

Abstract
Using the previously reported novel spirodiketopiperazine scaffold, the design and synthesis of orally available CCR5 antagonists was undertaken. Compounds possessing 3 carboxylic acid function in the appropriate position showed improved oral exposure (AUC) relative to the initial chemical leads without reduction in the antagonist activity. The optimized compound 40 was found to show potent anti-HIV activity. Full details of structure-activity relationship (SAR) study are presented.
Keywords: CCR5, Chemokine, Anti HIV
1. Introduction
Despite a tremendous effort to develop antiretroviral therapy, millions of people in the world are still suffering from Acquired Immune Deficiency Syndrome (AIDS).1 Although several classes of antiretroviral medications are currently available, challenges remain with anti-HIV therapies due to a gradual spread of drug-resistant strains, severe side effects, and expensive therapeutic costs among others. Hence there is a growing need for novel treatments for AIDS patients who are treatment-experienced and have become resistant to one or multiple classes of antiretroviral agents.2 These issues require new anti-HIV drugs to have a different mode of action from conventional drugs. CCR5 antagonists work by inhibiting HIV entry into target cells and are one of the most promising approaches to treat AIDS because of their ability to inhibit a previously untargeted step in the HIV-1 replication cycle.3 Maraviroc is currently the only approved CCR5 antagonist on the market.4
We previously reported the discovery of spirodiketopiperazine derivatives 1a and 1b (Fig. 1), as structurally novel CCR5 antagonist leads generated from a combinatorial library targeting GPCRs.5,7 However, it was unfortunate that both 1a and 1b showed poor oral exposure (AUC) in rodents (Table 7). Based on the analysis of their PK data, large clearance (CL) and tissue distribution values (Vss) were assumed to be one of the plausible reasons for poor AUC. Herein we report full details of the discovery of a new chemical lead 40 with an improved PK profile.
Figure 1.
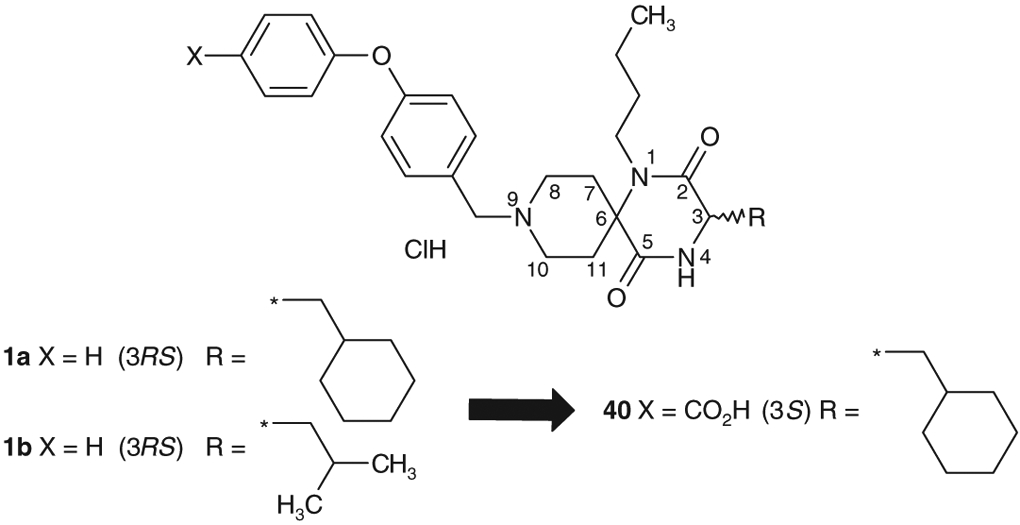
Molecular design of orally active chemical leads.
Table 7.
Pharmacokinetic data for 1a, 1b, 36, 37, and 40 in rat (n = 3)
| Compds | 30 mg/kg, po |
3 mg/kg, iv |
||||||
|---|---|---|---|---|---|---|---|---|
| Cmaxa (ng/mL) | T1/2 (min) | AUCa (ng h/mL) | BA (%) | AUC (ng) | T1/2 (min) | CL (mL/min/mL) | Vss (mL/kg) | |
| 1a | 16.7 | 103 | 74.4 | 1.9 | 400 | 19.9 | 113 | 2542 |
| 1b | 100 | 75.7 | 290 | 1.3 | 372 | 13 | 137 | 2349 |
| 36 | 100 | 205 | 195 | NDb | NDb | NDb | NDb | NDb |
| 37 | 1900 | 72.7 | 5453 | NDb | NDb | NDb | NDb | NDb |
| 4 | 7200 | 48.4 | 10,532 | 34.1 | 3091 | 11.1 | 16 | 145 |
Cmax and AUC are normalized to a dose of 30 mg/kg.
ND: not determined.
2. Results and discussion
2.1. Chemistry
As shown in Schemes 1 and 2, compounds 1a, 1b, 2–13, 15–18, 21–23, 25, 26, and 28 were efficiently synthesized using solid phase synthesis.5 The Ugi four-component condensation using the Rink-isonitrile resin is outlined in Scheme 1a. A mixture of 1-N-allyloxy-carbonyl piperidone 41a, an optional alkyl amine 42, an optional N-Boc-amino acid 43, and the Rink-isonitrile resin 44 in THF/MeOH (1:1) was shaken at 65 °C to afford 45. Palladium catalyzed removal of the N-allyloxycarbonyl moiety of 45 was followed by reductive amination with an optional aryl aldehyde to produce 46. Removal of the Boc protecting group of 46 followed by heating under acidic conditions resulted in cyclization accompanied by removal of the supporting-resin to afford spirodiketopiperazines 1a, 1b, 2–13, 15–18, 23, 25, 26, and 28. Compound 14 was prepared by alkaline hydrolysis of 15. 2-Butynylamine 42f was prepared from 2-butynyl alcohol as described in Scheme 1b. O-Methanesulfonylation of 47 followed by the substitution reaction with potassium phthalimide afforded 48, deprotection of which with hydrazine hydrate produced 42f in good yield. As described in Scheme 2, compounds 21 and 22 were also synthesized from 1-Cbz-piperidone 41b and 1-(6-phenylhexyl)-4-piperidone 41c, respectively, by the solid phase synthesis.
Scheme 1.
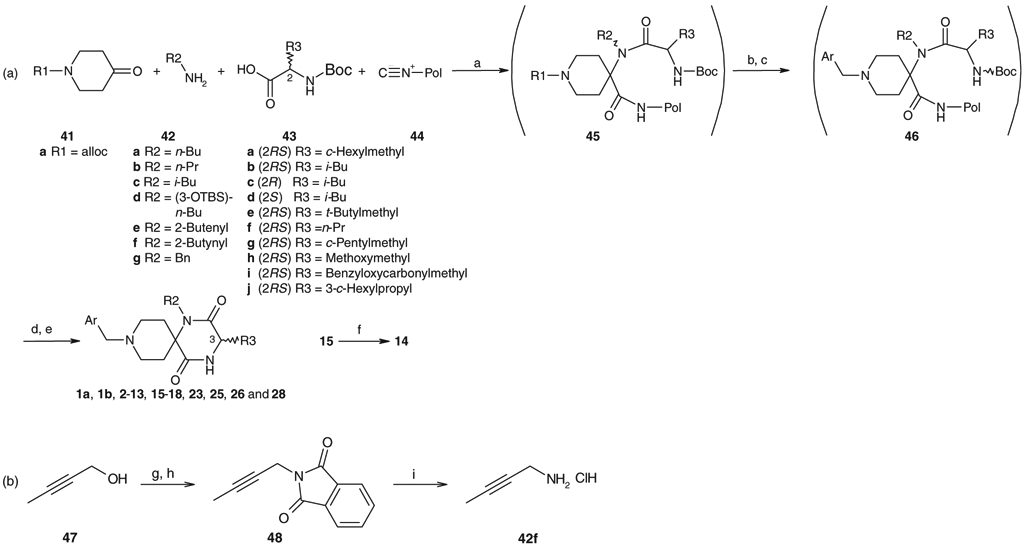
Solid phase synthesis of spirodiketopiperazines 1a, 1b, 2–18, 23, 25, 26, and 28. Reagents and conditions: (a) THF/MeOH (1:1), 65 ”C; (b) Pd(PPh3)4, Bu3SnH, AcOH/CH2Cl2, rt; (c) Ar-CHO, NaBH(OAc)3, AcOH/DMF; (d) TFA/CH2Cl2 (1:1); (e) AcOH/toluene, 90 °C and then 4 N HCl/AcOEt; (f) 2 N NaOH, MeOH; (g) MsCl, Et3N, CH2Cl2; (h) potassium phthalimide, DMF, 70 °C; (i) NH2NH2·H2O, EtOH, then concd HCl, reflux.
Scheme 2.

Synthesis of 21 and 22. Reagents and conditions: (a) THF/MeOH (1:1), 65 °C; (b) TFA/CH2Cl2 (1:1); (c) AcOH/toluene, 90 °C and then 4 N HCl/AcOEt.
Solution phase synthesis of compounds 24, 27, and 29–40 was carried out by reductive alkylation of 52a and 52b with an optional aryl aldehyde, respectively, as described in Scheme 3. Compounds 52a and 52b were prepared by solution phase synthesis. The Ugi four-component condensation of 1-benzyl-4-piperidone 41d, n-butylamine 42a, N-Boc-1-cyclohexylalanine 43a and 2-(4-morpholinyl)-ethylisonitrile 50 afforded spirodiketopiperazine 51a. Catalytic hydrogenation of 51a followed by the acid treatment provided 52a as a hydrochloride salt. Compound 52b was prepared by the same procedures as described above using 43k instead of 43a as one of the starting materials. Reductive alkylation of 52b with an optional aldehyde produced 24, 27, and 29–40, respectively. Commercially unavailable aryl aldehydes 54, 56, 58, 59, 62, and 64 were prepared as outlined in Scheme 4.
Scheme 3.
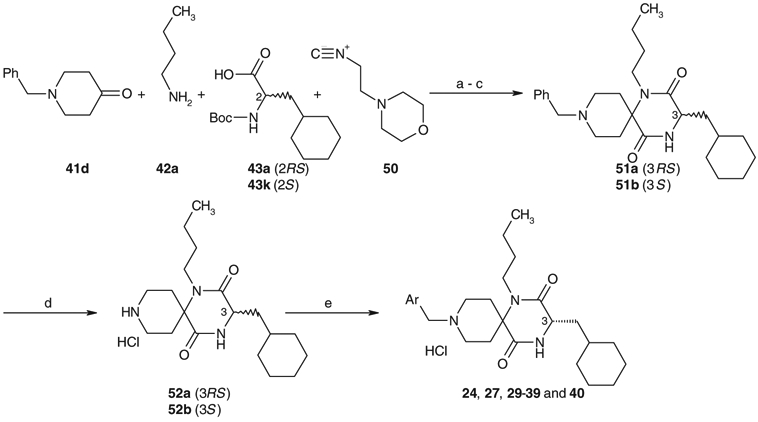
Solution phase synthesis of spirodiketopiperazines, 24, 27, and 29–40. Reagents and conditions: (a) MeOH, 55 °C; (b) concd HCl, 55 °C; (c) AcOH/toluene, 80 °C; (d) H2, Pd(OH)2/C, EtOH, 50 °C, then 4 N HCl/AcOEt (50–90% in four steps); (e) Ar-CHO, NaBH(OAc)3, AcOH, DMF and then 4 N HCl/AcOEt (50–90%).
Scheme 4.
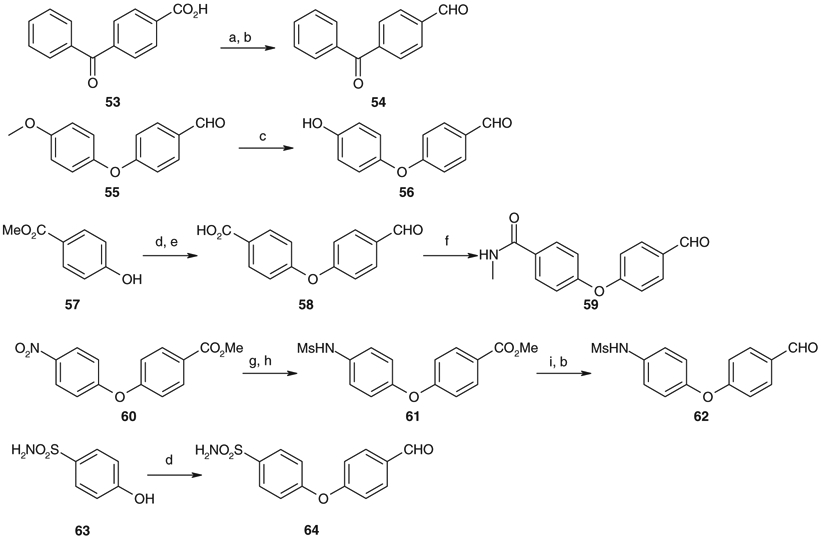
Synthesis of aryl aldehydes 54, 56, 58, 59, 62, and 64. Reagents and conditions: (a) i-BuOCOCl, Et3N, THF then NaBH4, H2O; (b) MnO2, DME; (c) BBr3, CH2Cl2; (d) 4-fluorobenzaldehyde, K2CO3, DMA, 150 °C; (e) NaOH, MeOH; (f) EDC, HOBt, MeNH2, DMF; (g) H2, 5% Pd/C, AcOEt; (h) MsCl, pyridine, THF; (i) DIBAL, THF.
Synthesis of 9-N-(4-phenoxy)phenylethyl analog 19 is described in Scheme 5. 9-N-Alkylation of 52b with polymer-supported 4-phenoxyphenylethyl tosylate, which was prepared by O-tosylation of 4-phenoxyphenylethanol 65 with polystyrene-supported tosyl chloride in pyridine, followed by the treatment with hydrogen chloride afforded 19.
Scheme 5.
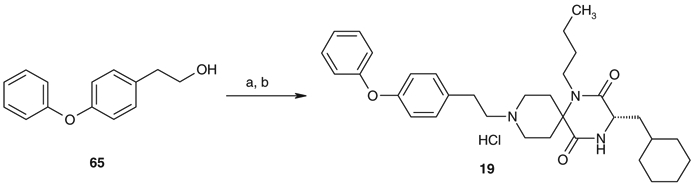
Synthesis of 19. Reagents and conditions: (a) polystyrene-supported tosyl chloride, pyridine, CH2Cl2; (b) 52b, i-Pr2NEt, MeCN and then 4 N HCl/AcOEt (48% in two steps).
As described in Scheme 6.9-N-phenyl analog 20 was prepared by the substitution reaction of ethyl 4-fluorobenzoate with 52a in the presence of potassium carbonate in DMSO followed by the acid treatment.
Scheme 6.
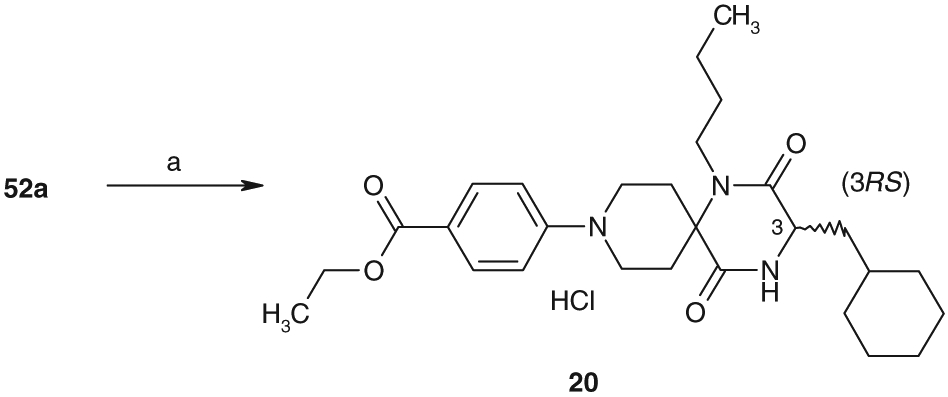
Synthesis of 20. Reagents and conditions: ethyl 4-fluorobenzoate, K2CO3, DMSO and then 4 N HCl/AcOEt (27%).
2.2. CCR5 antagonist activity
Compounds listed in Tables 1-6 were evaluated for their inhibitory activities against calcium mobilization of human CCR5 overexpressed CHO cells (hCCR5/CHO) stimulated with MIP-1α and with IC50 values as the mean of two experiments.6
Table 1.
Effect of the 1-N-substituent on activity profiles
 |
|||
|---|---|---|---|
| Compds | R | IC50 (nM) Ca assay |
T1/2 in rat liver microsomesa (min) |
| 2 | 500 | NTb | |
| 3 |  |
330 | 15 |
| 4 |
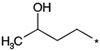 >3000 >3000 |
12 | |
| 5 | 69 | 14 | |
| 6 | 54 | 15 | |
| 7 | 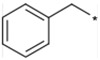 |
300 | NTb |
| 1b | 94 | 12 | |
The data show T1/2 after incubating with the 0.2 mg/mL rat liver microsomes.
NT: not tested.
Table 6.
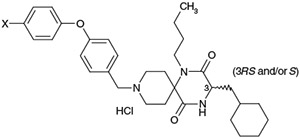
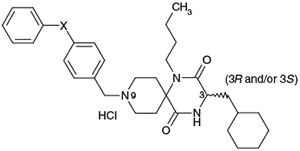
Effect of the p-substituent of the biphenyl ether residue on activity profiles
 |
|||||
|---|---|---|---|---|---|
| Compds | X | C3 Configuration (3R and/or 3S) |
IC50 (nM) Ca assay |
Stability in rat liver microsomesa remaining% |
cLog Pb |
| 33 | F− | (3S) | 92 | 53 | 7.80 |
| 34 | Me− | (3S) | 79 | 48 | 8.16 |
| 35 | MeO− | (3S) | 130 | 34 | 7.58 |
| 36 | HO− | (3S) | 42 | 22 | 6.99 |
| 37 | MeHNC(O)− | (3S) | 33 | 4 | 6.38 |
| 38 | MeSO2HN− | (3S) | 40 | 15 | 6.47 |
| 39 | H2NO2S− | (3S) | 28 | 24 | 5.82 |
| 40 | HO2C− | (3S) | 13 | 22 | 5.13 |
| 1a | H | (3RS) | 28 | 33 | 7.66 |
The data show the remaining% 15 min after incubating with the 0.5 mg/mL rat liver microsomes.
Calculated by ADMET predictor (ver. 4.0).
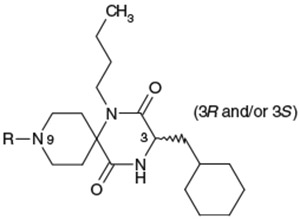
As described in our previous paper,5 the 1,4,9-triazaspiro[5.5]undecane-2,5-dione (spirodiketopiperazine) is an attractive scaffold to use to identify small molecule ligands of GPCRs because of the following three reasons. (1) This spirodiketopiperazine is a simple heterocyclic scaffold in which diversity can be introduced at up to four positions, and can be prepared from readily available α-amino acids. (2) Very few compounds with this spirodiketopiperazine scaffold have been reported at the starting point of this project. (3) The predicted 3D structure of spirodiketopiperazine suggested that utilization of three substituents on this template might have a similar orientation to three side chains on the type I β-turn structure of the protein.
Compounds 1a and 1b blocked the infectivity and replication of laboratory and clinical strains of HIV as well as highly drug-resistant HIV variants with minimal cytotoxicity.6a Despite their promising activity profiles, both 1a and 1b had very poor AUCs after oral dosing in rat. Pharmacokinetic analysis after iv dosing indicated that they exhibited rapid clearance (CL) and a large distribution volume (Vss). A large clearance value (CL) after iv dosing suggests that they are metabolically unstable which may explain their poor AUC and bioavailability (BA). Moreover, the large distribution volume (Vss) of analogs 1a and 1b after iv dosing is unfavorable as anti-HIV-1 drugs exert their effects in the blood. Thus, our synthetic effort was focused on increasing oral exposure (AUC), reduction of in vivo clearance (CL) and distribution volume (Vss). The optimization of chemical leads 1a and 1b was initiated by evaluating each of the diversity sites as described in Tables 1-6.
Optimization of the 1-N-alkyl residue was carried out with the aim of further increasing the in vitro activities and metabolic stability (Table 1). Replacement of the n-butyl residue of 1b with n-propyl, i-butyl, 3-hydroxybutyl, 2-butenyl, 2-butynyl and benzyl residues afforded 2–7, respectively. 1-N-(n-Propyl) analog 2 exhibited less potent activity relative to 1b. Analog 3 possessing a branched 1-N-alkyl residue, showed less potent activity relative to 1b. 1-N-(3-Hydroxybutyl) analog 4 had a remarkable reduction in its antagonist activity. Thus, a hydrophilic OH group was found to be unacceptable at this site. Compounds 5 and 6 possessing 1-N-(2-butenyl) and 1-N-(2-butynyl) residues, showed nearly equipotent activity as 1b. 1-N-Benzyl analog 7 showed significantly reduced potency. As a result, shortening and branching of the 1-N-(n-butyl) residue as illustrated by 2 and 3 negatively impacted the activity. After substantial chemical modification, lipophilic groups such as n-butyl, 2-butenyl and 2-butynyl residues, which are relatively linear and have a limited length (C4), were found to be the most optimal 1-N-substituent. The stability in rat liver microsomes of compounds 1b and 3–6 was investigated to estimate their metabolic stability. All the analogs tested were not as metabolically stable as 1b. Although this hydrophobic 1-N-residue was considered to be one of sites that were metabolically vulnerable, it seemed to be difficult to improve metabolic stability only by chemical modification of this moiety.
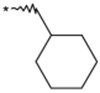
To identify an optimal C3-substituent, synthesis and evaluation of analogs 8–16 was carried out using the easily available optically active S-form or racemic RS-form as the starting amino acids. This was possible because there were no significant differences in antagonist activities between the enantiomers 8 (3S) and 9 (3R), both of which showed nearly equipotent activities relative to 1b. Analogs 10–12 possessing lipophilic C3-substituents exhibited equipotent activities relative to 1b. Analogs 13–15 possessing relatively hydrophilic substituents such as methoxymethyl, carboxymethyl, and benzyloxycarbonylmethyl tended to show reduced activities. Cyclohexylethyl analog 16, which is a C2 homolog of 1a, exhibited a remarkable decrease in its activity. Based on these results, only lipophilic C3-substituents are acceptable for the receptor pocket as illustrated by 1a, 1b and 8–16 while their length is strictly limited as illustrated by the result of 16. Analogs 8–12, which showed nearly equipotent activities relative to 1a and 1b, did not show any improvement in stability in liver microsomes as shown in Table 2.
Table 2.
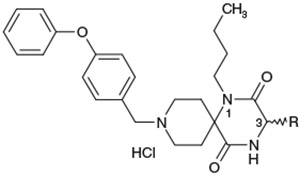
Effect of the 3-substituent on activity profiles
 |
||||
|---|---|---|---|---|
| Compds | R | C3 Configuration (R and/or S) |
IC50 (nM) Ca assay |
T1/2 in rat liver microsomesa (min) |
| 8 | 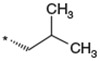 |
(3S) | 84 | 7.1 |
| 9 | 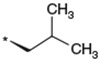 |
(3R) | 130 | NTb |
| 10 | 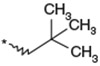 |
(3RS) | 79 | 12 |
| 11 | (3RS) | 50 | 22 | |
| 12 | 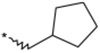 |
(3RS) | 52 | 20 |
| 13 | (3RS) | 320 | 16 | |
| 14 | (3RS) | >3000 | NTb | |
| 15 | 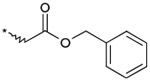 |
(3RS) | 590 | NTb |
| 16 | 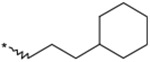 |
(3RS) | >3000 | 23 |
| 1a | 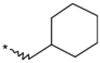 |
(3RS) | 28 | 24 |
| 1b | 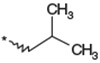 |
(3RS) | 94 | 12 |
The data show T1/2 after incubating with the 0.2 mg/mL rat liver microsomes.
NT: not tested.
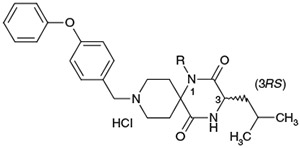
Further structural optimization of the 9-N-substituent was explored to provide additional increases in activity and metabolic stability. Results are summarized in Tables 3-5. To identify the optimal structure of the 9-N-substituent on the piperidine moiety of the lead structures 1a and 1b, compounds listed in Table 3 were synthesized and evaluated. To confirm the structural requirement of the 9-N-arylalkyl moiety, compounds 17 to 22 were synthesized and evaluated. 9-N-Phenylmethyl analogs 17 and 18 tended to show reduced activities relative to 1b and 1a, respectively. Replacement of the 9-N-{(4-phenoxy)phenylmethyl} residue with 9-N-{(4-phenoxy)phenylethyl} afforded 19 with a remarkable reduction in activity. Based on this result, the 9-N-phenylmethyl moiety was found to be required for the antagonist activity. The less basic aniline type analog 20 (calculated pKa 4.12) and non-basic analog 21 (calculated pKa 2.52) also showed remarkable reduction in activity. Recovery of the antagonist activity for 9-N-{6-(phenyl)hexyl} analog 22 relative to that of 17 strongly suggested another interaction site with the receptor. As a result, the 9-N-{(4-phenoxy)phenylmethyl} moiety was concluded to be the optimal 9-N-substituent.
Table 3.
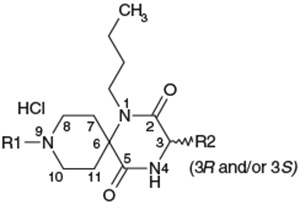
Effect of the 9-N-substituent on activity profiles
 |
||||||
|---|---|---|---|---|---|---|
| Compds | R1 | R2 | C3 Configuration (3R and/or 3S) |
IC50 (nM) Ca assay |
T1/2 in rat liver microsomesa (min) |
Calculated pKab |
| 17 | 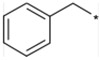 |
 |
(3RS) | 900 | NTc | 7.54 |
| 18 |  |
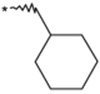 |
(3RS) | 120 | 8.3 | 7.53 |
| 19 | 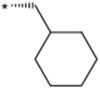 |
(3S) | >3000 | NTc | 7.51 | |
| 20 | 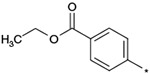 |
 |
(3RS) | >3000 | NTc | 4.12 |
| 21 | 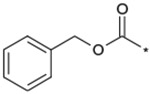 |
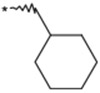 |
(3RS) | 3000 | NTc | 2.52 |
| 22 | 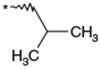 |
(3RS) | 270 | NTc | 8.24 | |
| 1a | 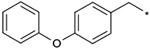 |
 |
(3RS) | 28 | 24 | 7.15 |
| 1b |  |
 |
(3RS) | 94 | 12 | 7.18 |
The data show T1/2 after incubating with the 0.2 mg/mL rat liver microsomes.
Calculated by ADMET predictor (ver. 4.0).
NT: not tested.
Table 5.
Effect of chemical modification of the linker X on activity profiles
 |
||||
|---|---|---|---|---|
| Compds | X | C3 Configuration (3R and/or 3S) |
IC50 (nM) Ca assay |
Stability in rat liver microsomesa remaining% |
| 29 | 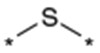 |
(3S) | 170 | 38 |
| 30 | 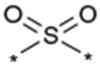 |
(3S) | 86 | 19 |
| 31 | 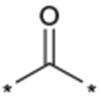 |
(3S) | 81 | 23 |
| 32 | 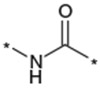 |
(3S) | 57 | 29 |
| 1a | 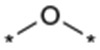 |
(3RS) | 28 | 33 |
The data show the remaining% 15 min after incubating with the 0.5 mg/mL rat liver microsomes.
The effects of the 9-N-substitution with heteroarylmethyl moieties on the activity profiles were investigated. Results are summarized in Table 4. Analogs 23–28 possess lower cLog P values relative to 1a. 9-N-{2-(phenoxy)pyridine-5-ylmethyl} analog 23 exhibited nearly equipotent in vitro activities relative to 1a although it showed less stability in rat liver microsomes. Accordingly, the benzylic phenyl of 1a could be replaced by a more hydrophilic pyridine moiety without loss of the in vitro activities. 9-N-{4-(pyrid-3-yl)phenylmethyl} analog 24 exhibited slightly less potent activity relative to 1a. Five-membered heteroarylmethyl analogs 25–28 were also synthesized and evaluated. Unexpectedly, 9-N-(3,5-dimethyl-1-phenylpyrazol-4-ylmethyl) analog 25 was found to show agonist-like activity in the calcium assay. 9-N-(2-Phenylimidazol-4-ylmethyl) analog 26, 9-N-(2-phenyl-5-methyloxazole-4-ylmethyl) analog 27, and 9-N-(2-phenyl-4-thiazolylmethyl) analog 28 exhibited weaker antagonist activity than 1a. Unfortunately, their stabilities in rat liver microsomes were not improved using this approach.
Table 4.
Effect of the 9-N-substituent with heteroaryl moieties on activity profiles
 |
|||||
|---|---|---|---|---|---|
| Compds | R | C3 Configuration (3R and/or 3S) | IC50 (nM) Ca assay | Stability in rat liver microsomesa remaining% | cLog Pb |
| 23 | 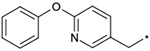 |
(3RS) | 21 | 10 | 6.16 |
| 24 | 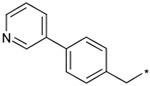 |
(3S) | 71 | 29 | 6.16 |
| 25 | 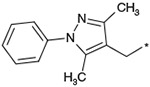 |
(3RS) | 35 | 23 | 6.05 |
| 26 | 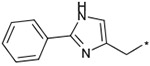 |
(3RS) | 160 | NTc | 5.49 |
| 27 | 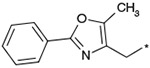 |
(3S) | 450 | NTc | 5.55 |
| 28 | 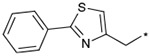 |
(3RS) | 570 | NTc | 6.00 |
| 1a | 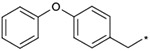 |
(3RS) | 28 | 33 | 7.66 |
The data show the remaining% 15 min after incubating with the 0.5 mg/mL rat liver microsomes.
Calculated by ADMET predictor (ver. 4.0).
NT: not tested.
As described in Table 5, the SAR of the linker X which connects the two phenyl moieties of the 9-N-{4-phenoxyphenylmethyl} moiety of 1a was investigated. Results are summarized in Table 5. Replacement of the ether oxygen of 1a with a sulfur atom afforded the corresponding sulfide analog 29 with less potent activity and an increased stability in rat liver microsomes. The corresponding sulfone analog 30 exhibited less potent activity relative to 1a. Replacement of the ether oxygen of 1a with a carbonyl provided 31 which also had slightly less potent activity relative to 1a. Replacement of the ether oxygen of 1a with an amide moiety afforded 32 with slightly less potent activity. Accordingly, an ether oxygen was selected as the best linker to connect the two phenyls of the 9-N-{4-(phenoxy)phenylmethyl} residue.
The structure activity relationship (SAR) study described in Tables 3-5 strongly suggests that concurrent conversion of the activity and stability in rat liver microsomes by chemical modification of the 9-N-substituent from 9-N-[4-phenoxy]phenylmethyl] moiety is difficult. Accordingly, stepwise optimizations of this cite which had one of the most promising activity profiles was attempted to improve its PK profiles as described in Tables 6 and 7.
The effects of a p-substitution at the predicted metabolic site8 of the terminal phenyl moiety of 1a on the stability in rat liver microsomes were investigated. As shown in Table 6, introduction of p-methyl and p-methoxy groups as electron-donating substituents into the terminal phenyl moiety of 1a afforded 4-(p-methylphenoxy)phenylmethyl and 4-(p-methoxyphenoxy)phenylmethyl analogs 34 and 35, respectively. Both had a tendency to be slightly less potent relative to 1a, while demethylation of 35 afforded the analog 36 which had slightly more potent activity relative to the corresponding methoxy analog 35. Introduction of a p-fluoro group as an electron-withdrawing group instead of utilizing the electron-donating p-methyl and p-methoxy groups afforded 33 with slightly less potent activity relative to 1a. Introduction of N-methyl aminocarbonyl, methanesulfonylamino, aminosulfonyl and hydroxycarbonyl groups as the electron-withdrawing and hydrophilic p-substituent afforded 37, 38, 39, and 40, respectively, with nearly equipotent in vitro activities. The stability of these test compounds in the rat liver microsomes was investigated but in vitro data did not indicate a significant improvement in their metabolic stability. However, analogs possessing hydrophilic substituents are predicted to have the potential of improved physicochemical properties such as solubility. PK studies of analogs 36, 37, and 40, which possess relatively good in vitro potency and hydrophilic substituents such as hydroxyl, N-methyl aminocarbonyl and hydroxycarbonyl residues, were carried out.9
2.3. Pharmacokinetics
PK data obtained after single-dose oral administration of the initial chemical leads 1a, 1b and the representative compounds 36, 37, 40 to rats, are presented in Table 7. As described previously, the initial leads 1a and 1b showed very poor bioavailability (1.9% and 1.3%, respectively). Other PK values such as the maximum plasma concentration (Cmax), plasma elimination half-life (T1/2) and AUC were also very poor. The probable reasons for such poor PK values for 1a and 1b were the large clearance (CL= 113 and 137 mL/min/kg, respectively) and the large distribution volumes (Vss = 2542 and 2349 mL/kg, respectively) which are unfavorable for compounds such as anti-HIV drugs which require transport in the blood to achieve efficacy.
With the expectation of improved PK profiles, analogs 36, 37, and 40 possessing hydrophilic substituents were investigated for their pharmacokinetics. N-Methyl carboxy amide analog 37 tended to show improved PK values in its Cmax and oral AUC relative to 1a and 1b, while the phenol analog 36 did not. Interestingly, benzoic acid analog 40 showed significantly improved PK values in its Cmax (7200 ng/mL), oral exposure (AUC = 10,532 ng h/mL) and bioavailability (BA = 34%) after oral dosing. The marked reduction in clearance (CL = 16 mL/min/kg) and distribution volume (Vss = 145 mL/kg) after iv dosing was considered to be the most plausible reason for the improved AUC and BA. Additionally, remarkable improvement of solubility and Caco-2 permeability of 40 relative to 1a and 1b was estimated to be other possible reasons as shown in Table 8. The marked reduction of the CL of 40 strongly suggested in vivo metabolic stabilization, though in vitro studies did not indicate a significant improvement in metabolic stability.
Table 8.
In vitro pharmacokinetic data for 1a, 1b, 36, 37, and 40
| Compds | Caco-2 papp(×10−6cm/s) |
Solubility (μM) | cLog Pa | |
|---|---|---|---|---|
| A to B | B to A | |||
| 1a | 0.15 | 0.27 | <5 | 7.66 |
| 1b | 1.51 | 2.23 | 14 | 6.46 |
| 36 | 0.33 | 0.14 | <5 | 6.99 |
| 37 | 3.51 | 3.91 | 6 | 6.38 |
| 40 | 11.0 | 16.8 | 29 | 5.13 |
Calculated by ADMET predictor (ver. 4.0).
2.4. Anti-HIV activity
The anti-HIV activities of compounds 39 and 40, both potent CCR5 antagonists, were evaluated in the p24 assay. Results are shown in Table 9 and compared with 1b. Compounds 39 and 40 were found to show more potent anti-HIV activity in the p24 assay relative to 1b. Especially 40, which has the most favorable PK profiles among the compounds tested, potently inhibited not only the replication of laboratory and primary (R5) HIV-1 strains but also that of various multidrug-resistant monocyte/macrophage tropic (R5) HIV-1 strains.6 Compound 40 was inactive against T cell tropic (X4) HIV-1. These results support the hypothesis that spirodiketopiperazines such as 1b, 39, and 40 possess potent anti-HIV activities through their antagonist effects on CCR5.
Table 9.
Anti-HIV activity of representative compounds
| Compds | Mean IC50 + SD (nM) |
|
|---|---|---|
| Anti-HIV-1 activity in p24 assay HV-1Ba-L (R5) | CCR5 Ca assay | |
| 1b | 160 ± 40 | 94 |
| 39 | 10 | 28 |
| 40 | 39 ± 34.4 | 13 |
| Zidovudinea | 7 ± 4 | – |
| Nelfinavirb | 12 ± 8 | – |
Zidovudine is a reverse transcriptase inhibitor.
Nerfinavir is a HIV-1 protease inhibitor.
3. Conclusions
In conclusion, starting with the initial hit compounds 1a and 1b, which showed unfavorable PK profiles, we identified 40 which showed significant improvement in its bioavailability (BA) and oral exposure (AUC) without reduction of its antagonist activity by introducing a carboxylic acid function into the p-position of the terminal phenyl moiety. Although the role of the carboxylic acid function is still unclear, significant reduction of CL and Vss was considered to be the most plausible reason for the increased Cmax, AUC and BA of 40. Its improved physicochemical properties, through the increase of hydrophilicity, were thought to contribute to the improved Cmax, AUC and BA after oral dosing. N-Methyl aminocarbonyl analog 37 also tended to show improved PK values including Cmax and AUC after its oral dosing. As such, introduction of hydrophilic hydroxycarbonyl and N-methyl aminocarbonyl functions into the p-position of the terminal phenoxy moiety was found to be effective not only to block the predicted metabolic deactivation but also to improve PK profiles. Compound 40 showed more potent activity than 1b, one of the initial hit compounds, in the p24 assay (the BAL strain of HIV6). Further optimization of 40 to improve its activity and PK profile to provide CCR5 antagonists suitable for clinical use is ongoing.
4. Experimental
4.1. Chemistry
4.1.1. General methods
Analytical samples were homogeneous as confirmed by thin layer chromatography (TLC), and afforded spectroscopic results consistent with the assigned structures. Proton nuclear magnetic resonance spectra (1H NMR) were recorded on a Varian Gemini-200 or a MERCURY-300 spectrometer with tetramethylsilane as an internal standard. The chemical shift values δ are reported in ppm and coupling constants (J) in Hertz (Hz). Fast atom bombardment (FAB) and electron ionization (EI) mass spectra were obtained with a JEOL JMS-700 spectrometer. Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectra were obtained on Perceptive Voyager Elete. Atmospheric pressure chemical ionization (APCI) mass spectra were determined by Hitachi M-1200H spectrometer. IR spectra were measured on a JASCO FTIR-430 spectrometer. Elemental analyses were performed with a Perkin-ElmerPE2400 series II CHNS/O Analyzer and were only indicated as the elements within ±0.4% of the theoretical values unless otherwise noted. Column chromatography was carried out on silica gel [Merck Silica Gel 60 (0.063–0.200 mm), Fuji Silysia BW235 or Fuji Silysia FL60D]. TLC was performed on silica gel (Merck TLC, Silica Gel 60 F254).
4.1.2. A typical procedure for the Ugi four-component condensation using Rink-isonitrile resin. (1a, 1b, 2–13, 15–18, 23, 25, 26, and 28)
4.1.2.1. 1-Butyl-3-(cyclohexylmethyl)-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (1a).
The Rink-isonitrile resin 44 (0.45 mmol/g, 500 mg, 0.23 mmol) was washed with THF/MeOH (1:1, 4 mL × 2). To a suspension of the resin in THF/MeOH (1:1, 4 mL) were added N-allyloxycarbonyl-4-piperidone 41a (206 mg, 1.13 mmol), n-butylamine 42a (82 mg, 1.13 mmol) and N-Boc-dl-cyclohexylalanine 43a (305 mg, 1.13 mmol). The mixture was shaken for 16 h at 65 °C. After cooling to room temperature, the resin was collected by filtration and successively washed with THF/MeOH (1:1, 4 mL × 3) and then CH2Cl2 (4 mL × 3). To a suspension of the resin in CH2Cl2 (4 mL) were successively added acetic acid (135 mg, 2.25 mmol), tetrakis(triphenylphosphine)palladium(0) (52.0 mg, 0.045 mmol) and tributyl tin hydride (327 mg, 1.13 mmol). The mixture was shaken for 4 h at room temperature. The resin was collected by filtration and successively washed with CH2Cl2 (4 mL × 4) and DMF (4 mL × 4). To a suspension of the resin in 1% acetic acid in DMF (4 mL) were successively added 4-phenoxybenzaldehyde (223 mg, 1.13 mmol) and then sodium triacetoxyborohydride (238 mg, 1.13 mmol). The mixture was shaken for 6 h at room temperature. The resin was collected by filtration and successively washed with MeOH (4 mL × 2), DMF (4 mL × 4), and then CH2Cl2 (4 mL × 4). The resin was suspended in 25%TFA in CH2Cl2 (4 mL) at 0 °C. The mixture was allowed to warm up to room temperature and then stirred for 30 min. The resin was collected by filtration and rinsed with CH2Cl2 (4 mL × 3), toluene (4 mL × 3), and 1.25 M acetic acid in toluene (4 mL). The resin was suspended in 1.25 M acetic acid in toluene (4 mL). The suspension was shaken for 24 h at 90 °C. After cooling to room temperature, the resin was collected by filtration and washed with CHCl3/MeOH (1:1, 4 mL). The combined filtrate and washings were evaporated. The resulting residue was purified by column chromatography on silica gel with a gradient of AcOEt/MeOH (from 1:0 to 10:1). The resulting residue after evaporation was treated with 4 N HCl in AcOEt and then washed with Et2O to afford the title compound 1a (23 mg, 70% yield) as a white powder. TLC Rf 0.73 (CHCl3/MeOH, 10:1); 1H NMR (200 MHz, CD3OD) δ 7.74–7.56 (m, 1H), 7.53 (d, J = 8.8 Hz, 2H), 7.40 (m, 2H), 7.18 (m, 1H), 7.10–7.00 (m, 3H), 4.33 (s, 2H), 4.04 (dd, J = 7.4, 4.8 Hz, 1H), 3.80 (m, 2H), 3.60–3.35 (m, 4H), 2.43 (m, 2H), 2.17 (m, 2H), 1.90–1.60 (m, 7H), 1.60–1.45 (m, 2H), 1.45–1.30 (m, 2H), 1.30–1.15 (m, 4H), 1.10–0.80 (m, 5H); IR (KBr) 3434, 3210, 3064, 2926, 2851, 2664, 2558, 1672, 1590, 1509, 1489, 1418, 1373, 1241, 1173, 1118, 1072, 1048 cm−1; MS (FAB, Pos) m/z 518 (M+H)+; Elemental Anal. Calcd for C32H43N3O3·HCl·0.5H2O: C, 68.25; H, 8.05; N, 7.46. Found: C, 68.23; H, 7.88; N, 6.77.
4.1.2.2. 1-Butyl-3-isobutyl-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (1b).
The title compound was synthesized in 48% yield according to the same procedures as described for the preparation of 1a using N-Boc-dl-leucine for 43b. TLC Rf 0.63 (CHCl3/MeOH, 10:1); 1H NMR (200 MHz, CD3OD) δ 7.54 (d, J = 8.8 Hz, 2H), 7.40 (m, 2H), 7.18 (m, 1H), 7.11–7.00 (m, 4H), 4.33 (s, 2H), 4.01 (dd, J = 7.6, 4.8 Hz, 1H), 3.80 (m, 2H), 3.60–3.35 (m, 4H), 2.43 (m, 2H), 2.18 (m, 2H), 1.80 (m, 1H), 1.70 (m, 1H), 1.54 (m, 2H), 1.37 (m, 3H), 1.00–0.90 (m, 9H); IR (KBr) 3440, 3221, 3066, 2957, 2871, 2559, 1673, 1590, 1509, 1489, 1419, 1371, 1329, 1242, 1172 cm−1; MS (FAB. Pos., Glycerin+m-NBA) m/z 478 (M+H)+, 183 (base peak); Elemental Anal. Calcd for C29H39N3O3·HCl·H2O: C, 65.46; H, 7.96; N, 7.90. Found: C, 65.67; H, 7.89; N, 7.83.
4.1.2.3. 3-Isobutyl-9-(4-phenoxybenzyl)-1-propyl-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (2).
The title compound was synthesized in 87% yield according to the same procedures as described for the preparation of 1a using n-propylamine 42b and N-Boc-dl-leucine 43b. TLC Rf 0.61 (CHCl3/MeOH, 10:1); 1H NMR (200 MHz, CD3OD) δ 7.55 (m, 2H), 7.40 (m, 2H), 7.18 (m, 1H), 7.05 (m, 4H), 4.33 (s, 2H), 4.01 (dd, J = 7.6, 4.8 Hz, 1H), 3.79 (m, 2H), 3.60–3.30 (m, 4H), 2.46 (m, 2H), 2.17 (m, 2H), 1.95–1.40 (m, 5H), 0.94 (m, 9H); IR (KBr) 3439, 3220, 3066, 2959, 2872, 2663, 2561, 1672, 1590, 1509, 1489, 1418, 1370, 1330, 1241, 1200, 1172, 1072 cm−1; MS (APCI, Pos) m/z 464 (M+H)+; Elemental Anal. Calcd for C28H37N3O3·HCl·H2O: C, 64.59; H, 7.74; N, 8.07. Found: C, 64.38; H, 7.67; N, 8.07.
4.1.2.4. 1,3-Diisobutyl-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (3).
The title compound was synthesized in 62% yield according to the same procedures as described for the preparation of 1a using i-butylamine 42c and N-Boc-dl-leucine 43b. TLC Rf 0.50 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.50 (d, J = 8.7 Hz, 2H), 7.39 (dd, J = 8.7, 7.5 Hz, 2H), 7.17 (t, J = 7.5 Hz, 1H), 7.13–7.04 (m, 4H), 4.28 (s, 2H), 4.4 (dd, J = 8.1, 4.2 Hz, 1H), 3.81–3.54 (m, 2H), 3.52–3.21 (m, 4H), 2.46–2.11 (m, 4H), 2.00–1.57 (m, 4H), 0.94 (d, J = 6.3 Hz, 6H), 0.90 (d, J = 6.3 Hz, 3H), 0.90 (d, J = 6.3 Hz, 3H); IR (KBr) 2958, 2516, 1676, 1590, 1510, 1489, 1410, 1242, 1199, 1172, 1097, 1071 cm−1; MS (APCI, Pos) m/z 478 (M+H)+; Elemental Anal. Calcd for C29H39N3O3·HCl: C, 67.75; H, 7.84; N, 8.17. Found: C, 67.35; H, 7.91; N, 8.06.
4.1.2.5. 1-(3-Hydroxybutyl)-3-isobutyl-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (4).
The title compound was synthesized in 31% yield according to the same procedures as described for the preparation of 1a using 3-t-butyldimethylsilylhydroxybutylamine 42d and N-Boc-dl-leucine 43b. TLC Rf 0.49 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.54 (d, J = 8.5 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.18 (t, J = 7.5 Hz, 1H), 7.04 (m, 4H), 4.33 (s, 2H), 4.02 (m, 1H), 3.80 (m, 3H), 3.51 (m, 4H), 2.46 (m, 2H), 2.19 (m, 2H), 1.85–1.57 (m, 5H), 1.17 (d, J = 6.0 Hz, 3H), 0.94 (d, J = 9.0 Hz, 6H); IR (KBr) 3405, 2960, 1675, 1590, 1510, 1489, 1421, 1242, 1172, 1048 cm−1; MS (APCI, Pos) m/z 494 (M+H)+; HRMS Calcd 494.3019, Obsd 494.3025.
4.1.2.6. 1-[(2E)-2-Butenyl]-3-isobutyl-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (5).
The title compound was synthesized in 29% yield according to the same procedures as described for the preparation of 1a using 2-butenylamine 42e and N-Boc-dl-leucine 43b. TLC Rf 0.32 (CHCl3/MeOH, 20:1); 1H NMR (300 MHz, CD3OD) δ 7.52 (d, J = 8.7 Hz, 2H), 7.44–7.35 (m, 2H), 7.22–7.14 (m, 1H), 7.06 (d, J = 8.7 Hz, 2H), 7.10–7.00 (m, 2H), 5.75–5.60 (m, 1H), 5.52–5.38 (m, 1H), 4.33 (s, 2H), 4.15–3.93 (m, 2H), 4.03 (dd, J = 7.8, 4.5 Hz, 1H), 3.88–3.66 (m, 2H), 3.55–3.42 (m, 2H), 2.52–2.35 (m, 2H), 2.28–2.08 (m, 2H), 1.90–1.57 (m, 3H), 1.65 (dd, J = 6.3, 1.5 Hz, 3H), 0.95 (d, J = 6.6 Hz, 3H), 0.94 (d, J = 6.6 Hz, 3H); IR (KBr) 2955, 2512, 1673, 1590, 1509, 1490, 1417, 1241 cm−1; MS (APCI, Pos) m/z 476 (M+H)+; Elemental Anal. Calcd for C29H37N3O3·HCl: C, 68.02; H, 7.48; N, 8.21.Found: C, 66.1; H, 7.51; N, 8.00.
4.1.2.7. 1-(2-Butynyl)-3-isobutyl-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (6).
The title compound was synthesized in 23% yield according to the same procedures as described for the preparation of 1a using 2-butynylamine hydrochloride 42f and N-Boc-dl-leucine 43b. TLC Rf 0.70 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.51 (d, J = 8.7 Hz, 2H), 7.39 (dd, J = 8.7, 7.2 Hz, 2H), 7.18 (t, J = 7.2 Hz, 1H), 7.09–7.00 (m, 4H), 4.33 (br s, 2H), 4.28–4.10 (m, 2H), 4.05 (dd, J = 7.8, 4.5 Hz, 1H), 3.86–3.70 (m, 2H), 3.56–3.43 (m, 2H), 2.59–2.40 (m, 2H), 2.34–2.15 (m, 2H), 1.89–1.57 (m, 6H), 0.94 (d, J = 6.6 Hz, 3H), 0.93 (d, J = 6.6 Hz, 3H); IR (KBr) 3432, 2956, 1675, 1590, 1509, 1489, 1413, 1242, 1173 cm−1; MS (APCI, Pos) m/z 474 (M+H)+; HRMS Calcd 474.2757, Obsd 474.2753.
4.1.2.8. 1-Benzyl-3-isobutyl-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (7).
The title compound was synthesized in 50% yield according to the same procedures as described for the preparation of 1a using benzylamine 42g and N-Boc-dl-leucine 43b. TLC Rf 0.66 (CHCl3/MeOH, 10:1); 1H NMR (200 MHz, CD3OD) δ 7.50 (d, J = 8.4 Hz, 2H), 7.45–7.12 (m, 8H), 7.10–6.98 (m, 4H), 4.82 (m, 2H), 4.29 (s, 2H), 4.18 (dd, J = 8.0, 4.6 Hz, 1H), 3.73 (m, 2H), 3.42 (m, 2H), 2.65–2.30 (m, 2H), 2.20–2.05 (m, 2H), 2.00–1.60 (m, 3H), 0.98 (d, J = 6.2 Hz, 6H); IR (KBr) 3405, 3194, 3063, 2954, 2871, 2661, 2508, 2462, 1681, 1614, 1589, 1511, 1488, 1470, 1455, 1412, 1361, 1331, 1307, 1240, 1200, 1176, 1155, 1130, 1069 cm−1; MS (MALDI, Pos) m/z 512 (M+H)+; Elemental Anal. Calcd for C32H37N3O3·Cl·H: C, 70.12; H, 6.99; N, 7.67. Found: C, 69.64; H, 7.03; N, 7.63.
4.1.2.9. (3S)-1-Butyl-3-isobutyl-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (8).
The title compound was synthesized in 13% yield according to the same procedures as described for the preparation of 1a using N-Boc-l-leucine 43c. −2.11 (c 0.95, MeOH);TLC Rf 0.29 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.54 (d, J = 8.7 Hz, 2H), 7.42–7.36 (m, 2H), 7.18 (m, 1H), 7.05 (d, J = 8.7 Hz, 2H), 7.05–7.02 (m, 2H), 4.33 (s, 2H), 3.98 (dd, J = 8.1, 4.5 Hz, 1H), 3.86–3.72 (m, 2H), 3.53–3.37 (m, 4H), 2.47–2.36 (m, 2H), 2.24–2.12 (m, 2H), 1.80–1.30 (m, 7H), 0.95 (t, J = 7.2 Hz, 3H), 0.95 (d, J = 6.3 Hz, 3H), 0.93 (d, J = 6.3 Hz, 3H); IR (KBr) 3445, 2956, 2565, 1676, 1590, 1509, 1489, 1418, 1329, 1242, 1172, 1073, 1049 cm−1; MS (APCI, Pos) m/z 478 (M+H)+; HRMS Calcd 478.307, Obsd 478.3069.
4.1.2.10. (3R)-1-Butyl-3-isobutyl-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (9).
The title compound was synthesized in 14% yield according to the same procedures as described for the preparation of 1a using N-Boc-d-leucine 43d. +2.25 (c 1.15, MeOH); TLC Rf 0.29 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.54 (d, J = 8.7 Hz, 2H), 7.42–7.36 (m, 2H), 7.18 (m, 1H), 7.05 (d, J = 8.7 Hz, 2H), 7.05–7.02 (m, 2H), 4.32 (s, 2H), 4.01 (dd, J = 7.8, 4.8 Hz, 1H), 3.85–3.72 (m, 2H), 3.50–3.39 (m, 4H), 2.52–2.38 (m, 2H), 2.24–2.11 (m, 2H), 1.84–1.20 (m, 7H), 0.95 (t, J = 7.2 Hz, 3H), 0.95 (d, J = 6.3 Hz, 3H), 0.93 (d, J = 6.3 Hz, 3H); IR (KBr) 3444, 2957, 2565, 1676, 1590, 1509, 1489, 1418, 1242, 1172 cm−1; MS (APCI, Pos) m/z 478 (M+H)+; HRMS Calcd 478.307, Obsd 478.3069.
4.1.2.11. (3S)-1-Butyl-3-neopentyl-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (10).
The title compound was synthesized in 51% yield according to the same procedures as described for the preparation of 1a using N-Boc-dl-t-butylalanine 43e. TLC Rf 0.52 (CHCl3/MeOH, 20:1); 1H NMR (300 MHz, CD3OD) δ 7.52 (d, J = 9.0 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.18 (t, J = 7.5 Hz, 1H), 7.04 (m, 4H), 4.33 (s, 2H), 4.01 (dd, J = 7.2, 3.3 Hz, 1H), 3.82 (m, 1H), 3.71 (m, 1H), 3.50 (m, 2H), 3.43 (m, 2H), 2.38 (m, 2H), 2.24 (m, 2H), 2.00 (dd, J = 14.0, 3.3 Hz, 1H), 1.55 (dd, J = 14.0, 7.2 Hz, 1H), 1.50 (m, 2H), 1.36 (m, 2H), 0.99 (s, 9H), 0.95 (t, J = 7.0 Hz, 3H); IR (KBr) 2957, 2506, 1678, 1590, 1510, 1489, 1419, 1370, 1285, 1243, 1174, 1115 cm−1; MS (APCI, Pos) m/z 492 (M+H)+; HRMS Calcd 492.3226, Obsd 492.3226.
4.1.2.12. 1-Butyl-9-(4-phenoxybenzyl)-3-propyl-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (11).
The title compound was synthesized in 52% yield according to the same procedures as described for the preparation of 1a using N-Boc-dl-norvaline 43f. TLC Rf 0.36 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.51 (d, J = 8.7 Hz, 2H), 7.39 (dd, J = 8.7, 7.5 Hz, 2H), 7.18 (t, J = 7.5 Hz, 1H), 7.10–7.00 (m, 4H), 4.33 (s, 2H), 4.04 (dd, J = 5.7, 4.5 Hz, 1H), 3.93–3.66 (m, 2H), 3.55–3.31 (m, 4H), 2.47–2.09 (m, 4H), 1.92–1.68 (m, 2H), 1.61–1.21 (m, 6H), 1.01–0.90 (m, 6H); IR (KBr) 3436, 2958, 2872, 2550, 1673, 1590, 1509, 1489, 1419, 1243, 1172, 1071, 954, 873, 787, 693 cm−1; MS (APCI, Pos) m/z 464 (M+H)+; HRMS Calcd 464.2913, Obsd 464.2915.
4.1.2.13. 1-Butyl-3-(cyclopentylmethyl)-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (12).
The title compound was synthesized in 53% yield according to the same procedures as described for the preparation of 1a using N-Boc-cyclopnetyl-dl-alanine 43g. TLC Rf 0.66 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.52 (d, J = 8.5 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.18 (t, J = 7.5 Hz, 1H), 7.05 (m, 4H), 4.34 (s, 2H), 4.00 (t, J = 6.0 Hz, 1H), 3.82 (m, 2H), 3.49 (m, 2H), 3.39 (m, 2H), 2.37 (m, 2H), 2.17 (m, 2H), 1.96 (m, 1H), 1.81 (m, 4H), 1.58 (m, 6H), 1.38 (m, 2H), 1.17 (m, 2H), 0.95 (t, J = 7.0 Hz, 3H); IR (KBr) 3433, 3199, 2953, 2870, 2499, 1681, 1589, 1510, 1488, 1419, 1373, 1336, 1240, 1199, 1174, 1114, 1070, 1049 cm−1; MS (APCI, Pos) m/z 504 (M+H)+; HRMS Calcd 504.3226, Obsd 504.3231.
4.1.2.14. 1-Butyl-3-(methoxymethyl)-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (13).
The title compound was synthesized in 59% yield according to the same procedures as described for the preparation of 1a using N-Boc-O-methyl-dl-serine 43h. TLC Rf 0.48 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.51 (d, J = 8.7 Hz, 2H), 7.39 (dd, J = 8.7, 7.2 Hz, 2H), 7.17 (t, J = 7.2 Hz, 1H), 7.09–6.99 (m, 4H), 4.30 (s, 2H), 4.07 (t, J = 3.0 Hz, 1H), 3.91 (m, 1H), 3.77 (dd, J = 9.0, 3.0 Hz, 1H), 3.67 (m, 1H), 3.58–3.39 (m, 4H), 3.31 (s, 3H), 3.26 (m, 1H), 2.48–2.13 (m, 4H), 1.65 (m, 1H), 1.53–1.28 (m, 3H), 0.95 (t, J = 7.5 Hz, 3H); IR (KBr) 3424, 2931, 2551, 1662, 1590, 1509, 1489, 1423, 1371, 1242, 1198, 1116, 1075 cm−1; MS (APCI, Pos) m/z 466 (M+H)+; Elemental Anal. Calcd for C27H35N3O4·Cl·H: C, 64.59; H, 7.23; N, 8.37. Found: C, 63.45; H, 7.12; N, 8.21.
4.1.2.15. Benzyl [1-butyl-2,5-dioxo-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undec-3-yl]acetate hydrochloride (15).
The title compound was synthesized in 36% yield according to the same procedures as described for the preparation of 1a using N-Boc-dl-aspartic acid β-benzyl ester 43i. TLC Rf 0.74 (CHCl3/MeOH, 9:1); 1H NMR (300 MHz, CD3OD) δ 7.52 (d, J = 7.0 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.33 (m, 5H), 7.18 (t, J = 7.5 Hz, 1H), 7.05 (m, 4H), 5.12 (s, 2H), 4.33 (s, 2H), 4.31 (m, 1H), 3.88 (m, 1H), 3.66 (m, 1H), 3.50–3.35 (m, 4H), 3.08 (dd, J = 17.7, 4.8 Hz, 1H), 2.86 (dd, J = 17.7, 3.0 Hz, 1H), 2.34 (m, 2H), 2.25 (m, 2H), 1.50 (m, 2H), 1.36 (m, 2H), 0.94 (t, J = 7.5 Hz, 3H); IR (KBr) 3735, 3412, 1736, 1675, 1489, 1423, 1242, 1175 cm−1; MS (APCI, Pos) m/z 570 (M+H)+; HRMS Calcd 570.2968, Obsd 570.297.
4.1.2.16. 1-Butyl-3-(3-cyclohexylpropyl)-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (16).
The title compound was synthesized in 39% yield according to the same procedures as described for the preparation of 1a using N-Boc-dl-3-cyclohexylpropylalanine 43j. TLC Rf 0.76 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.53–7.49 (m, 2H), 7.42–7.36 (m, 2H), 7.18 (m, 1H), 7.10–7.02 (m, 4H), 4.32 (s, 2H), 4.04 (t, J = 4.8 Hz, 1H), 3.87 (m, 1H), 3.71 (m, 1H), 3.56–3.40 (m, 3H), 3.35 (m, 1H), 2.48–2.12 (m, 4H), 1.86–1.10 (m, 19H), 0.95 (t, J = 7.5 Hz, 3H), 0.95 (m, 2H); IR (KBr) 2923, 2564, 1679, 1591, 1510, 1490, 1245, 1172 cm−1; MS (APCI, Pos) m/z 546 (M+H)+; HRMS Calcd 546.3696, Obsd 546.3703.
4.1.2.17. 9-Benzyl-1-butyl-3-isobutyl-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (17).
The title compound was synthesized in 59% yield according to the same procedures as described for the preparation of 1a using N-Boc-dl-leucine 43b and benzaldehyde for 4-phenoxybenzaldehyde. TLC Rf 0.54 (CHCl3/MeOH, 10:1); 1H NMR(300 MHz, CD3OD) δ 7.64–7.44 (m, 5H), 4.36 (s, 2H), 4.01 (dd, J = 7.8, 4.8 Hz, H), 3.77 (m, 2H), 3.55–3.35 (m, 4H), 2.60–2.30 (m, 2H), 2.17 (m, 2H), 1.95–1.75 (m, 1H), 1.75–1.60 (m, 2H), 1.60–1.45 (m, 2H), 1.45–1.20 (m, 2H), 1.10–0.80 (m, 9H); IR (KBr) 3435, 3230, 2957, 2871, 2505, 2454, 1680, 1647, 1459, 1413, 1370, 1326, 1147 cm−1; MS (MALDI, Pos) m/z 386 (M+H)+, 91; Elemental Anal. Calcd for C23H35N3O2·HCl: C, 65.46; H, 8.6; N, 9.96. Found: C, 65.09; H, 8.63; N, 9.88.
4.1.2.18. 1-Butyl-3-(cyclohexylmethyl)-9-(4-methoxybenzyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dionehydrochloride (18).
The title compound was synthesized in 59% yield according to the same procedures as described for the preparation of 1a using 4-methoxy-benzaldehyde for 4-phenoxybenzaldehyde. TLC Rf 0.63 (CHCl3/MeOH, 10:1); 1H NMR (200 MHz, CD3OD) δ 7.47 (d, J = 8.8 Hz, 2H), 7.03 (d, J = 8.8 Hz, 2H), 4.29 (s, 2H), 4.04 (dd, J = 7.6, 4.8 Hz, 1H), 3.83 (s, 3H), 3.74 (m, 2H), 3.55–3.35 (m, 4H), 2.41 (m, 2H), 2.15 (m, 2H), 1.85–1.55 (m, 7H), 1.55–1.42 (m, 3H), 1.42–1.30 (m, 3H), 1.30–1.10 (m, 2H), 1.08–0.80 (m, 5H); IR (KBr) 3436, 3221, 2926, 2851, 2666, 2560, 2362, 1672, 1613, 1585, 1517, 1448, 1419, 1373, 1305, 1255, 1182, 1031 cm−1; MS (FAB, Pos) m/z 456 (M+H)+, 121; Elemental Anal. Calcd for C27H41N3O3·HCl·1.5H2O: C, 62.47; H, 8.74; N, 8.09. Found: C, 62.59; H, 8.35; N, 7.90.
4.1.2.19. 1-Butyl-3-(cyclohexylmethyl)-9-[(6-phenoxypyridin-3-yl)methyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione dihydrochloride (23).
The title compound was synthesized in 15% yield according to the same procedures as described for 1a using 6-phenoxynicotinaldehyde for 4-phenoxybenzaldehyde. TLC Rf 0.67 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 8.31 (s, 1H), 8.07 (d, J = 8.3 Hz, 1H), 7.44 (t, J = 7.5 Hz, 2H), 7.26 (t, J = 7.5 Hz, 1H), 7.14 (d, J = 7.5 Hz, 2H), 7.06 (d, J = 8.3 Hz, 1H), 4.39 (s, 2H), 4.04 (dd, J = 7.8, 4.6 Hz, 1H), 3.90–3. 76 (m, 2H), 3.52–3.38 (m, 4H), 2.58–2.36 (m, 2H), 2.25–2.11 (m, 2H), 1.80–1.42 (m, 10H), 1.42–1.17 (m, 5H), 1.05–0.85 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H); IR (KBr) 3449, 2926, 2564, 1671, 1479, 1260, 1200 cm−1; MS (APCI, Pos) m/z 519 (M+H)+; HRMS Calcd 519.3335, Obsd 519.3337.
4.1.2.20. 1-Butyl-3-(cyclohexylmethyl)-9-[(3,5-dimethyl-1-phenyl-1H-pyrazol-4-yl)methyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione dihydrochloride (25).
The title compound was synthesized in 33% yield according to the same procedures as described for the preparation of 1a using 3,5-dimethyl-1-phenylpyrazole-4-carboxaldehyde for 4-phenoxybenzaldehyde. TLC Rf 0.35 (CHCl3/MeOH, 20:1); 1H NMR (300 MHz, CD3OD) δ 7.63–7.48 (m, 5H), 4.33 (s, 2H), 4.05 (dd, J = 7.8, 4.5 Hz, 1H), 3.95–3.74 (m, 2H), 3.67–3.56 (m, 2H), 3.48 (m, 2H), 2.72–2.58 (m, 2H), 2.45 (s, 3H), 2.41 (s, 3H), 2.30–2.07 (m, 2H), 1.84–1.10 (m, 15 H), 1.02–0.92 (m, 2H), 0.96 (t, J = 7.2 Hz, 3H); IR (KBr) 3426, 2926, 1670, 1421 cm−1; MS (APCI, Pos) m/z 520 (M+H)+; HRMS Calcd 520.3652, Obsd 520.3651.
4.1.2.21. 1-Butyl-3-(cyclohexylmethyl)-9-[(2-phenyl-1H-imidazol-4-yl)methyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dionedihydrochloride (26).
The title compound was synthesized in 77% yield according to the same procedures as described for the preparation of 1a using 2-phenyl-1H-imidazole-4-carboxaldehyde for 4-phenoxybenzaldehyde. TLC Rf 0.25 (CHCl3/MeOH, 10:1); 1H NMR (200 MHz, CD3OD) δ 8.04–7.92 (m, 3H), 7.74–7.62 (m, 3H), 4.58 (s, 2H), 4.05 (dd, J = 7.4, 4.8 Hz, 1H), 3.88 (m, 2H), 3.65 (m, 2H), 3.50 (m, 2H), 2.68 (m, 2H), 2.19 (m, 2H), 1.90–1.60 (m, 6H), 1.60–1.45 (m, 3H), 1.45–1.30 (m, 3H), 1.30–1.10 (m, 3H), 1.10–0.80 (m, 5H); IR (KBr) 3410, 2927, 2854, 2699, 2574, 1781, 1671, 1644, 1554, 1448, 1421, 1373, 1349, 1308, 1257, 1178, 1096, 1052, 1001 cm−1; MS (FAB, Pos) m/z 492 (M+H)+, 336; Elemental Anal. Calcd for C29H41N5O2·2HCl: C, 61.69; H, 7.68; N, 12.4. Found: C, 55.71; H, 7.32; N, 10.71.
4.1.2.22. 1-Butyl-3-(cyclohexylmethyl)-9-[(2-phenyl-1,3-thiazol-4-yl)methyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dionehydrochloride (28).
The title compound was synthesized in 51% yield according to the same procedures as described for the preparation of 1a using 2-phenyl-1,3-thiazole-4-carboxaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.62 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 8.03–8.00 (m, 2H), 7.87 (s, 1H), 7.52–7.49 (m, 3H), 4.54 (s, 2H), 4.04 (dd, J = 7.6, 4.8 Hz, 1H), 4.04–3.87 (m, 2H), 3.70–3.58 (m, 2H), 3.51–3.39 (m, 2H), 2.58–2.38 (m, 2H), 2.26–2.13 (m, 2H), 1.7 8–1.43 (m, 9H), 1.40–1.15 (m, 6H), 1.10–0.90 (m, 5H); IR (KBr) 3426, 3298, 3190, 3128, 3086, 2991, 2955, 2927, 2854, 2653, 2512, 2452, 1678, 1645, 1481, 1462, 1450, 1415, 1371, 1351, 1336, 1314, 1146, 1003 cm−1; MS (APCI, Pos) m/z 509 (M+H)+; HRMS Calcd 509.295, Obsd 509.295.
4.1.3. [1-Butyl-2,5-dioxo-9-(4-phenoxybenzyl)-1,4,9-triazaspiro[5.5]undec-3-yl]acetic acid hydrochloride (14)
To a stirred solution of the compound 15 (173 mg, 0.303 mmol) in MeOH (5 mL) was added 2 M NaOH (2 mL). After being stirred for 3 h at room temperature, the reaction mixture was acidified with 2 M HCl and then extracted with AcOEt. The organic layer was washed with water, brine, dried over Na2SO4 and concentrated in vacuo. The resulting residue was triturated with Et2O to yield the title compound (127 mg, 56%) as a white powder. TLC Rf 0.51 (CHCl3/MeOH/AcOH, 20:4:1); 1H NMR (300 MHz, CD3OD) δ 7.55–7.53 (m, 2H), 7.42–7.36 (m, 2H), 7.20–7.15 (m, 1H), 7.07–7.02 (m, 4H), 4.33 (s, 2H), 4.27 (t, J = 4.5 Hz, 1H), 3.96–3.90 (m, 1H), 3.72–3.66 (m, 1H), 3.54–3.38 (m, 4H), 2.97 (dd, J = 18.0, 4.8 Hz, 1H), 2.79 (dd, J = 18.0, 4.8 Hz, 1H), 2.50–2.36 (m, 3H), 2.27–2.16 (m, 1H), 1.62–1.48 (m, 2H), 1.41–1.30 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H); IR (KBr) 3485, 3214, 2960, 2717, 2579, 1664, 1589, 1419, 1241, 1076, 1051, 1005 cm−1; MS (APCI, Neg) m/z 478 (M−H)−; HRMS Calcd 480.2498, Obsd 480.2499.
4.1.4. Benzyl 1-butyl-3-(cyclohexylmethyl)-2,5-dioxo-1,4,9-triazaspiro[5.5]undecane-9-carboxylate (21)
To a suspension of the Rink-isonitrile resin 44 (0.75 mmol/g, 10.0 g, 7.5 mmol) resin in THF/MeOH (1:1, 200 mL) were added N-benzyloxycarbonyl-4-piperidone 41b (5.24 g, 22.5 mmol), n-butylamine 42a (2.22 mL, 22.5 mmol) and N-Boc-dl-cyclohexylalanine 43a (6.50 g, 22.5 mmol). The mixture was shaken for 16 h at 65 °C. After cooling to room temperature, the resin was collected by filtration and successively washed with THF (80 mL × 3), MeOH (80 mL × 3) and then CH2Cl2 (80 mL × 3). The resin was treated with 50% trifluoroacetic in CH2Cl2 (100 mL). The mixture was shaken for 30 min at room temperature. The resin was collected by filtration and successively washed with CH2Cl2 (70 mL × 4) and toluene (100 mL). The resin was suspended in 1.25 M acetic acid in toluene (100 mL). The suspension was shaken for 18 h at 90 °C. After cooling to room temperature, the resin was collected by filtration and washed with CHCl3/MeOH (1:1, 100 mL × 3). The combined filtrate and washings were evaporated. The resulting residue was purified by column chromatography on silica gel with a gradient of CHCl3/MeOH (40:1) to afford the title compound 21 (209 mg, 5.9% yield) as a white powder. TLC Rf 0.46 (CHCl3/MeOH, 20:1); 1H NMR (300 MHz, CDCl3) δ 7.40–7.29 (m, 5H), 5.98 (br s, 1H), 5.15 (s, 2H), 4.14 (br s, 2H), 4.00 (m, 1H), 3.65 (br s, 1H), 3.43 (br s, 1H), 3.26 (m, 2H), 2.03–1.81 (m, 4H), 1.80–1.60 (m, 5H), 1.60–1.10 (m, 10H), 1.10–0.85 (m, 5H); IR (KBr) 3449, 2925, 2852, 1675, 1418, 1352, 1281, 1239, 1179, 1153, 1105, 1018 cm−1; MS (APCI, Pos) m/z 470 (M+H)+.
4.1.5. 1-Butyl-3-isobutyl-9-(6-phenylhexyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (22)
The Rink-isonitrile resin (0.45 mmol/g, 500 mg, 0.225 mmol) was washed with THF/MeOH (1:1) (4 mL × 2). To a suspension of the resin in THF/MeOH (1:1) (4 mL) were successively added 1-(6-phenylhexyl)-4-piperidone 41c (292 mg, 1.125 mmol), n-butylamine 42a (82 mg, 1.125 mmol), and N-Boc-dl-leucine 43b (260 mg, 1.125 mmol). The mixture was shaken for 16 h at 65 °C. After cooling to room temperature, the mixture was filtrated. The collected resin was washed with THF/MeOH (1:1) (4 mL × 3), and CH2Cl2 (4 mL × 3). The resin was then added 25% TFA in CH2Cl2 (4 mL) at 0 °C. The mixture was allowed up to room temperature, and then stirred for 30 min. After filtration, the resin was washed with CH2Cl2 (4 mL × 3), toluene (4 mL × 3), and 1.25 M acetic acid in toluene (4 mL). The suspension of the resin in 1.25 M acetic acid in toluene was agitated for 24 h at 90 °C. After cooling to room temperature, the mixture was filtrated. The resin was washed with CHCl3/MeOH (1:1) (4 mL × 2). The filtrate and washings were concentrated under reduced pressure. The residue was purified by column chromatography over silica gel with a gradient of AcOEt/MeOH from 1:0 to 10:1 to give the title compound (35% yield) as a white powder. TLC Rf 0.62 (CHCl3/MeOH, 10:1); 1H NMR (200 MHz, CD3OD) δ 7.30–7.06 (m, 5H), 4.02 (dd, J = 7.8, 4.8 Hz, 1H), 3.70 (m, 2H), 3.56 (m, 2H), 3.43 (m, 2H), 3.11 (m, 2H), 2.63 (t, J = 7.8 Hz, 2H), 2.46 (m, 2H), 2.18 (m, 2H), 1.95–1.50 (m, 9H), 1.50–1.25 (m, 6H), 0.97 (m, 9H); IR (KBr) 3447, 3199, 2934, 2869, 2663, 2502, 2440, 1673, 1455, 1418, 1372, 1329, 1152, 1086, 1003 cm−1; MS (MALDI, Pos) m/z 456 (M+H)+; Elemental Anal. Calcd for C28H45N3O2·HCl·0.4H2O: C, 67.35; H, 9.45; N, 8.41. Found: C, 67.67; H, 9.39; N, 8.42.
4.1.6. A typical procedure for the solution phase Ugi four-component condensation
4.1.6.1. (3S)-9-Benzyl-1-butyl-3-cyclohexylmethyl-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (52b).
To a stirred solution of 1-benzyl-4-piperidone 41d (49 g, 260 mmol), n-butylamine 42a (19 g, 260 mmol) and N-Boc-l-cyclohexylalanine 43k (80 g, 260 mmol) in MeOH (1.3 L) was added 2-(4-morpholinyl)-ethylisonitrile 50 (36 g, 260 mmol). After being stirred at 55 °C overnight, the reaction mixture was treated with concd HCl (260 L) with cooling. The reaction mixture was stirred at 55 °C for another 2 h, evaporated, treated with Na2CO3 and extracted with AcOEt. The combined organic layers were washed with brine, dried (Na2SO4) and evaporated to give deprotected Ugi product as a yellow oil, which was dissolved in AcOH/toluene (1.25 M, 1.3 L) and stirred at 80 °C for 1 h. The reaction mixture was cooled to room temperature, diluted with AcOEt and washed twice with a small amount of water. The organic layer was washed with aqueous NaHCO3, brine, dried (Na2SO4) and evaporated to afford N-benzylpiperidinodiketopiperazine 51b as an oil (91.3 g). This compound was used to next step without further purification. Debenzylation was carried out by the catalytic hydrogenation at an atmospheric pressure of the resulting oily product 51b (91.3 g, 210 mmol) in EtOH (1.2 L) in the presence of 20% Pd(OH22/C (15 g) for 3 h at 50 °C. Catalyst was removed by filtration through a pad of Celite. The filtrate was treated with 4 N HCl in AcOEt (130 mL) and evaporated. The resulting powder was washed with t-butyl methyl ether to afford 52b as a white powder (70 g, 89% yield in four steps). −37.5 (c 1.04, MeOH); TLC Rf 0.08 (CHCl3/MeOH/AcOH, 9:1:0.1); 1H NMR (CD3OD) δ 4.05 (dd, J = 7.8, 4.8 Hz, 1H), 3.84–3.68 (m, 2H), 3.46–3.34 (m, 4H), 2.40–2.04 (m, 4H), 1.83–1.46 (m, 10H), 1.39 (sextet, J = 7.5 Hz, 2H), 1.33–1.15 (m, 3H), 1.05–0.86 (m, 2H), 0.97 (t, J = 7.2 Hz, 3H).
4.1.6.2. (3RS)-9-Benzyl-1-butyl-3-cyclohexylmethyl-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (52a).
Compound 52a was prepared from N-Boc-dl-cyclohexylalanine 43a according to the same procedure as described for the preparation of 52b.
TLC Rf 0.65 (CHCl3/MeOH/NH3 aq, 20:5:1); 1H NMR (CD3OD) δ 4.00 (dd, J = 7.8, 4.5 Hz, 1H), 3.46–3.24 (m, 4H), 3.03–2.92 (m, 4H), 2.08–1.08 (m, 19H), 1.05–0.84 (m, 5H).
4.1.7. General procedure for the preparation of 1-butyl-3-(cyclohexylmethyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione 9-N-aryl analogs
4.1.7.1. (3S)-1-Butyl-3-(cyclohexylmethyl)-9-[(6-phenylpyridin-3-yl)methyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione dihydrochloride (24).
To a stirred solution of 52b (100 mg, 0.27 mol), 4-(3-pyridyl)benzaldehyde (59 mg, 0.32 mmol) and 1 drop of acetic acid in DMF (2 mL) was added sodium triacetoxyborohydride (114 mg, 0.54 mmol). After being stirred overnight, the reaction mixture was evaporated. The resulting residue was purified by column chromatography on silica gel (AcOEt/MeOH from 1:0 to 10:1) and treated with 4 N HCl in AcOEt (2 mL) to give the title compound in 50% yield. TLC Rf 0.50 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 9.14 (m, 1H), 8.75 (m, 1H), 8.36 (m, 1H), 8.02–7.99 (m, 2H), 7.68–7.62 (m, 3H), 4.63 (s, 2H), 4.05 (dd, J = 7.5, 4.5 Hz, 1H), 4.02–3.94 (m, 2H), 3.64–3.42 (m, 4H), 2.72–2.56 (m, 2H), 2.25–2.06 (m, 2H), 1.80–1.10 (m, 15H), 1.00–0.86 (m, 5H); IR (KBr) 3408, 3017, 2925, 2852, 2648, 2494, 2426, 1681, 1666, 1636, 1605, 1452, 1427, 1387, 1374, 1347, 1331, 1314, 1276 cm−1; MS (APCI, Pos) m/z 503 (M+H)+; HRMS Calcd 503.3386, Obsd 503.3394.
4.1.7.2. (3S)-1-Butyl-3-(cyclohexylmethyl)-9-[(5-methyl-2-phenyl-1,3-oxazol-4-yl)methyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (27).
The title compound was synthesized in 76% yield according to the same procedures as described for the preparation of 24 using 5-methyl-2-phenyl-1,3-oxazole-4-carboxaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.48 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 8.04–8.00 (m, 2H), 7.51–7.49 (m, 3H), 4.34 (s, 2H), 4.04 (dd, J = 7.8, 4.8 Hz, 1H), 3.98–3.82 (m, 2H), 3.70–3.60 (m, 2H), 3.44–3.38 (m, 2H), 2.52 (s, 3H), 2.50–2.36 (m, 2H), 2.28–2.12 (m, 2H), 1.80–1.12 (m, 15H), 1.00–0.86 (m, 5H); IR (KBr) 3407, 3182, 3131, 3084, 2956, 2926, 2855, 2658, 2554, 2443, 1681, 1664, 1652, 1559, 1485, 1471, 1450, 1413, 1372, 1337, 1313, 1287, 1146, 1096, 1082, 1068, 1050 cm−1; MS (APCI, Pos) m/z 507 (M+H)+; Elemental Anal. Calcd for C30H42N4O3·HCl: C, 66.34; H, 7.98; N, 10.32. Found: C, 66.06; H, 8.17; N, 10.05.
4.1.7.3. (3S)-1-Butyl-3-(cyclohexylmethyl)-9-[4-(phenylsulfanyl)benzyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (29).
The title compound was synthesized in 49% yield according to the same procedures as described for the preparation of 24 using 4-phenylsulfanyl-benzaldehyde for 4-(3-pyridyl)benz-aldehyde. TLC Rf 0.74 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.50–7.37 (m, 7H), 7.29 (d, J = 8.4 Hz, 2H), 4.31 (s, 2H), 4.03 (dd, J = 7.5, 7.8 Hz, 1H), 3.84–3.70 (m, 2H), 3.50–3.32 (m, 4H), 2.56–2.38 (m, 2H), 2.24–2.05 (m, 2H), 1.81–1.06 (m, 15H), 1.02–0.84 (m, 5H); IR (KBr) 3425, 3207, 3073, 2957, 2924, 2872, 2850, 2656, 2549, 2436, 1679, 1647, 1601, 1582, 1494, 1472, 1445, 1419, 1373, 1340, 1333, 1312, 1288, 1270, 1146, 1116, 1096, 1081, 1049 cm−1; MS (APCI, Pos) m/z 534 (M+H)+; HRMS Calcd 534.3154, Obsd 534.3159.
4.1.7.4. (3S)-1-Butyl-3-(cyclohexylmethyl)-9-[4-(phenylsulfonyl)-benzyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (30).
The title compound was synthesized in 57% yield according to the same procedures as described for the preparation of 24 using 4-(benzenesulfonyl)benzaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.77 (AcOEt/MeOH, 9:1); 1H NMR (300 MHz, CD3OD) δ 8.08 (d, J = 8.4 Hz, 2H), 8.02–7.96 (m, 2H), 7.80 (d, J = 8.4 Hz, 2H), 7.70–7.55 (m, 3H), 4.43 (s, 2H), 4.02 (dd, J = 7.8, 4.8 Hz, 1H), 3.89–3.73 (m, 2H), 3.49–3.34 (m, 4H), 2.48–2.33 (m, 2H), 2.23–2.04 (m, 2H), 1.82–1.14 (m, 15H), 1.03–0.85 (m, 5H); IR (KBr) 3362, 3202, 3065, 2925, 2851, 2516, 2420, 1672, 1476, 1469, 1447, 1416, 1372, 1346, 1309, 1156, 1106, 1071 cm−1; MS (APCI, Pos) m/z 566 (M+H)+; HRMS Calcd 566.3053, Obsd 566.3055.
4.1.7.5. (3S)-9-(4-Benzoylbenzyl)-1-butyl-3-(cyclohexylmethyl)-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (31).
The title compound was synthesized in 43% yield according to the same procedures as described for the preparation of 24 using 4-benzoyl-benzaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.68 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.87 (d, J = 8.4 Hz, 2H), 7.82–7.74 (m, 4H), 7.67 (t, J = 8.4 Hz, 1H), 7.57–7.51 (m, 2H), 4.48 (s, 2H), 4.04 (dd, J = 7.8, 4.8 Hz, 1H), 3.84–3.78 (m, 2H), 3.58–3.38 (m, 4H), 2.58–2.40 (m, 2H), 2.30–2.10 (m, 2H), 1.82–1.14 (m, 15H), 1.02–0.86 (m, 5H); IR (KBr) 3434, 3370, 3209, 3061, 2925, 2851, 2659, 2517, 2422, 1660, 1612, 1598, 1577, 1469, 1447, 1418, 1372, 1347, 1317, 1278, 1179, 1148, 1115, 1097, 1074 cm−1; MS (APCI, Pos) m/z 530 (M+H)+; HRMS Calcd 530.3383, Obsd 530.3375.
4.1.7.6. 4-{[(3S)-1-Butyl-3-(cyclohexylmethyl)-2,5-dioxo-1,4,9-triazaspiro[5.5]undec-9-yl]methyl}-N-phenylbenzamide hydrochloride (32).
The title compound was synthesized in 38% yield according to the same procedures as described for the preparation of 24 using 4-(phenylcarbamoyl)benzaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.25 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 8.07 (d, J = 8.1 Hz, 2H), 7.73–7.67 (m, 2H), 7.71 (d, J = 8.1 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.17 (t, J = 7.5 Hz, 1H), 4.45 (s, 2H), 4.05 (dd, J = 7.8, 4.8 Hz, 1H), 3.92–3.72 (m, 2H), 3.58–3.36 (m, 4H), 2.50–2.08 (m, 4H), 1.84–1.08 (m, 15H), 0.96 (t, J = 7.8 Hz, 3H), 0.96 (m, 2H); IR (KBr) 3362, 3250, 3061, 2925, 2851, 2662, 2552, 2424, 1669, 1599, 1575, 1540, 1509, 1498, 1493, 1469, 1442, 1420, 1372, 1321, 1261, 1147, 1113, 1098, 1076 cm−1; MS (APCI, Pos) m/z 545 (M+H)+; HRMS Calcd 545.3492, Obsd 545.3499.
4.1.7.7. (3S)-1-Butyl-3-(cyclohexylmethyl)-9-[4-(4-fluorophenoxy)benzyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (33).
The title compound was synthesized in 87% yield according to the same manner as described for the preparation of 24 using 4-(4-fluorophenoxy)benzaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.53 (CHCl3/MeOH, 20:1); 1H NMR (300 MHz, CD3OD) δ 7.53 (d, J = 8.7 Hz, 2H), 7.18–7.00 (m, 6H), 4.33 (s, 2H), 4.04 (dd, J = 7.5, 4.5 Hz, 1H), 3.87–3.69 (m, 2H), 3.55–3.32 (m, 4H), 2.52–2.32 (m, 2H), 2.28–2.08 (m, 2H), 1.83–1.12 (m, 15H), 1.06–0.83 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H); IR (KBr) 3432, 3194, 3134, 3071, 2923, 2852, 2656, 2553, 2426, 1678, 1655, 1648, 1614, 1498, 1473, 1446, 1420, 1375, 1332, 1314, 1250, 1214, 1192, 1173, 1146 cm−1; MS (APCI, Pos) m/z 536 (M+H)+; HRMS Calcd 536.3288, Obsd 536.3287.
4.1.7.8. (3S)-1-Butyl-3-(cyclohexylmethyl)-9-[4-(4-methylphenoxy)benzyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (34).
The title compound was synthesized in 58% yield according to the same procedures as described for the preparation of 24 using 4-(4-methylphenoxy)benzaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.71 (AcOEt); 1H NMR (300 MHz, CD3OD) δ 7.50 (d, J = 8.7 Hz, 2H), 7.19 (d, J = 8.7 Hz, 2H), 7.02 (d, J = 8.7 Hz, 2H), 6.92 (d, J = 8.7 Hz, 2H), 4.32 (s, 2H), 4.04 (dd, J = 7.5, 4.5 Hz, 1H), 3.87–3.69 (m, 2H), 3.55–3.42 (m, 2H), 3.42–3.34 (m, 2H), 2.49–2.30 (m, 2H), 2.33 (s, 3H), 2.30–2.08 (m, 2H), 1.82–1.10 (m, 15H), 1.05–0.85 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H); IR (KBr) 3368, 3206, 3066, 3033, 2925, 2851, 2661, 2514, 2427, 1670, 1657, 1604, 1502, 1478, 1467, 1449, 1420, 1366, 1347, 1331, 1315, 1243, 1210, 1172, 1113, 1101 cm−1; MS (APCI, Pos) m/z 532 (M+H)+; HRMS Calcd 532.3539, Obsd 532.3541.
4.1.7.9. (3S)-1-Butyl-3-(cyclohexylmethyl)-9-[4-(4-methoxyphenoxy)benzyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (35).
The title compound was synthesized in 70% yield according to the same procedures as described for the preparation of 24 using 4-(4-methoxyphenoxy)benzaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.67 (AcOEt); 1H NMR (300 MHz, CD3OD) δ 7.49 (d, J = 8.4 Hz, 2H), 7.02–6.92 (m, 6H), 4.31 (s, 2H), 4.03 (dd, J = 7.5, 4.5 Hz, 1H), 3.86–3.69 (m, 2H), 3.79 (s, 3H), 3.54–3.30 (m, 4H), 2.50–2.30 (m, 2H), 2.28–2.06 (m, 2H), 1.83–1.10 (m, 15H), 1.05–0.83 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H); IR (KBr) 3417, 3204, 3069, 2925, 2851, 2656, 2496, 2426, 1681, 1615, 1500, 1475, 1464, 1447, 1420, 1374, 1345, 1315, 1300, 1230, 1199, 1173, 1113, 1098, 1035 cm−1; MS (APCI, Pos) m/z 548 (M+H)+; HRMS Calcd 548.3488, Obsd 548.3481.
4.1.7.10. (3S)-1-Butyl-3-(cyclohexylmethyl)-9-[4-(4-hydroxyphenoxy) benzyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (36).
The title compound was synthesized in 45% yield according to the same procedures as described for the preparation of 24 using 4-(4-hydroxyphenoxy)benzaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.54 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.47 (d, J = 8.4 Hz, 2H), 6.97 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 9.0 Hz, 2H), 6.80 (d, J = 9.0 Hz, 2H), 4.30 (s, 2H), 4.03 (dd, J = 7.5, 4.5 Hz, 1H), 3.83–3.72 (m, 2H), 3.49–3.34 (m, 4H), 2.38 (m, 2H), 2.23–2.10 (m, 2H), 1.78–1.16 (m, 15H), 1.02–0.92 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H); IR (KBr) 3363, 3199, 2924, 2851, 2670, 2566, 1674, 1638, 1503, 1468, 1448, 1420, 1373, 1346, 1315, 1227, 1196, 1172 cm−1; MS (APCI, Pos) m/z 534 (M+H)+; HRMS Calcd 534.3332, Obsd 534.3333.
4.1.7.11. 4-(4-{[(3S)-1-Butyl-3-(cyclohexylmethyl)-2,5-dioxo-1,4,9-triazaspiro[5.5]undec-9-yl]methyl}phenoxy)-N-methylbenzamide hydrochloride (37).
The title compound was synthesized in 68% yield according to the same procedures as described for the preparation of 24 using 4-(4-N-methylaminocarbonylphenoxy)benzaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.25 (AcOEt/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.85 (d, J = 8.7 Hz, 2H), 7.62 (d, J = 8.7 Hz, 2H), 7.15 (d, J = 8.7 Hz, 2H), 7.08 (d, J = 8.7 Hz, 2H), 4.37 (s, 2H), 4.05 (dd, J = 7.5, 4.5 Hz, 1H), 3.90–3.68 (m, 2H), 3.58–3.36 (m, 4H), 2.92 (s, 3H), 2.58–2.36 (m, 2H), 2.28–2.06 (m, 2H), 1.84–1.10 (m, 15H), 1.06–0.84 (m, 2H), 0.96 (t, J = 7.2 Hz, 3H); IR (KBr) 3263, 2927, 2547, 1674, 1600, 1499, 1417, 1314, 1245, 1175, 1112, 1049, 1006 cm−1; MS (APCI, Pos) m/z 575 (M+H)+; Elemental Anal. Calcd for C34H46N4O4·HCl: C, 66.81; H, 7.75; N, 9.17. Found: C, 64.8; H, 7.98; N, 8.92.
4.1.7.12. N-[4-(4-{[(3S)-1-Butyl-3-(cyclohexylmethyl)-2,5-dioxo-1,4,9-triazaspiro[5.5]undec-9-yl]methyl}phenoxy)phenyl]methanesulfonamide hydrochloride (38).
The title compound was synthesized in 47% yield according to the same procedures as described for the preparation of 24 using 4-(4-methansulfonylaminophenoxy)benzaldehyde for 4-(3-pyridyl)benzaldehyde. TLC Rf 0.42 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.53 (d, J = 9.0 Hz, 2H), 7.29 (d, J = 9.0 Hz, 2H), 7.08–7.00 (m, 4H), 4.33 (s, 2H), 4.03 (dd, J = 7.5, 4.8 Hz, 1H), 3.85–3.72 (m, 2H), 3.54–3.36 (m, 4H), 2.95 (s, 3H), 2.48–2.34 (m, 2H), 2.25–2.08 (m, 2H), 1.80–1.14 (m, 15H), 0.98–0.88 (m, 5H); IR (KBr) 3361, 3237, 3108, 3057, 2925, 2851, 2663, 2580, 1677, 1632, 1502, 1477, 1449, 1420, 1402, 1389, 1330, 1302, 1276, 1254, 1248, 1218, 1171, 1150, 1109 cm−1; MS (APCI, Pos) m/z 611 (M+H)+; HRMS Calcd. 611.3267, Obsd 611.3267.
4.1.7.13. 4-(4-{[(3S )-1-Butyl-3-(cyclohexylmethyl)-2,5-dioxo-1,4,9-triazaspiro[5.5]undec-9-yl]methyl}phenoxy)benzenesulfonamide hydrochloride (39).
The title compound was synthesized in 40% yield according to the same procedures as described for the preparation of 24 using 4-(4-aminosulfonylphenoxy)benzaldehyde for4-(3-pyridyl)benzaldehyde. TLC Rf 0.33 (CHCl3/MeOH, 10:1); 1H NMR(300MHz, DMSO-d6) δ (300 MHz,) 11.03 (br s, 1H), 8.42 br s, 1H), 7.82 (d, J = 8.7 Hz, 2H), 7.71 (d, J = 8.7 Hz, 2H), 7.33 (br s, 2H), 7.16 (d, J = 8.7 Hz, 4H), 4.38–4.23 (m, 2H), 3.91 (m, 1H), 3.61–3.23 (m, 6H), 2.58–2.30 (m, 2H), 2.18–1.91 (m, 2H), 1.76–1.00 (m, 15H), 0.98–0.71 (m, 5H); MS (APCI, Pos) m/z (APCI, Pos) 597 (M+H)+.
4.1.7.14. 4-(4-{[(3S)-1-Butyl-3-(cyclohexylmethyl)-2,5-dioxo-1,4,9-triazaspiro[5.5]undec-9-yl]methyl}phenoxy)benzoic acid hydrochloride (40).
The title compound was synthesized in 60% yield according to the same procedures as described for the preparation of 24 using 4-(4-formylphenoxy)benzoic acid for 4-(3-pyridyl)benzaldehyde. −27.7 (c 1.03, MeOH); TLC Rf 0.37 (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 8.03 (d, J = 8.7 Hz, 2H), 7.63 (d, J = 8.7 Hz, 2H), 7.16 (d, J = 8.7 Hz, 2H), 7.07 (d, J = 8.7 Hz, 2H), 4.37 (s, 2H), 4.04 (dd, J = 7.5, 4.5 Hz, 1H), 3.90–3.70 (m, 2H), 3.56–3.35 (m, 4H), 2.59–2.38 (m, 2H), 2.27–2.05 (m, 2H), 1.83–1.08 (m, 15H), 1.05–0.83 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H); IR (KBr) 2925, 1673, 1598, 1502, 1417, 1243, 1160 cm−1; MS (APCI, Pos) m/z 562 (M+H)+; Elemental Anal. Calcd for C33H43N3O5·HCl: C, 66.26; H, 7.41; N, 7.02. Found: C, 64.8; H, 7.64; N, 6.98.
4.1.8. (3S)-1-Butyl-3-(cyclohexylmethyl)-9-[2-(4-phenoxyphenyl)ethyl]-1,4,9-triazaspiro[5.5]undecane-2,5-dione hydrochloride (19)
A solution of 4-phenoxyphenethyl alcohol 65 (214 mg, 1 mmol) in pyridine/CH2Cl2 (1:1,4 mL) was added to chlorosulfonated polystyrene resin (305 mg, 0.5 mmol). The mixture was shaken for 5 h at room temperature. The resin was collected by filtration and successively washed with CH2Cl2 (4 mL), DMF (4 mL), DMF/H2O (3:1, 4 mL), THF (4 mL), CH2Cl2 (4 mL) and CH3CN (4 mL). To a suspension of the resin in MeCN (5 mL) were added N,N-diisopropylamine (271 mg, 2.1 mmol) and then compound 52b (112 mg, 0.3 mmol). The mixture was shaken for 18 h at 70 °C. After cooling to room temperature, the resin was collected by filtration and washed with MeCN. The combined filtrate and washings were evaporated. The resulting residue was purified by column chromatography on silica gel AcOEt/MeOH (from 1:0 to 10:1). The resulting residue after evaporation was treated with 4 N HCl in AcOEt (2 mL) and then washed with Et2O to afford 19 (81 mg, 48% yield) as a powder. TLC Rf 0.54 (AcOEt/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 7.37–7.29 (m, 4H), 7.11(t, J = 7.2 Hz, 1H), 6.97–6.95 (m, 4H), 4.06 (d, J = 7.5, 4.5 Hz, 1H), 3.88–3.77 (m, 2H), 3.65 (m, 2H), 3.46–3.36 (m, 4H), 3.13–3.07 (m, 2H), 2.48 (m, 2H), 2.28–2.14 (m, 2H), 1.80–1.21(m, 15H), 0.98 (t, J = 7.0 Hz, 3H), 0.99–0.91 (m, 2H); IR (KBr) 3364, 3195, 3065, 2924, 2851, 2661, 2525, 2421, 1671, 1589, 1508, 1489, 1470, 1449, 1418, 1373, 1347, 1333, 1317, 1238, 1200, 1169, 1148, 1073 cm−1; MS (APCI, Pos) m/z 532 (M+H)+; HRMS Calcd 532.3539, Obsd 532.3536.
4.1.9. Ethyl 4-[1-butyl-3-(cyclohexylmethyl)-2,5-dioxo-1,4,9-triazaspiro[5.5]undec-9-yl]benzoate hydrochloride (20)
To a solution of compound 52a (186 mg, 0.501 mmol) in MeCN (2.5 mL) were added ethyl 4-fluorobenzoate (164 mg, 0.975 mmol) and then K2CO3 (141 mg, 1.02 mmol). After being stirred for 12 h at 100 °C, the reaction mixture was treated with DMSO (0.5 mL). Stirring was continued for another 12 h at 140 °C. The reaction mixture was cooled to room temperature, diluted with H2O and extracted with t-butyl methyl ether. The combined organic layers were washed with brine, dried (MgSO2) and evaporated. The resulting residue was purified by column chromatography on silica gel (hexane/AcOEt, from 4:1 to 3:1). The resulting residue after evaporation was treated with 4 N HCl in AcOEt (2 mL) and washed with t-butyl methyl ether to afford the title compound 20 (67 mg, 26% yield). TLC Rf 0.27 (hexane/AcOEt, 2:1); 1H NMR (300 MHz, CD3OD) δ 8.13 (d, J = 8.7 Hz, 2H), 7.59 (d, J = 8.7 Hz, 2H), 4.37 (q, J = 7.2 Hz, 2H), 4.31–4.15 (m, 2H), 4.07 (dd, J = 7.5, 4.5 Hz, 1H), 3.85–3.75 (m, 2H), 3.47–3.38 (m, 2H), 2.67–2.50 (m, 2H), 2.30–2.12 (m, 2H), 1.85–1.46 (m, 10H), 1.44–1.19 (m, 5H), 1.38 (t, J = 7.2 Hz, 3H), 1.05–0.88 (m, 2H), 0.95 (t, J = 7.2 Hz, 3 H); IR (KBr) 3214, 3085, 2959, 2927, 2851, 2630, 2463, 2401, 1726, 1675, 1658, 1608, 1469, 1459, 1448, 1417, 1407, 1374, 1315, 1307, 1277, 1188, 1177, 1146, 1109, 1020, 1006 cm−1; MS (APCI, Pos) m/z 484 (M+H)+; HRMS Calcd 484.3175, Obsd 484.317.
4.1.10. 2-Butynylamine hydrochloride (42f)
4.1.10.1. 2-Butynylphthalimide (48).
To a stirred solution of 2-butynylalchol 47 (5,0 g, 71.3 mmol) in triethylamine (14.9 mL 107 mmol) and CH2Cl2 (210 mL) was slowly added methanesulfonyl chloride (6.1 mL, 78.5 mmol) at 0 °C under argon atmosphere. After being stirred for 1 h, the reaction mixture was quenched with water and extracted with AcOEt. The organic layer was washed with 1 N HCl, saturated NaHCO3 aq, brine, dried over Na2SO4, and evaporated. To a stirred solution of the resulting residue in DMF (80 mL) was added potassium phthalimide (11.4 g, 61.5 mmol). After being stirred for 1.5 h at 70 °C, the reaction mixture was poured into water (400 mL) and extracted with AcOEt. Precipitates were removed by filtration and washed with diethyl ether. The filtrate was extracted with AcOEt, and the organic layer was washed with 1 N NaOH and brine, dried over Na2SO4 and evaporated. The resulting solid was triturated with diethyl ether and dried in vacuo to yield 48 (10.4 g, 73% in two steps). TLC Rf 0.56 (hexane/AcOEt, 2:1); 1H NMR (200 MHz, DMSO-d6) δ 7.90–7.77 (m, 4H), 4.30 (q, J = 2.4 Hz, 2H), 1.74 (t, J = 2.4 Hz, 3H).
4.1.10.2. 2-Butynylamine hydrochloride (42f).
To a stirred suspension of 48 (10.3 g, 51.7 mmol) in EtOH (600 mL) was added hydrazine hydrate (7.6 mL, 155 mmol). After being stirred for 1.5 h at 100 °C, the mixture was cooled to room temperature, quenched with concd HCl (25 mL) and evaporated. The resulting residue was treated with 5 N NaOH and extracted with CH2Cl2 repeatedly. The combined organic layers were washed with brine, dried over Na2SO4. After the addition of 4 N HCl/AcOEt (20 mL), the solution was evaporated. The resulting solid was washed with diethyl ether to yield 42f (5.25 g, 96%). TLC Rf 0.23 (CHCl3/MeOH/AcOH, 20:4:1); 1H NMR (200 MHz, DMSO-d6) δ 8.70–8.15 (br, 3H), 3.62 (br, 2H), 1.83 (t, J = 2.4 Hz, 3H).
4.1.11. 4-Benzoylbenzaldehyde (54)
To a stirred solution of 4-benzoylbenzencalboxylic acid 53 (590 mg, 2.61 mmol) in triethylamine (0.44 mL, 3.13 mmol) and THF (7 mL) was added i-butylchloroformate (0.40 mL, 3.13 mmol) at −78 °C. After being stirred at room temperature for 30 min, triethylamine hydrochloride salt was removed by filtration. The filtrate was added to the suspension of sodium borohydride (296 mg, 7.83 mmol) in water at 0 °C. After being stirred at room temperature for overnight, the mixture was concentrated. The resulting residue was extracted with AcOEt. The organic layer was washed with water, brine, dried over Na2SO4 and evaporated. To a stirred solution of the resulting residue in DME (20 mL) was added MnO2 (682 mg, 7.84 mmol). After being stirred at 100 °C for overnight, additional MnO2 (2.02 g, 23.2 mmol) was added to the solution. The reaction mixture was stirred at 110 °C for 4 h and then cooled to room temperature. MnO2 was removed by filtration through the pad of Celite, and the filtrate was evaporated. The resulting residue was purified by column chromatography on silica gel to yield 54 (277 mg, 51%). TLC Rf 0.35 (hexane/AcOH, 4:1); 1H NMR (300 MHz, CD3OD) δ 10.14 (s, 1H), 7.94–7.92 (m, 2H), 7.83–7.80 (m, 2H), 7.67–7.61 (m, 1H), 7.54–7.49 (m, 2H).
4.1.12. 4-(4-Hydroxyphenoxy)benzaldehyde (56)
To a stirred solution of 55 (3.69 g, 16.1 mmol) in CH2Cl2 (80 mL) was slowly added 1.0 M boron tribromide/CH2Cl2 solution (35 mL, 0.35 mmol) at 0 °C. After being stirred for 2 h, the reaction mixture was quenched with water and extracted with diethyl ether. The organic layer was washed with water, brine, dried over Na2SO4 and evaporated. The resulting residue was purified by column chromatography on silica gel to yield the title compound 56 (3.06 g, 86%). TLC Rf 0.28 (hexane/AcOEt, 2:1); 1H NMR (300 MHz, CDCl3) δ 9.90 (s, 1H), 7.83 (d, J = 9.0 Hz, 2H), 7.01 (d, J = 9.0 Hz, 2H), 6.98 (d, J = 9.0 Hz, 2H), 6.89 (d, J = 9.0 Hz, 2H), 5.47 (br s, 1H). MS (APCI, Pos) 257 (M+H)+.
4.1.13. 4-(4-Formylphenoxy)benzoic acid (58)
A mixture of methyl 4-hydroxybezoate 57 (25.2 g, 16.5 mmol), 4-fluorobenzaldehyde (18.6 g, 14.9 mmol) and K2CO3 in DMF (150 mL) was stirring under reflux for 2 h. After being cooled to room temperature, the reaction mixture was quenched with water and extracted with AcOEt. The organic layer was washed with water, brine, dried over MgSO4 and evaporated. The resulting solid was triturated with n-hexane and dried to yield the methyl ester of 58 (27.8 g, 72%). To a stirred solution of the methyl ester in methanol (400 mL) was added 2 N NaOH (108 mL, 54 mmol). After being stirred for 2 h at 50 °C, the reaction mixture was cooled to room temperature and quenched with 2 M HCl (110 mL) and water (200 mL). The precipitates were collected by filtration and dried to yield 58 (23.4 g, 89%). TLC Rf 0.20 (hexane/AcOEt, 1:1); 1H NMR δ (300 MHz, CDCl3) 9.98 (s, 1H), 8.16 (d, J = 8.7 Hz, 2H), 7.92 (d, J = 8.7 Hz, 2H), 7.17 (d, J = 8.7 Hz, 2H), 7.13 (d, J = 8.7 Hz, 2H).
4.1.14. 4-(4-N-Methylaminocarbonylphenoxy)benzaldehyde (59)
To a stirred solution of compound 58 (9.8 g, 40.5 mmol) in DMF were added HOBt (6.56 g, 48.6 mmol), EDC hydrochloride (9.32 g, 48.6 mmol) and a solution of methylamine in THF (2.0 M, 41 mL, 81 mmol). After being stirred for 1.5 h, the reaction mixture was poured into water (500 mL), acidified with 2 N HCl (10 mL) and extracted with AcOEt. The organic layer was washed with saturated NaHCO3 aq, brine, dried over Na2SO4, and evaporated. The resulting solid was triturated with t-butyl methyl ether to yield 59 (9.23 g, 89%). TLC Rf 0.55 (CHCl3/MeOH = 10:1); 1H NMR δ (300 MHz, CDCl3) 9.95 (s, 1H), 7.89 (d, J = 8.7 Hz, 2H), 7.81 (d, J = 9.0 Hz, 2H), 7.13–7.10 (m, 4H), 3.03 (d, J = 4.8 Hz, 3H); MS (APCI, Pos) 256 (M+H)+.
4.1.15. 4-(4-Methansulfonylaminophenoxy)benzaldehyde (62)
4.1.15.1. Methyl-4-(4-methansulfonylaminophenoxy)benzoate (61).
A suspension of 60 (25.5 g, 93.5 mmol) and 5% Pd–C (1.3 g) in AcOEt (300 mL) was stirred overnight under hydrogen atmosphere. The reaction mixture was filtered through a pad of Celite and the filtrate was evaporated. To the resulting residue in THF (300 mL) and pyridine (22.4 mL, 277 mmol) was added methanesulfonyl chloride (10.7 mL, 139 mmol) at 0 °C. After being stirred for overnight at room temperature, the reaction mixture was quenched with water and extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4 and evaporated. The resulting solid was triturated with t-butyl methyl ether to yield 61 (29.0 g, 98%). TLC Rf 0.5 (hexane/AcOEt, 1:1); 1H NMR (300 MHz, CD3OD) δ 8.01 (d, J = 8.7 Hz, 2H), 7.26 (d, J = 9.0 Hz, 2H), 7.26 (s, 1H), 7.05 (d, J = 9.0 Hz, 2H), 6.98 (d, J = 8.7 Hz, 2H), 3.91 (s, 3H), 3.04 (s, 3H).
4.1.15.2. 4-(4-Methansulfonylaminophenoxy)benzaldehyde (62).
To a stirred solution of 61 (29 g, 90.3 mmol) in THF (300 mL) was added 1.0 M diisobutyl aluminum hydride/n-hexane solution (270 mL, 270 mmol) at 0 °C. After being stirred for 2 h, the reaction mixture was treated with additional diisobutyl aluminum hydride (1.0 M, 90 mL, 90 mmol) and stirred for 20 min at 0 °C. The reaction mixture was quenched with saturated Na2SO4 aq and the resulting precipitates were removed by filtration. The filtrate was evaporated and the resulting residue was dissolved in CH2Cl2/DME (150:150). MnO2 (40 g, 482 mmol) was added to the solution The reaction mixture was stirred for overnight and then filtered through a pad of Celite and the filtrate was concentrated in vacuo to yield 62 (26.0 g, 99%). TLC Rf 0.46 (hexane/AcOEt); 1H NMR (300 MHz, CDCl3) δ 9.92 (s, 1H), 7.85 (d, J = 8.7 Hz, 2H), 7.28 (d, J = 8.7 Hz, 2H), 7.25 (s, 1H), 7.10–7.05 (m, 4H), 3.05 (s, 3H).
4.1.16. 4-(4-Aminosulfonylphenoxy)benzaldehyde (64)
A suspension of 63 (4.33 g, 25 mmol), 4-fluorobenzaldehyde (2.68 mL, 25 mmol), and K2CO3 (6.9 g, 50 mmol) in DMA (20 mL) was stirring under reflux for 2 h. After being cooled to room temperature, the mixture was quenched with water and extracted with AcOEt. The organic layer was washed with water, brine, dried over Na2SO4 and evaporated. The resulting solid was recrystallized from AcOEt/hexane to yield the title compound 64 (1.22 g, 18%). TLC Rf (CHCl3/MeOH, 10:1); 1H NMR (300 MHz, CD3OD) δ 9.93 (s, 1H), 7.96 (d, J = 8.7 Hz, 2H), 7.95 (d, J = 8.7 Hz, 2H), 7.21 (d, J = 8.7 Hz, 2H), 7.19 (d, J = 8.7 Hz, 2H); MS (APCI, Neg) 276 (M−H)−.
4.2. Biology
4.2.1. Stability study of liver microsomes
The test substance (5 μL:10 mmol/L in DMSO) was diluted with 195 μL of 50% acetonitrile in water to make a 250 μmol/L solution of the test substance.
Phosphate buffer (0.1 mol/L, 245 μL) containing 0.2 mg/mL or 0.5 mg/mL rat liver microsomes and NADPH-Co-factor* was added to a reaction container, pre-warmed to 37 °C in a water bath, and incubated for 5 min. The reaction was initiated with the addition of 5 μL of the test substance solution (in 0.975% acetonitrile with 0.05% DMSO, final concentration of 5 μmol/L). A 20 μL aliquot was taken from the mixture immediately after the start of the reaction, and transferred to 180 μL of acetonitrile containing the internal standard (warfarin) to terminate the reaction. A 20 μL aliquot of the mixture was stirred with 180 μL of 50% acetonitrile on a plate with a filter for deproteinization, and filtered by suction. The filtrate was used as the standard sample.
After a 15 min incubation of the above mixture, a 20 μL aliquot was transferred to 180 μL of acetonitrile containing the internal standard (warfarin) to terminate the reaction. A 20 μL aliquot of the mixture was stirred with 180 μL of 50% acetonitrile on a plate with a filter for deproteinization, and filter by suction. The filtrate was used as the reaction sample.
*NADPH-Co-factor: Dilute solutions A and B in the NADPH-regenerating system (BD-Bioscience) 20- and 100-fold, respectively, with 0.1 mol/L phosphate buffer (NADP+ 2.6 mmol/L).
4.2.1.1. Determination of test substance concentration and data processing.
One microliter of aliquot was injected into an LC-MS/MS system. The percent residue (%) is calculated by dividing the peak area ratio (i.e., test substance/I.S.) for the reaction sample by the peak area ratio for the standard sample and multiplying by 100.
4.2.2. Primary screening of solubility
The test substance (10 mmol/L DMSO) was diluted with acetonitrile. Acetonitrile containing the internal standard (warfarin) was added to prepare samples for calibration curves at 0.1, 0.4, and 2 μmol/L.
The test substance (5 μL:10 mmol/L DMSO) was added to 495 μL of JP Solution 2. The solution was stirred at room temperature for 5 h, and transferred onto a filter plate, and filtered by centrifugation at 3000 rpm, 24 °C for 15 min. The filtrate (20 μL) was diluted with acetonitrile, and additional acetonitrile containing the internal standard to prepare a sample solution.
The sample solution (1 μL) was injected into an LC-MS/MS system for quantification (range: 0.1–2 μmol/L). The solubility was determined as 50 times of the observed value. The solubility was reported as <5 μmol/L or 100 μmol/L if the observed value is outside the range.
4.2.3. Caco-2 membrane permeability assay
Caco-2 cells were grown on a 12-well Costar Transwell plate (with a collagen-coated fine porous polycarbonate membrane) until a confluent monolayer is formed. Buffer for permeability assay was prepared using 10 mM HEPES and Hank’s balanced salt solution containing 15 mM glucose with the pH being adjusted at 7.3–7.5. A test substance solution was prepared at 10 mM the assay buffer. The test substance solution was added to the apical side of Caco-2 cell monolayer at a final concentration of 10 μM, and then the plate was incubated in a humidified incubator (5% CO2, 37 °C). Two hours later, 200- and 50 μL aliquots were taken from the receiver chambers. The measurements should be performed in duplicate. The concentration of test substance in the samples was determined by LC–MS/MS.
The apparent permeability coefficient (Papp) was calculated according to the following equation:
dCr/dt: slope of compound accumulation in the receiver compartment over time (μM/s)
Vr: volume of the receiver compartment (cm3)
A: area of the cell monolayer (1.13 cm2 for 12-well transwell plate)
C0: initial normality of the buffer (μM)
4.2.4. Pharmacokinetic (PK) studies
Single dose pharmacokinetics was studied in rats. Formulation for intravenous injection was prepared using SWI (sterile water for injection) containing 30% HP-β-CD (w/v). Formulation for oral dosing was prepared using SWI containing 1% sucrose fatty acid ester (w/v). Test compounds (3 mg/kg) were dosed intravenously to the fasted male rats (n = 3). Test compounds (30 mg/kg) were dosed orally to the fasted male rats (n = 3). After dosing, blood samples (250 μL) were collected from the jugular vein using a heparinized syringe at the selected time points (iv: pre-dosing, 2, 5, 15, and 30 min; po: 1, 2, 4, 6, and 8 h, respectively). The blood samples were ice-chilled and then centrifuged at 12,000 rpm for 2 min at room temperature to obtain plasma, which was preserved at −70 °C in a freezer. The AUC, Cmax, Tmax, T1/2, Vss, and CL were obtained by measuring the time course of the plasma concentration of the test compounds. Bioavailability (BA) was calculated according to the following equation:
References and notes
- 1.Hoffmann C; Mulcahy F Overview of Antiretroviral Agents In HIV Medicine 2006; Hoffmann C, Rockstroh JK, Kamps BS, Eds.; Flying Publisher; (FlyingPublisher.com): Paris, 2006; pp 94. [Google Scholar]
- 2.(a) Schwarz MK; Wells TNC Nat. Rev. Drug Disc 2002, 1, 347. [DOI] [PubMed] [Google Scholar]; (b) Gerard C; Rollins BJ Nat. Immunol 2001, 2, 108. [DOI] [PubMed] [Google Scholar]; (c) Pirotte B; Tullio P; Nguyen QA; Somwes F; Fraikin P; Florence X; Wahl P; Hansen JB; Lebrun P J. Med. Chem 2010, 53, 147. [DOI] [PubMed] [Google Scholar]
- 3.(a) Feng Y; Broder CC; Kennedy PE; Berger EA Science 1996, 272, 8728629022 [Google Scholar]; (b) Deng HK; Liu R; Ellmeier W; Choe S; Unutmaz D; Burkhart M; Marzio PD; Marmon S; Sutton RE; Hill CM; Davis CB; Peiper SC; Schall TJ; Littman DR; Landau NR Nature 1996, 381, 661. [DOI] [PubMed] [Google Scholar]; (c) Dragic T; Litwin V; Allaway GP; Martin SR; Huang Y; Nagashima KA; Cayanan C; Maddon PJ; Koup RA; Moore JP; Paxton WA Nature 1996, 381, 667. [DOI] [PubMed] [Google Scholar]; (d) Alkhatib G; Combadiere C; Broder CC; Feng Y; Kennedy PE; Murphy PM; Berger EA Science 1996, 272, 1955. [DOI] [PubMed] [Google Scholar]; (e) Berger EA; Murphy PM; Farber JM Annu. Rev. Immunol 1999, 17, 657. [DOI] [PubMed] [Google Scholar]; (f) Caldwell DJ; Evans JD Expert Opin. Pharmacother 2008, 9, 3231. [DOI] [PubMed] [Google Scholar]
- 4.(a) Leonard JT; Roy K Curr. Med. Chem 2006, 13, 91. [DOI] [PubMed] [Google Scholar]; (b) Price DA; Armour D; Groot M; Leishman D; Napier C; Perros M; Stammen BL; Wood A Bioorg. Med. Chem. Lett 2006, 16, 4633. [DOI] [PubMed] [Google Scholar]
- 5.Habashita H; Kokubo M; Hamano S; Hamanaka N; Toda M; Shibayama S; Tada H; Sagawa K; Fukushima D; Maeda K; Mitsuya H J. Med. Chem 2006, 49, 4140. [DOI] [PubMed] [Google Scholar]
- 6.(a) Maeda K; Yoshimura K; Shibayama S; Habashita H; Tada H; Sagawa K; Miyakawa T; Aoki M; Fukushima D; Mitsuya H J. Biol. Chem 2001, 276, 35194. [DOI] [PubMed] [Google Scholar]; (b) Maeda K; Nakata H; Koh Y; Miyakawa T; Ogata H; Takaoka Y; Shibayama S; Sagawa K; Fukushima D; Moravek J; Koyanagi Y; Mitsuya H J. Virol 2004, 78, 8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Nishizawa R; Nishiyama T; Hisaichi K; Matsunaga N; Minamoto C; Habashita H; Takaoka Y; Toda M; Shibayama S; Tada H; Sagawa K; Fukushima D; Maeda K; Mitsuya H Bioorg. Med. Chem. Lett 2007, 17, 727. [DOI] [PubMed] [Google Scholar]; (b) Nishizawa R; Nishiyama T; Hisaichi K; Hirai K; Habashita H; Takaoka Y; Tada H; Sagawa K; Shibayama S; Maeda K; Mitsuya H; Nakai H; Fukushima D; Toda M Bioorg. Med. Chem. Lett 2010, 20, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalfutkar AS; Gardner I; Obach RS; Shaffer CL; Callegari E; Henne KR; Mutlib AE; Dalvie DK; Lee JS; Nakai Y; O’Donnell JP; Boer J; Harriman SP Curr. Drug Metab 2005, 6, 161. [DOI] [PubMed] [Google Scholar]
- 9.Further PK studies of 38 and 39 were not carried out because of the poor AUC after their oral cassette dosing.