

SUMMARY
An important organizing feature of the CNS is that individual neurons receive input from many different sources. Independent regulation of synaptic input is critical for the function and adaptive responses of the nervous system, but the underlying molecular mechanisms are not well understood. We identify the leucine-rich repeat (LRR)-containing protein NGL-2 (Lrrc4) as a key regulator of input-specific synapse development in the hippocampus. Using genetic deletion and shRNA-mediated knockdown, we demonstrate a role for NGL-2 in regulating the strength of synaptic transmission and spine density specifically at Schaffer collateral synapses in the stratum radiatum (SR) in CA1. NGL-2 protein is restricted to SR and spine regulation requires NGL-2’s LRR and PDZ-binding domains. Finally, loss of NGL-2 disrupts cooperative interactions between distal and proximal synapses in CA1 pyramidal cells. These results demonstrate that NGL-2 is critical for pathway-specific synapse development and functional integration of distinct inputs.
INTRODUCTION
The developing brain faces the challenge of wiring up billions of synapses that can vary significantly in their anatomy and functional properties depending on the identity of the pre- and postsynaptic cell types. In area CA1 of the hippocampus, CA3 Schaffer collateral (SC) axons target proximal dendrites, while temporoammonic (TA) axons from entorhinal cortex (EC) target the distal dendrites. Furthermore, the relative organization of these two classes of excitatory synaptic input has important consequences for how the dendrite processes incoming information to generate a specific output (Remondes and Schuman, 2002; Spruston, 2008). Such convergence of distinct classes of inputs onto a given cell is a general theme of the CNS. In order to generate specific patterns of connectivity between varieties of cell types, the brain must have precise control over the formation of each class of synapse, but the molecular mechanisms underlying this organization are only beginning to be understood. In this study, we demonstrate that the leucine-rich repeat (LRR)-containing protein netrin-G ligand-2 (NGL-2/Lrrc4) is critical for input-specific synapse development in CA1 pyramidal cells.
The family of LRR proteins has recently generated attention for its role as synaptic organizer proteins (de Wit et al., 2011). For instance, LRRTM1 and LRRTM2 (de Wit et al., 2009; Ko et al., 2009; Linhoff et al., 2009; Siddiqui et al., 2010) were identified as synaptogenic proteins that interact with presynaptic neurexins. The family of LRR-containing proteins is large (Dolan et al., 2007) and many of these proteins have striking expression patterns throughout the brain, suggesting that they might regulate specific subsets of synapses. Thus, LRR family diversity may play an important role in generating the large variety of synapses and precise connectivity seen in the vertebrate brain. To date though, most studies of these proteins have been carried out in vitro, in which it is difficult to identify classes of synapses, so our understanding of how they regulate specific synapses in the intact brain remains limited. In order to understand how members of the LRR family of proteins might contribute to the development of specific synaptic connections, it is critical to examine the role of LRR proteins in vivo. In this study, we explore the role of the LRR-containing protein NGL-2 in specifically regulating the differentiation and function of Schaffer collateral synapses in hippocampal area CA1.
NGL-2 is an LRR-containing synaptic protein that interacts with PSD-95 (Kim et al., 2006). NGL-2 along with NGL-1 and NGL-3 comprise an LRR subfamily and each member has a known interaction with a presynaptic binding partner. NGL-1 and NGL-2 have isoform-specific interactions with axonal glycosylphosphatidylinositol (GPI)-anchored netrin-G1 and netrin-G2, respectively (Kim et al., 2006; Lin et al., 2003), while NGL-3 interacts with the leukocyte common antigen-related (LAR) protein (Woo et al., 2009b). NGL-2 was found to be synaptogenic and to regulate structural and functional excitatory synapse development in vitro (Kim et al., 2006). Although NGL mRNA is expressed widely (Kim et al., 2006), mRNA expression of their unique presynaptic binding partners is limited to discrete brain areas (Kwon et al., 2010; Nakashiba et al., 2002; Yin et al., 2002). In the hippocampus, NGL-1 and netrin-G1 proteins are restricted to stratum lacunosum moleculare (SLM), whereas NGL-2 and netrin-G2 are restricted to stratum radiatum (SR) (Niimi et al., 2007; Nishimura-Akiyoshi et al., 2007), suggesting that these ligand-receptor pairs interact in distinct dendritic domains of CA1 pyramidal neurons. The laminar NGL expression patterns become diffuse in Netrin-G knockout mice (Nishimura-Akiyoshi et al., 2007), suggesting that axonal Netrin-Gs may restrict NGLs to specific dendritic compartments.
Here we investigate the role of NGL-2 in regulating specific classes of synapses onto CA1 pyramidal cells. CA1 neurons receive inputs from entorhinal cortex and CA3 neurons in distinct dendritic domains. Whereas temporoammonic axons from the entorhinal cortex make synapses onto the distal dendrites of CA1 neurons in the SLM, CA3 Schaffer collateral axons provide more proximal input to CA1 neurons in the SR. We find that NGL-2 expression in CA1 neurons selectively regulates the strength of excitatory transmission at synapses in the SR, without affecting transmission in the SLM. Loss of NGL-2 results in a decrease in the frequency of miniature excitatory postsynaptic currents (mEPSCs) as well as a complementary decrease in spine density in CA1 neurons that is restricted to dendrites in the SR. Structure-function analyses revealed that both the LRR domain and the PDZ-binding domain of NGL-2 are involved in mediating the pathway-specific effects of NGL-2. Consistent with these cellular effects, loss of NGL-2 disrupts cooperative interactions between distal and proximal synapses, resulting in impaired CA1 pyramidal cell spiking. Our study reveals a critical function for NGL-2 in regulating pathway-specific synapse development, which affects the integration of parallel excitatory inputs in CA1 neurons.
RESULTS
Analysis of NGL-2 in the Developing Hippocampus
To determine whether NGL-2 and its binding partner netrin-G2 were expressed in the developing brain, we carried out in situ hybridization on sections from rat brain at postnatal day 7 (P7) and P14. NGL-2 was expressed widely throughout the neocortex and hippocampus, while its presynaptic interactor netrin-G2 was expressed in discrete cell populations during the synaptogenic period between P7 and P14 (Figure 1A). Importantly, netrin-G2 was expressed in CA3 but not in neurons in layer 3 of entorhinal cortex that project to CA1 (see Figure S1B available online). Interestingly, NGL-1 was highly expressed in all layers of hippocampus and neocortex, but netrin-G1 displayed a restricted expression pattern (Figure S1A). In contrast to netrin-G2, netrin-G1 was highly expressed in layer 3 of the entorhinal cortex but was absent from CA3. (Figure S1B). Importantly, all of these mRNA expression patterns are consistent with reported protein expression patterns (Nishimura-Akiyoshi et al., 2007). Thus, we hypothesized that NGL-2 might specifically regulate the development of CA3-CA1 synapses.
Figure 1. Loss of NGL-2 Does Not Disrupt Gross Morphological Development or Axon Targeting in the Hippocampus.
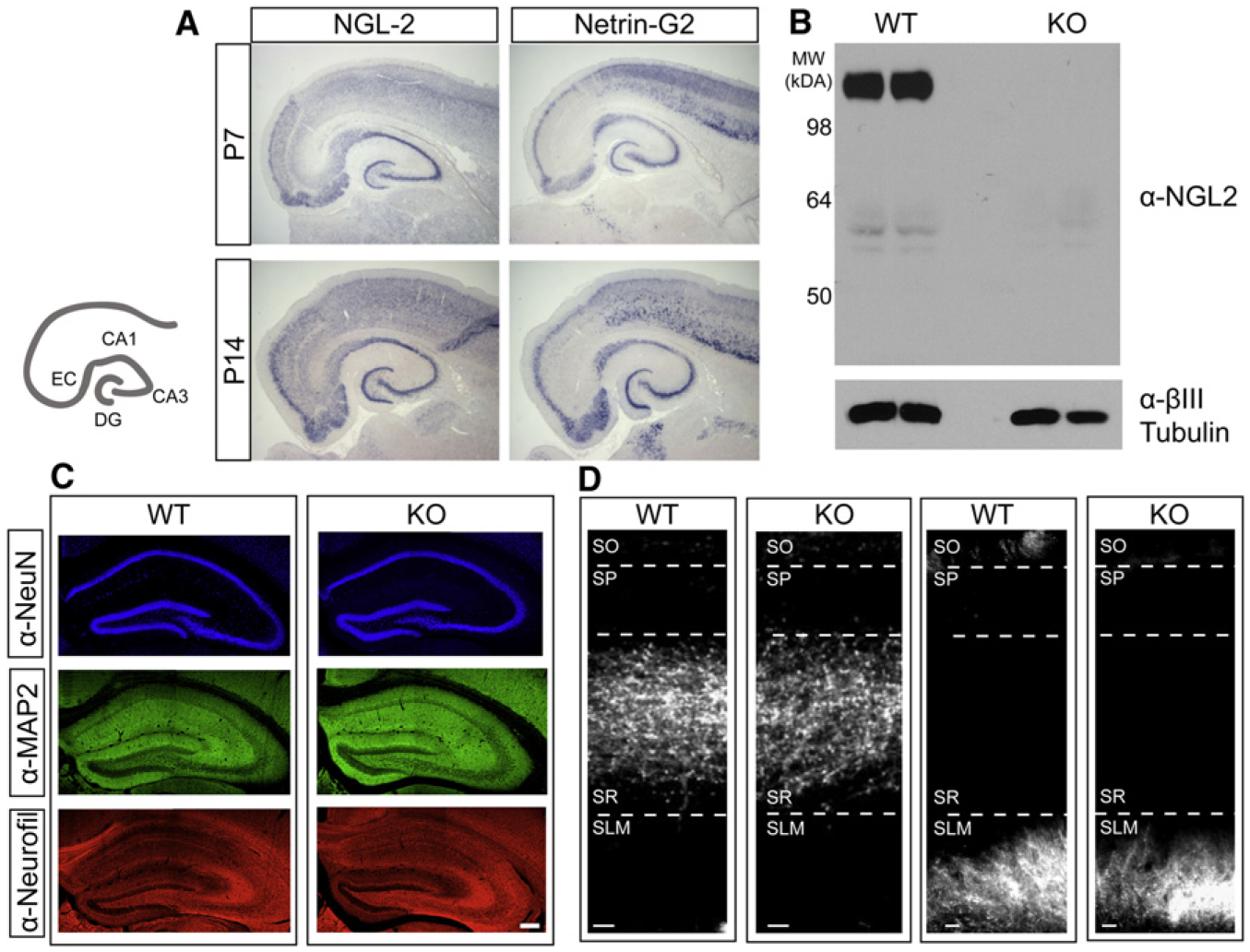
(A) Cartoon illustrates hippocampal and parahippocampal areas of interest. In situ hybridizations with antisense NGL-2 and netrin-G2 probes in horizontal sections from P7 (top) and P14 (bottom) rat brain.
(B) Western blot for NGL-2 and βIII-tubulin protein lysed from crude membranes prepared from wild-type and NGL-2 knockout littermate brains.
(C) Immunohistochemistry using antibodies against NeuN, MAP2, and Neurofilament in coronal sections from WT and NGL-2 KO mouse hippocampus. Scale bar represents 50 μm.
(D) DiI crystals were placed in CA3 (left) or in the entorhinal cortex (right) of WT and NGL-2 KO mice. Labeled axons were imaged in coronal sections from the hippocampus after 2.5–4 weeks. Scale bar represents 20 μm. See also Figure S1.
To examine the role of endogenous NGL-2 in regulating the development and function of hippocampal synapses, we obtained and analyzed NGL-2 knockout mice in which the entire coding exon of the NGL-2 gene was deleted (Zhang et al., 2008). To confirm loss of NGL-2 protein in knockout mice, we prepared crude membrane lysates from P25 wild-type (WT) and NGL-2 knockout (KO) mice and analyzed them by SDS-PAGE, followed by detection with a mouse monoclonal anti-NGL-2 antibody that targets a portion of the C-terminal domain (aa 550–662). A strong band was detected near 98 kDa in the WT brains but was absent from the KO brains (Figure 1B), confirming loss of NGL-2 protein.
To determine whether the cytoarchitecture of the hippocampus remained intact in the absence of NGL-2, we performed immunohistochemical analysis using antibodies to label neuronal nuclei (NeuN), dendrites (MAP2), or axons (Neurofilament). We found that gross hippocampal anatomy was comparable between wild-type and knockout mice (Figure 1C).
Because netrin-G2 is specifically expressed in Schaffer collateral axons (Nishimura-Akiyoshi et al., 2007), we wanted to determine whether loss of NGL-2 affected axon targeting to CA1. To do so, we placed DiI crystals in CA3 or in the entorhinal cortex in fixed wild-type and NGL-2 knockout brains (Figure 1D). The hippocampus was sectioned and imaged to determine whether axons invading CA1 maintained their laminar targeting. In both wild-type and knockout conditions, Schaffer collateral axons were restricted to the stratum radiatum and temporoammonic axons from EC were restricted to the stratum lacunosum moleculare (Figure 1D), indicating that NGL-2 does not affect laminar axon targeting in CA1. Based on the unique expression pattern of netrin-G2 and the fact that NGL-2 does not affect axon guidance, we initiated a series of experiments to determine whether NGL-2 regulates the development of specific subsets of synapses in CA1.
NGL-2 Selectively Regulates Excitatory Transmission at Schaffer Collateral Synapses in CA1 Stratum Radiatum
To determine whether NGL-2 has a general or specific role in regulating synapses, we recorded field excitatory postsynaptic potentials (fEPSPs) in CA1 in acute slices prepared from P13–P16 NGL-2 KO mice and wild-type littermates. Recording and stimulating electrodes were placed in the SR and SLM (Figure 2A). We were confident that we were stimulating the pathways in isolation because stimulation of SC axons caused a downward deflection (sink) in the SR field recording, while stimulation of TA axons caused an upward deflection (source) and vice-versa for the SLM field recordings (data not shown). Dendritic field responses were recorded in each pathway at three to five different stimulation intensities. Remarkably, we found that normalized SR field responses in NGL-2 null mice were significantly reduced compared to controls (Figure 2B), but SLM responses were not affected (Figure 2C), indicating that NGL-2 exerts a pathway-specific effect on synaptic transmission in CA1 neurons.
Figure 2. NGL-2 Regulates the Strength of Transmission and Spine Density at Subsets of Excitatory Synapses in CA1.
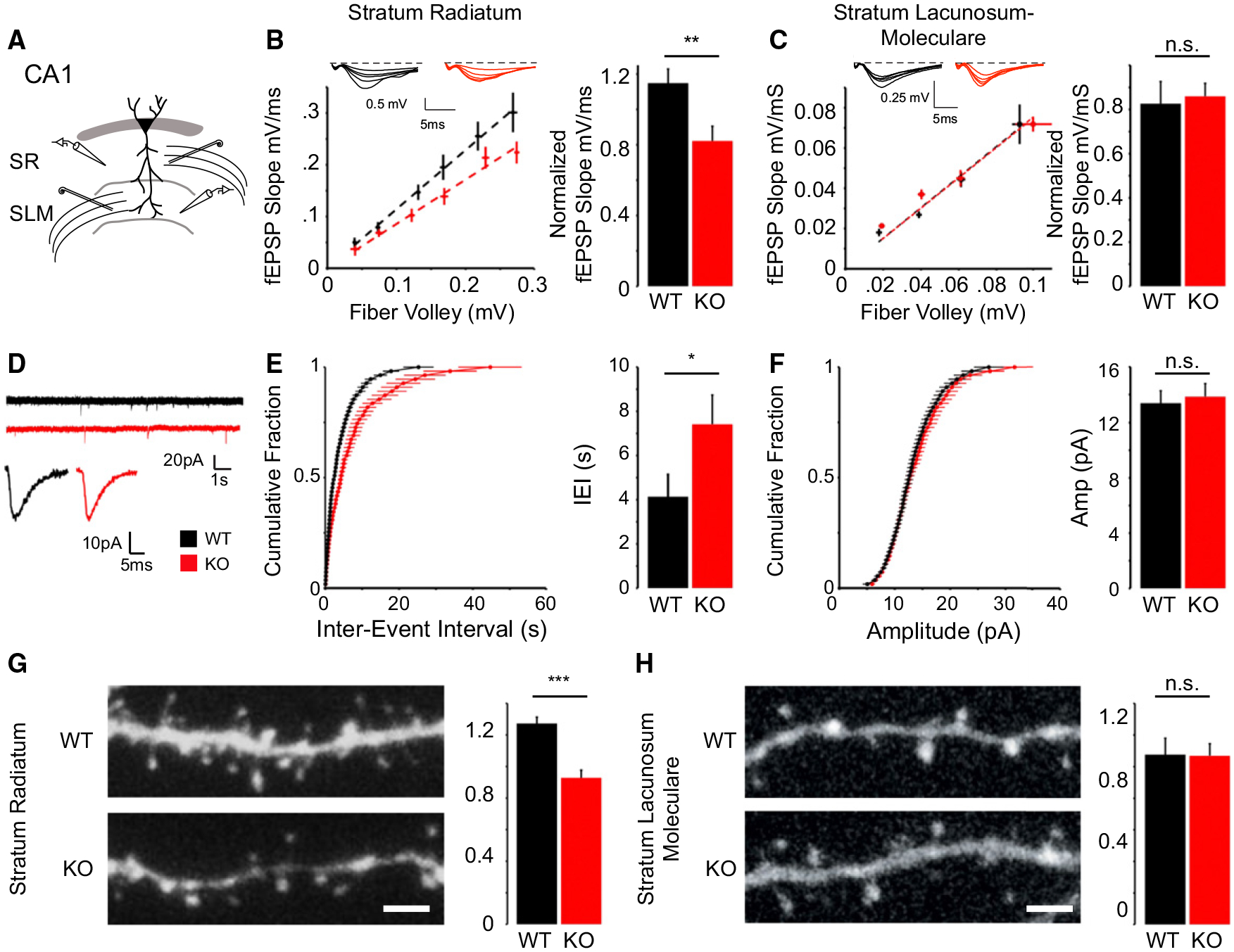
(A) Schematic of field recording configuration in CA1. Bipolar stimulating electrodes were placed in the stratum radiatum and stratum lacunosum moleculare. Glass recording electrodes filled with ACSF were placed nearby in the same layer to measure the fiber volley and field response in each pathway.
(B) NGL-2 knockout mice have a decreased normalized fEPSP slope in stratum radiatum (WT = 1.14 ± 0.05, n = 15; KO = 0.82 ± 0.08; n = 16; p < 0.01, Student’s t test).
(C) There is no difference between the normalized fEPSP slope in SLM (WT = 0.82 ± 0.10, n = 10; KO = 0.86 ± 0.05, n = 7; p = 0.79, Student’s t test).
(D) Example recordings of miniature excitatory postsynaptic currents in wild-type and NGL-2 knockout mice shown on compressed (top) and expanded (bottom) timescales.
(E and F) KO animals have a significantly greater interevent interval (WT = 4.26 ± 0.59 s, n = 17; KO = 7.41 ± 13 s, n = 13; p < 0.05, Student’s t test) (E) but mEPSC amplitude is unchanged (WT = 13.37 ± 0.89 pA, n = 17; KO = 13.85 ± 0.93 pA, n = 13; p = 0.71, Student’s t test) (F).
(G) NGL-2 KO mice exhibit reduced spine density in CA1 dendrites spanning stratum radiatum (WT: 1.27 ± 0.39 protrusions/μm, n = 32; KO: 0.927 ± 0.04 protrusions/μm, n = 16; p < 0.001, Student’s t test).
(H) NGL-2 KO mice exhibit no significant difference in spine density in SLM compared to WT littermates (WT: 1.07 ± 0.10 protrusions/μm, n = 8; KO: 1.07 ± 0.95 protrusions/μm, n = 11; p = 0.95, Student’s t test). All summary statistics are reported as mean ± SEM.
To determine whether NGL-2 regulates the function of individual synapses, we recorded mEPSCs from CA1 pyramidal cells in acute slices prepared from wild-type and NGL-2 knockout mice (Figure 2D). Voltage-clamp recordings at 70mV in the presence of tetrodotoxin (TTX) indicated that loss of NGL-2 caused a significant decrease in frequency of mEPSCs (Figure 2E) without affecting mEPSC amplitude (Figure 2F). Thus, NGL-2 appears not to affect the postsynaptic response of individual synapses but more likely acts by regulating synapse density or release probability in the stratum radiatum, which would affect mEPSC frequency.
Since excitatory synapses tend to form on spine heads in CA1 (Fiala et al., 1998), we analyzed spine density in wild-type and knockout mice to determine whether there was an anatomical correlate to the reduction in mEPSC frequency we observed. To do so, we filled CA1 neurons in fixed sections with fluorescent dye and analyzed spine density in dendritic segments in SR and SLM. We found that the NGL-2 knockout mice exhibited a specific decrease in spine density in SR (Figure 2G) but no change relative to WT in SLM (Figure 2H). In combination with our functional data, these findings demonstrate that NGL-2 specifically regulates spine and synapse density in stratum radiatum.
To investigate whether loss of NGL-2 alters the probability of release, we measured short-term plasticity properties at synapses in both the SR and the SLM of wild-type and knockout mice. Whole-cell recordings were performed from CA1 pyramidal cells clamped at −70mV while stimulating electrodes were placed in the SR and SLM (Figure 3A). Trains of five stimuli were delivered at 5, 10, and 20 Hz. No difference in the normalized amplitude of EPSCs throughout the train or in the facilitation ratio between the first and the fifth peaks was detected between wild-type and knockout mice for any interval in either pathway (Figures 3B and 3D), suggesting that NGL-2 does not regulate the probability of release. Together with the change in mEPSC frequency, these data support the hypothesis that NGL-2 primarily acts postsynaptically to regulate synapse density.
Figure 3. Loss of NGL-2 Does Not Affect AMPA/NMDA Ratio or Short-Term Facilitation in CA1.
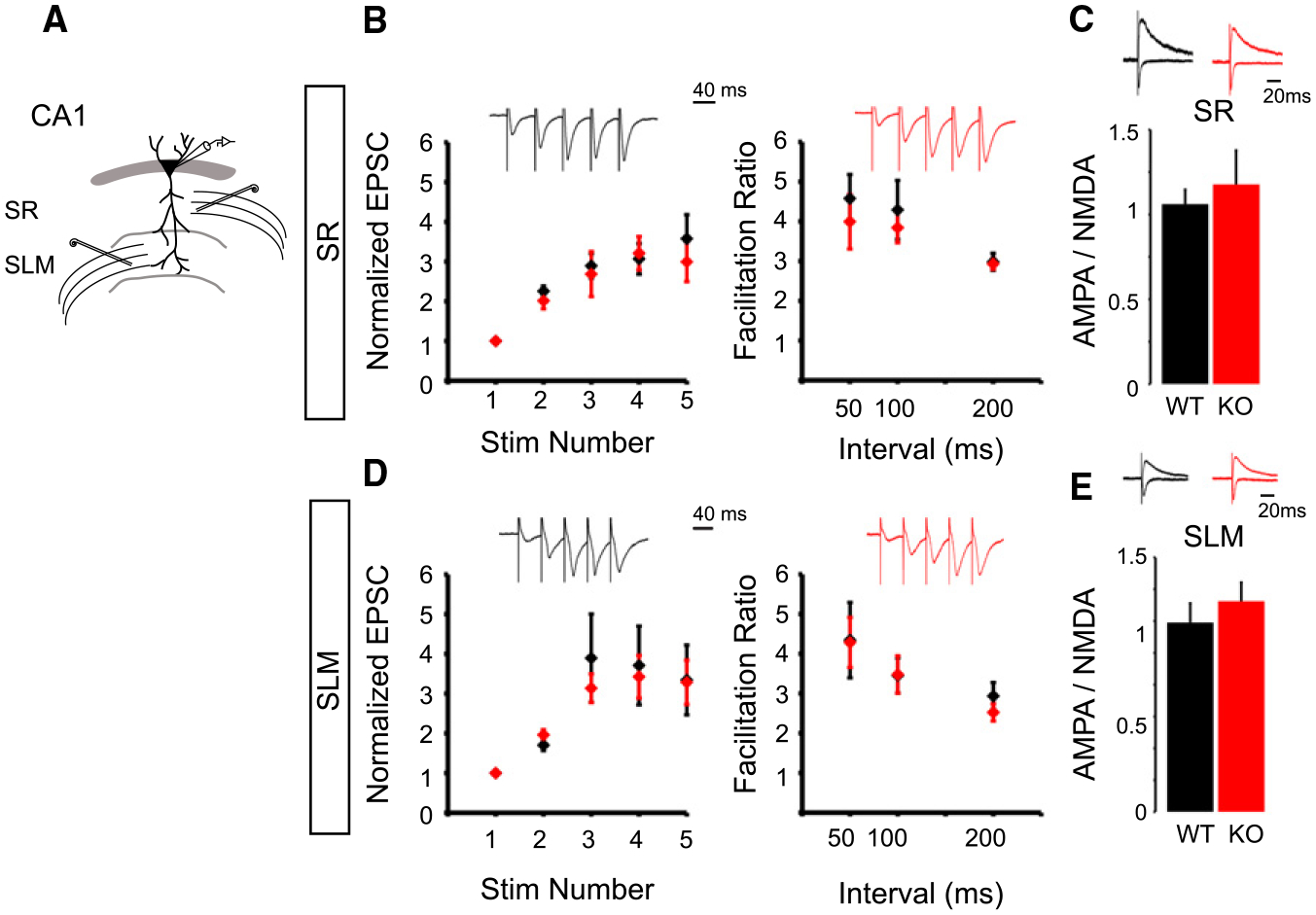
(A) Schematic of CA1 dual pathway recording experiments. Whole-cell recordings were performed from CA1 pyramidal cells in WT and NGL-2 KO littermates while stimulating the SR and SLM in an alternating manner.
(B) Loss of NGL-2 does not affect short-term facilitation in CA1. Trains of five pulses were delivered to SR or SLM at 5, 10, or 20 Hz. Example traces show trains at 20 Hz. At 20 Hz, there was no difference in the amplitude of the normalized EPSCs in a train of five stimuli in WT versus KO (EPSC 2:WT= 2.25 ± 0.13, n = 19;KO= 2.01 ± 0.20, n = 13; p = 0.30; EPSC 3: WT = 2.89 ± 0.31, n = 8; KO = 2.80 ± 0.51, n = 7; p = 0.88; EPSC 4: WT = 3.06 ± 0.38, n = 8; KO = 3.20 ± 0.76, n = 6; p = 0.86; EPSC 5: WT = 3.57 ± 0.60, n = 8; KO = 2.99 ± 0.68, n = 6; p = 0.53, Student’s t test). There was no difference in the facilitation ratio of EPSC5/EPSC1 in 5, 10, or 20 Hz trains in WT versus KO (50 ms: WT = 4.57 ± 0.60, n = 8; KO = 4.10 ± 0.59, n = 7; p = 0.58; 100 ms: WT = 4.28 ± 0.74, n = 10; KO = 3.84 ± 0.37, n = 10; p = 0.59; 200 ms: WT = 2.98 ± 0.21, n = 12; KO = 2.93 ± 0.14, n = 9; p = 0.85, Student’s t test).
(C) Loss of NGL-2 also does not affect AMPA/NMDA ratio in SR (WT = 0.85 ± 0.06, n = 21; KO = 0.92 ± 0.16, n = 16; p = 0.57, Student’s t test).
(D) NGL-2 does not regulate short-term facilitation at SLM synapses in CA1. At 20 Hz, there was no difference in the amplitude of the normalized EPSCs in a train of five pulses (EPSC 2: WT = 1.70 ± 0.13, n = 18; KO = 1.96 ± 0.13, n = 18; p = 0.18; EPSC 3: WT = 3.89 ± 1.19, n = 6; KO = 3.13 ± 0.40, n = 7; p = 0.53; EPSC 4: WT = 4.33 ± 0.91, n = 8; KO = 3.42 ± 0.61, n = 7; p = 0.43; EPSC 5: WT = 3.34 ± 0.94, n = 6; KO = 3.28 ± 0.63, n = 7; p = 0.95, Student’s t test). NGL-2 also does not affect the facilitation ratio in 5, 10, or 20 Hz trains of stimuli to the SLM (50 ms: WT = 4.34 ± 0.94, n = 6; KO = 4.28 ± 0.63, n = 7, p = 0.95; 100 ms: WT = 3.45 ± 0.44, n = 8; KO = 3.47 ± 0.46, n = 7; p = 0.97; 200 ms: WT = 2.93 ± 0.34, n = 8; KO = 2.52 ± 0.21, n = 8; p = 0.32, Student’s t test).
(E) SLM AMPA/NMDA ratio was unaffected in the NGL2 KO (WT = 1.19 ± 0.12, n = 18; KO = 1.32 ± 0.12, n = 19; p = 0.42, Student’s t test). All summary statistics are reported as mean ± SEM.
To determine whether NGL-2 regulates the complement of AMPA- and NMDA-type glutamate receptors at synapses, we measured the ratio of AMPA to NMDA receptor-mediated currents at synapses in the SR and SLM. In these experiments, we performed whole-cell recordings from CA1 pyramidal cells while stimulating axons in SR and SLM in an alternating manner (Figure 3A). We clamped the membrane potential at −70mV to isolate AMPA receptor-mediated currents and then depolarized the cell to +40mV to measure the compound EPSC. We analyzed the amplitude of the NMDA receptor-mediated EPSC 50 ms after the stimulus artifact, at which time the fast AMPAR-mediated component had decayed and the remaining current could be attributed to NMDARs. No change was detected between wild-type and NGL-2 knockout mice (Figures 3C and 3E), indicating that NGL-2 does not affect the ratio of AMPA to NMDA receptor-mediated transmission.
NGL-2 Cell Autonomously Regulates Synaptic Transmission at Stratum Radiatum Synapses onto CA1 Pyramidal Neurons
While the analysis of NGL-2 null mice provided clear genetic evidence for a role for NGL-2 in regulating synaptic transmission at individual synapses, it did not conclusively reveal whether NGL-2 expressed in CA1 pyramidal cells was responsible for this effect since the mouse we used was a global knockout. To determine whether NGL-2 regulates the strength of synaptic transmission and synapse density in a cell-autonomous manner, we cloned an shRNA targeting NGL-2 (Kim et al., 2006) into a lentiviral vector that contained enhanced green fluorescent protein (EGFP) driven by the CaMKII promoter (Dittgen et al., 2004). shNGL2 caused a strong reduction in the expression of mycNGL2 protein in HEK293T cells. By contrast, expression of the shRNA-resistant construct mycNGL2*, which has two silent point mutations in the shRNA-targeting region, was unaffected (Figure 4A). In addition, shNGL2 did not affect the expression of mycNGL1, indicating that NGL-2 knockdown was effective and target sequence specific (Figure 4A).
Figure 4. shNGL2 Causes a Reduction in AMPAR- and NMDAR-Mediated Currents Selectively in the Stratum Radiatum in CA1.
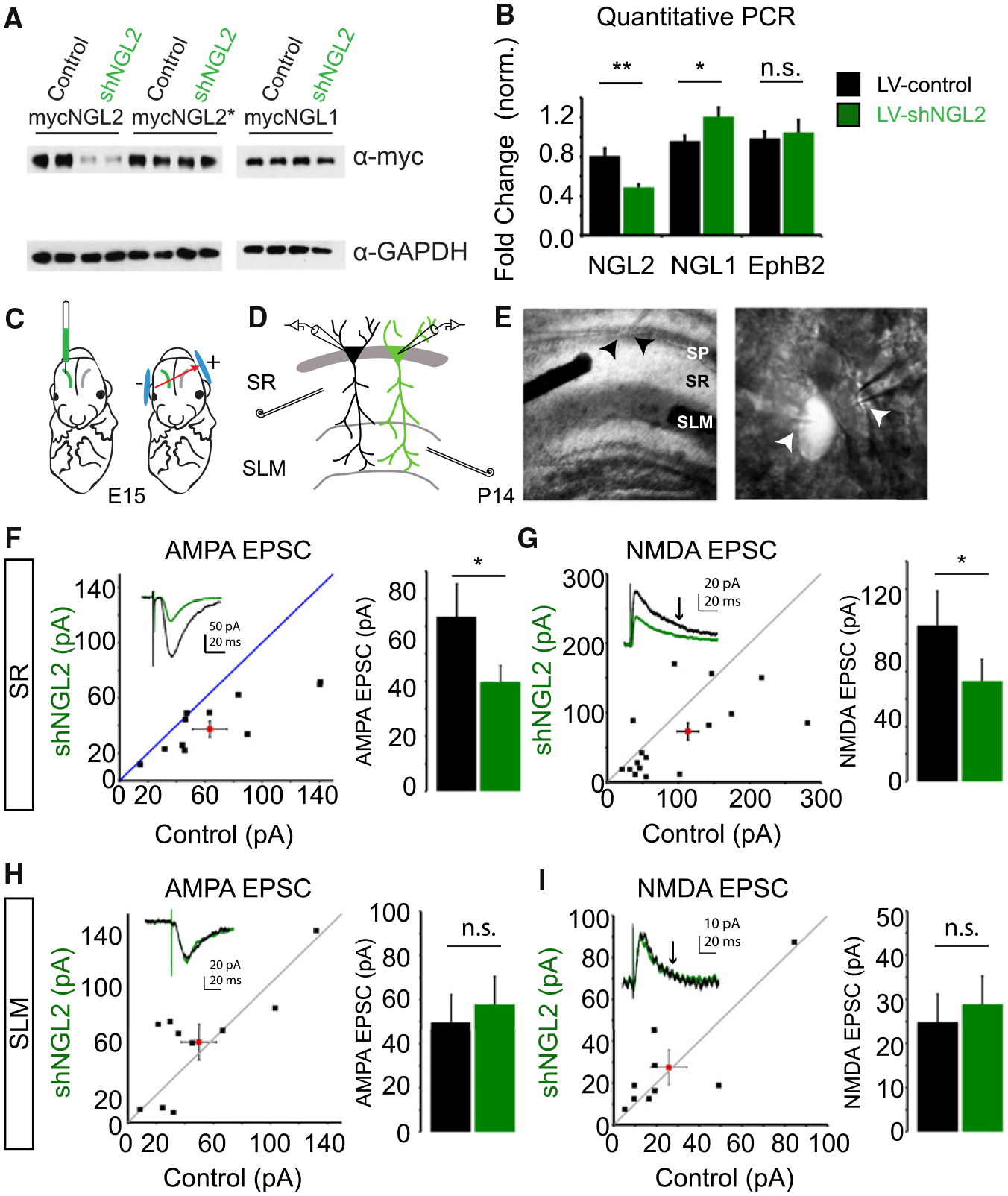
(A) Control and shNGL-2 plasmids were coex-pressed with mycNGL2, mycNGL2*, and mycNGL1 in HEK293T cells. NGL expression was analyzed by western blot.
(B) LV-control virus or LV-shNGL2 virus was expressed in cultured cortical neurons from DIV6–DIV9. mRNA was isolated and analyzed by qPCR. mRNA levels normalized to GAPDH for NGL2 (LV-control virus 0.96 ± 0.07, n = 3; LV-shNGL2 0.48 ± 0.02, n = 3; p < 0.01, Student’s t test), NGL1 (LV-control 0.80 ± 0.05, n = 3; LV-shNGL2 1.20 ± 0.090, n = 3; p < 0.05, Student’s t test), and EphB2 (LV-control 0.98 ± 0.06, n = 3; LV-shNGL2 1.04 ± 0.12, n = 3; p = 0.49, Student’s t test).
(C) Control plasmid or shNGL2 plasmids containing cytosolic GFP were electroporated into E15 embryos.
(D) At P12–P16, acute slices were prepared for paired recordings from neighboring electroporated (shNGL2) and unelectroporated (control) neurons. Whole-cell voltage-clamp recordings were performed while stimulating the SR and SLM in an alternating manner.
(E) Example acute slice and pair of recorded neighboring neurons. Arrowheads indicate recording electrode location.
(F) shNGL2 significantly reduces the amplitude of AMPAR-mediated EPSCs at SR synapses (control 63.89 ± 12.03 pA, n = 12; shNGL2 39.08 ± 5.88 pA, n = 12, p < 0.05, Student’s t test).
(G) shNGL2 significantly reduces the amplitude of NMDAR-mediated EPSCs at SR synapses (control 113.42 ± 25.11 pA, n = 17; shNGL2 73.01 ± 615.62 pA, n = 17, p < 0.05, Student’s t test).
(H) shNGL2 has no effect on the amplitude of AMPAR-mediated currents at SLM synapses (control 49.77 ± 12.47 pA, n = 10; shNGL2 57.80 ± 12.72 pA, n = 10; p = 0.34, Student’s t test).
(I) shNGL2 has no effect on the amplitude of NMDAR-mediated currents at SLM synapses (control 25.76 ± 8.46 pA, n = 9; shNGL2 27.51 ± 8.35 pA, n = 9, p = 0.73, Student’s t test). All summary statistics are reported as mean ± SEM. See also Figure S2.
To ensure that shNGL2 effectively knocked down endogenous NGL-2, we infected cultured cortical neurons with a control virus (LV-control) or a virus expressing shNGL2 (LV-shNGL2) and performed quantitative PCR (qPCR) analysis to assess the levels of NGL-2 mRNA. LV-shNGL2 caused a significant reduction in the level of NGL-2 mRNA. LV-shNGL2 also caused a small increase in the level of NGL-1 mRNA and no change in the level of EphB2, a non-NGL family transsynaptic protein (Figure 4B). Since shNGL2 does not directly affect NGL-1 levels (Figure 4A), the increase in NGL-1 levels may be a homeostatic response to the reduction in levels of NGL-2.
To further confirm the specificity of the shRNA, we performed postnatal injections of LV-shNGL2 into the CA1 region of NGL-2 KO mice such that the intended target of the shRNA was not present. In this case, if the shRNA only acts on NGL-2 mRNA, there should be no effect on excitatory synaptic transmission. To test this, we performed simultaneous whole-cell recordings from CA1 pyramidal cells that were infected with LV-shNGL2 and neighboring control cells in the NGL-2 knockout background. We measured the amplitudes of both AMPAR- and NMDAR-mediated EPSCs while stimulating shared inputs in SR. We found that the shRNA had no effect on the amplitudes of AMPAR-mediated (Figure S2A) or NMDAR-mediated (Figure S2B) EPSCs in the NGL-2 KO, confirming that our shRNA does not cause off-target effects that lead to changes in excitatory synaptic transmission in CA1.
To examine the consequences of postsynaptic NGL-2 knockdown on excitatory synaptic transmission, we used in utero electroporation to deliver an shNGL2 plasmid to a subset of CA1 pyramidal cells (Figure 4C) and prepared acute slices from electroporated mice at P12–P16. Electroporated neurons were identified by GFP epifluorescence. We performed whole-cell recordings from neighboring electroporated and unelectroporated neurons while stimulating SR and SLM synapses in an alternating manner (Figures 4D and 4E). Again, cells were voltage clamped at −70mV to measure AMPAR-mediated EPSCs and then depolarized to +40mV to measure the NMDAR-mediated EPSCs 50 ms after the stimulus onset. NGL-2 knockdown caused a decrease in AMPAR-mediated currents (Figure 4F) and a similar decrease in NMDAR-mediated currents (Figure 4G) in the stratum radiatum. In contrast, NGL-2 knockdown had no effect on AMPAR- or NMDAR-mediated currents in the SLM (Figures 4H and 4I), suggesting that NGL-2 acts postsynaptically to specifically regulate Schaffer collateral synapses in CA1.
Expression of shNGL2 had no effect on the ratio of AMPAR- to NMDAR-mediated currents in either SR or SLM (Figures S2C and S2D), further indicating that NGL-2 does not preferentially regulate AMPA- or NMDA-type glutamate receptors. Furthermore, a control plasmid expressing only GFP had no effect on AMPAR- or NMDAR-mediated currents or on the AMPA/NMDA ratio in stratum radiatum (AMPA: control 61.68 ± 18.16 pA, n = 5; GFP 61.00 ± 18.13 pA, n = 5; p = 0.69; NMDA: control 44.85 ± 13.94 pA, n = 6; GFP 63.85 ± 23.32 pA, n = 6; p = 0.119; AMPA/NMDA: control 1.70 ± 0.30, n = 5; GFP 1.61 ± 0.39, n = 5, p = 0.75, paired Student’s t test, data not shown) demonstrating that in utero electroporation of a control plasmid does not affect synaptic transmission in stratum radiatum.
NGL-2 Selectively Regulates Spine Density in Stratum Radiatum
Since NGL-2 affects synaptic transmission selectively in the SR pathway but does not affect the properties of individual synapses, we sought to determine whether NGL-2 exerts a cell-autonomous and pathway-specific effect on synapse density. We investigated the role of postsynaptic NGL-2 in regulating spine density by knocking down NGL-2 in a subset of CA1 pyramidal cells. We electroporated the GFP-containing shNGL2 or control plasmids into embryonic day 15 (E15) mouse embryos (Figure 5A). Animals were perfused at P13–P15, the brains were sectioned and immunostained for GFP (Figure 5B), and spine density was analyzed on secondary apical dendrites in the stratum radiatum (Figure 5C). Consistent with the electro-physiological experiments, we found that NGL-2 knockdown caused a significant decrease in spine density on CA1 dendrites in stratum radiatum as compared to the GFP control (Figures 5D and 5G). To determine whether the effect on spine density was selective to the dendritic segment traversing the SR, we also measured CA1 spine density on secondary apical dendrites in the SLM and found that shNGL2 expression did not affect spine density in this domain (Figures 5D and 5H). Thus, postsynaptic knockdown of NGL-2 selectively affects spine density in the stratum radiatum without affecting spine density in the SLM, indicating that a major role of NGL-2 is to regulate synapse density in the SR pathway.
Figure 5. NGL-2 Regulation of SR Spine Density Requires Its LRR and PDZ-Binding Domains.
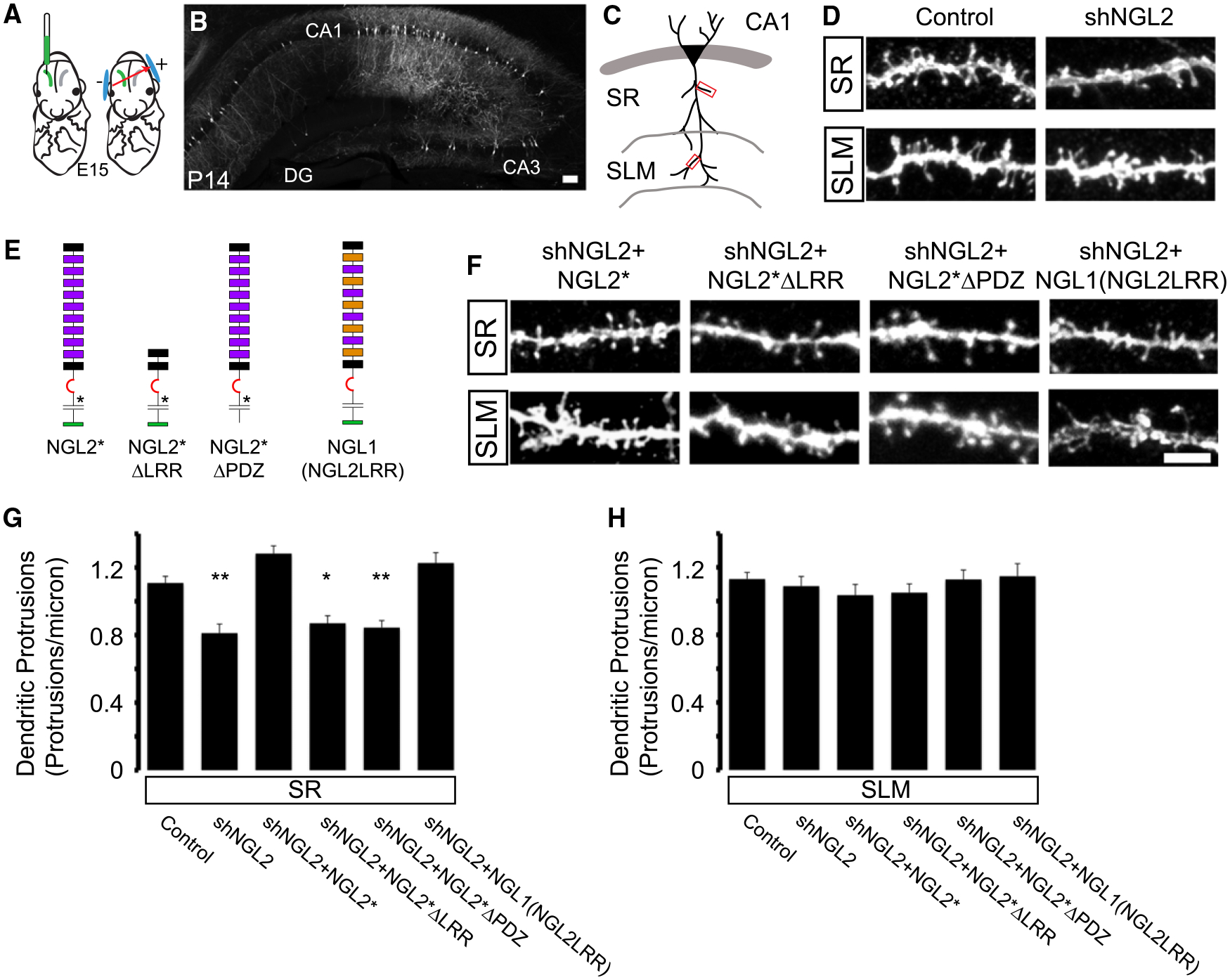
(A) We used control or shNGL2 plasmids that contained cytosolic GFP for CA1-targeted in utero electroporation at E15.
(B) Mice were perfused at P13–P15 and coronal sections were immunostained for GFP. Scale bar represents 50 μm.
(C) GFP-labeled secondary dendrites were imaged in stratum radiatum (SR) or stratum lacunosum moleculare (SLM) using a confocal microscope.
(D) Example images from secondary dendrites in SR (top) and SLM (bottom) electroporated with control or shNGL2 plasmids.
(E) Schematic of NGL-2 shRNA-resistant expression constructs that were coelectroporated with shNGL2 for rescue experiments (NGL2*, NGL2*ΔLRR, NGL2*ΔPDZ, NGL1(NGL2LRR)).
(F) Example images from secondary dendrites in SR (top) and SLM (bottom) that were coelectroporated with shNGL2+rescue plasmids. Scale bar represents 5 μm.
(G) Quantification of SR spine density demonstrates that shRNA-mediated reduction in spine density can be rescued by coelectroporating NGL2* or NGL1(NGL2LRR), but not NGL2*ΔLRR or NGL2*ΔPDZ (control 1.10 ± 0.03 protrusions/μm, n = 18; shNGL2 0.81 ± 0.05 protrusions/μm, n = 13; shNGL2+NGL2* 1.28 ± 0.04 protrusions/μm, n = 22; shNGL2+NGL2*ΔLRR 0.87 ± 0.04 protrusions/μm, n = 14; shNGL2+NGL2*ΔPDZ 0.84 ± 0.04 protrusions/μm, n = 15; shNGL2+NGL1(NGL2LRR) 1.22 ± 0.06 protrusions/μm, n = 19). One-way ANOVA, p < 0.0001. Tukey-Kramer post hoc comparisons to control are shown on graph (*p < 0.05, **p < 0.01).
(H) Quantification of SLM spine density demonstrates that neither shNGL2 nor the rescue conditions affect spine density in SLM (control 1.13 ± 0.03 protrusions/μm, n = 17; shNGL2 1.08 ± 0.05 protrusions/μm, n = 13; shNGL2+NGL2* 1.03 ± 0.05 protrusions/μm, n = 16; shNGL2+NGL2*ΔLRR 1.05 ± 0.05 protrusions/μm, n = 14; shNGL2+NGL2*ΔPDZ 1.12 ± 0.05 protrusions/μm, n = 18; shNGL2+NGL1(NGL2LRR) 1.14 ± 0.07 protrusions/μm, n = 16). One-way ANOVA, p = 0.65. All summary statistics are reported as mean ± SEM. See also Figure S3 and Table S1.
NGL-2 Regulation of Spine Density Requires the LRR and PDZ-Binding Domains and Relies on the Specific Interaction with Netrin-G2
To identify the domains of NGL-2 that mediate its synaptic effects, we coelectroporated shNGL2 with an shRNA-insensitive full-length NGL2* or domain deletion mutants and quantified spine density in the SR and SLM (Figures 5E–5H). Expressing NGL2* rescued spine density back to control levels on dendrites in the SR (Figures 5F and 5G). We observed no change in spine density in the SLM (Figures 5F and 5H), which is consistent with the targeting of NGL-2 to SR synapses.
To determine whether the LRR and PDZ-binding domains of NGL-2 contribute to the spine effects of NGL-2, we generated shRNA-resistant deletion mutants NGL2*ΔLRR and NGL2*ΔPDZ (Figure 5E). Like the full-length rescue construct, both mutants are insensitive to shNGL2 (Figure S3B) and reach the surface of HEK293T cells (Figure S3C). Unlike the full-length NGL2*, neither NGL2*ΔLRR nor NGL2*ΔPDZ could rescue the shNGL2-mediated decrease in spine density in SR (Figures 5F and 5G). Furthermore, neither mutant had an effect in SLM (Figures 5F and 5H). Thus, both the LRR and PDZ-binding domains are required for NGL-2-mediated regulation of spine density in CA1.
To further explore the roles of the LRR and PDZ-binding domains in excitatory synapse formation, we overexpressed these mutants, full-length NGL2* or EGFP control in cultured hippocampal neurons and analyzed excitatory synapse density by staining for excitatory synapse markers PSD-95 and VGlut1 (Figure S3A). We found that full-length NGL2* and NGL2*ΔPDZ both caused a significant increase in synapse density relative to EGFP control levels, while overexpressing NGL2*ΔLRR resulted in no change relative to EGFP control levels (Figure S3A). These data indicate that the LRR domain is critical for promoting excitatory synapse formation in vitro and confirm that the lack of rescue we observed in vivo is not taking place due to dominant-negative effects of the domain deletion mutant proteins.
To try to further understand the role of the specific interaction with netrin-G2, we obtained a mutant protein that we termed NGL1(NGL2LRR), in which 20 residues of the NGL-1 LRR domain have been swapped for NGL-2 residues (Seiradake et al., 2011). These mutations cause NGL1(NGL2LRR) to bind to its normal receptor, netrin-G1, with very low affinity and instead to bind netrin-G2 with high affinity (Seiradake et al., 2011). We coelectroporated shNGL2 and NGL1(NGL2LRR) into a subset of CA1 pyramidal cells and analyzed spine density in SR and SLM. We found that NGL1(NGL2LRR) could fully rescue the spine density in SR (Figures 5F and 5G) but had no effect on spine density in SLM (Figures 5F and 5H), indicating that the interaction between NGL-2 and netrin-G2 is critical for driving spine formation in SR.
The LRR Domain of NGL-2 Is Required for Proper Subcellular Localization to Spines in SR
The specific effect of NGL-2 manipulations on SR synapses suggested that NGL-2 might be localized to the dendritic domain of CA1 neurons where SR synapses form. To address this possibility, we coelectroporated GFP-tagged NGL-2 with pCAG-tdTomato as a cytosolic marker. At P14, brains were perfused, sectioned, stained for GFP, and segments of dendrites were imaged in SR and SLM (Figure 6A). The GFP signal was visible in spines and in the dendritic shaft in a pattern that was predominantly restricted to SR (Figure 6A, top), consistent with an earlier report (Nishimura-Akiyoshi et al., 2007). We quantified the intensity of the GFP signal normalized to the intensity of the tdTomato signal and found more NGL-2 in SR (Figure 6A, bottom). These data indicate that localization of NGL-2 to a restricted domain of CA1 pyramidal cells could account for the synapse-specific effect of NGL-2.
Figure 6. The LRR and PDZ-Binding Domains Are Critical for Proper Subcellular Localization of NGL-2.
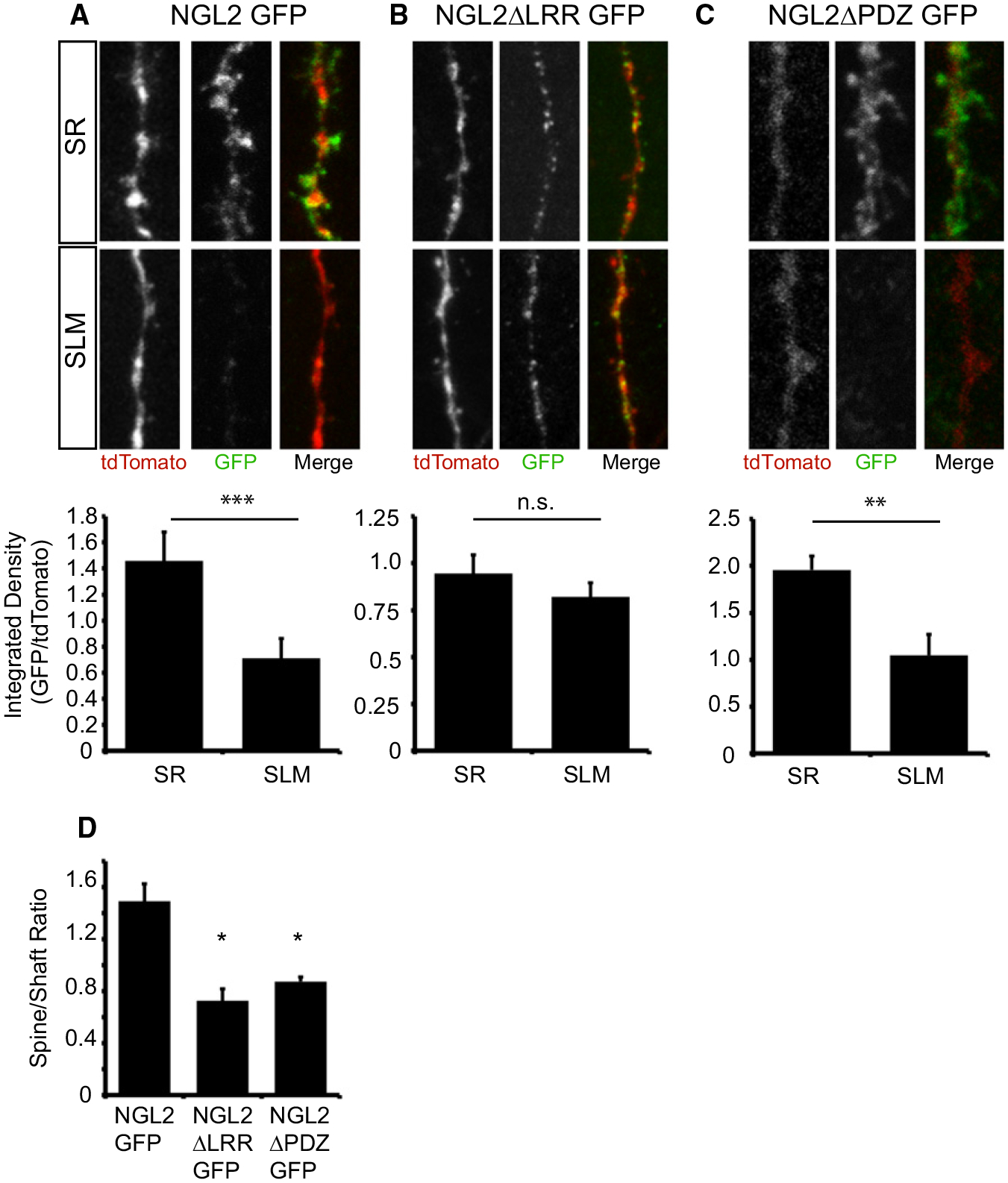
(A) Example image of secondary dendrites in SR and SLM from CA1 cells that were coelectroporated with NGL2-GFP and pCAG-tdTomato (top). Quantification of GFP/tdTomato immunofluorescence demonstrates that NGL2-GFP is preferentially localized to stratum radiatum (SR: 1.45 ± 0.21, n = 7; SLM: 0.71 ± 0.15, n = 8, p < 0.01, Student’s t test) (bottom).
(B) Example image of secondary dendrites in SR and SLM from CA1 cells that were coelectroporated with NGL2ΔLRR-GFP and pCAG-tdTomato (top). Quantification of GFP/tdTomato immunofluorescence demonstrates that NGL2ΔLRR-GFP is evenly distributed throughout SR and SLM (SR: 0.94 ± 0.09, n = 8; SLM: 0.82 ± 0.07, n = 9; p = 0.32, Student’s t test) (bottom).
(C) Example image of secondary dendrites in SR and SLM from CA1 cells that were coelectroporated with NGL2ΔPDZ-GFP and pCAG-tdTomato (top). Quantification of GFP/tdTomato immunofluorescence demonstrates that NGL2ΔPDZ-GFP is preferentially localized to SR (SR: 1.95 ± 0.14, n = 8; SLM: 1.04 ± 0.22, n = 8; p < 0.01, Student’s t test).
(D) NGL2ΔLRR-GFP exhibits impaired spine targeting. Quantifying the spine/shaft ratio of GFP fluorescence indicates that this ratio is decreased for the NGL2ΔLRR domain deletion mutant (NGL2GFP: 1.43 ± 0.13, n = 7; NGL2ΔLRR: 0.83 ± 0.20, n = 7; NGL2ΔPDZ: 0.87 ± 0.03, n = 8; p < 0.01, one-way ANOVA, *p < 0.05, post hoc Tukey test).
To test whether this localization depends on the interaction with netrin-G2, we generated a GFP-tagged LRR domain deletion mutant version of NGL-2 because the LRR domain is required for binding to Netrin-G2 (Seiradake et al., 2011). We coelectroporated this mutant with pCAG-tdTomato and analyzed the pattern of GFP immunofluorescence within CA1 dendrites. NGL2ΔLRR-GFP was expressed in a punctate pattern throughout the entire length of the CA1 pyramidal cell apical dendrites (Figure 6B, top). We quantified GFP expression levels in SR and SLM and found no significant difference between these regions (Figure 6B, bottom). In contrast to full-length NGL-2, we noticed that NGL2ΔLRR-GFP seemed to be restricted to the dendritic shaft, so we quantified a spine-to-shaft ratio of the GFP signal for the full-length protein and the LRR domain deletion mutant. We found that the mutant had a significant reduction in the spine/shaft ratio of the GFP signal (Figure 6D). Together, these data suggest that the LRR domain is required for subcellular localization in SR and for proper targeting to spines.
We also generated a GFP-tagged PDZ-binding domain deletion mutant since this domain probably mediates NGL-2 binding to PSD-95 and may also be important for proper spine targeting (Kim et al., 2006). We found that this mutant was preferentially targeted to SR (Figure 6C) but had reduced spine targeting (Figure 6D), which is consistent with what was reported in vitro (Kim et al., 2006). This suggests that NGL2*ΔPDZ failed to rescue CA1 spine density because it has impaired spine targeting.
NGL-2 Regulates the Functional Integration of SR and SLM Inputs in CA1 Pyramidal Cells
The SR and SLM pathways convey distinct information to CA1 neurons, which need to be integrated to generate a spike output. Whereas Schaffer collaterals from CA3 send indirect information from EC via a trisynaptic pathway and target proximal portions of CA1 dendrites in SR, TA axons carry sensory information directly from EC via a monosynaptic pathway and target distal CA1 dendrites in the SLM. CA1 pyramidal cells must integrate spatial information from the entorhinal cortex and contextual information from CA3 to generate the spike output of the hippocampus. Several studies have demonstrated that cooperative interactions between SLM and SR inputs can modulate both plasticity and spiking in CA1 (Dudman et al., 2007; Remondes and Schuman, 2004). Specifically timed trains of stimuli in the SLM can gate spike output from CA1 pyramidal cells, which project back to deep layers of entorhinal cortex. When the SLM train begins 20–80 ms before an SR EPSP, spike probability is greatly enhanced, which is probably due to temporal summation of the two inputs (Remondes and Schuman, 2002). This delay is consistent with the delay between the monosynaptic and trisynaptic pathways reaching CA1, which has been reported in vivo (Yeckel and Berger, 1990).
Our finding that NGL-2 regulates synaptic transmission specifically in the SR suggested that loss of NGL-2 might impair the ability of the SR and SLM synaptic inputs to cooperatively drive the output of CA1 pyramidal cells. To explore this possibility, we prepared acute hippocampal slices from WT or NGL-2 KO mice aged postnatal days 12–16. We performed whole-cell current-clamp recordings from CA1 pyramidal cells and simultaneous dendritic field recordings in SR. We used bipolar stimulating electrodes in SR and SLM to activate the two pathways independently (Figure 7A). Schaffer collateral stimulation elicited field responses that consisted of a TTX-sensitive fiber volley (FV) and a DNQX and APV-sensitive EPSP (Figure 7B). We stimulated the SLM and SR pathways at an intensity that reliably elicited an EPSP but never a spike. Stimulation of SC caused a downward deflection (sink) in the SR field, while stimulation of the TA pathway caused a small upward deflection (source) (Figure 7C), indicating that we could independently stimulate the two pathways in our configuration. We then delivered a train of ten stimuli at 100 Hz to the TA pathway followed by a single SC stimulus 20, 40, 60, or 80 ms after initiating the TA train. We found that in this paradigm, SC and SLM stimuli that were subthreshold when delivered alone were able to produce a spike when paired (Figure 7D), indicating that this spike-enhancing phenomenon originally observed in adult rats (Remondes and Schuman, 2002) also occurs in developing mice.
Figure 7. NGL-2 Regulates SLM-Mediated Gating of SR-Elicted Spikes in CA1 Pyramidal Cells.
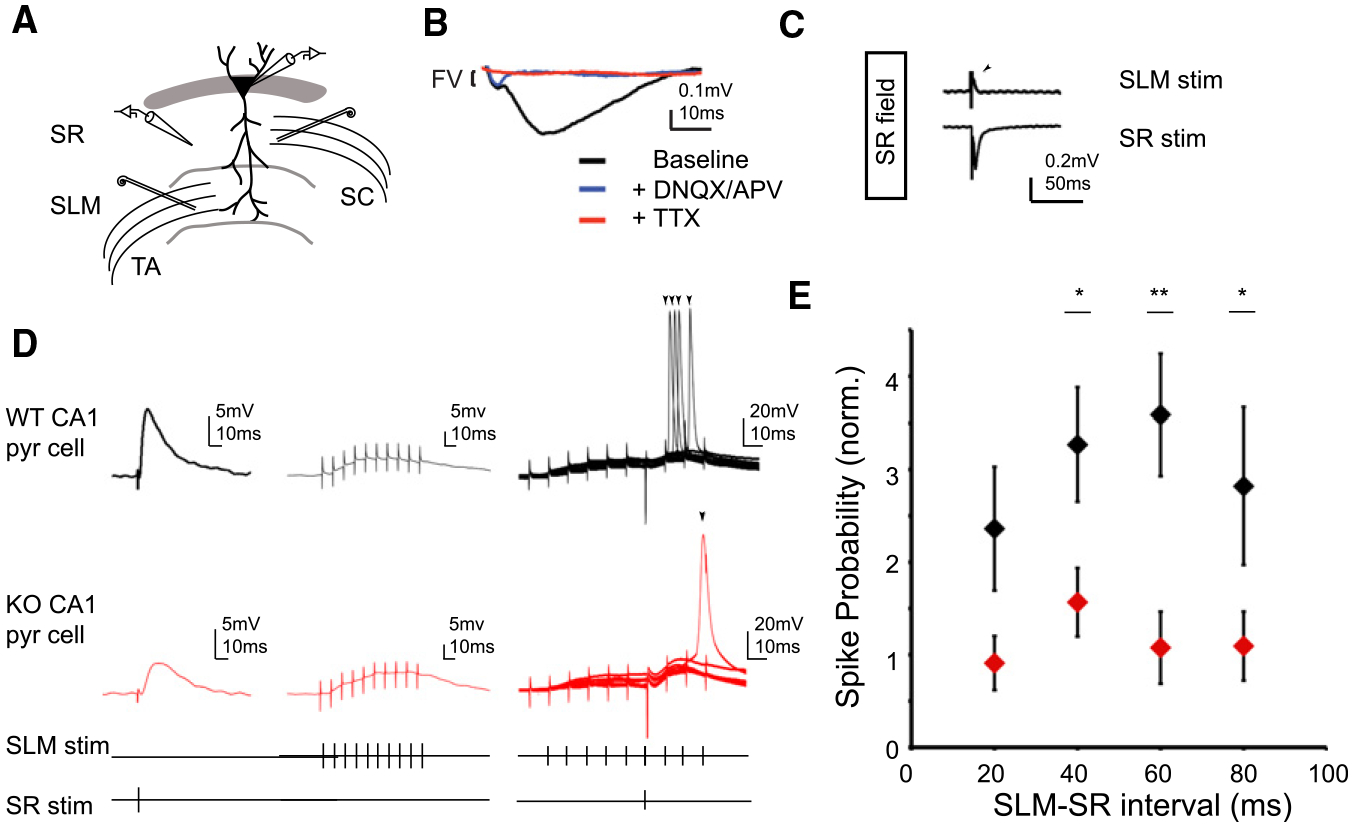
(A) Schematic of recording configuration. Whole-cell current-clamp recordings from CA1 pyramidal cells and extracellular field recordings in stratum radiatum were performed in NGL-2 KO mice and WT littermates (P12–P16). Bipolar stimulating electrodes were placed in SR and SLM.
(B) Example SR field recording. Washing in DNQX and APV abolishes the field response, isolating a TTX-sensitive fiber volley.
(C) Isolation of SC and TA field potentials. SLM stim appears as a source in SR field (top, arrowhead), while SR field stim appears as a sink (bottom).
(D) Example recordings from WT (top, black) and KO (middle, red) CA1 cells. Subthreshold EPSPs were elicited by delivering single stimuli to TA and SC axons (bottom). A train of ten stimuli at 100 Hz was then delivered to the TA pathway followed by a single SC stimulus 60 ms after the start of the TA stimulus (right). This stimulus pattern elicited spikes on some trials. Example images show eight overlaid sweeps in each condition.
(E) Spike probability normalized to fiber volley amplitude was reduced at all measured SLM-SR intervals NGL-2 KO animals (20 ms: WT = 2.36 ± 0.66, n = 9; KO = 0.67 ± 0.05, n = 5; p = 0.10; 40 ms: WT = 3.26 ± 0.72, n = 12; KO = 1.54 ± 0.36, n = 14; p < 0.05; 60 ms: WT = 3.58 ± 0.65, n = 11; KO = 0.97 ± 0.31, n = 11; p < 0.01; 80 ms: WT = 2.81 ± 0.84, n = 8; KO = 0.94 ± 0.26, n = 10; p < 0.05; Student’s t test). All summary statistics reported as mean ± SEM. See also Figure S4.
During the dual stimulation protocol, we interleaved sweeps during which we only stimulated one pathway to ensure that the single stimuli remained subthreshold throughout the duration of the experiment. We quantified normalized spike probability by dividing the number of sweeps in which the cell fired an action potential by the total number of dual stimulation sweeps and then dividing this value by the amplitude of the FV recorded in SR. This value represents the spike probability for a given number of stimulated axons. We found that the normalized spike probability was significantly reduced in NGL-2 KO animals when the SLM-SR interval was 40, 60, and 80 ms, and there was a similar trend when the interval was 20 ms (Figure 7E). We quantified a normalized value for SR-evoked EPSP by dividing the recorded EPSP amplitude by the amplitude of the SR fiber volley. We found that this value was significantly reduced in NGL-2 KO mice (Figure S4A). There was no difference in peak amplitude of the TA-evoked EPSP, resting membrane potential, or input resistance between conditions (Figures S4B–S4D). Together, these data demonstrate that reduced SR synapse density resulting from loss of NGL-2 impairs cooperative interactions between SC and TA synapses in CA1 cells. Thus, the level of NGL-2 expression strongly influences the integrated output of CA1 neurons.
DISCUSSION
In the CNS, a postsynaptic neuron typically receives synaptic input from a variety of distinct sources, but the molecular mechanisms that give rise to the formation of these different classes of synapses are not well understood. Our study demonstrates that the postsynaptic adhesion molecule NGL-2 plays a critical role in regulating the Schaffer collateral synapses onto CA1 neurons without affecting other excitatory inputs. The synapse specificity of NGL-2 action appears to be mediated by selective localization of the protein to the domain of the apical dendrite where CA1 neurons receive Schaffer collateral inputs (Figure 8). NGL-2 belongs to an LRR protein subfamily that includes NGL-1 and NGL-3, which are all expressed widely throughout the CNS (Kim et al., 2006) but interact with different presynaptic receptors (Kim et al., 2006; Lin et al., 2003; Woo et al., 2009b) that are expressed in discrete neuronal populations (Kwon et al., 2010; Nakashiba et al., 2002; Yin et al., 2002). Thus, the targeting of NGL proteins to specific dendritic domains could be a central mechanism of regulating input-specific synapse development in the CNS.
Figure 8. Proposed Model of NGL-2 Regulation of Input Specificity in CA1.
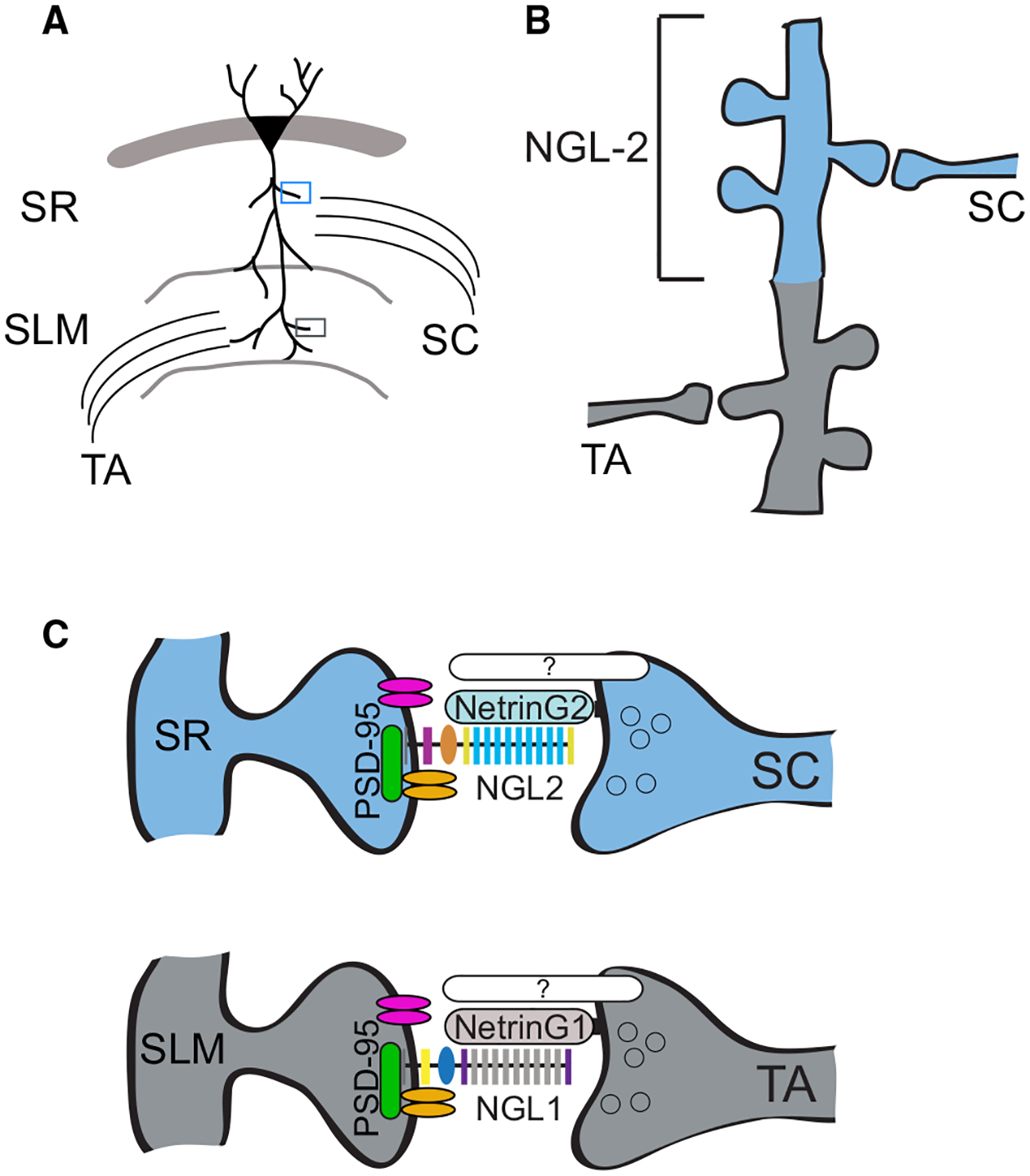
(A) Schematic of laminar excitatory pathways targeting CA1 pyramidal cell apical dendrites.
(B) NGL-2 is localized to CA1 dendrites in the stratum radiatum.
(C) NGL-2 regulates input-specific synapse development because it is restricted to SR by its interaction with Netrin-G2, which is specifically expressed in SC axons. NGL-2 interacts with PSD-95 and may recruit glutamate receptors to the nascent synapse. Distal synapses in CA1 have a complementary set of synaptic proteins, including NGL-1 and Netrin-G1.
Our conclusions are based on detailed analyses of the role of NGL-2 in the formation of synapses onto CA1 neurons. We found that NGL-2 knockout mice show a selective decrease in the strength of the SR fEPSP as well as an increase in the interevent interval of mEPSCs. NGL-2 knockdown also caused a decrease in spine density that was restricted to dendrites in the SR and required both the LRR domain and the PDZ-binding domain. Together, these findings suggest that NGL-2 specifically regulates synapse density in SR via both its transsynaptic interaction and its interaction with the postsynaptic density. As a result, loss of NGL-2 disrupts cooperative interactions between excitatory synaptic inputs in CA1 and pyramidal neuron spiking output.
Regulation of Specific Synapse Formation by NGL Proteins
We find that NGL-2 regulates the development of excitatory synapses onto CA1 pyramidal cells in a pathway-specific manner. How is this input specificity of NGL-2 function accomplished? A key factor appears to be the selective localization of NGL-2 to the SR domain in CA1. This is probably mediated by an interaction between the NGL-2 LRR domain and its presynaptic receptor, netrin-G2, which is expressed by SC axons (Nishimura-Akiyoshi et al., 2007). Seiradake et al. (2011) recently solved the crystal structures of netrin-G-NGL complexes and found that the laminin domain of netrin-G interacts with the LRR domain of NGL (Seiradake et al., 2011). Furthermore, loss of Netrin-Gs in afferent populations leads to mislocalization of NGLs (Nishimura-Akiyoshi et al., 2007), demonstrating the importance of transsynaptic interaction with netrin-G for localization to the SR domain. Consistent with these observations, we find that NGL2*ΔLRR cannot rescue SR spine density after knockdown of NGL-2 (Figures 5E–5G), while the netrin-G2-binding domain is sufficient to rescue spine density (Figures 5E–5G).
It is possible that NGL2*ΔLRR fails to rescue the spine defect because it is mislocalized or because the LRR domain directly mediates its spinogenic effect. We find that while full-length NGL2-GFP is preferentially localized to spines in SR, the NGL2ΔLRR-GFP fusion protein is expressed evenly throughout SR and SLM (Figure 6B). This diffuse localization is consistent with reports from the netrin-G2 KO mouse that suggested that specific interactions with netrin-G2 drive NGL-2 subcellular targeting to SR (Nishimura-Akiyoshi et al., 2007). While NGL2ΔLRR-GFP was present in SR, we found that it was not efficiently targeted to spines (Figure 6D), suggesting that the interactions between NGL-2 and netrin-G2 are required to localize NGL-2 to spines in SR, where it then specifically regulates spine formation. Consistent with this interpretation, Kim et al. (2006) demonstrated that full-length NGL-2 can induce presynaptic differentiation in vitro, but NGL-2 lacking the extracellular domain cannot (Kim et al., 2006).
Still, while the LRR domain is responsible for NGL-2’s interaction with netrin-G2 (Seiradake et al., 2011), direct clustering of GPI-anchored netrin-G2 does not induce clustering of presynaptic machinery (Kim et al., 2006). Since many transsynaptic organizers exhibit both forward and reverse signaling (de Wit et al., 2011), it is possible that NGL-2 regulates synapse density via a transsynaptic interaction with an additional coreceptor that recruits the components of a functional presynaptic terminal. Thus, netrin-G2 could serve as an adaptor between NGL-2 and an additional presynaptic organizer protein. Such a scenario would be similar to the interaction between EphA and GPI-anchored Ephrin-A, which requires an interaction with coreceptor p75(NTR) for proper retinotopic axon mapping (Lim et al., 2008). Netrin-Gs undergo extensive alternative splicing, most of which occurs in the EGF domains (Nakashiba et al., 2002; Yin et al., 2002), which do not directly interact with NGLs (Seiradake et al., 2011). This suggests that different netrin-G splice variants could potentially recruit different coreceptors that coordinate presynaptic differentiation.
We also find that the PDZ-binding domain of NGL-2 plays a key role in input-specific regulation of synapse development. Like NGL2*ΔLRR, NGL2*ΔPDZ also fails to rescue shNGL2-mediated reduction in spine density (Figures 5E–5G), suggesting that its interaction with PSD-95 is critical for its role in synapse formation. In support of this notion, we found that NGL2ΔPDZ GFP exhibited impaired trafficking to spines in vivo (Figures 6C and 6D). This is consistent with experiments in vitro that demonstrated that NGL-2 and PSD-95 show interdependent trafficking to synapses (Kim et al., 2006). When we overexpressed NGL2*ΔPDZ in vitro, we found that it was as effective as the full-length protein at increasing synapse density beyond control levels (Figure S3A). One possible explanation for the lack of effect of the PDZ-binding domain deletion could be if NGL2ΔPDZ dimerizes with the endogenous protein, the cis-binding partner might compensate for the deficits derived from the truncation, as has been suggested in the case of other synaptic organizer proteins (Shipman et al., 2011).
Although the specific sequence of events that leads to the formation of SR synapses in CA1 is not known, one possibility is that netrin-G2 on SC axons recruits NGL-2 to the stratum radiatum, and NGL-2 subsequently recruits PSD-95 to the nascent synapse and perhaps PSD-95 helps to maintain NGL-2 in the postsynaptic density. Consistent with our findings that the PDZ-binding domain of NGL-2 is critical for its role in regulating synapse number, the NGL-2 KO mouse has a similar phenotype to the PSD-95 KO, which also exhibits decreased mEPSC frequency in CA1 pyramidal cells, while mEPSC amplitude is unaffected (Béïque et al., 2006).
PSD-95 regulates both the number and glutamate receptor content of excitatory synapses (El-Husseini et al., 2000). Furthermore, glutamate receptors themselves play an important role in spine and synapse formation (Passafaro et al., 2003; Ripley et al., 2011; Ultanir et al., 2007). Thus, NGL-2 might regulate synapse formation by indirectly recruiting glutamate receptors to a nascent synapse via PSD-95. Alternatively, it is possible that NGL-2 directly recruits glutamate receptor subunits. NGLs coprecipitate with NMDA receptor subunits (Kim et al., 2006) and another LRR superfamily member, LRRTM2, has been shown to coprecipitate with GluR2 via its LRR domain (de Wit et al., 2009), suggesting it may have a direct interaction. Thus, NGL-2 might regulate postsynaptic development by recruiting glutamate receptors directly or via its interaction with PSD-95.
In addition to NGL-2, NGL-1 and NGL-3 also interact with PSD-95 (Kim et al., 2006). The NGLs exhibit approximately 60% sequence homology in their extracellular domains (Woo et al., 2009a) and, based on mRNA localization, they are probably expressed in many of the same cells (Kim et al., 2006). If this is the case, why does the CNS need multiple NGLs in a given postsynaptic neuron? Due to their interactions with discrete presynaptic partners, we would suggest that NGLs are responsible for controlling the distribution or relative numbers of synapses in regions of dendrites targeted by different afferent pathways. In this scenario, having multiple NGLs would allow the developing CNS to genetically control synapse density in an input-specific manner. Consistent with this idea, we find that NGL1(NGL2LRR) can rescue the shNGL2 spine phenotype, indicating that the netrin-G1/G2 binding specificity is critical in determining the pathway-restricted role of NGL proteins in spine formation in vivo. Notably, the sequences of the cytoplasmic domains are highly divergent (Woo et al., 2009a). The role of the intracellular molecular dissimilarity remains unclear, but it is possible that the different intracellular domains recruit distinct intracellular signaling cascades to confer divergent functional properties to specific subsets of synapses. A side-by-side comparison of the roles of full-length NGLs within the same cells, as well as further analysis of the consequences of deleting or swapping the C-terminal regions, will provide crucial insight to this issue.
Role of NGL-2 in Integration of SR and SLM Synaptic Inputs to CA1
Functional interactions between different classes of synaptic inputs can powerfully affect the output of neurons. In CA1, the SLM synapses may play a modulatory role in CA1 (Dudman et al., 2007). Depending on the timing relative to SR input, SLM bursts can either enhance or suppress spike probability in CA1 (Remondes and Schuman, 2002). Additionally, given different stimulation protocols, SLM bursts can also suppress, enhance, or induce SR long-term potentiation (Dudman et al., 2007; Remondes and Schuman, 2002). Importantly, our study demonstrates that the pathway-specific reduction in synapse density resulting from loss of NGL-2 impairs cooperative interactions between SR and SLM synapses that typically facilitate CA1 spike output (Remondes and Schuman, 2002) (Figure 7), suggesting that NGL-2-mediated regulation of SR synapse density is critical for the function of CA1 within the hippocampal circuit.
We find that for a given SR fiber volley amplitude, which is related to the number of stimulated Schaffer collateral axons, CA1 pyramidal cells lacking NGL-2 are much less likely to spike when they receive coincident inputs from SR and SLM synapses (Figure 7E). What differs between genotypes is the amplitude of the SR EPSP for a given fiber volley amplitude (Figure S4A); the relative amplitude of the SR EPSP is diminished in the NGL-2 KO, which is consistent with our findings that NGL-2 regulates the strength of synaptic transmission and spine density selectively in the SR of CA1. Thus, our study indicates that as a result of the decreased strength of synaptic transmission at SR synapses, coincident SLM and SR synaptic input is less effective at driving spikes in CA1 pyramidal cells that lack NGL-2 (Figure 7E).
The parallel excitatory inputs from CA3 and EC to CA1 are both implicated in generating place fields and in formation of contextual and episodic memories (Brun et al., 2008; Nakashiba et al., 2008; Remondes and Schuman, 2004; Suh et al., 2011). Furthermore, mice that have impaired plasticity in CA1 have contextual memory deficits (Tsien et al., 1996) and disrupted place field coding properties (McHugh et al., 1996). Since interactions between SR and SLM synapses are involved in plasticity in CA1 (Dudman et al., 2007; Remondes and Schuman, 2002), the relationship between these two classes of synapses is probably critical for proper CA1 function. Thus, the deficit in functional integration of inputs to CA1 in the NGL-2 knockout (Figure 7) may lead to impairments observable at the level of CA1-dependent behaviors.
In conclusion, our study demonstrates a role for the LRR-containing protein NGL-2 in specifically regulating the number of SC-CA1 synapses. Loss of NGL-2 impairs cooperative interactions between distal and proximal inputs onto CA1 pyramidal cells, implicating NGL-2 in establishing precise circuits that are critical for navigation and contextual memory. Similar dendritic integration phenomena have been observed in the neocortex, where layer V pyramidal cells also receive distinct inputs to different dendritic compartments and it has been hypothesized that these inputs could coactivate to enable coincidence detection, or the distal inputs might modulate responses to proximal inputs (Spruston, 2008). NGLs along with many other synaptic organizing proteins are expressed widely throughout the neocortex. In the case of NGLs, their presynaptic receptors netrin-Gs and LAR have unique expression patterns that implicate these complexes at distinct sets of synapses throughout the brain (Kim et al., 2006; Lin et al., 2003); thus, interactions involving NGL proteins might be critical for establishing specific circuits throughout the CNS.
EXPERIMENTAL PROCEDURES
DNA Constructs
pEGFPN-1 containing full-length mouse NGL-2 (DQ177325) and NGL2* (A1494G and T1497C; no AA change) with a Myc epitope (EQKLISEEDL) between residues 44 and 45 and pSilencer shNGL-2 were gifts from Eunjoon Kim (KAIST). Myc-NGL-2 and myc-NGL2* were both subcloned into the pEF-BOS (Mizushima and Nagata, 1990) vector downstream of the elongation factor promoter. shNGL-2 was subcloned into the pSUPER/Neo vector (Oligoengine) downstream of the H1 promoter. The H1 promoter and shNGL-2 were then subloned into the PacI site of FCK(0.4)GW (a gift from Dr. Pavel Osten, Cold Spring Harbor Laboratory) lentiviral backbone upstream of the CamKII promoter, which contains a 0.4 kb fragment of mouse CamKII protomoter-driving EGFP (Dittgen et al., 2004). FCK(0.4)GW was used as a control. NGL-2 deletion constructs were as follows: NGL2*ΔLRR (aa 79–287 deleted from full-length mouse NGL2*) and NGL2*ΔPDZ (aa 1–648 of full-length mouse NGL2*). NGL-2-GFP fusion was generated by sequentially subcloning NGL-2 cDNA obtained from Open biosystems (Thermo Fisher Scientific) in frame with GFP into the pEF-BOS vector downstream of the elongation factor promoter. All constructs were sequenced to verify integrity. NGL1(NGL2LRR) (originally termed pCA NGL1r123-mVenus) was a gift from Elena Seiradake and Alexandru Radu Aricescu.
In Situ Hybridization
In situ hybridizations were performed as described (Pasterkamp et al., 1999), using 20 μm horizontal P7 and P14 rat brain cryosections.
Detection of NGL-2 Protein Isolated from Brain Lysate
P28 mice were deeply anesthetized with isofluorane, decapitated, and brains were harvested, flash frozen, and stored at −80°C. Crude membranes were isolated by homogenizing each brain in 5 mL homogenization buffer (0.32 mM sucrose, 4 mM HEPES [pH 7.5], and protease inhibitors) using a Dounce homogenizer. Homogenate was spun at 3,000 rpm for 10 min at 4°C. Supernatant (S1) was collected and spun at 10,000 × g for 15 min at 4°C. Each pellet (P2) was resuspended in homogenization buffer and spun at 10,000 × g for 15 min at 4°C. Pellets (P2′) were lysed in RIPA buffer (150 mM NaCl, 20 mM Tris-HCl [pH 7.5], 1% Triton-X, 0.5 M EDTA, protease inhibitors) and rocked for 30 min at 4°C. Samples were centrifuged at 10,000 × g for 20 min at 4°C, and supernatant was removed and mixed with sample buffer for analysis by western blot. Western blots were probed with mouse anti-NGL2 (Clone N50/35, NeuroMab) and rabbit anti-βIII tubulin (Abcam).
Immunohistochemistry
P14 WT and KO littermate mice were given a lethal dose of sodium pentobarbital and perfused with PBS, followed by 4% paraformalydehyde (PFA) in PBS. We cut 100 μm coronal sections with a vibrating microtome (Vibratome), then blocked them in PBS containing 3% bovine serum albumin and 0.2% Triton X-100 (Sigma) for 1 hr at room temperature, and then immunostained them using standard procedures. See Supplemental Experimental Procedures for more information.
DiI Labeling of Axon Pathways
P7 mice were given a lethal dose of sodium pentobarbital and perfused with PBS, followed by 4% PFA in PBS. DiI crystals (Invitrogen) were placed in CA3 or EC of fixed brains. Brains were maintained at 37°C in a solution containing 2% PFA and 0.05% sodium azide in PBS for 2.5 (CA3) or 4 (EC) weeks. Brains were then sectioned using a vibrating microtome (Vibratome), mounted on slides in Fluorogel (Electron Microscopy Sciences), and imaged immediately on an Olympus FV300 confocal microscope.
Electrophysiology
At P12–P16, the brain was removed and placed in ice-cold carbogenated slicing artificial cerebrospinal fluid (ACSF) (83 mM NaCl, 2.5 mM KCl, 1 mM NaH2PO4, 26.2 mM NaHCO3, 22 mM glucose, 72 mM sucrose, 0.5 CaCl2, and 3.3 mM MgSO4). We cut 300 μm sagittal sections on a Leica VT1200 vibratome. Slices were allowed to recover at 31°C for 40 min and then at room temperature for 30 min to 6 hr. Slices were then placed in carbogenated recording ACSF (119 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1 mM NaH2PO4, 1.5 mM MgSO4, 2.5 mM CaCl2, and 11 mM glucose) that contained 100 mM picrotoxin (Tocris). In most experiments, a small cut was made to separate CA3 from CA1 to prevent recurrent excitation from contaminating the recording. Signals were recorded with a 5× gain, low-pass filtered at 2 kHz and digitized at 10 kHz (Molecular Devices Multiclamp 700B) and analyzed with pClamp 10 (Molecular Devices).
Whole-cell recordings were made using 3–5 MΩ pipettes filled with an internal solution that contained 150 mM potassium-D-Gluconate, 1.5 mM MgCl2, 5 mM HEPES, and 1 mM EGTA (current clamp) or 123 mM Cs-gluco-nate, 8 mM NaCl, 1 mM CaCl2, 10 mM EGTA, 10 mM HEPES, and 10 mM glucose, pH 7.3 with CsOH, 280–290 mOsm (voltage clamp). Series resistance (Rs) and input resistance (Rin) were monitored throughout the experiment by measuring the capacitive transient and steady-state deflection in response to a −5mV test pulse, respectively. Field recordings were obtained using a 1–2 MΩ pipette filled with ACSF. See Supplemental Experimental Procedures for more information.
Intracellular Injection of Alexa 594 Hydrazide
P14 mice were given a lethal dose of sodium pentobarbital and intracardially perfused with PBS, followed by 4% PFA in PBS. Brains were postfixed for 1 hr, 100 μm coronal sections were cut with a vibrating microtome (Vibratome), and then sections were postfixed for 15 min. Penetrating microelectrodes were pulled from borosilicate capillary glass with filament (1 mm outer diameter/ −0.58 mm inner diameter) and backfilled with a solution containing KCl (200 mM) and Alexa 594 hydrazide (10 mM) (Invitrogen). Slices were mounted on a glass slide under PBS and CA1 neurons were filled via iontophoresis using visual guidance. Sections were postfixed for 5 min and then mounted in Fluorogel (Electron Microscopy Sciences). Secondary apical dendrites were imaged on a Leica SP5 confocal microscope. Dendritic protrusions were counted in z stacks in NIH ImageJ and the length of dendritic segments measured with the Simple Neurite Tracer plugin blind to genotype.
Lentivirus Production
Lentivirus was produced as described earlier (de Wit et al., 2009). See Supplemental Experimental Procedures for more information.
Quantitative PCR
At 10 days in vitro (DIV), mRNA was isolated from cortical cultures using Trizol (Invitrogen) and cDNA was synthesized using iScript cDNA synthesis kit (BioRad). Quantitative PCR was performed in an Applied Biosystems PRISM 7900HT Fast Real-Time PCR system with SYBR green PCR master mix. The relative abundance of each cDNA was determined by using a standard curve generated from 10-fold serial dilutions of cDNA from rat hippocampal neurons that were infected with control lentivirus. These values were normalized to GAPDH cDNA levels. See Supplemental Experimental Procedures for more information.
In Utero Electroporation
Timed-pregnant CD-1 white mice (Charles River, E15) were anaesthetized with 3% isoflurane. The abdomen was swabbed with iodine. A small vertical incision was made in the skin and abdominal wall and embryos were gently exposed. Each embryo was injected with 1–2 μl of DNA solution and 0.01% Fast Green using a pressure-controlled bevelled glass pipette (Drummond, WPI Microbeveller). After each injection, the embryos were moistened with PBS and voltage steps via tweezertrodes (BTX, 5 mm round, platinum, BTX electroporator) were applied at a 45° angle with respect to the interaural line to target CA1. Voltage was 36V for five pulses at 1 Hz, each pulse lasting 50 ms, as described previously (Navarro-Quiroga et al., 2007). The embryos were returned to the abdomen, which was sutured, followed by suturing of the skin. The procedure typically lasted 20 min.
Imaging and Analysis of Dendritic Spines from Electroporated Animals
At P13–P15, electroporated mice were transcardially perfused with 4% PFA in PBS, brains removed and postfixed overnight in the same solution, and 100 μm coronal sections were cut on a vibratome. Sections in which CA1 pyramidal neurons visibly expressed GFP were then immunostained for GFP, imaged, and analyzed blind to transfection condition. See Supplemental Experimental Procedures for more information.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Ghosh Laboratory, Jeffry Isaacson, and Peter Scheiffele for valuable discussion and comments on the manuscript. We would also like to thank Eunjoon Kim (NGL-2, shNGL2) and Alexandru Radu Aricescu and Elena Seiradake (NGL1r123-mVenus) for plasmids and Phu Huynh for technical assistance. This work was supported by NIH grants R01NS067216 and R01NS064124 (A.G.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures, one table, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2012.10.013.
REFERENCES
- Béïque J-C, Lin D-T, Kang M-G, Aizawa H, Takamiya K, and Huganir RL (2006). Synapse-specific regulation of AMPA receptor function by PSD-95. Proc. Natl. Acad. Sci. USA 103, 19535–19540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun VH, Leutgeb S, Wu H-Q, Schwarcz R, Witter MP, Moser EI, and Moser M-B (2008). Impaired spatial representation in CA1 after lesion of direct input from entorhinal cortex. Neuron 57, 290–302. [DOI] [PubMed] [Google Scholar]
- de Wit J, Sylwestrak E, O’Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR 3rd, Comoletti D, Taylor P, and Ghosh A (2009). LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron 64, 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J, Hong W, Luo L, and Ghosh A (2011). Role of leucine-rich repeat proteins in the development and function of neural circuits. Annu. Rev. Cell Dev. Biol 27, 697–729. [DOI] [PubMed] [Google Scholar]
- Dittgen T, Nimmerjahn A, Komai S, Licznerski P, Waters J, Margrie TW, Helmchen F, Denk W, Brecht M, and Osten P (2004). Lentivirus-based genetic manipulations of cortical neurons and their optical and electro-physiological monitoring in vivo. Proc. Natl. Acad. Sci. USA 101, 18206–18211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan J, Walshe K, Alsbury S, Hokamp K, O’Keefe S, Okafuji T, Miller SFC, Tear G, and Mitchell KJ (2007). The extracellular Leucine-Rich Repeat superfamily; a comparative survey and analysis of evolutionary relationships and expression patterns. BMC Genomics 8, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudman JT, Tsay D, and Siegelbaum SA (2007). A role for synaptic inputs at distal dendrites: instructive signals for hippocampal long-term plasticity. Neuron 56, 866–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE-D, Schnell E, Chetkovich DM, Nicoll RA, and Bredt DS (2000). PSD-95 involvement in maturation of excitatory synapses. Science 290, 1364–1368. [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, and Harris KM (1998). Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J. Neurosci 18, 8900–8911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Burette A, Chung HS, Kwon S-K, Woo J, Lee HW, Kim K, Kim H, Weinberg RJ, and Kim E (2006). NGL family PSD-95-interacting adhesion molecules regulate excitatory synapse formation. Nat. Neurosci 9, 1294–1301. [DOI] [PubMed] [Google Scholar]
- Ko J, Fuccillo MV, Malenka RC, and Südhof TC (2009). LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron 64, 791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon S-K, Woo J, Kim S-Y, Kim H, and Kim E (2010). Trans-synaptic adhesions between netrin-G ligand-3 (NGL-3) and receptor tyrosine phosphatases LAR, protein-tyrosine phosphatase delta (PTPdelta), and PTPsigma via specific domains regulate excitatory synapse formation. J. Biol. Chem 285, 13966–13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim Y-S, McLaughlin T, Sung TC, Santiago A, Lee K-F, and O’Leary DDM (2008). p75(NTR) mediates ephrin-A reverse signaling required for axon repulsion and mapping. Neuron 59, 746–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JC, Ho W-H, Gurney A, and Rosenthal A (2003). The netrin-G1 ligand NGL-1 promotes the outgrowth of thalamocortical axons. Nat. Neurosci 6, 1270–1276. [DOI] [PubMed] [Google Scholar]
- Linhoff MW, Laurén J, Cassidy RM, Dobie FA, Takahashi H, Nygaard HB, Airaksinen MS, Strittmatter SM, and Craig AM (2009). An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron 61, 734–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh TJ, Blum KI, Tsien JZ, Tonegawa S, and Wilson MA (1996). Impaired hippocampal representation of space in CA1-specific NMDAR1 knockout mice. Cell 87, 1339–1349. [DOI] [PubMed] [Google Scholar]
- Mizushima S, and Nagata S (1990). pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res 18, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashiba T, Nishimura S, Ikeda T, and Itohara S (2002). Complementary expression and neurite outgrowth activity of netrin-G subfamily members. Mech. Dev 111, 47–60. [DOI] [PubMed] [Google Scholar]
- Nakashiba T, Young JZ, McHugh TJ, Buhl DL, and Tonegawa S (2008). Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science 319, 1260–1264. [DOI] [PubMed] [Google Scholar]
- Navarro-Quiroga I, Chittajallu R, Gallo V, and Haydar TF (2007). Long-term, selective gene expression in developing and adult hippocampal pyramidal neurons using focal in utero electroporation. J. Neurosci 27, 5007–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi K, Nishimura-Akiyoshi S, Nakashiba T, and Itohara S (2007). Monoclonal antibodies discriminating netrin-G1 and netrin-G2 neuronal pathways. J. Neuroimmunol 192, 99–104. [DOI] [PubMed] [Google Scholar]
- Nishimura-Akiyoshi S, Niimi K, Nakashiba T, and Itohara S (2007). Axonal netrin-Gs transneuronally determine lamina-specific subdendritic segments. Proc. Natl. Acad. Sci. USA 104, 14801–14806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passafaro M, Nakagawa T, Sala C, and Sheng M (2003). Induction of dendritic spines by an extracellular domain of AMPA receptor subunit GluR2. Nature 424, 677–681. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Giger RJ, Ruitenberg MJ, Holtmaat AJ, De Wit J, De Winter F, and Verhaagen J (1999). Expression of the gene encoding the chemorepellent semaphorin III is induced in the fibroblast component of neural scar tissue formed following injuries of adult but not neonatal CNS. Mol. Cell Neurosci 13, 143–166. [DOI] [PubMed] [Google Scholar]
- Remondes M, and Schuman EM (2002). Direct cortical input modulates plasticity and spiking in CA1 pyramidal neurons. Nature 416, 736–740. [DOI] [PubMed] [Google Scholar]
- Remondes M, and Schuman EM (2004). Role for a cortical input to hippocampal area CA1 in the consolidation of a long-term memory. Nature 431, 699–703. [DOI] [PubMed] [Google Scholar]
- Ripley B, Otto S, Tiglio K, Williams ME, and Ghosh A (2011). Regulation of synaptic stability by AMPA receptor reverse signaling. Proc. Natl. Acad. Sci. USA 108, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiradake E, Coles CH, Perestenko PV, Harlos K, McIlhinney RA, Aricescu AR, and Jones EY (2011). Structural basis for cell surface patterning through NetrinG-NGL interactions. EMBO J. 30, 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman SL, Schnell E, Hirai T, Chen BS, Roche KW, and Nicoll RA (2011). Functional dependence of neuroligin on a new non-PDZ intracellular domain. Nat. Neurosci 14, 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui TJ, Pancaroglu R, Kang Y, Rooyakkers A, and Craig AM (2010). LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J. Neurosci 30, 7495–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N (2008). Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci 9, 206–221. [DOI] [PubMed] [Google Scholar]
- Suh J, Rivest AJ, Nakashiba T, Tominaga T, and Tonegawa S (2011). Entorhinal cortex layer III input to the hippocampus is crucial for temporal association memory. Science 334, 1415–1420. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, and Tonegawa S (1996). The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87, 1327–1338. [DOI] [PubMed] [Google Scholar]
- Ultanir SK, Kim J-E, Hall BJ, Deerinck T, Ellisman M, and Ghosh A (2007). Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc. Natl. Acad. Sci. USA 104, 19553–19558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J, Kwon S-K, and Kim E (2009a). The NGL family of leucine-rich repeat-containing synaptic adhesion molecules. Mol. Cell. Neurosci 42, 1–10. [DOI] [PubMed] [Google Scholar]
- Woo J, Kwon S-K, Choi S, Kim S, Lee J-R, Dunah AW, Sheng M, and Kim E (2009b). Trans-synaptic adhesion between NGL-3 and LAR regulates the formation of excitatory synapses. Nat. Neurosci 12, 428–437. [DOI] [PubMed] [Google Scholar]
- Yeckel MF, and Berger TW (1990). Feedforward excitation of the hippocampus by afferents from the entorhinal cortex: redefinition of the role of the trisynaptic pathway. Proc. Natl. Acad. Sci. USA 87, 5832–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Miner JH, and Sanes JR (2002). Laminets: laminin- and netrin-related genes expressed in distinct neuronal subsets. Mol. Cell. Neurosci 19, 344–358. [DOI] [PubMed] [Google Scholar]
- Zhang W, Rajan I, Savelieva KV, Wang C-Y, Vogel P, Kelly M, Xu N, Hasson B, Jarman W, and Lanthorn TH (2008). Netrin-G2 and netrin-G2 ligand are both required for normal auditory responsiveness. Genes Brain Behav. 7, 385–392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.