

Abstract
We present a method to synthesize mRNAs from synthetic DNA templates that produce biologically active proteins. To illustrate utility, we constructed five unique synthetic DNA templates, produced mRNAs and demonstrated biologic activity of their translated proteins. Examples include secreted luciferase, enhanced green fluorescence protein, IL-4, and IL-12A and IL-12B to form active IL-12. We propose that this method offers a cost- and time-saving alternative to plasmid-based cloning.
Keywords: : alternative to molecular cloning, cell transfection, chemical DNA synthesis, protein translation, synthetic mRNA from DNA template
METHOD SUMMARY
In vitro transcription reactions were performed starting with a double-stranded DNA fragment with the bacteriophage SP6 promoter. A 5′ CAP and 3′ poly (A) tail were added via enzymatic reactions to these single-stranded RNAs to produce functional mRNAs. Synthetic mRNAs were transfected into HeLa cells and expression of translated proteins determined.
Since the beginning of modern molecular biology, plasmid-based cloning has been an essential component of both basic and medical research [1,2]. A limitation is that plasmid-based cloning is a complicated, multistep process. DNA needs to be isolated and plasmid-based vectors prepared. DNA fragments are ligated into plasmid-based vectors and transformed into bacteria. Finally, bacteria are plated and screened to identify the correct clone expressing the DNA construct of interest. The entire process takes many days, if not weeks, and even then there is no guarantee that the correct clone will be identified. Not only is the process time consuming, it is also expensive. Vectors, restriction enzymes, ligases, Taq polymerase, competent cells and bacterial plates are just some of the components required, and these can cost thousands of dollars.
With advances in in vitro transcription (IVT) technology from either plasmid-based DNA templates or DNA PCR amplified from plasmids, as well as in delivery vehicles including polymeric- and lipid-based nanoparticles, interest has increased in the use of RNA molecules, including mRNA molecules as therapeutics for human disease [3–8]. Examples include loss-of-function mutations such as adenosine deaminase deficiency, ornithine transcarbamylase deficiency, phenylketonuria, hemophilia B and cystic fibrosis. The use of mRNAs is also gaining in interest as an alternative to attenuated viruses, inactivated viruses or protein subunits in development of vaccines [9,10]. Examples of different mRNA candidates for either replacement therapies for diseases arising from inactivating mutations or as vaccine candidates have all advanced to various stages of preclinical or clinical development as therapeutics for their target diseases. In fact, one of five of the confirmed COVID-19 vaccine candidates that had reached the stage of clinical development by the end of April 2020 employed an mRNA-based platform for vaccination against this novel coronavirus [11]. mRNAs are also attractive candidates for a variety of therapeutic applications due to their low toxicity, low immunogenicity, cost of production and other factors compared with more traditional platforms.
For the preceding reasons, the use of mRNAs as therapeutic modalities may increase substantially in the future. Improvements in costs and ease of mRNA synthesis from DNA templates or via chemical synthetic methods, mRNA stability, efficiency of translation or lowered immunogenicity may advance therapeutic use of mRNAs. Recent advances in the chemical synthesis of DNA have significantly lowered the cost and increased the lengths of DNA that can be purchased commercially. It is now possible to purchase synthetic double-stranded DNA fragments ranging in lengths from 100 to 3000 base pairs (bp) or greater for very reasonable prices. Due to these advances, we developed a simple process to produce mRNAs from these commercially available double-stranded DNA fragments that offers a faster and significantly cheaper method to produce functional mRNAs than can be achieved by conventional cloning methods. These mRNAs can be transfected into human cells and produce functional proteins.
Methods
Cell culture
HeLa cells (ATCC, CCL-2) were cultured in RPMI 1640 (Gibco, cat. no. 21870) supplemented with 10% (vol/vol) fetal calf serum (Atlanta Biologicals, cat. no. S12450), 100 U/ml of penicillin, 100 μg/ml of streptomycin and 2 mM of L-glutamine (final concentration, Gibco, cat. no. 15140). Cells were cultured at 37°C in the presence of 5% CO2 in humidified air.
Human peripheral blood mononuclear cells (PBMCs) were isolated from healthy volunteers and stimulated with soluble mouse anti-human CD28 (1 μg ml-1; 555725; BD Biosciences) and plate-bound anti-CD3 (10 μg ml-1; OKT3 clone, American Type Culture Collection), 1 × 106 cells ml-1, under TH1 polarizing conditions (IL-12 [10 ng ml-1]), TH2 polarizing conditions (IL-4 [10 ng ml-1] or nonpolarizing conditions; no additional cytokines [12]. Culture fluids were harvested after 5 days and analyzed for expression of IFN-γ or IL-13 by ELISA. The study was approved by the institutional review board at Vanderbilt University Medical Center. Written informed consent was obtained at the time of blood sample collection.
Constructs
Double-stranded DNA molecules (gBlocks) were ordered from Integrated DNA Technologies. DNA constructs were designed with the SP6 RNA promoter sequence, 5′ untranslated region (UTR), Kozak sequence and ATG initiation sequence at the 5′ end followed by the gene insert. A 3′ UTR and short poly (A) sequence were added at the 3′ end of the double-stranded DNA molecule. Samples were resuspended in RNase/DNase-free distilled water (Invitrogen, cat. no. 10977) to a concentration of 20 ng/μl. Samples were heated at 50°C for 20 min as per manufacturer's instructions and stored at -20°C.
RNA transcription
RNA transcription was performed using the Megascript SP6 kit (Invitrogen, cat. no. AM1330) according to manufacturer's instructions. Briefly, 8 μl of DNA construct was heated for 5 min at 65°C and cooled. Then 2 μl of ATP, CTP, GTP UTP, 10× reaction buffer and SP6 RNA polymerase were added. The reaction was incubated overnight at 37°C. The next day, 1 μl of Turbo DNAse was added and incubated for 30 min. The reaction was stopped by addition of 30 μl of dH20 and 30 μl of lithium chloride precipitation solution. The reaction was incubated for 30 min at -20°C, followed by centrifugation at 4°C at 14,000 RPM, 20,000×g for 15 min. The supernatant was removed and pellet washed with 1 ml of 70% ethanol. The centrifugation step was repeated once under the same conditions. The 70% ethanol was removed and the pellet briefly air-dried prior to suspension in 50 μl of nuclease-free water. Absorbance was determined at 260 nm to quantitate yields of RNA and stored at -80°C. Yields typically totaled approximately 10–70 μg.
5′ capping reaction
The capping reaction was performed using the Vaccinia Capping System (New England Biolabs, cat. no. 2080S) following manufacturer's instructions, and capping efficiency is estimated to exceed 95%. Briefly, 10 μg of RNA was combined with nuclease-free water to a final volume of 15 μl and heated to 65°C for 5 min. The tube was cooled on ice for 5 min. The following kit ingredients were added: 2 μl of 10× capping buffer, 1 μl of 10-mM GTP, 1 μl of 2-mM S-adenosylmethionine and 2 μl of Vaccinia capping enzyme. This reaction was incubated for 30 min at 37°C [13–15].
RNA cleanup
After the capping reaction, the RNA was cleaned up using the RNeasy MinElute Cleanup Kit (Qiagen, cat. no. 74204) following manufacturer's instructions.
Poly (A) tailing reaction
The addition of a poly (A) tail was performed using E. coli Poly (A) polymerase (New England Biolabs, cat. no. M276S), following manufacturer's instructions. Briefly, up to 10 μg of capped RNA was diluted to 15 μl using nuclease-free water. To the RNA, 2 μl of 10× E. coli Poly (A) polymerase reaction buffer, 2 μl of 10-mM ATP and 1 μl of E. coli Poly (A) polymerase (5 units) were added and incubated for 30–60 min at 37°C. The capped and tailed RNA was purified using the RNA cleanup protocol described earlier and quantitated. Analysis of the synthetic mRNA by agarose gel electrophoresis demonstrated that the efficiency of poly (A) tail addition was >95% (Figure 1).
Figure 1. . Analysis of addition of poly (A) tail to synthetic RNAs by agarose gel electrophoresis.

From left to right, the 1-kb ladder and the indicated synthetic RNAs before (-) and after (+) enzymatic addition of the poly (A) tail.
eGFP: Enhanced green fluorescent protein.
Transfections
Transfections were performed using Lipofectamine RNAiMAX Transfection Reagent (ThermoFisher Scientific, cat. no. 13778150) according to manufacturer's instructions. All dilutions were performed in Opti-MEM medium (ThermoFisher Scientific, cat. no. 31985070). Briefly, on day 0, HeLa cells were plated at 100,000 cell/ml and in either 96- or 6-well plates at volumes of either 100 μl or 3 ml, respectively. On day 1, 1.5 μl of RNAiMAX was diluted into 25 μl of Opti-MEM. This was scaled depending upon the number of samples, dilutions and culture volumes. Mature mRNAs were diluted in Opti-MEM at varying concentrations before addition to cell cultures. Culture after transfection was typically 24 h. Transfection experiments were performed a minimum of three-times. Unpaired t-tests with Welch's correction were employed to determine statistical significance.
Results & discussion
We designed synthetic double-stranded DNAs with the bacteriophage SP6 promoter at the 5′ end followed by a synthetic 5′ UTR, a Kozak sequence, transcriptional start site, complementary DNA to the mRNA of interest and a 3′ UTR (obtained from Integrated DNA Technologies) [16–22]. We performed IVT reactions to synthesize single-stranded RNA. Yields of RNA from 100 ng of DNA were typically 100–300 μg. The 5′ cap [20] and poly (A) tail were added to the RNA (Figure 2) [19]. Purified mRNAs were transfected into target cells and protein expression and function determined as outlined in the following.
Figure 2. . Schematic describing production of synthetic genes and mature mRNAs.

Synthetic genes were designed with, from 5′ to 3′, the SP6 bacteriophage promoter, a 5′ untranslated region (UTR), Kozak sequence, coding sequence of interest and 3′ UTR and transcribed into RNA. A 5′ Cap and poly (A) tail were enzymatically added to the purified single-stranded RNA to yield the mature mRNA.
Using the preceding process, we designed a synthetic gene to express a secreted form of luciferase [23]. The coding sequence we inserted into the synthetic gene was 621 bp. Our rationale for choosing the luciferase gene was that luciferase is an oxidoreductase enzyme. The enzymatic reaction requires molecular oxygen and reduced flavin to catalyze light emission. We assumed that accurate transcription, translation, folding and entry into secretory pathways would all be required to produce secreted luciferase that could be detected in cell culture supernatant fluids. Varying amounts of the synthetic luciferase mRNA were transfected into HeLa cells. After 24 h, culture fluids were harvested and luciferase activity measured. We found abundant expression of luciferase protein activity in cultures transfected with 100 ng/well of the synthetic luciferase mRNA (Figure 3A).
Figure 3. . Functional proteins generated from synthetic genes.
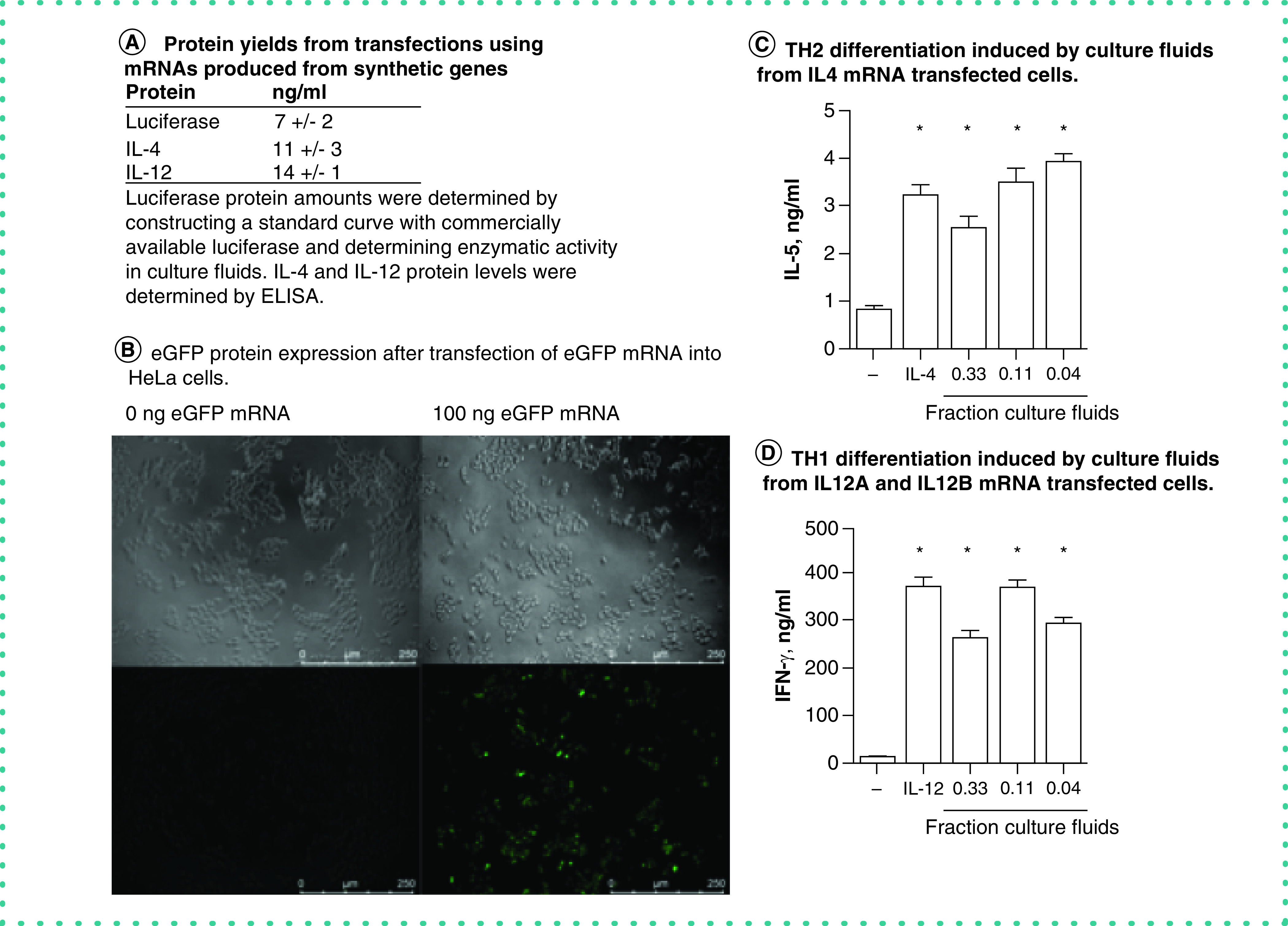
(A) Protein yields produced by HeLa cells transfected with the indicated mRNAs produced from synthetic genes; secreted luciferase (determined by measuring luminescence using a TD20/20 luminometer [Quanti-luc, rep-qlc1, Invivogen]), IL-4 and IL-12 (determined by ELISA). (B) Expression of eGFP by HeLa cells transfected with eGFP mRNA determined by fluorescence microscopy. (C) The indicated fractions of supernatant fluids from HeLa cell cultures transfected with IL-4 mRNA or purified recombinant IL-4 were added to peripheral blood mononuclear cell cultures stimulated with anti-CD3 and anti-CD28. After 5 days of culture, supernatant fluids were harvested and IL-5 protein levels were determined by ELISA. (D) The indicated fractions of supernatant fluids from HeLa cell cultures transfected with IL-12A and IL-12B mRNAs or purified recombinant IL-12 were added to peripheral blood mononuclear cell cultures stimulated with anti-CD3 and anti-CD28. After 5 days of culture, supernatant fluids were harvested and IFN-γ protein levels were determined by ELISA.
*p < 0.05 compared with mock-transfected cultures.
Using the same process, the next synthetic gene we prepared was eGFP [24]. The coding sequence we inserted into the synthetic gene was 718 bp. Our rationale was that for emission of green fluorescence from cells, eGFP must be accurately transcribed, translated, form a homodimer and remain in the intracellular space. We transfected 100 ng of eGFP mRNA into HeLa cells and determined eGFP protein expression by fluorescence microscopy. We found abundant expression of green fluorescence in eGFP mRNA-transfected HeLa cells but not in mock-transfected HeLa cells (Figure 3B). After transfection of cultures with the eGFP mRNA, we found that ∼50% of cells were eGFP+, suggesting that transfection efficiency was at least 50%. We conclude that the synthetic eGFP gene was accurately transcribed in the IVT reaction, translated and folded properly inside cells to produce active eGFP protein.
IL-4 and IL-12 are cytokines that play critical roles in both the innate and adaptive arms of the immune response. Perhaps most notably, IL-4 directs differentiation of naive T cells into effector T-helper 2 (TH2) cells capable of producing the cytokines, IL-4, IL-5 and IL-13, critical to control extracellular parasite infections by the adaptive arm of the immune system, and IL-12 directs the differentiation of naive T cells into effector T-helper 1 (TH1) cells to enable their expression of IFN-γ, a critical cytokine required for protection against an array of intracellular pathogens [25–27]. We designed an IL-4 synthetic gene. The IL-4 coding sequence was 456 nt. The IL-4 synthetic mRNA, prepared by the preceding method, was transfected into HeLa cells (100 ng/culture) and culture fluids harvested after 24 h. IL-4 protein levels in mock-transfected and synthetic IL-4 mRNA-transfected cultures were determined by ELISA (Figure 3A). Biological activity of IL-4 produced from the IL-4 mRNA was determined in a TH2 differentiation assay [12]. Briefly, human PBMCs were treated with anti-CD3 and anti-CD28 to stimulate T-cell proliferation and either purified IL-4 or culture fluids from HeLa cells transfected with IL-4 mRNA. Culture fluids from PBMC cultures were harvested after 5 days and IL-5 levels determined by ELISA as a measure of TH2 differentiation. We found that levels of IL-4 produced from IL-4 mRNA induced substantial TH2 differentiation as determined by the ability of naive human T cells to differentiate into effector TH2 cells that express IL-5 protein (Figure 3A & C). Active IL-12 is composed of a heterodimeric protein consisting of IL-12 p35 and IL-12 p40 subunits, also named IL-12A and IL-12B, respectively. We designed both IL-12A and IL-12B synthetic genes using the preceding processes. The IL-12A coding insert was 759 bp, and the IL-12B coding insert was 984 bp. Both IL-12A and IL-12B mRNAs were simultaneously transfected into HeLa cells, 100 ng/well. Culture fluids were harvested after 24 h. IL-12 protein levels were determined by ELISA (Figure 3A). Ability to induce TH1 differentiation was determined by stimulating human PBMCs with anti-CD3 and anti-CD28 and either purified IL-12 or culture fluids from HeLa cells transfected with IL-12A and IL-12B mRNAs. Culture fluids from PBMC cultures were harvested after 5 days and IFN-γ levels determined by ELISA as a measure of TH1 differentiation. We found that culture fluids from IL-12A and IL-12B mRNA-transfected HeLa contained abundant levels of IL-12 protein, whereas IL-12 protein was undetectable in mock-transfected HeLa culture fluids (Figure 3A). We also found that culture fluids from IL-12A and IL-12B mRNA-transfected HeLa cells were potent inducers of PBMC TH1 differentiation as determined by the ability of these culture fluids to induce expression of IFN-γ by PBMC cultures (Figure 3D). Thus, these results indicate that IL-4 synthetic genes and IL-12A and IL-12B synthetic genes were efficiently transcribed into mRNA and formed biologically active proteins capable of inducing TH2 or TH1 differentiation, respectively.
We also varied the amount of luciferase synthetic mRNA transfected into cultures and assayed the presence of luciferase in the cultured fluid. We found that the yield of luciferase was proportional to the amount of transfected luciferase mRNA over a range of 1–100 ng/culture (Figure 4A). In contrast, transfection of luciferase synthetic RNA without addition of the 5′ CAP and poly (A) tail did not result in production of detectable luciferase protein, indicating that these steps were necessary to produce a functional mRNA. We also compared the yield of luciferase protein as a function of time after transfection with synthetic luciferase mRNA. We found that levels of secreted luciferase steadily increased over at least a 72-h period (Figure 4B). Thus, production of luciferase protein was proportional to both the amount of transfected synthetic mRNA and time of culture.
Figure 4. . Synthetic mRNA amount and time dependence upon luciferase protein expression.

(A) The indicated amounts of luciferase mRNA (o) or un-capped and un-poly (A)-tailed luciferase RNA (nanograms) were transfected into HeLa cells. Culture supernatant fluids were harvested after 24 h and assayed for luciferase activity. (B) HeLa cells were transfected with 100 ng of luciferase synthetic mRNA. Culture supernatant fluids were harvested at the indicated times and assayed for luciferase activity, expressed as relative light units, Y-axis. Results are expressed as the mean ± standard error of three independent experiments.
RLU: Relative luminescence unit.
In conclusion, we present a simple, inexpensive and rapid method to prepare synthetic genes and mature mRNAs that can be efficiently introduced into cells and translated into functional proteins without the need for plasmid-based cloning (Table 1). Other RNAs, such as long noncoding RNAs, can also be prepared using this method. These synthetic genes may have many cell-based applications, such as structure–function studies or studies akin to site-directed mutagenesis studies, gain or recovery of function studies. These mRNAs produced from synthetic genes may also have applications in vivo. For example, with newer delivery vehicles, such as nanoparticles, these mRNAs may have medical applications such as gene therapy or vaccine development without the need for plasmid-based vectors or viral delivery vehicles.
Table 1. . Cost and time estimates for synthetic RNA versus plasmid-based cloning methods.
| Synthetic mRNA | Vendor | Reagent costs ($) | Cost for 1 ($) | Hands-on time (h) | Total time (h) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 1 | Design gBLOCK with SP6 promoter | IDT | 75–3000 | 150.00 | 1 | 2 weeks | |||||
| Day 2 | In vitro transcription/LiCl ppt | Megascript | 8.6/rxn | 8.60 | 2 | 14 | |||||
| Day 3 | Add 5′ cap | NEB | 3.5/rxn | 3.50 | 0.5 | 2 | |||||
| Day 3 | Add poly (A) tail | NEB | 3.6/rxn | 3.60 | 0.5 | 1 | |||||
| Day 3 | RNA cleanup 2× | Qiagen | 7.38/rxn | 14.76 | 1 | 1 | |||||
| Totals | 180.46 | 5 | 18 | ||||||||
| Traditional plasmid-based cloning | |||||||||||
| Day 1 | Design gBLOCK with restriction sites | IDT | 75.00–3000 | 150.00 | 1 | 2 weeks | |||||
| Day 1 | Purchase expression vector | NEB | 100/clone | 100.00 | 1 | 48 | |||||
| Day 2 | Digest vector and gBLOCK | NEB | 0.05/u | 0.10 | 0.5 | 1 | |||||
| Day 2 | Run on gel | Sigma | 1.97/g | 1.97 | 0.5 | 1 | |||||
| Day 2 | Gel extraction + cleanup | Qiagen | 2.3/rxn | 2.30 | 3 | 3 | |||||
| Day3 | Ligation | Thermo Fisher | 1.18/rxn | 1.18 | 0.5 | 0.5 | |||||
| Day3 | Transformation | Thermo Fisher | 19.92/rxn | 19.92 | 1 | 1 | |||||
| Day3 | Out grow | Media | Negligible | 0.5 | 18 | ||||||
| Day3 | Plating | Media | 2/plate | 10.00 | 1 | 2 | |||||
| Day 4 | Colony screening | Taq | 0.5/U | 10.00 | 1 | 18 | |||||
| Day 4 | Run on gel | Sigma | 1.97/g | 3.94 | 0.5 | 2 | |||||
| Day 4 | Pick PCR positives, start cultures | Media | Negligible | 1 | 18 | ||||||
| Day 5 | Make mini preps | Qiagen | 1.81/rxn | 23.60 | 4 | 3 | |||||
| Day 5 | Digest with RE | NEB | 0.05/U | 1.00 | 0.5 | 1 | |||||
| Day 5 | Run on gel | Sigma | 1.97/g | 3.94 | 1 | 2 | |||||
| Day 6 | Send clone for sequencing validation | Genewiz | 4/rxn | 20.00 | 1 | 18 | |||||
| Day 7 | Start cultures for Maxi preps. | Media | negligible | 0.5 | 18 | ||||||
| Day 8 | Prepare DNA for transfections | Qiagen maxi | 24.5/rxn | 24.50 | 4 | 18 | |||||
| Totals | 372.45 | 22.5 | 172.5 | ||||||||
| gBLOCK fragment price vs nucleotide length | |||||||||||
| Base pair | 500 | 750 | 1000 | 1250 | 1500 | 1750 | 2000 | 2250 | 2500 | 2750 | 3000 |
| $ | 89 | 129 | 149 | 209 | 249 | 289 | 329 | 399 | 449 | 499 | 549 |
IDT: Integrated DNA technology; NEB: New England Biolabs.
An alternate method to the use of gBlock DNA fragments for RNA synthesis includes the use of RT-PCR with a forward primer containing a promoter and appropriate reverse primer to amplify a cDNA from a given mRNA present in a biological sample. The cDNA would probably need to be purified and sequenced to ensure the anticipated product was in hand for its intended use. RNA could be synthesized from the recovered cDNA, 5′ CAP and poly A tail added. Using this RT-PCR-based approach also assumes availability of the desired RNA, which may be difficult in certain experimental settings such as study of extinct or rare species, study of cells or organs not readily available such as human brain tissue or other human vital organs. Other limitations of RT-PCR may include study of hybrid or other proteins forms that do not exist in nature. Of course, there is the added cost of the thermal cycler and reagents to perform the RT-PCR reactions. We have performed a version of this method by designing PCR primers to amplify new DNA from the gBlock DNA fragments using the newly synthesized DNA for IVT followed by addition of a 5′ CAP and poly (A) tail and found this a satisfactory approach. An alternate method is chemical synthesis of the desired RNA followed by addition of the 5′ CAP and poly (A) tail to produce the desired synthetic mRNA. However, there are limitations to this method because of costs and lengths of RNA strands that can be synthesized using currently available technology.
In general terms, addition of the 5′ CAP reduces mRNA degradation and aids binding of mRNAs to the ribosome [13–15]. Similarly, the poly (A) tail reduces mRNA degradation and allows export of mRNAs to the cytoplasm and stimulates protein translation. We find no detectable translation of synthetic mRNAs in the absence of the 5′ CAP and poly (A) tail. In addition, eukaryotic cells express sensors termed pattern recognition receptors that detect pathogen-associated molecular patterns [28]. One of these endogenous sensors present in the cytosol is the DExD/H-box helicase, RIG-I, that recognizes the 5′ triphosphate present on nascent cytosolic single- and double-stranded RNAs produced after infection by RNA viruses as part of their normal life cycle and triggers a strong inflammatory response mediated by activation of proinflammatory transcription factors, interferon regulatory factors and NF-κB [29,30]. Thus, addition of the 5′ CAP to synthetic mRNAs prior to introduction into the cytosol also prevents activation of these strong inflammatory responses.
We clearly show that the synthetic mRNA system described here has advantages in terms of cost, ease of synthesis and required time commitment versus standard plasmid-cloning methods for production of mRNAs and functional proteins in human cells. Other advantages may include ability to easily synthesize mRNA sequence variants for structure function studies or ability to express proteins for study when mRNA is not readily available for plasmid-based cloning or all that is known is the DNA sequence, such as from an extinct species. We did not totally optimize all facets of this synthetic mRNA system to maximize, for example, protein production, so direct comparison between yields of protein using this synthetic mRNA system to more traditional systems is not possible, as this was not our primary goal. Further optimization to maximize mRNA stability and protein production and increase overall systems efficiencies using this system is warranted given the increasing importance of IVT techniques to laboratory-based research and the use of mRNAs as therapeutics or in vaccine development.
Future perspective
Time and cost, as well as the likelihood of producing novel scientific results, factor into decisions in the laboratory on whether or not to embark on a given experimental approach. Application of the simplified method to produce synthetic mRNAs and other classes of RNA described here will minimize time and cost factors that enter into this decision-making process, thus creating additional avenues of investigation into both RNA and protein biology that less likely would have been using plasmid-based cloning approaches.
Footnotes
Author contributions
All authors contributed to the methodology, formal analysis, and writing review and editing. Conceptualization and writing of the original draft were performed by JT Tossberg and TM Aune. The investigation was performed by JT Tossberg and TM Esmond. Supervision, project administration and funding acquisition were performed by TM Aune.
Financial & competing interests disclosure
This work was supported by NIH grants R01AI44924, R21AI124766 and R21AI144193. Vanderbilt University has filed a provisional patent application for the methods and approaches described in this article. TM Aune and JT Tossberg are listed as co-inventors. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest
- 1.Fritsch EF, Sambrook J, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, NY, USA: (1982). [Google Scholar]
- 2.Lodish H, Berk A, Zipursky SL. et al. Molecular Cell Biology. WH Freeman & Company, MD, USA: (1986). [Google Scholar]
- 3.Hadas Y, Sultana N, Youssef E. et al. Optimizing modified mRNA in vitro synthesis protocol for heart gene therapy. Mol. Ther. Methods Clin. Dev. 14, 300–305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berraondo P, Martini PGV, Avila MA, Fontanellas A. Messenger RNA therapy for rare genetic metabolic diseases. Gut 68(7), 1323–1330 (2019). [DOI] [PubMed] [Google Scholar]; • Potential of mRNAs as therapeutics for monogenic diseases.
- 5.Kwon H, Kim M, Seo Y. et al. Emergence of synthetic mRNA: in vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 156, 172–193 (2018). [DOI] [PubMed] [Google Scholar]; • Emergence of synthetic RNAs as therapeutic modalities.
- 6.Wilson JT, Keller S, Manganiello MJ. et al. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano 7(5), 3912–3925 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pradere U, Brunschweiger A, Gebert LFR, Lucic M, Roos M, Hall J. Chemical synthesis of mono- and bis-labeled pre-microRNAs. Angew. Chem. Int. 52(46), 12028–12032 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Kadekar S, Nawale GN, Karlsson K, Alander C, Oommen OP, Varghese OP. Synthetic design of asymmetric miRNA with an engineered 3′ overhang to improve strand selection. Mol. Ther. Nucl. Acids. 16, 597–604 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang CL, Maruggi G, Shan H, Li JW. Advances in mRNA vaccines for infectious diseases. Front. Immunol. 10, 594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines – a new era in vaccinology. Nat. Rev. Drug Discov. 17(4), 261–279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Potential of mRNA vaccines is described.
- 11.Le TT, Andreadakis Z, Kumar A. et al. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 19(5), 305–306 (2020). [DOI] [PubMed] [Google Scholar]; • mRNA vaccines for COVID-19 pandemic.
- 12.Gibbons HR, Shaginurova G, Kim LC, Chapman N, Spurlock CF, III, Aune TM. Divergent lncRNA GATA3-AS1 regulates GATA3 transcription in T-helper 2 cells. Front. Immunol. 9, 2512 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banerjee AK. 5′-terminal cap structure in eucaryotic messenger ribonucleic acids. Microbiol. Rev. 44(2), 175–205 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen Y, Yue Z, Shatkin AJ. Mammalian capping enzyme binds RNA and uses protein tyrosine phosphatase mechanism. Proc. Natl Acad. Sci. USA 95(21), 12226–12231 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu C, Das K, Tyminski JR. et al. Structure of the guanylyltransferase domain of human mRNA capping enzyme. Proc. Natl Acad. Sci. USA 108(25), 10104–10108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown JE, Klement JF, McAllister WT. Sequences of three promoters for the bacteriophage SP6 RNA polymerase. Nucleic Acids Res. 14(8), 3521–3526 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell. Mol. Life Sci. 69(21), 3613–3634 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guhaniyogi J, Brewer G. Regulation of mRNA stability in mammalian cells. Gene 265(1–2), 11–23 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Richter JD. Cytoplasmic polyadenylation in development and beyond. Microbiol. Mol. Biol. Rev. 63(2), 446–456 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shatkin AJ. Capping of eucaryotic mRNAs. Cell 9(4 Pt 2), 645–653 (1976). [DOI] [PubMed] [Google Scholar]
- 21.Wu X, Brewer G. The regulation of mRNA stability in mammalian cells: 2.0. Gene 500(1), 10–21 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozak M. The scanning model for translation: an update. J. Cell Biol. 108(2), 229–241 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wurdinger T, Badr C, Pike L. et al. A secreted luciferase for ex vivo monitoring of in vivo processes. Nat. Methods 5(2), 171–173 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang G, Gurtu V, Kain SR. An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells. Biochem. Biophys. Res. Commun. 227(3), 707–711 (1996). [DOI] [PubMed] [Google Scholar]
- 25.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 28, 445–489 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 7, 145–173 (1989). [DOI] [PubMed] [Google Scholar]
- 27.O'Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327(5969), 1098–1102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 20, 197–216 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Goubau D, Schlee M, Deddouche S. et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514(7522), 372–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinrich MJ, Purcell CA, Pruijssers AJ. et al. Endogenous double-stranded Alu RNA elements stimulate IFN-responses in relapsing remitting multiple sclerosis. J. Autoimmun. 100, 40–51 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Detection of endogenous double-stranded RNAs that activate innate immunity.