on an EVOS fluorescence microscopy imaging system (ThermoFisher) quantification. For DNA staining, cells were washed twice with PBS and then incubated for 30 min at 37°C with PBS containing 10% FBS and 1 µM of Hoechst and then imaged. For localization quantification, five images were taken of each well and the number of GFP-positive cells were counted in each of the five frames.
Ethics statement and approvals
Ethical approval for whole-exome sequencing (WES) was received from the Uppsala Ethical Review Board and informed written consent was obtained from the parents.
Whole-exome sequencing
Exome enrichment was performed using SureSelect (Agilent) version 4 following the manufacturer's protocol and samples were sequenced on Illumina HiSeq Sequencer. The sequencing was performed to achieve at least 30× coverage of the captured regions.
Sequence reads were mapped to human reference genome Hg19 as previously described (Halvardson et al. 2016). Analysis of the data was performed to identify disease-causing variants under recessive and de novo dominant models. Identified variants were verified by Sanger sequencing using standard protocols, and subsequently classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al. 2015). Patients’ data have been submitted to DECIPHER. (https://decipher.sanger.ac.uk/).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the participating family for their cooperation. Sequencing was performed using the SciLifeLab National Genomics Infrastructure at Uppsala Genome Center and the Uppsala SNP & Seq Facility. Computational analyses were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX). We thank members of the Fu laboratory for helpful discussions on the manuscript. This work was supported by National Science Foundation CAREER award 1552126 to D.F. and the Sävstaholm Society to ACT. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions: J.R., M.P., J.H., F.H., E.K., B.P., R.B., S.M.K., L.F., and A.T. collected the data. J.R., L.F., A.T., and D.F wrote the manuscript. J.R., L.F., A.T., and D.F. contributed to experimental design.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.076380.120.
REFERENCES
- Alazami AM, Hijazi H, Al-Dosari MS, Shaheen R, Hashem A, Aldahmesh MA, Mohamed JY, Kentab A, Salih MA, Awaji A, et al. 2013. Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J Med Genet 50: 425–430. 10.1136/jmedgenet-2012-101378 [DOI] [PubMed] [Google Scholar]
- Angelova MT, Dimitrova DG, Dinges N, Lence T, Worpenberg L, Carre C, Roignant JY. 2018. The emerging field of epitranscriptomics in neurodevelopmental and neuronal disorders. Front Bioeng Biotechnol 6: 46 10.3389/fbioe.2018.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik S, Alexander R. 2020. Oxford textbook of the psychiatry of intellectual disability. Oxford University Press. [Google Scholar]
- Bornelov S, Selmi T, Flad S, Dietmann S, Frye M. 2019. Codon usage optimization in pluripotent embryonic stem cells. Genome Biol 20: 119 10.1186/s13059-019-1726-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewe JM, Fuller BL, Lentini JM, Kellner SM, Fu D. 2017. TRMT1-catalyzed tRNA modifications are required for redox homeostasis to ensure proper cellular proliferation and oxidative stress survival. Mol Cell Biol 37: e00214-17 10.1128/MCB.00214-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hattab AW, Saleh MA, Hashem A, Al-Owain M, Asmari AA, Rabei H, Abdelraouf H, Hashem M, Alazami AM, Patel N, et al. 2016. ADAT3-related intellectual disability: further delineation of the phenotype. Am J Med Genet A 170A: 1142–1147. 10.1002/ajmg.a.37578 [DOI] [PubMed] [Google Scholar]
- Elias Y, Huang RH. 2005. Biochemical and structural studies of A-to-I editing by tRNA:A34 deaminases at the wobble position of transfer RNA. Biochemistry 44: 12057–12065. 10.1021/bi050499f [DOI] [PubMed] [Google Scholar]
- Gerber AP, Keller W. 1999. An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science 286: 1146–1149. 10.1126/science.286.5442.1146 [DOI] [PubMed] [Google Scholar]
- Grosjean H, Keith G, Droogmans L. 2004. Detection and quantification of modified nucleotides in RNA using thin-layer chromatography. Methods Mol Biol 265: 357–391. 10.1385/1-59259-775-0:357 [DOI] [PubMed] [Google Scholar]
- Guy MP, Phizicky EM. 2014. Two-subunit enzymes involved in eukaryotic post-transcriptional tRNA modification. RNA Biol 11: 1608–1618. 10.1080/15476286.2015.1008360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halvardson J, Zhao JJ, Zaghlool A, Wentzel C, Georgii-Hemming P, Mansson E, Ederth Savmarker H, Brandberg G, Soussi Zander C, Thuresson AC, et al. 2016. Mutations in HECW2 are associated with intellectual disability and epilepsy. J Med Genet 53: 697–704. 10.1136/jmedgenet-2016-103814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss M, Reichle VF, Kellner S. 2017. Observing the fate of tRNA and its modifications by nucleic acid isotope labeling mass spectrometry: NAIL-MS. RNA Biol 14: 1260–1268. 10.1080/15476286.2017.1325063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y. 2012. Quantification of adenosine-to-inosine editing of microRNAs using a conventional method. Nat Protoc 7: 1426–1437. 10.1038/nprot.2012.073 [DOI] [PubMed] [Google Scholar]
- Kim J, Malashkevich V, Roday S, Lisbin M, Schramm VL, Almo SC. 2006. Structural and kinetic characterization of Escherichia coli TadA, the wobble-specific tRNA deaminase. Biochemistry 45: 6407–6416. 10.1021/bi0522394 [DOI] [PubMed] [Google Scholar]
- Motorin Y, Muller S, Behm-Ansmant I, Branlant C. 2007. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol 425: 21–53. 10.1016/S0076-6879(07)25002-5 [DOI] [PubMed] [Google Scholar]
- Patel DR, Apple R, Kanungo S, Akkal A. 2018. Intellectual disability: definitions, evaluation and principles of treatment. Pediatric Medicine 1: 11 10.21037/pm.2018.12.02 [DOI] [Google Scholar]
- Pereira M, Francisco S, Varanda AS, Santos M, Santos MAS, Soares AR. 2018. Impact of tRNA modifications and tRNA-modifying enzymes on proteostasis and human disease. Int J Mol Sci 19: 3738 10.3390/ijms19123738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafels-Ybern A, Attolini CS, Ribas de Pouplana L. 2015. Distribution of ADAT-dependent codons in the human transcriptome. Int J Mol Sci 16: 17303–17314. 10.3390/ijms160817303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafels-Ybern A, Torres AG, Grau-Bove X, Ruiz-Trillo I, Ribas de Pouplana L. 2017. Codon adaptation to tRNAs with inosine modification at position 34 is widespread among Eukaryotes and present in two Bacterial phyla. RNA Biol 15: 500–507. 10.1080/15476286.2017.1358348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafels-Ybern A, Torres AG, Camacho N, Herencia-Ropero A, Roura Frigole H, Wulff TF, Raboteg M, Bordons A, Grau-Bove X, Ruiz-Trillo I, et al. 2019. The expansion of inosine at the Wobble position of tRNAs, and its role in the evolution of proteomes. Mol Biol Evol 36: 650–662. 10.1093/molbev/msy245 [DOI] [PubMed] [Google Scholar]
- Ragone FL, Spears JL, Wohlgamuth-Benedum JM, Kreel N, Papavasiliou FN, Alfonzo JD. 2011. The C-terminal end of the Trypanosoma brucei editing deaminase plays a critical role in tRNA binding. RNA 17: 1296–1306. 10.1261/rna.2748211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos J, Fu D. 2019. The emerging impact of tRNA modifications in the brain and nervous system. Biochim Biophys Acta Gene Regul Mech 1862: 412–428. 10.1016/j.bbagrm.2018.11.007 [DOI] [PubMed] [Google Scholar]
- Ramos J, Han L, Li Y, Hagelskamp F, Kellner SM, Alkuraya FS, Phizicky EM, Fu D. 2019. Formation of tRNA Wobble inosine in humans is disrupted by a millennia-old mutation causing intellectual disability. Mol Cell Biol 39: e00203-19 10.1128/MCB.00203-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi Chaleshtori AR, Miyake N, Ahmadvand M, Bashti O, Matsumoto N, Noruzinia M. 2018. A novel 8-bp duplication in ADAT3 causes mild intellectual disability. Hum Genome Var 5: 7 10.1038/s41439-018-0007-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TG, Batz L, Bonet L, Carl U, Holzapfel G, Kiem K, Matulewicz K, Niermeier D, Schuchardt I, Stanar K. 2013. Development of the Twin-Strep-tag(R) and its application for purification of recombinant proteins from cell culture supernatants. Protein Expr Purif 92: 54–61. 10.1016/j.pep.2013.08.021 [DOI] [PubMed] [Google Scholar]
- Sharkia R, Zalan A, Jabareen-Masri A, Zahalka H, Mahajnah M. 2019. A new case confirming and expanding the phenotype spectrum of ADAT3-related intellectual disability syndrome. Eur J Med Genet 62: 103549 10.1016/j.ejmg.2018.10.001 [DOI] [PubMed] [Google Scholar]
- Spears JL, Rubio MA, Gaston KW, Wywial E, Strikoudis A, Bujnicki JM, Papavasiliou FN, Alfonzo JD. 2011. A single zinc ion is sufficient for an active Trypanosoma brucei tRNA editing deaminase. J Biol Chem 286: 20366–20374. 10.1074/jbc.M111.243568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas E, Lewis AM, Yang Y, Chanprasert S, Potocki L, Scott DA. 2019. Novel missense variants in ADAT3 as a cause of syndromic intellectual disability. J Pediatr Genet 8: 244–251. 10.1055/s-0039-1693151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AG, Batlle E, Ribas de Pouplana L. 2014. Role of tRNA modifications in human diseases. Trends Mol Med 20: 306–314. 10.1016/j.molmed.2014.01.008 [DOI] [PubMed] [Google Scholar]
- Torres AG, Pineyro D, Rodriguez-Escriba M, Camacho N, Reina O, Saint-Leger A, Filonava L, Batlle E, Ribas de Pouplana L. 2015. Inosine modifications in human tRNAs are incorporated at the precursor tRNA level. Nucleic Acids Res 43: 5145–5157. 10.1093/nar/gkv277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AG, Wulff TF, Rodriguez-Escriba M, Camacho N, Ribas de Pouplana L. 2018. Detection of inosine on transfer RNAs without a reverse transcription reaction. Biochemistry 57: 5641–5647. 10.1021/acs.biochem.8b00718 [DOI] [PubMed] [Google Scholar]
- Tsutsumi S, Sugiura R, Ma Y, Tokuoka H, Ohta K, Ohte R, Noma A, Suzuki T, Kuno T. 2007. Wobble inosine tRNA modification is essential to cell cycle progression in G(1)/S and G(2)/M transitions in fission yeast. J Biol Chem 282: 33459–33465. 10.1074/jbc.M706869200 [DOI] [PubMed] [Google Scholar]
- Tuorto F, Parlato R. 2019. rRNA and tRNA bridges to neuronal homeostasis in health and disease. J Mol Biol 431: 1763–1779. 10.1016/j.jmb.2019.03.004 [DOI] [PubMed] [Google Scholar]
- Unger FM, Takemura S. 1973. A comparison between inosine- and guanosine-containing anticodons in ribosome-free codon-anticodon binding. Biochem Biophys Res Commun 52: 1141–1147. 10.1016/0006-291X(73)90619-0 [DOI] [PubMed] [Google Scholar]
- Wolf J, Gerber AP, Keller W. 2002. tadA, an essential tRNA-specific adenosine deaminase from Escherichia coli. EMBO J 21: 3841–3851. 10.1093/emboj/cdf362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff TF, Arguello RJ, Molina Jordan M, Roura Frigole H, Hauquier G, Filonava L, Camacho N, Gatti E, Pierre P, Ribas de Pouplana L, et al. 2017. Detection of a subset of posttranscriptional transfer RNA modifications in vivo with a restriction fragment length polymorphism-based method. Biochemistry 56: 4029–4038. 10.1021/acs.biochem.7b00324 [DOI] [PubMed] [Google Scholar]
- Zhou W, Karcher D, Bock R. 2014. Identification of enzymes for adenosine-to-inosine editing and discovery of cytidine-to-uridine editing in nucleus-encoded transfer RNAs of Arabidopsis. Plant Physiol 166: 1985–1997. 10.1104/pp.114.250498 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.