

Abstract
Nitrogen (N2) fixation to produce bio-available ammonia (NH3) is essential to all life but is a challenging transformation to catalyse owing to the chemical inertness of N2. Transition metals can, however, bind N2 and activate it for functionalization. Significant opportunities remain in developing robust and efficient transition metal catalysts for the N2 reduction reaction (N2RR). One opportunity to target in catalyst design concerns the stabilization of transition metal diazenido species (M-NNH) that result from the first N2 functionalization step. Well-characterized M-NNH species remain very rare, likely a consequence of their low N–H bond dissociation free energies (BDFEs). In this essay, we discuss the relationship between the BDFEN–H of a given M-NNH species to the observed overpotential and selectivity for N2RR catalysis with that catalyst system. We note that developing strategies to either increase the N–H BDFEs of M-NNH species, or to avoid M-NNH intermediates altogether, are potential routes to improved N2RR efficiency.
Keywords: Nitrogen fixation, Catalysis, Inorganic chemistry, Proton transfer-electron transfer, Thermochemistry
Graphical Abstract
The reduction of N2 to NH3 (N2RR) is a globally significant reaction that is challenging due to the inertness of N2. Transition metals can activate N2 and mediate catalytic N2RR but challenges remain with respect to catalytic efficiency in terms of overpotential and selectivity. We discuss the role of the N–H bond dissociation free energy (BDFE) of metal diazenidos (M-NNH), the first intermediates of N2RR, plays in determining N2RR efficiency.

1. Introduction
Despite the abundance of nitrogen (N2) in the atmosphere, its bioavailability limits growth in many environments.[1] Thus, the fixation of N2 into its most common bioavailable form, ammonia (NH3), is essential to all life and is among the most important (and fascinating) chemical transformations on the planet. N2-to-NH3 conversion (commonly called the nitrogen reduction reaction and abbreviated as N2RR) is challenging owing to the chemical inertness of N2 with respect to proton transfer (PT), electron transfer (ET), and hydrogen atom transfer (Figure 1).
Figure 1:

(A) Properties of free N2 highlighting its recalcitrance to initial functionalization (e−, H+, and H• affinity), which are similar to those of the noble gases, and its strong N≡N triple bond (BDE = bond dissociation enthalpy).[2,8,9] (B) N2 terminally binds transition metals via σ-donation of its lone and acceptance of π-backdonation from the metal center. Binding induces a dipole on N2 and weakens the N≡N bond, priming it for conversion to NNH. Subsequent reductive protonation steps lead to the formation of NH3. (C) Comparison of the standard (1 M or 1 atm, 298 K, 0 V vs NHE) free energy for the formation of NH3 from both H2 (ΔG°f) and H+/e− (ΔG°(N2RR)).[10]
Its high stability is due to a very strong N≡N triple bond and the absence of a dipole (distinct from isoelectronic CO).[2] N2 can be rendered far more chemically reactive by binding to a transition metal. The σ-lone pair donation from N2 to the metal and corresponding π-backdonation from the metal into the empty π* orbitals of N2 can weaken the N≡N bond and induce a dipole, priming the N2 ligand for functionalization.[3] Nonetheless, the first functionalization step to form a metal diazenido (M-NNH) intermediate remains a significant challenge in catalytic N2RR. In accord with their highly reactive nature, very few M-NNH species have been reliably characterized.[4–7] In this short perspective, we draw attention to the significance of the M-NNH intermediate in terms of overall catalytic N2RR efficiency with respect to overpotential and likely also selectivity. We formulate a semi-quantitative relationship between the stability of the M-NNH species, assessed via its N–H bond dissociation free energy (BDFEN-H), and the net overpotential required for catalytic N2RR (Figure 1).
2. Bonding in M-N2Hy Species
Chatt and coworkers were the first to synthesize well-defined M-NxHy coordination complexes and to demonstrate stoichiometric ammonia generation via the protonation of N2 complexes, with particular emphasis on bis(phosphine)-supported N2 complexes of the group 6 metals Mo and W (e.g., (depe)2W(N2)2, depe = diethylphosphinoethane; Figure 2).[4,11] Later, other groups established that such systems could serve as precursors for electrosynthetic (but not electrocatalytic) NH3 formation.[12,13] Most recently, catalytic N2RR in the presence of SmI2 and H2O or ethylene glycol has been established using these types of phosphine-supported Mo complexes.[14]
Figure 2:

(left) Force constants for the W–N and N–N normal modes in a PT series derived from (depe)2W(N2)2 ([W] ≡ (depe)2W). (right) Force constants for the N–N bond in free N2, NNH2, and N2H4 provided as standards for N–N triple, double, and single bonds respectively.[15,17]
Detailed vibrational spectroscopy and analysis by Tuczek and coworkers mapped the evolution of the W–N and N–N bonding as a function of the protonation state (e.g., N2 vs NNH vs NNH2 vs NNH3) in a family of these foundational group 6 systems. Their studies underscore the challenge of stabilizing a M-NNH intermediate.[15–17] To summarize their findings, the N≡N bond in free N2 is characterized by a force constant (f) of 22.4 mdyn•Å−1. The formal reduction to isodiazene (NNH2) generates a double bond (f = 11.7) and, ultimately, reduction to hydrazine (N2H4) yields a single bond (f = 4.3, Figure 2). To compare, in (depe)2W(N2)2, backdonation from the zerovalent W center reduces the N–N bond order, evinced by the reduction of fN–N to 16.4 mdyn•Å−1 and an increase in W–N bond character (fW–N = 2.9 mdyn•Å−1). Monoprotonation to form (depe)2W(F)(NNH) significantly attenuates the N–N π-bonding (fN–N = 8.3 mdyn•Å−1), with W–N bonding increasing only slightly (fW–N = 4.5 mdyn•Å−1). Further protonation to yield [(depe)2W(F)(NNH2)]+ decreases the N–N bonding only negligibly (ΔfN–N = −1 mdyn•Å−1), which is compensated for by a further increase in the W–N bonding (ΔfW–N = +1.5 mdyn•Å−1).[15] Lastly, in [(depe)2W(F)(NNH3)]2+, its formal W–N triple bond has a fW–N = 7.3 mdyn•Å−1, and its formal N–N single bond has a fN–N = 6.0 mdyn•Å−1 (Figure 2).[17]
If we compare the first, second, and third PT steps, the first PT causes a much more significant reduction in the N–N bonding then the next two. Indeed, after the first step the N–N force constant is already significantly less than that of the N=N double bond in free isodiazene (NNH2). However, the increase in the W–N force constant for each PT is qualitatively similar. Thus, there is a relatively uncompensated loss of N–N π-bonding following the first PT step to generate the M-NNH, and, hence, we can expect the resultant N–H bond in the resulting diazenido species to be homolytically weak. Indeed, whereas N–H bonds in amines typically have BDFEs between 90 and 100 kcal•mol−1,[8] density functional theory (DFT) studies on the BDFEN–H of several diazenido species have consistently predicted values less than 50 kcal•mol−1.[18–21] This is an important value to keep in mind, because below 50 kcal•mol−1 N–H (and other X-H) bonds are thermodynamically prone to bimolecular reactions that release H2 (BDFE(H2) = 102.3 kcal•mol−1 in MeCN).[8]
Thus, although a transition metal can significantly stabilize an NNH species via coordination compared to its free state (calculated gas-phase BDEN–H(NNH) = −4 kcal•mol−1),[8] M-NNH species remain susceptible to the hydrogen evolution reaction (HER, vide infra). DFT calculations[19–21] and fundamental chemical intuition suggest that reductive protonation steps that occur after N–N bond cleavage are comparatively easier. Therefore, in N2RR reactions proceeding from terminal N2 complexes, the most challenging functionalization step is likely to be formation of the M-NNH intermediate. What is more, we suggest that the necessary overpotential for catalytic N2RR with a particular catalyst can be roughly estimated from the BDFEN–H of the catalytically relevant diazenido intermediate. To explore this idea further, we test here the validity of this approach for two cases of N2RR catalysts (Mo and Fe) that proceed through an M-NNH intermediate.
3. M-NNH Intermediates and Catalytic N2RR
In 2003, Schrock and coworkers published the first example of a well-defined, organometallic N2RR catalyst.[22] Their triamidoamine Mo system, (HIPTN3N)Mo(N2) ([HIPTN3N]3− = [HIPTNCH2CH2)3N]3−, HIPT = 3,5-(2,4,6-iPr3C6H2)2C6H3)), is capable of forming NH3 catalytically (63%) concomitant with H2 formation (33%) in the presence of excess Cp*2Cr (−1.45 V vs Fc+/0 in MeCN) and [Lut-H]+ ([Lut-H]+ = 2,6-dimethylpyridinium, pKa = 14.13 in MeCN).[22,23] Later, Schrock and coworkers established that lower but still catalytic yields of NH3 (3.6 equiv NH3 per Mo) could be obtained using the same acid and the weaker reductant Cp2Co (−1.33 V vs Fc+/0 in MeCN, Figure 3).[24] As articulated by Mayer and coworkers,[8,25] the equation (Eq 1), in which CG is a solvent dependent constant (CG = 54.9 kcal•mol−1 in MeCN), that is typically used to determine BDFEs can also be used to predict an effective BDFE (BDFEeff) for any acid/reductant pair.[8] Such estimates are inherently limited by the lack of available data with respect to E°, pKa, and CG values in the types of non-polar solvents typically used in N2RR catalysis. However, since net H• transfer reactions do not change the overall charge of an intermediate, solvent-dependence is anticipated to be moderate and likely does not significantly affect the estimates presented here.
| (1) |
Figure 3:

(A) Lowest established overpotential catalytic conditions, using [LutH]+ and Cp2Co as the acid/reductant pair, for N2RR by a (HIPTN3N)Mo species ([Mo]).[24] (B) Hess’s law scheme showing the thermodynamic equivalence between pKa and E° for the separated acid/reductant pair and the BDFEeff.[8,25] (C) Protonation equilibrium between [(HIPTN3N)Mo(N2)]−/(HIPTN3N)Mo(NNH) and [DBU-H]+/DBU in THF, which allows the pKa to be estimated.[5] (D) The redox couple for [(HIPTN3N)Mo(N2)]0/− in THF.[23] (E) Decay pathway[5] and estimated BDFE of (HIPTN3N)Mo(NNH).
By comparing the BDFEeff value of the Cp2Co/[LutH]+ pair (43.6 kcal•mol−1) to the strength of an H• derived from H2,[8] one can derive the excess energy used relative to the Gibbs free energy of formation for NH3 (ΔΔGf(NH3), Eq 2) in this reaction. Since the reduction of N2 with H2 to form NH3 is nearly thermoneutral (i.e., ΔGf(NH3) ~ 0 kcal•mol−1, Figure 1), this analysis provides a good estimate of the net overpotential needed to drive N2RR.[18,19,26,27] For the present case, the deduced ΔΔGf(NH3) of 23 kcal•mol−1 provides an experimental lower limit for N2RR by the Schrock Mo catalyst; in accord with this notion, their efforts to perform catalysis using weaker acids, such as [Et3NH]+ (pKa = 18.82 in MeCN),[23] proved unsuccessful.[22]
| (2) |
An estimate of the BDFEN–H of the parent diazenido species in this same system, (HIPTN3N)Mo(NNH), can be ascertained from experimental data previously reported by Schrock and coworkers (Figure 3).[5,18,23] In particular, [(HIPTN3N)Mo(N2)]−/(HIPTN3)Mo(NNH) were shown to be in a PT equilibrium with [DBU-H]+/DBU (DBU = 1,8-diazaobicyclo[5.4.]undec-7-ene) in THF (pKa([DBU-H]+) = 18.5 in THF).[28,29] The [(HIPTN3N)Mo(N2)]0/− redox couple is −1.81 V vs Fc+/0 in THF.[23] From these observations, the BDFEN–H(Mo-NNH) can be approximated as 45 kcal•mol−1 (CG in THF ~ 61 kcal•mol−1).[30] This value accords both with computational studies[18,19] and its solution instability; (HIPTN3N)Mo(NNH) slowly decays via formal β-hydride elimination to release N2 and form (HIPTN3N)Mo(H) (Figure 3).27 One can then predict a ΔΔGf(NH3) for N2RR catalysis by (HIPTN3N)Mo, using as a basis the BDFEN–H((HIPTN3N)Mo(NNH)) and comparing this to BDFE(H2) (Eq 3). This analysis leads to the prediction of a minimum ΔΔGf(NH3) for (HIPTN3N)Mo to be 20 kcal•mol−1, which compares quite well with the 23 kcal•mol−1 value deduced above using the BDFEeff for Cp2Co/[LutH]+.
| (3) |
In 2013, our lab at Caltech reported the first example of a molecular iron catalyst for N2RR, (iPrP3B)Fe (iPrP3B = tris(o-diisopropylphosphinophenyl)-borane). This was the first non-Mo system to be identified for catalytic N2RR and was initially shown to be active using HBArF4 and KC8 at low temperature in diethyl ether.[31,32] It was later shown that this same catalyst was competent for N2RR with Cp*2Co (E° = −1.91 V vs Fc+/0 in MeCN) and substituted anilinium acids (Figure 4).[18] A comparison of the N2RR efficacy versus the strength of the acid revealed that the weakest anilinium acid for which catalysis could be observed (7.3 equiv NH3 per Fe at the loading tested) was [PhNH3]+ (pKa = 10.62 in MeCN).[33] Efforts to achieve catalysis with milder reductants, such as Cp*2Cr or Cp2Co, were unsuccessful.[18] Using the above E° and pKa values for Cp*2Co and [PhNH3]+, respectively, and Eqs 1 and 2, allows one to deduce a BDFEeff = 25.4 kcal•mol−1, and hence an experimental ΔΔGf(NH3) of 75 kcal•mol−1 for this acid/reductant pair.
Figure 4:
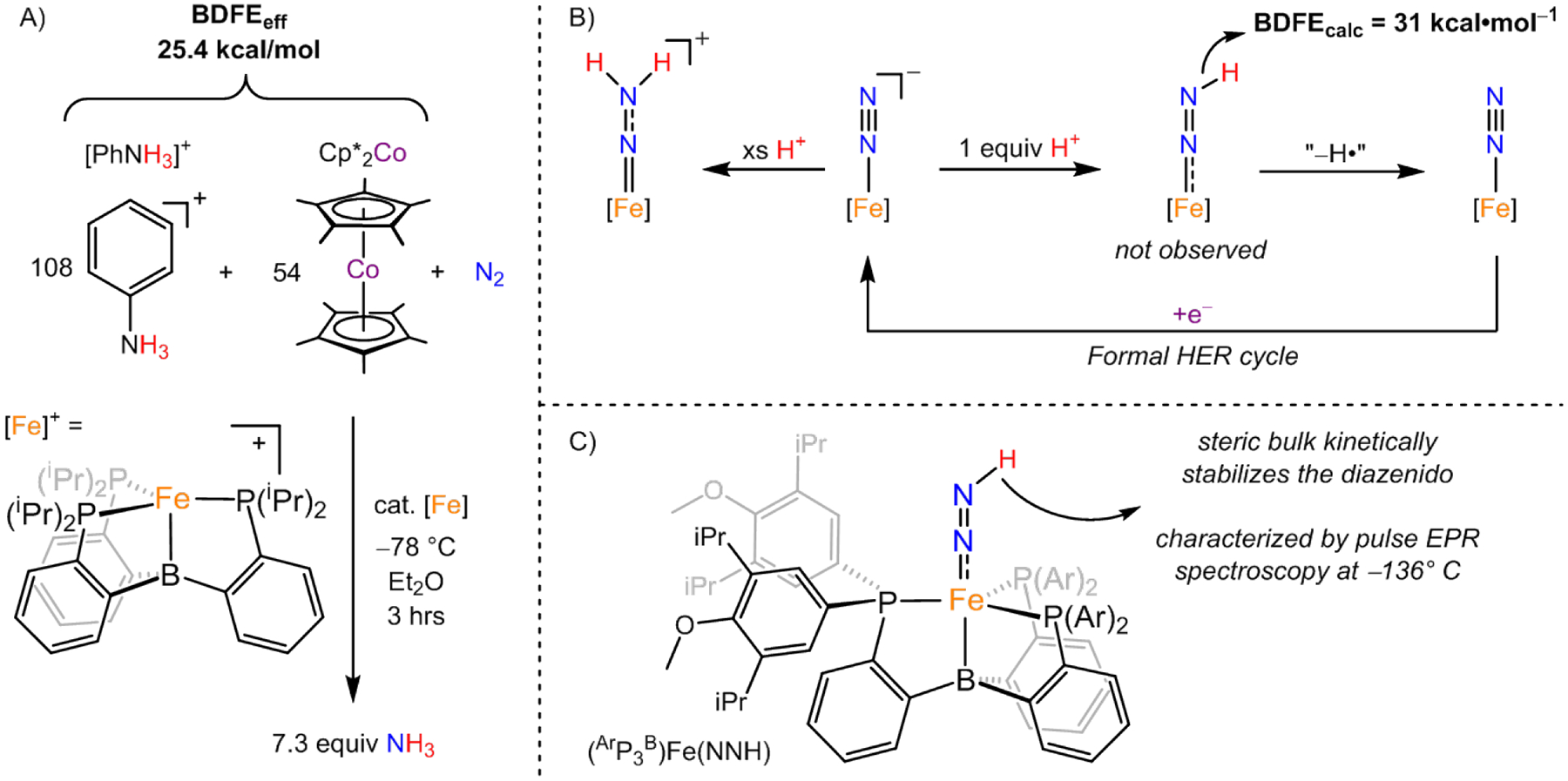
(A) Lowest established overpotential conditions, using [PhNH3]+ and Cp*2Co as the acid/reducant pair, for N2RR catalysis observed with (iPrP3B)Fe.[18,33] (B) Double protonation of [(iPrP3B)Fe(N2)]− leads to formation of the cationic isodiazene complex.[34,35] Efforts to form the Fe-NNH diazenido via single protonation lead to formal H• loss (observed as 0.5 equiv H2), consistent with its low BDFEN–H.[18,20,34] Reduction of the resultant (iPrP3B)Fe(N2) closes a cycle for HER. (C) Bulkier (ArP3B)Fe platform allows for kinetic stabilization (at −136 °C) of an Fe-NNH diazenido species, as elucidated by pulse electron paramagnetic resonance (EPR) spectroscopy.[7]
Efforts to synthesize (iPrP3B)Fe(NNH) from [(iPrP3B)Fe(N2)]− via monoprotonation lead instead to the instantaneous formation of H2 and oxidation at Fe, even at very low temperature,[34] highlighting that the Fe-NNH species is a potential source of H2 formation during catalytic N2RR. However, the intermediacy of (iPrP3B)Fe(NNH) in N2RR is supported by the double protonation of [(iPrP3B)Fe(N2)]− in the presence of excess acid to afford the isodiazene intermediate, [(iPrP3B)Fe(NNH2)]+.[33,34] Additionally, direct spectroscopic characterization of a related diazenido species, (ArP3B)Fe(NNH) (ArP3B = tris(o-di-(3,5-diisopropyl-4-methoxy)phenylphosphinophenyl)-borane), was accomplished at cryogenic temperatures (−136 °C) with a ligand featuring bulkier phosphine substituents.[7] While the solution instability of (RP3B)Fe(NNH) species has to date precluded the experimental determination of a BDFEN–H, DFT calculations predict a BDFEN–H of ~31 kcal•mol−1. This theoretical approach has been calibrated with experimentally derived BDFE values derived from similar iron complexes.[18,20] Using this BDFEN–H in Eq 3 leads to a predicted minimum ΔΔGf(NH3) of 60 kcal•mol−1 for the (iPrP3B)Fe-catalyst system.
4. Conclusion
Note that in both of the case studies presented here, the experimentally observed minimum ΔΔGf(N2RR) is higher than that estimated from the BDFEN–H of the M-NNH species. This “extra” driving force might be necessary because steps that are not related to N2 functionalization, such as NH3/N2 exchange, are instead thermodynamically limiting. Alternatively, as most N2RR catalysts are also HER catalysts,[23,32,33,36] this excess driving force may be necessary to form (and consume) reactive intermediates at a sufficient rate to prevent their decay to H2.[20,21] For our iron catalyst system, it may remain to be discovered that a modestly weaker acid (or reductant) is compatible with N2RR, such that an overpotential closer to the predicted ΔΔGf(NH3) of 60 kcal•mol−1 can be achieved. Our view is that strategies to improve not only the thermodynamic stability but also the kinetic stability of the key M-NNH intermediate will offer a means of achieving N2RR catalysis at lower overpotentials and with higher selectivities. These efforts should be complemented by further development of N2RR catalysts that can circumvent such an M-NNH intermediate, such as those recently proposed for Mo by Nishibayashi and coworkers[27,37] and for Ti by Liddle and coworkers.[38] Given the global importance of nitrogen fixation, and the rich catalytic landscapes that are at play in the growing number of synthetic catalyst systems, there are exciting opportunities to improve mechanistic understanding and develop more robust and efficient catalyst systems.[39]
Supplementary Material
Acknowledgements
This research was supported by the National Institutes of Health (GM-070757). M.J.C. thanks the Resnick Sustainability Institute at Caltech for a graduate research fellowship.
References
- [1].Smil V, Sci. Am 1997, 277, 76–81. [Google Scholar]
- [2].Bazhenova TA, Shilov AE, Coord. Chem. Rev 1995, 144, 69–145. [Google Scholar]
- [3].Burford RJ, Fryzuk MD, Nat. Rev. Chem 2017, 1, 1–13. [Google Scholar]
- [4].Chatt J, Pearman AJ, Richards RL, J. Chem. Soc., Dalton Trans 1976, 15, 1520–1524. [Google Scholar]
- [5].Yandulov DV, Schrock RR, Rheingold AL, Ceccarelli C, Davis WM, Inorg. Chem 2003, 42, 796–813. [DOI] [PubMed] [Google Scholar]
- [6].Yandulov DV, Schrock RR, Can. J. Chem 2005, 83, 341–357. [Google Scholar]
- [7].Nesbit MA, Oyala PH, Peters JC, J. Am. Chem. Soc 2019, 141, 8116–8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Warren JJ, Tronic TA, Mayer JM, Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ruscic B, Feller D, Peterson KA, Theor. Chem. Acc 2013, 133, 1415–1426. [Google Scholar]
- [10].Lindley BM, Appel AM, Krogh-Jespersen K, Mayer JM, Miller AJM, ACS Energy Lett 2016, 1, 698–704. [Google Scholar]
- [11].Chatt J, Heath GA, Richards RL, J. Chem. Soc., Dalton Trans 1974, 19, 2074–2082. [Google Scholar]
- [12].Pickett CJ, Talarmin J, Nature 1985, 317, 652–653. [Google Scholar]
- [13].Becker JY, Avraham S, J. Electroanal. Chem 1990, 280, 119–127. [Google Scholar]
- [14].Ashida Y, Arashiba K, Tanaka H, Egi A, Nakajima K, Yoshizawa K, Nishibayashi Y, Inorg. Chem 2019, 58, 8927–8932. [DOI] [PubMed] [Google Scholar]
- [15].Lehnert N, Tuczek F, Inorg. Chem 1999, 38, 1659–1670. [DOI] [PubMed] [Google Scholar]
- [16].Lehnert N, Tuczek F, Inorg. Chem 1999, 38, 1671–1682. [DOI] [PubMed] [Google Scholar]
- [17].Horn KH, Lehnert N, Tuczek F, Inorg. Chem 2003, 42, 1076–1086. [DOI] [PubMed] [Google Scholar]
- [18].Chalkley MJ, Del Castillo TJ, Matson BD, Roddy JP, Peters JC, ACS Cent. Sci 2017, 3, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bezdek MJ, Pappas I, Chirik PJ, in Nitrogen Fixation (Ed.: Nishibayashi Y), Springer International Publishing, Cham, 2017, pp. 1–21. [Google Scholar]
- [20].Matson BD, Peters JC, ACS Cat 2018, 8, 1448–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Benedek Z, Papp M, Oláh J, Szilvási T, Inorg. Chem 2019, 58, 7969–7977. [DOI] [PubMed] [Google Scholar]
- [22].Yandulov DV, Schrock RR, Science 2003, 301, 76–78. [DOI] [PubMed] [Google Scholar]
- [23].Weare WW, Dai X, Byrnes MJ, Chin JM, Schrock RR, Müller P, Proc. Natl. Acad. Sci. U.S.A 2006, 103, 17099–17106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ritleng V, Yandulov DV, Weare WW, Schrock RR, Hock AS, Davis WM, J. Am. Chem. Soc 2004, 126, 6150–6163. [DOI] [PubMed] [Google Scholar]
- [25].Waidmann CR, Miller AJM, Ng C-WA, Scheuermann ML, Porter TR, Tronic TA, Mayer JM, Energy Environ. Sci 2012, 5, 7771–7780. [Google Scholar]
- [26].Pappas I, Chirik PJ, J. Am. Chem. Soc 2016, 138, 13379–13389. [DOI] [PubMed] [Google Scholar]
- [27].Ashida Y, Arashiba K, Nakajima K, Nishibayashi Y, Nature 2019, 568, 536–540. [DOI] [PubMed] [Google Scholar]
- [28].Garrido G, Rosés M, Ràfols C, Bosch E, J. Solution Chem 2008, 37, 689–700. [Google Scholar]
- [29].Kaljurand I, Kütt A, Sooväli L, Rodima T, Mäemets V, Leito I, Koppel IA, J. Org. Chem 2005, 70, 1019–1028. [DOI] [PubMed] [Google Scholar]
- [30].Quist DA, Ehudin MA, Schaefer AW, Schneider GL, Solomon EI, Karlin KD, J. Am. Chem. Soc 2019, 141, 12682–12696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Anderson JS, Rittle J, Peters JC, Nature 2013, 501, 84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Del Castillo TJ, Thompson NB, Peters JC, J. Am. Chem. Soc 2016, 138, 5341–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chalkley MJ, Del Castillo TJ, Matson BD, Peters JC, J. Am. Chem. Soc 2018, 140, 6122–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Anderson JS, Cutsail GE, Rittle J, Connor BA, Gunderson WA, Zhang L, Hoffman BM, Peters JC, J. Am. Chem. Soc 2015, 137, 7803–7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Thompson NB, Oyala PH, Dong HT, Chalkley MJ, Zhao J, Alp EE, Hu M, Lehnert N, Peters JC, Inorg. Chem 2019, 58, 3535–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kuriyama S, Arashiba K, Nakajima K, Tanaka H, Kamaru N, Yoshizawa K, Nishibayashi Y, J. Am. Chem. Soc 2014, 136, 9719–9731. [DOI] [PubMed] [Google Scholar]
- [37].Arashiba K, Eizawa A, Tanaka H, Nakajima K, Yoshizawa K, Nishibayashi Y, Bull. Chem. Soc. Jpn 2017, 90, 1111–1118. [Google Scholar]
- [38].Doyle LR, Wooles AJ, Jenkins LC, Tuna F, McInnes EJL, Liddle ST, Angew. Chem. Int. Ed 2018, 57, 6314–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen JG, Crooks RM, Seefeldt LC, Bren KL, Bullock RM, Darensbourg MY, Holland PL, Hoffman B, Janik MJ, Jones AK, et al. , Science 2018, 360, 873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.